
Abstract
Photorespiration is a costly cellular process that reduces photosynthetic efficiency. While mitigating photorespiratory losses could boost crop yields, the interconnection of photorespiration with other processes is increasingly recognized. Its high carbon turnover generates mitochondrial one-carbon (C1) metabolites, including formate, but their contribution to cellular C1 metabolism has remained unclear. DNA methylation is an important epigenetic modification that depends on methyl groups provided by folate-mediated C1 metabolism. Here we show that photorespiration supplies C1 units for DNA methylation in Arabidopsis. We demonstrate that carbon from formate is incorporated into 5-methylcytosine through the C1-tetrahydrofolate synthase pathway, which operates predominantly during the day. Elevated CO2 that suppresses photorespiration alters the methylome, especially when the serine-derived C1 supply, which compensates for a blocked formate-derived supply, is compromised. These findings establish a metabolic link between photorespiration and epigenome stability and provide a framework for understanding methylome dynamics under rising CO2 levels and other environmental influences on photorespiration.
Similar content being viewed by others
Main
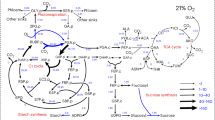
Plants undergo photorespiration because ribulose-1,5-bisphosphate carboxylase/oxygenase (Rubisco) poorly discriminates between CO2 and O2 (ref. 1). Changes in CO2 levels, temperature and light intensity alter carboxylation and oxygenation rates, making photorespiration highly dynamic2. Detoxification of the oxygenation product and carbon salvage through the photorespiratory pathway consume ATP and release CO2, thereby reducing net photosynthesis by up to 50% (ref. 3). Reducing oxygenation and improving photorespiratory efficiency can therefore increase crop yields3. However, photorespiration is tightly connected to other cellular processes, including folate-mediated one-carbon metabolism (FOCM)2,4,5.
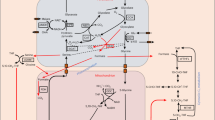
During photorespiration, glycine accumulates and is transported into mitochondria, where it is oxidized by the glycine decarboxylase complex (GDC), transferring a C1 unit to tetrahydrofolate (THF)6,7 (Fig. 1a). In parallel, mitochondrial serine hydroxymethyltransferase (SHMT) converts glycine to serine, regenerating THF8,9. A portion of the C1 units generated by the GDC supports mitochondrial FOCM, while surplus C1 is released by 10-formyl-THF deformylase (FDF) as formate to sustain flux and avoid the build-up of photorespiratory intermediates10. Mitochondrial formate dehydrogenase readily oxidizes accumulating formate to CO2 (ref. 11).

a, Cross-compartmental biochemical pathways of FOCM. (1a) and (1b) SHMT; (2) MTHFR; (3) methionine synthase; (4) S-adenosylmethionine (SAM) synthetase; (5) SAM-dependent methyltransferase, including DNA methyltransferases; (6) SAH hydrolase (SAHH); (7a) methylenetetrahydrofolate dehydrogenase and (7b) methenyltetrahydrofolate cyclohydrolase (MTHFD); (8) THFS; (9) 10-FDF; (10) GDC. Hcy, homocysteine. b, Representative pictures of 3-week-old mthfd1-1 (m), mthfd1-1 thfs double mutant (mt), thfs (t) and wild-type (WT) plants grown under LD conditions, and leaf area quantification from automated phenotyping (right). Scale bars, 1 cm. The data are presented as mean values ± s.d. (n = 8). c, Corrected total cell fluorescence (CTCF) from SDCpro-GFP expression quantified via confocal laser scanning microscopy. The box plots represent the median (centre lines), the 25th (bottom) and 75th (top) percentiles, and the minimum and maximum points within 1.5× the interquartile range (IQR) (n = 10). Lowercase letters represent significant differences (P < 