
Abstract
Seed weight and oil/protein content are critical agronomic traits that determine soybean yield and quality. However, the key genes controlling these traits and the underlying regulatory mechanisms remain poorly understood. Here, we performed a combination of genome-wide association study and quantitative trait loci (QTL) mapping with seed weight variations, and identified a Nuclear Factor-YA (NF-YA) gene on chromosome 14 that positively regulates seed weight and protein content while negatively regulating oil content without affecting other agronomic traits, designated as Seed Weight 14 (SW14). SW14 physically interacts with GmLEC1a/b, the soybean orthologs of the central regulator of plant seed development, Leafy Cotyledon1 (LEC1), to disrupt the formation of a non-canonical NF-Y complex comprising GmLEC1, GmNF-YC2, and GmbZIP67, thereby inhibiting the GmLEC1-mediated transcriptional activation involved in seed development process. Natural allelic variations in SW14 affect the stability of the SW14 protein, which in turn confers varied seed weight and oil/protein content in soybean. Further analysis demonstrates that the elite SW14H3 allele has undergone artificial selection during domestication and holds potential for improving yield in soybean. Collectively, our findings provide insights into the molecular basis that specifically regulates seed weight and quality, offering a potential strategy for overcoming tradeoff effects and facilitating high-yield breeding in soybean.
Similar content being viewed by others


Introduction
Soybean (Glycine max [L.] Merr.) is one of the most important crops that provides up to 30% of oils and 69% of proteins to the human consumption worldwide1. To meet the demands of an ever-growing global population, it is urgent to develop soybean varieties with higher production. Seed weight, a major agronomic trait, is likely one of the first traits selected during domestication to determine soybean yield and quality, and it is largely controlled by genetic factors2. Thus far, although over 300 quantitative trait loci (QTLs) associated with seed weight have been identified across 20 chromosomes in soybean (SoyBase, http://www.soybase.org/), the genes underlying these loci have seldom been isolated due to the complex soybean genome structure3. Therefore, exploring genes and elucidating the genetic and molecular basis underlying the seed weight trait could greatly contribute to improving soybean yield and quality.
To date, few genes controlling seed weight and oil/protein content have been isolated and characterized in soybean using forward genetic approaches, including GmSWEET10a/b4, Seed Thickness 1 (ST1)5, Mother of FT and TFL1 (GmMFT)6, Protein Oil Weight Regulator 1 (POWR1)7, and Fatty Acid 9 (FA9)8, whose natural allelic variations have contributed to the genetic improvement of soybean. However, the majority studies on these genes have focused only on seed traits, potentially causing tradeoffs among different yield-related traits, such as negative correlation between seed weight and seed number. These tradeoffs are often a consequence of linkage drags or pleiotropy9. Although pleiotropy has limited the successful application of cloned genes in breeding, the tradeoffs caused by pleiotropy are challenging to address through conventional breeding methods, thus constraining the greater achievements for improving crop yield9,10. Therefore, cloning excellent gene resources that regulate seed traits without affecting other agronomic traits should be an effective strategy to overcome these tradeoff effects and improve soybean yield in modern breeding.
During the past decades, a series of genes that regulate seed weight have been identified and functionally studied in plants, involving in multiple pathways such as the ubiquitin-proteasome system, mitogen-activated protein kinase (MAPK) cascade, G proteins, phytohormones, and transcription regulatory factors10. Among these, the Nuclear Factor-Y (NF-Y) transcription factors (TFs) consist of three subunits, including NF-YB, NF-YC, and NF-YA/other TFs. These subunits are traditionally understood to form a complete heterotrimeric complex that affects transcription11,12. Our initial understanding of NF-Y genes in seed development was derived from researches on Leafy Cotyledon1 (LEC1), which encodes an NF-YB subunit and acts as a central regulator of embryonic development, seed maturation, and storage reserve accumulation13,14, but the detailed function of LEC1 in soybean remains unclear. In recent years, increasing evidences suggest that other NF-Y family members play crucial roles in seed development control15. Nevertheless, NF-Y genes associated with soybean seed weight and quality have seldom been reported.
In this study, we have identified an NF-YA TF gene, Seed Weight 14 (SW14), that specifically regulates soybean seed traits, including seed weight and oil/protein content, without affecting other agronomic traits. SW14 can directly interact with soybean LEC1 (GmLEC1), a key regulator of seed development, and subsequently inhibit the formation and transactivating activity of the GmLEC1/GmNF-YC2/GmbZIP67 trimeric complex. Furthermore, the natural allele SW14H3 has been selected during soybean domestication and exhibits potential for increasing yield. These findings reveal insights into the molecular mechanisms controlling seed traits and provide a valuable genetic resource for improving yield and quality in soybean.
Results
Identification and positional cloning of SW14
To identify genetic loci that control seed weight in soybean, we phenotyped 320 accessions from previous resequencing panel16 in 2018 and 2019 (Supplementary Fig. 1 and Supplementary Data 2). Using a linear mixed model, we conducted a Genome-wide Association Study (GWAS) and identified three significant association loci for seed weight across the 2 years on chromosomes 10, 11, and 14, respectively (Fig. 1a, b). The locus on chromosome 14 has not been previously reported and exhibited higher association values than those on chromosome 10 and 11 (Fig. 1a, b and Supplementary Fig. 2), indicating that it makes a robust and important contribution to seed weight in soybean, we thus named this locus Seed Weight 14 (SW14) for further characterization.

GWAS scan for seed weight using data from the 320-accession panel grown over the 2018 (a) and 2019 (b) field seasons in Guangzhou (113°23′E, 23°16′N), China. Arrows indicate significant association loci for seed weight across the 2 years. The dashed horizontal lines represent the significance threshold determined by the Bonferroni correction. c QTL mapping of SW14 locus in the F2 segregation population derived from the cross between Yunchun 2014 carrying SW14H1 and Huachun 8 carrying SW14H3. The genetic distance (cM) and the name of markers of the linkage group is given on the left- and the right-hand side of the chromosome model, respectively. d Delimitation of the SW14 locus to a 383-kb region. Graphical genotypes of 7 recombinants carrying crossovers in the SW14 region (left panel), and phenotypic segregation patterns of progeny are shown in boxplot format (right panel), where the box representing the interquartile range, the central line indicating the median, and the whiskers showing the minimum or maximum value (n = 20, one plot indicates one plant). e Heat map of candidate genes located in the candidate region. The color key (blue to red) represents gene expression (fragments per kilobase per million mapped reads, FPKM). f Haplotypes detected in the coding region of SW14. g Comparison of seed weight between different haplotypes of SW14 over the 2018 and 2019 field seasons in Guangzhou, China. In the violin plot, the dashed lines indicate the median and dotted lines the interquartile range. Different lowercase letters indicate statistically significant differences (one-way ANOVA, P < 0.01). Source data are provided as a Source Data file.