0.05, one-way analysis of variance followed by post-hoc Tukey test). See Supplementary Table 8 for the P values. d, Hierarchically clustered heat map of mean scaled mCG ratios (z scores) of all DMRs (rows) from pairwise comparisons to the WT (left); distributions of mCG ratios in four main clusters, A1 (n = 9,959), A2 (n = 2,537), A3 (n = 245) and A4 (n = 268) (centre); and per cent overlaps of DMRs with hypervariable DNA methylation (HV) sites, CMT2-dependent methylated sites, RNA-directed DNA methylation (RdDM) sites, gbM sites and the remaining reference mthfd1-1 hypo-DMRs (Other m), as well as the remaining non-overlapping DMRs (Rest) per cluster (right). mCG ratios were computed from per-site C/T counts pooled across two biological replicates for each genotype. The box plots indicate the medians (centre lines), IQRs (boxes) and 1.5 × IQR (whiskers); the violin width reflects the kernel density. n indicates the number of DMRs. e, Heat map of mean (n = 3) normalized transcript levels (left) and mean mCG ratios (right) of all differentially expressed transposable elements (DETEs; rows) compared to the WT. The rows are hierarchically clustered by transcript level. The upper right plot shows the correlation between transcript levels and mCG ratios over DETEs per cluster. TSS, transcription start site; TTS, transcription termination site.
While activated C1 units are not exchanged between compartments, both serine and formate produced in mitochondria can supply cytosolic C1 metabolism: serine via cytosolic SHMT, generating 5,10-methylene-THF, and formate via 10-formyl-THF synthetase (THFS)4,8,9,12 (Fig. 1a). 5,10-methylene-THF directly serves for thymidylate synthesis (required for DNA replication) or is reduced to 5-methyl-THF for homocysteine-to-methionine recycling and production of the universal methyl donor S-adenosylmethionine (SAM)13,14. The THFS product, 10-formyl-THF, is converted to 5,10-methylene-THF via 5,10-methenyl-THF by 5,10-methylene-THF dehydrogenase/cyclohydrolase 1 (MTHFD1)12,15. The SHMT and THFS/MTHFD1 reactions operate reversibly12,15,16. The THFS/MTHFD1 pathway is essential for de novo purine biosynthesis in yeast and animals (through tri-functional C1-THF synthase)17, but its role in plant FOCM has been unclear12,18. We previously showed that mutations in MTHFD1 strongly impair DNA methylation in Arabidopsis, implicating the THFS/MTHFD1 branch in methylation reactions19.
In plants, distinct DNA methyltransferases catalyse methyl transfer from SAM to cytosine in CG, CHG and CHH sequence contexts (H represents A/C/T)20. The byproduct S-adenosylhomocysteine (SAH) is a competitive inhibitor and must be cleared by SAH hydrolase to sustain methylation21. Stable DNA methylation is essential for transposable element (TE) silencing and is faithfully maintained during replication22. Still, epimutations (that is, heritable DNA methylation changes) accumulate stochastically over generations and occasionally generate phenotypic variation23,24; climate and environmental stress, including drought and cold, associate with epigenetic variation and adaptation in natural populations and crops24,25,26. Although most stress-induced DNA methylation changes are somatic, they can act as cellular memory to support transient environmental adaptations27,28. How stress drives such epigenetic changes remains poorly understood.
Because SAM supply and SAH clearance directly affect DNA methylation, we dissected the cytosolic folate network and its coupling with photorespiration in Arabidopsis. This revealed that photorespiratory formate is reassimilated into FOCM to sustain DNA methylation and silencing, implying that environmental modulation of photorespiration may influence plant epigenetic regulation.
Results
Suppression of mthfd1 by thfs
To dissect cytosolic FOCM genetically, we used the hypomorphic mthfd1-1 mutant (hereafter mthfd1), which exhibits SAH accumulation and DNA hypomethylation19. This prompted us to ask why FOCM impairment in mthfd1 is not compensated by the SHMT4-dependent serine branch. To address this, we screened for mutants that modified the growth defects and derepression of the DNA-methylation-sensitive SDCpro-GFP reporter in mthfd1 (ref. 19). Unexpectedly, a loss-of-function mutation in THFS fully suppressed the mthfd1 phenotype (Extended Data Fig. 1a,b). Instead of phenocopying mthfd1, homozygous thfs single and mthfd1 thfs double mutants (mt) were indistinguishable from the wild type in leaf growth, GFP fluorescence, flowering time and root length (Fig. 1b,c and Extended Data Fig. 1c–e).
To exclude background genetic effects, we generated CRISPR–Cas9 THFS knockouts in the mthfd1 background and confirmed the restoration of global DNA methylation (Extended Data Fig. 2a–e). To verify that suppression was not specific to mthfd1-1, we crossed thfs with a plant heterozygous for the strong mthfd1-3 allele19 (Extended Data Fig. 1a). Double mutant offspring suppressed the DNA hypomethylation and dwarf phenotype of mthfd1-3 (Extended Data Fig. 2f). Together, these results indicate that the THFS/MTHFD1 branch of cytosolic FOCM is dispensable for growth and global DNA methylation under standard conditions—consistent with compensation by SHMT4.
To examine DNA methylation changes in detail, we performed whole genome bisulfite sequencing (WGBS) on leaf tissue of the wild type, mthfd1, thfs and mt (Supplementary Table 1). Principal component analysis of methylome profiles and genome tracks of CG, CHG and CHH methylation (mCG, mCHG and mCHH) showed that the widespread DNA hypomethylation across pericentromeric regions and chromosome arms of mthfd1 was largely suppressed in mt (Extended Data Fig. 3a,b). Consistently, analysis of differentially methylated regions (DMRs) and hierarchical clustering of CG DMRs identified a major cluster of mthfd1 hypo-DMRs fully suppressed in mt (A1), a partially suppressed cluster (A2) and two small clusters—one comprising hypo-DMRs shared by the mutants and the other DMRs that were hypermethylated in thfs and mt (Fig. 1d and Supplementary Table 2). Although dispensable for maintaining global DNA methylation levels, the THFS/MTHFD1 branch appeared to stabilize DNA methylation patterns at specific genomic regions. We therefore defined regional DNA methylation classes (detailed in Methods) and quantified their overlap with DMRs per cluster. Heterochromatic regions—defined as Chromomethylase 2 (CMT2) sites—were enriched in A1 and thus preferentially restored, whereas gene-body-methylated (gbM) regions were enriched in A2 (Fig. 1d and Extended Data Fig. 3c). Accordingly, mCG over gbM sites was significantly lower in mt than in the wild type (Wilcoxon rank-sum test; false discovery rate, <2 × 10−16), but not over CMT2 sites (FDR > 0.01) (Extended Data Fig. 3d). These regional patterns indicate that perturbations to FOCM differentially affect methylation in heterochromatin versus euchromatin.
TE expression analysis via mRNA sequencing (mRNA-seq) showed that thfs suppressed mthfd1-induced TE derepression genome-wide, with the exception of a small cluster comprising primarily autonomous Mutator-like DNA transposons (VANDAL21) that can antagonize DNA methylation by anti-silencing proteins29 (Fig. 1e and Extended Data Fig. 3e–g).
Photoperiod controls FOCM
We observed that the mthfd1 phenotype is sensitive to photoperiod—a major regulator of Arabidopsis development30—as growth under short days (SD) mitigated the growth defects exhibited under long days (LD) (Figs. 1b and 2a). Methylome comparisons confirmed that the hypomethylation seen in mthfd1 under LD was largely attenuated under SD (Fig. 2b and Extended Data Fig. 4a,b). Furthermore, shifting plants from SD to LD for 48 hours led to the upregulation of SDC and selected TEs previously found derepressed in mthfd1 (ATCOPIA28 and SADHU3)19, underscoring the dynamic influence of photoperiod on folate-mediated C1 supply for DNA methylation (Fig. 2c).