To isolate the causal gene of the SW14 locus, we firstly performed QTL mapping using a segregating F2 population from a cross between the small seed variety Yunchun 2014 (YC2014) and the big seed variety Huachun 8 (HC8) (Supplementary Fig. 3), and detected a seed weight QTL in region around the SW14 locus on chromosomes 14 (Fig. 1c and Supplementary Fig. 2). We next generated a residual heterozygous inbred population (n = 502) for fine mapping of the SW14 locus by recurrent selection for heterozygosity at SW14 with M11 and M16 markers from the F2 to F5 generations, obtaining 7 recombinants that delimit SW14 to a 383 kb genomic region between markers M12 and M15 (Fig. 1d).
Based on the annotated soybean genome Williams 82 (W82)3 and the resequencing data16, there are 11 predicted genes harboring nonsynonymous or frameshift mutation between YC2014 and HC8 located in the SW14 interval (Supplementary Data 3). Among these, Glyma.14G007700 and Glyma.14G010000 were highly expressed during seed development, while only Glyma.14G010000, an NF-YA TF gene12, exhibited higher expression in seed compared with other tissues (Fig. 1e). Haplotype analysis showed that two SNPs result in amino acids change of SW14 protein, which are conserved sites in leguminous plants (Fig. 1f and Supplementary Fig. 4). They were divided into three SW14 haplotypes in the natural population, and accessions with SW14H3 exhibited higher seed weight than those with SW14H1 and SW14H2, which displayed similar seed weight phenotypes (Fig. 1f, g and Supplementary Data 2). Consistently, the big seed variety HC8 harbors SW14H3, while the small seed variety YC2014 harbors SW14H1, respectively, indicating that the amino acid alterations of Pro80Ser and His87Tyr may cause distinct functions between SW14H1 and SW14H3 (Fig. 1f). In contrast, the haplotypes of Glyma.14G007700 corresponding to the two parents did not show significantly different seed weight in the natural population across the 2 years (Supplementary Fig. 5). Collectively, we considered Glyma.14G010000 as the most likely causal gene controlling seed weight and subsequently referred to SW14.
SW14 controls seed weight and quality in soybean
Due to the similar effects of the SW14H1 and SW14H2 haplotypes in seed weight in the natural population (Fig. 1g), we assessed the specific function of SW14 by comparing the SW14H1 and SW14H3 haplotypes in the text below. We firstly selected a set of SW14 near-isogenic lines (NILs) from the heterozygous SW14 locus by crossing YC2014 and HC8 parents, which harbor the SW14H1 haplotype and the SW14H3 haplotype, respectively. NIL-SW14H3 exhibited significant increases in 100-seed weight and protein content, but a decrease in oil content compared to NIL-SW14H1, without affecting the number of seeds per plant (Fig. 2a–e). However, the variations in SW14 have no effects on other agronomic traits of soybean (Supplementary Fig. 6).

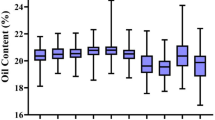
a Seed phenotypes of the near-isogenic lines (NILs) possessing homozygous H1 (NIL-SW14H1) and H3 (NIL-SW14H3) in Sanya (108°56′E, 18°09′N) field. Scale bar, 1 cm. Statistical analysis of the 100-seed weight (b), oil content (c), protein content (d), and seed number per plants (e) of NILs. Values represent means ± SD (n = 8, one plot indicates one plant). One-way ANOVA was used to generate the P values. f Seed phenotypes of Williams 82 (W82) and the sw14 mutant lines in Sanya field. Scale bar, 1 cm. Statistical analysis of the 100-seed weight (g), oil content (h), protein content (i), and seed number per plant (j) of W82 and the sw14 mutant lines. Values represent means ± SD (n = 15, one plot indicates one plant). k Volcano plot of differentially expressed genes in sw14#1/W82. Differential expression analysis was conducted using DESeq2 with the default Wald test (two-sided). P values were adjusted for multiple comparisons using the Benjamin–Hochberg method to control the false discovery rate. l Transcript levels of FA9, GmFAD3s, and GmOLEO1 in W82 and the sw14 mutant lines. GmACT11 was used as an internal control. Values represent means ± SD (n = 3 biological replicates). Different lowercase letters indicate statistically significant differences (one-way ANOVA, P < 0.01). Source data are provided as a Source Data file.
To further confirm whether SW14 controls soybean seed weight and oil/protein content, we generated three independent SW14 knockout mutants (sw14#1, sw14#2, and sw14#3) using CRISPR/Cas9 system in the W82 background (Supplementary Fig. 7 and Supplementary Data 4). All the sw14 mutant lines exhibited reduced 100-seed weight and protein content, and higher oil content (Fig. 2f–i and Supplementary Fig. 8a–d), but there was no significant difference in seed number per plant and other agronomic traits compared with the W82 wild type when they were repeatedly planted at different latitudes (Fig. 2j, Supplementary Fig. 8e–h and Supplementary Fig. 9). The above results suggest that SW14 functions as a specific regulator for seed weight and oil/protein content in soybean.
Next, we investigate how SW14 regulates soybean seed traits through RNA sequencing (RNA-seq) experiments using W82 and the sw14#1 mutant. The results revealed that 1597 genes were upregulated and 1573 genes downregulated in the sw14#1 mutant versus W82 (Fig. 2k and Supplementary Data 5). Among them, five genes known to regulate soybean seed traits were found to be upregulated in the sw14#1 mutant, as confirmed by RT-qPCR (Supplementary Data 5 and Fig. 2l). These genes included FA9, Fatty Acid Desaturase 3a (GmFAD3a), GmFAD3b, GmFAD3c, and OLEOSIN 1 (GmOLEO1), consistent with their roles in the regulation of seed weight and quality8,17,18. These results indicate that SW14 regulates the expression of these genes to control seed weight and oil/protein content in soybean.
Natural variations affect SW14 protein stability
To examine the expression pattern of SW14 during seed development, we first performed a reverse transcription quantitative PCR (RT-qPCR) analysis. The result showed that the transcript level of SW14 was slightly increased at 14 days after fertilization (DAF), peaked at approximately 21 DAF, then gradually decreased (Fig. 3a). An RNA in situ hybridization assay revealed that SW14 mRNA localized in all seed tissues, including the seed coat, endosperm, and embryo (Fig. 3b). Additionally, SW14 was found to be exclusively localized in the nucleus, similar to other plant NF-YA subunits12, and this nuclear localization pattern was not influenced by the two SW14 haplotypes. (Supplementary Fig. 10). These observations further confirm the pivotal role of SW14 as an NF-YA subunit in soybean seed development.

a RT-qPCR analysis of SW14 transcript levels in different tissues of W82. DAF, days after fertilization. GmACT11 was used as an internal control. Values represent means ± SD (n = 3 biological replicates). b RNA in situ hybridization of SW14 in 10 DAF developing seeds of W82. EP epidermis, ES endosperm, EM embryo. Scale bar, 100 μm. c Comparison of seeds of the indicated SW14 overexpression lines in Arabidopsis. Scale bar, 500 µm. d Statistical analysis of 1000-seed weight in (c). Values represent means ± SD (n = 15, one plot indicates one plant). Different lowercase letters indicate statistically significant differences (one-way ANOVA, P < 0.01). Protein expression analyses of SW14-FLAG in transgenic Arabidopsis plants (e), N. benthamiana leaves (f), and soybean hairy roots (g). Col-0 or the relevant empty vector (Empty) was used as a negative control. The immunoblots were probed with α-FLAG antibody. h Cell-free degradation assay of SW14. GST-SW14H1 and GST-SW14H3 were incubated with equal amounts of plant cell crude extracts from 21 days after fertilization (DAF) developing seeds at room temperature for 0–90 min. MG132 (50 μM) was added as indicated. The proteins were detected by α-GST antibody. The bottom panel shows staining with coomassie brilliant blue (CBB, e, f) or probing with an α-ACTIN antibody (g, h) as a loading control. Source data are provided as a Source Data file.
Given the facts that variations in SW14 protein are conserved sites in leguminous plants and affect seed weight and oil/protein content in soybean (Fig. 1 and Supplementary Fig. 4), we asked how different SW14 haplotypes cause the functional changes of SW14, as natural variations did not alter the protein’s subcellular localization (Supplementary Fig. 10). To uncover this, the intact SW14H1 and SW14H3 cDNAs driven by a strong and constitutively expressed CaMV 35S promoter were introduced into Arabidopsis ecotype Col-0 background. Phenotypic analysis showed that SW14 could positively regulate seed weight in Arabidopsis, and the SW14H3 plants produced larger seeds compared with the SW14H1 plants (Fig. 3c, d). Further analysis revealed that the SW14H1 and SW14H3 transgenic plants had similar transcript levels of SW14 (Supplementary Fig. 11). Interestingly, SW14 protein levels were remarkably higher in the SW14H3 plants than those in the SW14H1 plants (Fig. 3e and Supplementary Fig. 12a), indicating a probable function of natural variations of SW14 in regulating the stability of SW14 protein.
Next, we performed transient expression assays in N. benthamiana and transgenic soybean hairy roots to assess SW14 protein stability, using the above SW14 overexpression constructs. The accumulation levels of SW14H3 protein were always higher compared with the SW14H1 protein in both systems (Fig. 3f, g and Supplementary Fig. 12b, c). We next examined whether natural variations of SW14 affect the stability of SW14 protein using cell-free degradation assays. The results indicated that the degradation rate of SW14H3 was much slower than SW14H1, and addition of the proteasome inhibitor MG132 significantly inhibited SW14 degradation (Fig. 3h and Supplementary Fig. 12d). These results support that natural variations of SW14 play important role in the 26S proteasome-dependent degradation of SW14.
SW14 interacts with GmLEC1a and GmLEC1b
To further elucidate how SW14 regulates seed weight and oil/protein content, we attempted to identify its potential interaction proteins, using a yeast two-hybrid (Y2H) system with soybean seed cDNA library. Notably, the orthologs of the Arabidopsis LEC1, GmLEC1a (Glyma.07G268100) and GmLEC1b (Glyma.17G005600)19, interacted with SW14 in yeast cells (Fig. 4a). We subsequently confirmed the interaction between SW14 and GmLEC1a/b by split-luciferase (split-LUC) complementation assays in N. benthamiana leaves (Fig. 4b, c). Co-immunoprecipitation (Co-IP) assays indicated that GmLEC1a/b-FLAG fusion proteins could be immunoprecipitated with SW14-GFP but not with the negative control BFP-GFP (Fig. 4d, e), indicating the interaction of SW14 with GmLEC1a/b in vivo. These results demonstrate that SW14 interacts with GmLEC1.

a Yeast two-hybrid assays showing the interactions of SW14 protein with GmLEC1a and GmLEC1b. Transformed yeast cells were grown on SD/-LW (lacking Leu and Trp) and -LWHA (lacking Leu, Trp, His, and Ade) medium. b, c Split-LUC complementation imaging assay showing the interaction of SW14 with GmLEC1a and GmLEC1b in N. benthamiana leave. GUS-nLUC and cLUC-GUS served as negative controls. d, e Co-IP assay showing the interaction of SW14 with GmLEC1a and GmLEC1b in N. benthamiana leave. The BFP-GFP protein was used as a negative control. The precipitated proteins were detected by α-FLAG or α-GFP antibody. f Seed phenotypes of Tianlong 1 (TL1) and the Gmlec1a mutant lines in Sanya field. Scale bar, 1 cm. Statistical analysis of the 100-seed weight (g), oil content (h), and protein content (i) of TL1 and the Gmlec1a mutant lines. Values represent means ± SD (n = 8, one plot indicates one plant). Transcript levels of FA9 (j) and GmFAD3c (k) in TL1 and the Gmlec1a mutant lines. GmACT11 was used as an internal control. Values represent means ± SD (n = 3 biological replicates). Different lowercase letters indicate statistically significant differences (one-way ANOVA, P < 0.01). Source data are provided as a Source Data file.
To explore the function of GmLEC1 in controlling soybean seed weight and oil/protein content, we generated Gmlec1a mutants using CRISPR/Cas9 system in the Tianlong 1 (TL1) background. Two independent lines were identified with different frameshift mutations in GmLEC1a coding region, and named as Gmlec1a#1 and Gmlec1a#2 (Supplementary Fig. 13 and Supplementary Data 6). Compared with TL1, the Gmlec1a mutant lines showed a significant increase in 100-seed weight and protein content, along with a decrease in oil content (Fig. 4f–i). Furthermore, the expression levels of FA9 and GmFAD3c were downregulated in these mutants (Fig. 4j, k). Collectively, these findings suggest that GmLEC1 and SW14 play opposite roles in the regulation of soybean seed weight and oil/protein content by controlling the transcription of FA9 and GmFAD3c.
SW14 inhibits GmLEC1 function to control seed weight and quality
The LEC1-NF-YC dimer has been reported to interact with seed-specific bZIP TF bZIP67, to activate seed maturation genes such as the CRUCIFERIN C (CRC) and FAD3 in Arabidopsis20,21. Consistent with these studies, we found that the co-expression of GmLEC1a/GmNF-YC2 with GmbZIP67 greatly enhanced the activation levels of the FA9 and GmFAD3c promoters compared to those achieved with GmLEC1a/GmNF-YC2 or GmbZIP67 alone. However, the co-expression of SW14 significantly repressed the activation of these two promoters-driven luciferase gene (Fig. 5a, b), suggesting an inhibitory role of SW14 in GmLEC1a-mediated activation of the FA9 and GmFAD3c promoters.