a, Representative pictures of 4-week-old m, mt, t and WT plants grown under SD, as well as leaf area quantification from automated phenotyping. Scale bars, 1 cm. The data are presented as mean values ± s.d. (n = 10). b, Distribution of mCG ratios in MET1-targeted gbM regions (n = 420) and CMT2-targeted regions (n = 1,623) in rosette leaves of m and WT plants grown under SD. The values were computed from per-site C/T counts binned into 100-bp windows and pooled across two biological replicates for each genotype. The box plots indicate the medians (centre lines), IQRs (boxes) and 1.5 × IQR (whiskers); the violin width reflects the kernel density. n indicates the number of regions. c, Transcript levels (ΔΔCt) of three marker loci for transcriptional silencing (SADHU3, ATCOPIA28 and SDC) that were analysed via reverse transcription quantitative PCR (RT–qPCR). Plants were grown continuously under SD or shifted from SD to LD for 48 h before sampling at day 28 after germination. The data are presented as mean values ± s.d. (n = 3). The indicated P values are from two-sided Welch’s t-tests. d,e, Diurnal steady-state levels of target metabolites in leaves under LD (d) and SD (e). The grey shading depicts dark periods. The data are presented as mean values ± s.d. (n = 5). For glycine and homocysteine, values below the detection limit were treated as not available. The P values and sample sizes are provided in Supplementary Tables 12 and 13 for plants grown under LD and SD, respectively. FW, fresh weight. f,g, Steady-state levels of selected THF intermediates in leaves under LD (f) and SD (g). The grey shading depicts dark periods. The data are presented as mean values ± s.d. (n = 3). Values below the detection limit were treated as not available. The P values and sample sizes are provided in Supplementary Tables 12 and 13 for plants grown under LD and SD, respectively.
Diurnal profiling showed that, in mthfd1 leaves, homocysteine and SAH accumulate strongly in the morning (0–6 h after dawn/ZT6) and decline at night, when MTHFD1 appears dispensable (Fig. 2d). Whereas the wild type, thfs and mt accumulated methionine and SAM during the day, mthfd1 showed an attenuated and prolonged methionine rise (Fig. 2d). Sustained SAM levels indicate that DNA hypomethylation in mthfd1 is caused by SAH accumulation rather than SAM limitation (Fig. 2d). These metabolic defects in mthfd1 were mitigated under SD (Fig. 2e).
Among folate intermediates, 5-methyl-THF varied only modestly, and methylenetetrahydrofolate reductase (MTHFR) activity was similar across genotypes (Extended Data Fig. 4c,d). Homocysteine accumulation despite stable 5-methyl-THF in mthfd1 leaves indicates that bulk folate pools may obscure subcellular constraints and that steady-state 5-methyl-THF is a poor proxy for C1 flux through the methionine cycle19.
By contrast, 5-formyl-THF was consistently elevated in mthfd1 under LD, but not in other genotypes or under SD (Fig. 2f,g). 5-formyl-THF has no known function as a cofactor, but it can act as folate storage and an FOCM regulator; its inhibition of serine–glycine interconversion by SHMT is well established31,32,33. Accordingly, the accumulation of 5-formyl-THF in 5-fcl and fdf1,2 double mutants is associated with the build-up of photorespiratory glycine10,32. In comparison, mthfd1 exhibited elevated serine (but not glycine) during the day (Fig. 2d), suggesting that cytosolic 5-formyl-THF accumulation may inhibit SHMT4. Moreover, THF peaked at ZT12—consistent with simultaneous blocking of serine- and formate-derived C1 flux in mthfd1 under LD (Fig. 2f). Upregulation of SHMT4 in mthfd1 (Extended Data Fig. 4e) that mirrored methionine dynamics supports a compensatory response to C1 restriction, most apparent as homocysteine build-up. Homocysteine is an activator of 3-phosphoglycerate dehydrogenase (PGDH)34, the first enzyme of the phosphorylated serine biosynthesis pathway (PSBP), reinforcing that C1 supply via serine is upregulated in mthfd1.