a Schematic diagram depicts the constructs used in the transient expression assay shown in (b). b Transient expression assay indicating that SW14 inhibits the GmLEC1a/GmNF-YC2/GmbZIP67-mediated activation of FA9 and GmFAD3c promoters. LUC, luciferase; REN, Renilla LUC. A GUS fragment was used as a negative control. Values represent means ± SD (n = 3 biological replicates). Different lowercase letters indicate statistically significant differences (one-way ANOVA, P < 0.05). c Schematic representations of FA9 and GmFAD3c promoters. The regions analyzed by ChIP-qPCR are indicated. Red lines indicate the regions that were bound by GmLEC1 according to previous ChIP-seq data23. d ChIP analysis of GmLEC1a-FLAG binding to the FA9 and GmFAD3c promoters in W82 and sw14#1 mutant. The enrichment of a GmELF3b genomic fragment was used as a negative control. Values represent means ± SD (n = 3 biological replicates). e SW14 inhibits the formation of GmLEC1a/GmNF-YC2/GmbZIP67 complex in yeast. Transformed yeast cells were grown on SD/-LW and -LWHA medium. f Seed phenotypes of TL1, Gmlec1a#1, sw14TL, and Gmlec1a#1 sw14TL mutant lines in Guangzhou field. Scale bar, 1 cm. Statistical analysis of the 100-seed weight (g), oil content (h), and protein content (i) in (f). Values represent means ± SD (n = 6, one plot indicates one plant). Different lowercase letters indicate statistically significant differences (one-way ANOVA, P < 0.01). Source data are provided as a Source Data file.
We next performed chromatin immunoprecipitation (ChIP) qPCR to determine whether SW14 affects the binding affinity of GmLEC1 to the promoters of its target genes. To this end, we generated 35S:GmLEC1a-FLAG (GmLEC1a-FLAG) transgenic plant and crossed it with the sw14#1 mutant to obtain GmLEC1a-FLAG sw14#1 plant. Consistent with previous ChIP-seq data22, we observed significant enrichment of GmLEC1a-FLAG at several regions of the FA9 and GmFAD3c promoters, and the enrichment was higher in the sw14#1 background compared to the W82 background (Fig. 5c, d), while the level of GmLEC1a-FLAG protein was not altered between these two backgrounds (Supplementary Fig. 14), indicating that SW14 inhibits the binding of GmLEC1 to the FA9 and GmFAD3c promoters.
To investigate how SW14 inhibits the activity of the GmLEC1-mediated transcriptional activation, we detected the interaction of GmLEC1a with GmbZIP67 in the presence of GmNF-YC2 and/or SW14 in yeast. Similar to the formation of the canonical NF-YB/C/A complex23, the interaction between GmLEC1a and GmbZIP67 was only detected when GmNF-YC2 was present in yeast (Fig. 5e). However, this trimer interaction was markedly repressed by SW14 (Fig. 5e and Supplementary Fig. 15), suggesting that SW14 interferes with the assembly of the GmLEC1a/GmNF-YC2/GmbZIP67 complex. We also determined the genetic interaction between SW14 and GmLEC1 by knocking out SW14 in the TL1 background (sw14TL, Supplementary Fig. 16 and Supplementary Data 7) and crossing it with the Gmlec1a#1 mutant to generate Gmlec1a#1 sw14TL double mutant. Comparable phenotypes in 100-seed weight, protein content, and oil content between Gmlec1a#1 and Gmlec1a#1 sw14TL indicated that GmLEC1a is genetically epistatic to SW14 (Fig. 5f–i). Taken together, these findings suggest that SW14 controls seed weight and oil/protein content by inhibiting the function of GmLEC1.
Selection of the SW14 H3 allele during soybean domestication
Given that seed weigh is likely one of the first-selected traits that determines yield throughout the course of domestications2, we investigated whether SW14 alleles have been selected during soybean domestication using 1295 previously re-sequenced accessions, including 147 wild accessions, 574 landraces, and 574 cultivars24. We first compared the seed weight of the SW14 haplotypes and found that accessions with the SW14H3 haplotype had higher seed weight than those with the SW14H1 or SW14H2 haplotype (referred to as SW14H1&H2) in wild soybean, landraces, and cultivars (Fig. 6a–c). The analysis of the distribution of the SW14 haplotypes in different germplasm pools revealed that the proportion of the SW14H1&H2 haplotypes exhibited a continuously decreasing pattern from wild accessions to landraces and subsequently to cultivars, while the proportion of the SW14H3 haplotype increased, becoming predominant in landraces and cultivars (Fig. 6d). Notably, the proportion of the SW14H3 haplotype increased to 48.8% in cultivars, indicating that this allele has been selected during soybean post-domestication improvement (Fig. 6d). Meanwhile, a whole-genome selective sweep analysis revealed that SW14 is located within a selective sweep region, as inferred by negative Tajima’s D value and low SW14H3/SW14H1 plus H2 π-ratios (Fig. 6e), suggesting that SW14H3 was selected during soybean domestication.

Comparison of seed weight between different haplotypes of SW14 in wild soybean (a), landraces (b), and cultivars (c) grown in Guangzhou and Zhengzhou (112°42′E, 34°16′N) field. In the violin plot, the dashed lines indicate the median and dotted lines the interquartile range. d Haplotype frequency of SW14 in wild soybean, landraces, and cultivars. e Tajima’s D value and genetic variations (π) across the SW14 locus in 574 cultivars, 574 landraces, and 147 wild accessions. f, g Plot yield, seed oil content, and protein content of NIL-SW14H1 and NIL-SW14H3 in Guangzhou and Xiangyang (110°45′E, 31°14′N) field. The plot area is 5 m2. Values represent means ± SD (n = 3 biologically independent field plots). One-way ANOVA was used to generate the P values. Source data are provided as a Source Data file.
To evaluate if the introduction of the SW14H3 allele could improve the yield and quality performance of soybean, we compared the plot yield, seed oil content, and protein content of NIL-SW14H1 and NIL-SW14H3 in different regions, including Guangzhou (113°23′E, 23°16′N) and Xiangyang (110°45′E, 31°14′N). The results showed that the plot yield and seed protein content of NIL-SW14H3 were significantly greater than those of NIL-SW14H1, while the seed oil content of NIL-SW14H3 was lower in both regions (Fig. 6f, g). These findings suggest that the elite SW14H3 allele has potential for utilization in high-yield and high-quality breeding programs for soybean.
Discussion
Seed weight and oil/protein content are essential factors linking the breeding goals of achieving high yield and good quality in agricultural production. Although numerous QTLs have been identified to be responsible for seed weight, oil content, and protein content in soybean25, the genes underlying these QTLs and their regulatory mechanisms remain largely unknown. In this study, we demonstrated that SW14, an NF-YA encoding gene, exhibits preferential expression in developing seeds and functions as a repressor of seed trait-related gene expression by interacting with GmLEC1 proteins, the orthologs of the central seed development regulator LEC1, thereby genetically regulating both seed weight and quality in soybean. Notably, the negative role of SW14 contrasts with the positive regulatory function of its homolog GmNFYA in the regulation of oil content26,27, indicating functional divergence of the NF-YA family genes during this process. In addition, we propose that SW14 may also act as an activator to modulate gene expression, potentially through the formation of the NF-YB/C/A complex, as observed with GmNF-YA1628, or by affecting epigenetic modifications at target genes, similar to GmNFYA29. These mechanisms are worth further investigation in future studies.
NF-Y is a heterotrimeric TF complex composed of NF-YA, NF-YB, and NF-YC subunits, which are ubiquitous in all eukaryotes12. The NF-YB and NF-YC subunits can form a heterodimer in the cytosol that translocates to the nucleus, where they recruit NF-YA subunit to bind the CCAAT box with sequence specificity and modulate transcription11. However, a previous study reported that the NF-YA subunit can interfere with the LEC1/L1L-NF-YC2 activation in transient experiments20. In addition, NF-YA plays an opposing role to CONSTANS, which forms a trimer with NF-YB2/NF-YC3 during flowering30. These observations together indicate that the NF-YA subunit may affect the formation of non-canonical NF-Y complexes comprising NF-YB, NF-YC, and other TFs. In this study, we demonstrate that SW14, an NF-YA subunit, inhibits the assembly and transcriptional activation activity of the GmLEC1/GmNF-YC2/GmbZIP67 trimeric complex, revealing a regulatory mechanism among NF-Y subunits in plant seed development. This mechanism may extend to other plant development processes, such as flowering, hypocotyl elongation, and antiviral immunity, given the opposing functions of certain NF-Ys in these biological processes23,31,32,33,34. Furthermore, the partial repression of the GmLEC1a-mediated trimer complex and its transcriptional activation by SW14 suggests that additional repressors may exist to inhibit the assembly of the GmLEC1-mediated complex. This speculation is supported by recent studies that have reported the disruption of the NF-Y complex by factors other than NF-YA, including the WD40 domain-containing protein Early Heading Date 5 (Ehd5)35, the Jasmonate-zim-domain protein 8 (JAZ8)36, and the transcription factor SLR1-like 2 (SLRL2)37.
Gene pleiotropy, which arises from different spatial-temporal expression patterns or multiple biological functions of a single protein, often causes tradeoffs among multiple complex traits9, for example, higher seed weight is associated with decreased seed number, limiting the application of many genes for yield improvement because other unfavorable agronomic traits must be considered. Therefore, the identification of valuable genetic resources that can overcome these tradeoffs should be much higher priority in crop breeding. For instance, nature variations of OsMADS17 and Brassinosteroid-deficient Dwarf3 (BRD3) can overcome the tradeoff between grain number and grain weight in rice38,39. Here, we have identified that a seed-preferentially expressed factor SW14 interacts with the seed central regulator GmLEC1 to control seed traits without affecting other agronomic traits, especially seed number in soybean. Importantly, the elite SW14H3 allele, selected during soybean domestication, possesses potential application for improving yield by promoting SW14 protein accumulation, making it a valuable tool in soybean molecular breeding. However, the precise mechanisms underlying how nature variations in SW14 affect its stability, such as whether they influence the interaction of SW14 with E3 ligases or the degree of its ubiquitination, require further investigation.
Seed weight is an early domesticated trait in crops, including soybean, and is associated with increased oil content following seed enlargement2,7. Due to the inverse correlation between protein and oil content, protein content typically decreases during domestication. In other words, seed oil/protein content has been indirectly selected as it is pleiotropically controlled by a series of genes associated with a positive correlation between seed weight and oil content during soybean domestication. For example, genes such as GmSWEET10a/b4, POWR17, GmMFT6, and ST15 have been implicated in this process. However, the selection of natural variations in SW14 suggest that the seed weight trait can be domesticated independent of seed quality. Additionally, FA9, a positive regulator of oil content with a negative effect on seed weight and protein content, has been reported to be selected during soybean domestication and improvement8. These gene resources, controlling the inverse correlation between seed weight and oil content, can largely enrich genetic diversity and improve adaptation to the external environments in soybean.
Methods
Plant materials and growth conditions
The 320 previously sequenced accessions16 utilized for GWAS, and an F2 population (n = 280) and its progenies utilized for fine mapping, were grown under natural conditions from 2018 to 2021 in Guangzhou (113°23′E, 23°16′N; July to October) or Sanya (108°56′E, 18°09′N; November to March) to evaluate 100-seed weight. Each row was 1.5 m long with 0.5 m spacing between rows, and a plant spacing of 0.1 m.
NILs for the locus SW14 were selected from F6 generation of the cross Yunchun 2014× Huachun 8 using a molecular marker for SW14. The NILs, CRISPR/Cas9 knockout mutants, and transgenic plants used for phenotyping were planted under natural conditions from 2021 to 2024 in Sanya, Guangzhou, and Beijing (116°23′E, 39°54′N; May to October) as indicated in the text. For the field trials, the indicated lines were grown in a randomized complete block design with three replications for each field environment. In Sanya and Guangzhou, each row was 1.5 m long with 0.5 m spacing between rows, and a plant spacing of 0.1 m. In Beijing, each row was 3.0 m long with 0.5 m spacing between rows, and a plant spacing of 0.3 m. At the R8 stage, agronomic traits including plant height, node number, and branch number were measured. Seed traits were determined using uniform dry seeds after harvest.
To assess the effect of nature variations in SW14 on yield, a randomized complete block design with three replicates was implemented in Guangzhou and Xiangyang (110°45′E, 31°14′N; May to October). Field plots were arranged in 2 m long rows with 0.5 m spacing between each row, covering a total plot area of 5.0 m2. A plant density of 120,000 plants per hectare was maintained, and grain yield per plot was measured and calculated after harvest.
Arabidopsis plants used in this study are in the Col-0 background and grown at 22 °C under long-day conditions (16 h light/8 h dark photoperiod).
GWAS for the 100-seed weight
A total of 3,455,323 high-quality SNPs (MAF > 0.05) were used for GWAS assay in 320 accessions. Association analyses were performed by MLM implemented in efficient mixed model association expedited (EMMAX) software40. Kinship was derived from all these SNPs. The significant association threshold was determined by Bonferroni correction as 1/n (n, total SNP number), based on a previous report24. The significant association regions were manually verified from resequencing reads aligned against the Williams 82 (W82) genome with SAMtools41.
QTL analysis
Genomic DNA was extracted from leaves using the SurePlant DNA Kit (CWBIO, CW2298M) and used for indel marker amplification. Linkage map construction was carried out according to a previous report16, and the QTL analysis performed using MapQTL V5.0 software. Primer sequences of the markers for mapping are listed in Supplementary Data 1.
Multiple alignment analysis
Homologous SW14 protein sequences were downloaded from Phytozome (https://phytozome-next.jgi.doe.gov/). Amino acid sequences were aligned using the Jalview V2.11.1 with manually adjustments.