5-formyl-THF arises from 5,10-methenyl-THF via a side reaction of SHMT35 (Fig. 1a) and is reconverted by mitochondrial 5-FORMYLTETRAHYDROFOLATE CYCLOLIGASE (5-FCL)36. The arginine-to-glutamine substitution in mthfd1 is predicted to impair NADP+/NADPH binding, affecting the interconversion of 5,10-methenyl-THF to 5,10-methylene-THF—supported by diminished MTHFD1 activity in mthfd1 and mt (Extended Data Fig. 4d)—while maintaining the cyclohydrolase conversion between 10-formyl-THF and 5,10-methenyl-THF37,38. Accordingly, our data suggest that, in mthfd1-1, C1 flux through the THFS/MTHFD1 branch is diverted towards 5-formyl-THF. This folate sink is mended by blocking 10-formyl-THF synthesis, which we confirmed in thfs and mt (Fig. 2f and Extended Data Fig. 4d).
While THFS did not show diurnal regulation, the relative expression of MTHFD1 increased during the day (Extended Data Fig. 4e). Together, the profiles and genetic analyses are consistent with predominant daytime activity and increased engagement of the THFS/MTHFD1 branch under LD, assimilating formate into 10-formyl-THF and converting it via 5,10-methenyl-THF to 5,10-methylene-THF.
Formate serves as a C1 source
To confirm the assimilation of formate into cytosolic FOCM, we established [13C]formate labelling with targeted quantification by gas chromatography–mass spectrometry (Extended Data Figs. 5a,b and 10). We observed THFS- and MTHFD1-dependent labelling of 5-methylcytosine and methionine, with stronger incorporation during the day (Fig. 3a,c). Glycine and serine also showed label enrichment that peaked at the start of the photoperiod upon [13C]formate supplementation, but this was independent of THFS and MTHFD1 (Extended Data Fig. 5c), indicating that—unlike the THFS/MTHFD1-dependent labelling of methionine and 5-methylcytosine—glycine/serine labelling probably resulted from the oxidation of [13C]formate by formate dehydrogenase and the reassimilation of 13CO2 into 3-phosphoglycerate18. Short-term labelling with [2-13C]glycine indicated that photorespiratory glycine-to-serine conversion was unaffected in mthfd1 and thfs (Fig. 3b). Moreover, thymine was labelled from [13C]formate in a THFS- and MTHFD1-dependent way, demonstrating that 5,10-methylene-THF produced by MTHFD1 also contributes to de novo thymidylate synthesis (Fig. 3c). In contrast, adenine labelling was THFS- and MTHFD1-independent, indicating that the cytosolic folate network is not involved in de novo purine biosynthesis, which is plastid-localized39; this result aligns with the viability of mthfd1-3 thfs double mutants.

a, Label enrichment in methionine after [13C]formate supplementation at the beginning (left) or end (right) of the photoperiod. The grey shading depicts dark periods. The data are presented as mean values ± s.d. (n = 3). See Supplementary Table 16 for the P values. b, Label enrichment in serine after [2-13C]glycine supplementation at the beginning of the photoperiod. Dual labelling (M + 2) results from combined glycine cleavage and glycine-to-serine conversion in mitochondria. The grey shading depicts dark periods. The data are presented as mean values ± s.d. (n = 3). See Supplementary Table 17 for the P values. c, Label enrichment in targeted nucleobases after [13C]formate supplementation. Guanine was not quantified due to low recovery (Methods). The data are presented as mean values ± s.d. (n = 3). Lowercase letters represent significant differences between genotypes for each nucleobase (P < 0.05, one-way analysis of variance followed by post-hoc Tukey test). See Supplementary Table 18 for the P values. 5mC, 5-methylcytosine.