Plasmid construction and plant transformation
The coding sequence of GmLEC1a was amplified from W82 cDNA and inserted into the XbaI/BamHI restriction sites of pTF101 vector to generate the overexpression construct. To generate sw14 and Gmlec1 mutants, multiple sgRNAs targeting different positions of the candidate genes were designed using the CRISPR direct website (http://crispr.dbcls.jp/)42. Primers used are listed in Supplementary Data 1. All constructs were introduced into the Agrobacterium tumefaciens strain EHA101, and separately transformed into W82 or TL1 (all carrying SW14H1) via the cotyledon-node method43.
To overexpress different SW14 haplotypes in Arabidopsis, N. benthamiana leaves, and soybean hairy roots, the coding sequences of SW14H1 and SW14H3 were inserted into the pCAMBIA1350 vector. Primers used are listed in Supplementary Data 1. The floral dip method was used to generate the transgenic plants, and positive transgenic plants were selected on MS medium supplemented with hygromycin. Transgenic hairy roots were generated through Agrobacterium rhizogenes-mediated transformation44.
RNA in situ hybridization
RNA in situ hybridization was performed following a previously described protocol45. Briefly, seeds were fixed in RNase-free solution containing 5% acetic acid, 50% ethanol, and 3.7% formaldehyde at 4 °C. A 153-bp fragment specific to the SW14 cDNA was amplified with gene-specific primers containing T7 and SP6 RNA polymerase binding sites and integrated into the pSPT18 T-Easy vector (Roche, 11175025910). Primers used are listed in Supplementary Data 1. The digoxigenin-labeled antisense or sense probes were transcribed and labeled using a DIG RNA Labeling kit (Roche, 11175025910) according to the manufacturer’s protocol. Images were captured under a Leica stereomicroscope (DVM6).
Subcellular localization
To investigate the subcellular localization of different SW14 haplotypes, the coding region of SW14H1 was inserted into the PstI restriction site of the pGreen-35S: GFP vector to fuse in-frame with GFP. Primers used are listed in Supplementary Data 1. The above constructs were introduced into Arabidopsis mesophyll protoplasts. Fluorescence signals were captured using a confocal spectral microscope imaging system (TCS SP5; Leica).
Gene expression analysis and RNA-seq
For gene expression analysis in seeds, flowers were marked with color-coded thread after fertilization. The corresponding pods were harvested at the indicated days after fertilization (DAF), and the developing seeds were then dissected using a needle and immediately placed in liquid nitrogen. For SW14 gene expression analysis, various tissues were collected from 2-week-old seedlings or plants at flowering stage. All experiments were performed with three biological replicates, each of which is a pooled tissue from more than 10 individual plants. Total RNA was isolated with a Plant RNA Kit (Promega, LS1040) and reverse transcribed to cDNA using M-MLV Reverse Transcriptase (Promega, M1701). RT-qPCR was performed using ChamQ Universal SYBR qPCR Master Mix (Vazyme, Q711) on a Light Cycler 480 thermal cycler system (Roche). The relative quantification was calculated in triplicate and normalized to that of GmACT11 (as an internal control). Primers used are listed in Supplementary Data 1. For the RNA-seq analysis, total RNA was extracted from 21 DAF seeds of W82 and sw14#1 plants, with three biological replicates for each genotype. Differentially expressed genes were assessed using R package DESeq246 with a criterion of fold change ≥1.5 and an adjusted P < 0.01.
Cell-free protein degradation assay
The coding sequences of SW14H1 and SW14H3 were inserted into the BamHI/SalI restriction sites of pGEX-4T-1 (Pharmacia, 28954549). Primers used are listed in Supplementary Data 1. GST and GST fusion recombinant proteins were induced in Escherichia coli Rosetta cells and purified by Glutathione Sepharose Beads (GE Healthcare, 17-0756-01).
The cell-free protein degradation assay was performed as described47 using 21 DAF developing seeds. Total proteins were extracted with degradation buffer (25 mM Tris-HCl, pH 7.5, 10 mM NaCl, 10 mM MgCl2, 5 mM DTT, 100 mM CHX, and 10 mM ATP) and cell debris was removed by centrifugation at 20,000 × g for 15 min. 100 μL cell extracts (containing 500 μg total proteins) were incubated with equal amounts of recombinant GST-SW14H1, GST-SW14H3, or GST protein at 25 °C for the indicated time. Reactions were boiled in SDS loading buffer and then analyzed by immunoblotting with α-GST antibody (Tiangen, AB101-02, 1:10,000 dilution).
Yeast two-hybrid, three-hybrid, and four-hybrid assays
The coding sequence of SW14H1 was inserted into the EcoRI/BamHI restriction sites of pGBKT7 (Clontech, 631604). The coding sequences of GmLEC1a and GmLEC1b were inserted into the EcoRI/BamHI restriction sites of pGADT7 (Clontech, K1612-1). Primers used are listed in Supplementary Data 1. Yeast two-hybrid assays were performed according to the Yeastmaker Yeast Transformation System 2 (Clontech). Yeast AH109 cells were co-transformed with the bait and prey plasmids. All yeast cells were grown on SD/-LW medium for selection. Positive clones were selected in SD/-LWHA dropout plates to evaluate direct protein interactions following incubation at 30 °C.
To investigate the effect of GmNF-YC2 and SW14 on the interaction between GmLEC1a and GmbZIP67, the coding sequence of GmbZIP67 was inserted into the EcoRI/BamHI restriction sites of pGADT7, and the coding sequences of GmNF-YC2 and SW14 were inserted into the XhoI/XmaI restriction sites of pQH05, respectively. For the yeast three-hybrid assay, yeast AH109 cells were co-transformed with bait and prey plasmids in the presence of either pQH05 or pQH05-GmNF-YC2. All yeast cells were grown on SD/-LWH medium for selection, and positive clones were then spotted on SD/-LWHA medium for the interaction test. For yeast four-hybrid assays, PCR genotyping was performed to select the yeast single colonies containing both GmNF-YC2 and SW14 genes. Primers used are listed in Supplementary Data 1. These experiments were repeated at least three times with similar results.
Split-luciferase assay
The coding sequences of GmLEC1a and GmLEC1b were inserted into the KpnI/SalI restriction sites of pCAMBIA1300-nLUC, and the coding sequence for SW14H1 was inserted into the KpnI/SalI restriction sites of pCAMBIA1300-cLUC48. Primers used are listed in Supplementary Data 1. Agrobacterium tumefaciens strain GV3101 cells harboring the indicated constructs were mixed at a 1:1 ratio and introduced into N. benthamiana leaves. The luciferase was activated by 1 mM D-luciferin sodium salt substrate (Abcam, ab145164) after 2 to 3 days of infiltration. The luminescence imaging workstation (NightSHADE LB985; Berthold) was used to capture luciferase images. Split-luciferase experiment was repeated at least three biological replicates.
Co-IP assay
The coding sequence of SW14H1 was inserted into the pGreen-35S: GFP vector to generate 35S: SW14-GFP construct. The 35S:BFP-GFP construct is from a previous study49. The coding sequences of GmLEC1a and GmLEC1b were inserted into the XbaI/BamHI restriction sites of pTF101 vector to generate 35S: GmLEC1a/b-FLAG constructs. Primers used are listed in Supplementary Data 1. Pairwise constructs were co-transformed into N. benthamiana leaves. At 2 to 3 days after infiltration, the leaves were harvested for total protein extraction in co-immunoprecipitation buffer (50 mM HEPES [pH 7.5], 150 mM KCl, 10 mM ZnSO4, 5 mM MgCl2, 1% Triton X-100, and 0.05% SDS, 0.5 mM PMSF, proteinase inhibitor cocktail,). The total proteins were incubated with GFP trap beads (Chromotek, gtak-20) at 4 °C overnight, and rinsed three times with co-immunoprecipitation buffer. The precipitated proteins were boiled in 1× SDS loading buffer and detected by immunoblotting with α-GFP (TransGen, HT801-01, 1:5000 dilution) and α-FLAG (Sigma, F3165, 1:10,000 dilution) antibodies. Co-IP experiment was repeated three biological replicates.
Transient expression assay
To generate the pFA9:LUC and pGmFAD3c:LUC reporter constructs, 3 kb FA9 and 2 kb GmFAD3c promoters were cloned into the HindIII/BamHI restriction site of the pGreenII 0800-LUC vector, respectively. The Renilla Luciferase (REN) gene under the control of the 35S promoter in the pGreenII 0800-LUC vector was used as the internal control. The coding sequences of GmLEC1a, GmNF-YC2, GmbZIP67, and SW14 were cloned into the modified pDOE-01 vector used as effectors. Primers used are listed in Supplementary Data 1. The indicated combinations of effectors and reporters were transformed into N. benthamiana leaves. The LUC and REN activities were measured using the Dual-Luciferase Reporter Assay System Kit (Promega, E1910).
Chromatin immunoprecipitation (ChIP) qPCR assay
ChIP assays were performed as described previously23. Briefly, 21 DAF developing seeds were crosslinked with 1% formaldehyde (Sigma, F8775). Nuclei were isolated from the samples and sonicated to fragment chromatin with an average size of ~500 bp. Subsequently, the chromatin was immunoprecipitated with Protein G PLUS/ Protein A agarose (Millipore, 16-201) plus α-FLAG antibody. The precipitated DNA were purified and used for qPCR analysis with ChamQ Universal SYBR qPCR Master Mix using the primers shown in Supplementary Data 1. Relative enrichment fold was quantified by normalizing the amount of a target DNA fragment against that of a GmACT11 genomic fragment and then against the respective input DNA samples.
Genetic diversity analysis
SNPs from previous study24 with <10% missing data and MAF > 5% were utilized to analyze the genetic diversity (π) of SW14 locus. The pairwise genomic differentiation values for wild, landrace and cultivated soybean populations or soybean accessions harboring different natural variations were calculated using a—window pi2000—window-pi-step 1000 sliding window in VCFtools50. The values of Tajima’s D were calculated as the probability of the sequence departure of a neutrally evolved model using VCFtools24.
Statistical analyses
GraphPad Prism 8.0 and SPSS (version 19, IBM) were used for statistical analysis of the numerical data. The statistically significant differences between two groups or multiple samples were determined by one-way ANOVA. The figure legends provide details on the statistical tests utilized for each experiment.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The raw sequence data of RNA-seq was deposited in the Genome Sequence Archive (https://bigd.big.ac.cn/gsa) under GSA accession number CRA024467. All data supporting the findings of this work are provided within the paper and its Supplementary Information files. The Wm82 a2.v1 reference genome was download from Phytozome. Source data are provided with this paper.
References
Lam, H. M. et al. Resequencing of 31 wild and cultivated soybean genomes identifies patterns of genetic diversity and selection. Nat. Genet. 42, 1053–9 (2010).
Purugganan, M. D. & Fuller, D. Q. The nature of selection during plant domestication. Nature 457, 843–8 (2009).
Schmutz, J. et al. Genome sequence of the palaeopolyploid soybean. Nature 463, 178–83 (2010).
Wang, S. et al. Simultaneous changes in seed size, oil content and protein content driven by selection of SWEET homologues during soybean domestication. Natl. Sci. Rev. 7, 1776–1786 (2020).
Li, J. et al. Identification of ST1 reveals a selection involving hitchhiking of seed morphology and oil content during soybean domestication. Plant Biotechnol. J. 20, 1110–1121 (2022).
Cai, Z. et al. MOTHER-OF-FT-AND-TFL1 regulates the seed oil and protein content in soybean. N. Phytol. 239, 905–919 (2023).
Goettel, W. et al. POWR1 is a domestication gene pleiotropically regulating seed quality and yield in soybean. Nat. Commun. 13, 3051 (2022).
Qi, Z. et al. Natural variation in Fatty Acid 9 is a determinant of fatty acid and protein content. Plant Biotechnol. J. 22, 759–773 (2024).
Song, X. et al. Targeting a gene regulatory element enhances rice grain yield by decoupling panicle number and size. Nat. Biotechnol. 40, 1403–1411 (2022).
Ren, D., Ding, C. & Qian, Q. Molecular bases of rice grain size and quality for optimized productivity. Sci. Bull. 68, 314–350 (2023).
Myers, Z. A. & Holt, B. F. 3rd NUCLEAR FACTOR-Y: still complex after all these years?. Curr. Opin. Plant Biol. 45, 96–102 (2018).
Laloum, T., De Mita, S., Gamas, P., Baudin, M. & Niebel, A. CCAAT-box binding transcription factors in plants: Y so many? Trends Plant Sci. 18, 157–66 (2013).
Jo, L., Pelletier, J. M. & Harada, J. J. Central role of the LEAFY COTYLEDON1 transcription factor in seed development. J. Integr. Plant Biol. 61, 564–580 (2019).
Lotan, T. et al. Arabidopsis LEAFY COTYLEDON1 is sufficient to induce embryo development in vegetative cells. Cell 93, 1195–205 (1998).
Yuan, H. Y., Kagale, S. & Ferrie, A. M. R. Multifaceted roles of transcription factors during plant embryogenesis. Front. Plant Sci. 14, 1322728 (2023).
Li, X. et al. Overcoming the genetic compensation response of soybean florigens to improve adaptation and yield at low latitudes. Curr. Biol. 31, 3755–3767.e4 (2021).
Singh, A. K. et al. Silencing genes encoding Omega-3 fatty acid desaturase alters seed size and accumulation of Bean pod mottle virus in soybean. Mol. Plant Microbe Interact. 24, 506–515 (2011).