Photorespiration stabilizes DNA methylation
Given that non-enzymatic oxidation of photorespiratory glyoxylate and probably mitochondrial FDF1/2 activity are major physiological sources of formate10,40, we hypothesized that suppressing photorespiration would restrict C1 flux through the THFS/MTHFD1 branch and partially rescue the mthfd1 phenotype. To test this, we profiled DNA methylation in wild-type and mthfd1 plants grown in control air (cCO2, 394 ± 39 ppm) and in non-photorespiratory high CO2 (hCO2, 3,141 ± 53 ppm) conditions that rescue photorespiratory cat2-15 and shmt1 mutants41,42 (Extended Data Fig. 6a). In the wild type, hCO2 caused only minor genome-wide changes—that is, slight CG hypomethylation, no apparent changes in mCHG and weak CHH hypermethylation in pericentromeric regions (Extended Data Fig. 6b–e). This indicates that the C1 supply for DNA methylation is largely maintained when photorespiration is suppressed, consistent with a serine-derived C1 supply via SHMT4 compensating for reduced formate-derived C1 units. In contrast, mthfd1 showed pronounced mCG, mCHG and mCHH gains under hCO2 relative to cCO2 (Extended Data Fig. 6b–e). Clustering of CG DMRs delineated mthfd1 hypo-DMRs that strongly (B1) or weakly (B2) regained mCG under hCO2, as well as a wild-type-specific hCO2-sensitive cluster (B3) (Fig. 4a and Supplementary Table 3). Cluster B3 accounted for approximately 10% of the combined CG DMRs and showed a significant decrease in mean CG ratios in the wild type under hCO2 versus cCO2 (Wilcoxon rank-sum test, FDR < 6 × 10−76), revealing that suppressing photorespiration induces CG hypomethylation in the wild type in addition to mitigating methylation defects in mthfd1 (Fig. 4a). As with cluster A1 (Fig. 1d), B1 and B3, which were both enriched in CMT2 sites, exhibited greater mCG dynamics than gbM-enriched B2 (Fig. 4a–c and Extended Data Fig. 6f). As expected, hCO2 was less effective than thfs in restoring DNA methylation in mthfd1, probably because formate is also produced independently of photorespiration (for example, by the oxidation of methanol released during pectin demethylation) under hCO2 (ref. 43).

a, Hierarchically clustered heat map of mean scaled mCG ratios (z scores) of all DMRs (rows) compared to the WT under cCO2 (left); distributions of mCG ratios in four main clusters, B1 (n = 9,019), B2 (n = 13,430), B3 (n = 2,499) and B4 (n = 76) (centre); and per cent overlaps of DMRs with categorized DNA-methylated regions per cluster (right). mCG ratios were computed from per-site C/T counts pooled across two biological replicates for each genotype and CO2 condition. The box plots indicate the medians (centre lines), IQRs (boxes) and 1.5 × IQR (whiskers); the violin width reflects the kernel density. b, Complete-linkage clustering dendrogram (Euclidean distance) of genotypes and CO2 conditions based on mCG, shown above distributions of mCG ratios in union DMRs (covered across samples) from clusters A2 (n = 2,374) and B1 (n = 5,277). The cluster labels follow a and Fig. 1d. DMR mCG ratios were computed from per-site C/T counts pooled across two biological replicates for each genotype and CO2 condition. The box plots indicate the medians (centre lines), IQRs (boxes) and 1.5 × IQR (whiskers); the violin width reflects the kernel density. n indicates the number of DMRs. c, DMR overlaps between CMT2-dependent methylation (A1 and B1) and gbM (A2 and B2) enriched clusters. d, Volcano plot of TE expression in m under hCO2 compared to m under cCO2 (n = 3). The top eight TEs ranked by q value are indicated by subfamily names. e, Heat map of mean (n = 3) expression changes relative to the WT under cCO2 of mthfd1-1 DEGs encoding enzymes involved in FOCM, photorespiration and other related metabolic pathways (see row annotation; SMM, S-methylmethionine cycle). The rows were hierarchically clustered by scaled normalized transcript levels.