Zhang, D. et al. Artificial selection on GmOLEO1 contributes to the increase in seed oil during soybean domestication. PLoS Genet. 15, e1008267 (2019).
Zhang, D. et al. Plasticity and innovation of regulatory mechanisms underlying seed oil content mediated by duplicated genes in the palaeopolyploid soybean. Plant J. 90, 1120–1133 (2017).
Yamamoto, A. et al. Arabidopsis NF-YB subunits LEC1 and LEC1-LIKE activate transcription by interacting with seed-specific ABRE-binding factors. Plant J. 58, 843–56 (2009).
Mendes, A. et al. bZIP67 regulates the omega-3 fatty acid content of Arabidopsis seed oil by activating fatty acid desaturase3. Plant Cell 25, 3104–16 (2013).
Pelletier, J. M. et al. LEC1 sequentially regulates the transcription of genes involved in diverse developmental processes during seed development. Proc. Natl. Acad. Sci. USA 114, E6710–E6719 (2017).
Hou, X. et al. Nuclear factor Y-mediated H3K27me3 demethylation of the SOC1 locus orchestrates flowering responses of Arabidopsis. Nat. Commun. 5, 4601 (2014).
Lu, S. et al. Stepwise selection on homeologous PRR genes controlling flowering and maturity during soybean domestication. Nat. Genet. 52, 428–436 (2020).
Zhang, M. et al. Progress in soybean functional genomics over the past decade. Plant Biotechnol. J. 20, 256–282 (2022).
Lu, X. et al. The transcriptomic signature of developing soybean seeds reveals the genetic basis of seed trait adaptation during domestication. Plant J. 86, 530–44 (2016).
Lu, L. et al. A transcriptional regulatory module controls lipid accumulation in soybean. N. Phytol. 231, 661–678 (2021).
Yu, T. F. et al. The NF-Y-PYR module integrates the abscisic acid signal pathway to regulate plant stress tolerance. Plant Biotechnol. J. 19, 2589–2605 (2021).
Lu, L. et al. Nuclear factor Y subunit GmNFYA competes with GmHDA13 for interaction with GmFVE to positively regulate salt tolerance in soybean. Plant Biotechnol. J. 19, 2362–2379 (2021).
Siriwardana, C. L. et al. NUCLEAR FACTOR Y, Subunit A (NF-YA) proteins positively regulate flowering and act through FLOWERING LOCUS T. PLoS Genet. 12, e1006496 (2016).
Tan, X. et al. Two different viral proteins suppress NUCLEAR FACTOR-YC-mediated antiviral immunity during infection in rice. Plant Physiol. 195, 850–864 (2024).
Huang, M., Hu, Y., Liu, X., Li, Y. & Hou, X. Arabidopsis LEAFY COTYLEDON1 mediates postembryonic development via interacting with PHYTOCHROME-INTERACTING FACTOR4. Plant Cell 27, 3099–111 (2015).
Myers, Z. A. et al. NUCLEAR FACTOR Y, Subunit C (NF-YC) transcription factors are positive regulators of photomorphogenesis in Arabidopsis thaliana. PLoS Genet. 12, e1006333 (2016).
Mu, J., Tan, H., Hong, S., Liang, Y. & Zuo, J. Arabidopsis transcription factor genes NF-YA1, 5, 6, and 9 play redundant roles in male gametogenesis, embryogenesis, and seed development. Mol. Plant 6, 188–201 (2013).
Zhang, X. et al. The WD40 domain-containing protein Ehd5 positively regulates flowering in rice (Oryza sativa). Plant Cell 35, 4002–4019 (2023).
Li, X. et al. Jasmonate signaling pathway confers salt tolerance through a NUCLEAR FACTOR-Y trimeric transcription factor complex in Arabidopsis. Cell Rep. 43, 113825 (2024).
Wang, J. D. et al. ABA-mediated regulation of rice grain quality and seed dormancy via the NF-YB1-SLRL2-bHLH144 Module. Nat. Commun. 15, 4493 (2024).
Zhang, X. et al. Enhancing rice panicle branching and grain yield through tissue-specific brassinosteroid inhibition. Science 383, eadk8838 (2024).
Li, Y. et al. OsMADS17 simultaneously increases grain number and grain weight in rice. Nat. Commun. 14, 3098 (2023).
Fang, C. et al. Genome-wide association studies dissect the genetic networks underlying agronomical traits in soybean. Genome Biol. 18, 161 (2017).
Li, H. et al. The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–9 (2009).
Naito, Y., Hino, K., Bono, H. & Ui-Tei, K. CRISPRdirect: software for designing CRISPR/Cas guide RNA with reduced off-target sites. Bioinformatics 31, 1120–3 (2015).
Paz, M. M., Martinez, J. C., Kalvig, A. B., Fonger, T. M. & Wang, K. Improved cotyledonary node method using an alternative explant derived from mature seed for efficient Agrobacterium-mediated soybean transformation. Plant Cell Rep. 25, 206–13 (2006).
Kereszt, A. et al. Agrobacterium rhizogenes-mediated transformation of soybean to study root biology. Nat. Protoc. 2, 948–52 (2007).
Yu, B. et al. Photoperiod controls plant seed size in a CONSTANS-dependent manner. Nat. Plants 9, 343–354 (2023).
Love, M.I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Zhang, W. et al. Arabidopsis NF-YCs play dual roles in repressing brassinosteroid biosynthesis and signaling during light-regulated hypocotyl elongation. Plant Cell 33, 2360–2374 (2021).
Chen, H. et al. Firefly luciferase complementation imaging assay for protein-protein interactions in plants. Plant Physiol. 146, 368–76 (2008).
Zhang, C. et al. Gibberellin signaling modulates flowering via the DELLA-BRAHMA-NF-YC module in Arabidopsis. Plant Cell 35, 3470–3484 (2023).
Wang, M. et al. Asymmetric subgenome selection and cis-regulatory divergence during cotton domestication. Nat. Genet. 49, 579–587 (2017).
Acknowledgements
This work was supported by the National Key R&D Program of China (grant no. 2021YFF1001203 to B.L.), the National Natural Science Foundation of China (grant no. 32372188 to C.Z. and 32230078 to X.H.), and the Guangdong Basic and Applied Basic Research Foundation (grant no. 2024A1515013149 to C.Z.).
Author information
Authors and Affiliations
Contributions
X.H. and C.Z. designed and supervised the project. C.Z., W.L., C.T., M.K., H.W., S.L., H.L. and X.L. performed the experiments. C.Z., W.L., C.T., Y.M., B.L., F.K. and X.H. analyzed the data. C.Z. and X.H. wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Guo-Liang Jiang, Paola Vittorioso for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhang, C., Li, W., Tan, C. et al. Natural allelic variation in SW14 determines seed weight and quality in soybean. Nat Commun 16, 8070 (2025). https://doi.org/10.1038/s41467-025-63582-0
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-63582-0