Photorespiration drives expression changes in mthfd1
TE derepression in mthfd1 was broadly reduced under hCO2 (Extended Data Fig. 7a–c), but with TE-family-specific differences. Compared with all mthfd1 differentially expressed transposable elements (DETEs), CO2-insensitive mthfd1 DETEs, which remained highly expressed despite increased DNA methylation, were enriched for En-Spm DNA transposons (pairwise comparison of proportions, P = 3.3 × 10−8), whereas CO2-responsive mthfd1 DETEs were enriched for Gypsy family retrotransposons (such as ATHILA) (P = 1.1 × 10−4) (Fig. 4d and Extended Data Fig. 7d,e).
Consistent with its pleiotropic phenotype, mthfd1 exhibits broad gene expression changes compared with the wild type19. Under hCO2, these changes were attenuated, and the number of differentially expressed genes (DEGs) in mthfd1 decreased by 60% compared with cCO2; consistently, mthfd1 showed ~10-fold more CO2-responsive DEGs than the wild type (Extended Data Fig. 7f). Among the most dynamic DEGs in mthfd1 was GLN1;4, encoding a cytosolic glutamine synthetase catalysing ammonium assimilation that is inhibited by 5-formyl-THF binding35. Prompted by this observation, we tested for enrichment and found that 18 of 51 genes encoding reported 5-formyl-THF-binding proteins were among the hCO2-responsive mthfd1 DEGs (hypergeometric test, P = 1.4 × 10−5) (Extended Data Fig. 7g), suggesting that 5-formyl-THF accumulation may contribute to these expression changes in mthfd1. Consistent with the attenuation of mthfd1 defects under hCO2, SDC expression was reduced, and DNA methylation at the SDC promoter was increased (Extended Data Figs. 7g and 8).
Photorespiration and serine biosynthesis are transcriptionally upregulated in mthfd1
Querying a curated gene annotation list, we identified 42 DEGs involved in FOCM, photorespiration and associated metabolic pathways, 36 of which were differentially expressed in mthfd1 under cCO2 but not under hCO2 (Supplementary Tables 4 and 5). Hierarchical clustering of these 42 DEGs revealed strongly co-expressed members of the photorespiratory glycolate and associated ammonia recycling pathways1, including GLUTAMINE SYNTHETASE 2 (GS2); DiT1, encoding the plastidal dicarboxylate transporter for the subsequent glutamine oxoglutarate aminotransferase (GOGAT) reaction, which together with GS2 constitutes the N-assimilatory GS/GOGAT cycle; and genes encoding the T and H subunits of the GDC (Fig. 4e and Extended Data Fig. 9). Together with the P subunit, which was also among the cCO2-upregulated mthfd1 DEGs (Extended Data Fig. 7g), the H subunit is known to limit the rate of glycine cleavage42. Additional cCO2-upregulated mthfd1 DEGs included key enzymes of assimilatory nitrate reduction; two of the three PSBP enzymes, including homocysteine-activated PGDH3; and several FOCM components, including SHMT4 and FDF2 (Fig. 4e). The patterns align with published transcriptome changes in shmt1 and PSBP mutants5 and suggest that the photorespiratory glycolate pathway and the PSBP (the two principal routes for serine production) are upregulated in mthfd1 under photorespiratory conditions, probably in response to perturbed C1 and sulfur-amino-acid homeostasis.
Discussion
Our study reveals a previously unrecognized link between photorespiration and DNA methylation: photorespiratory formate feeds the cytosolic folate cycle via THFS and MTHFD1, supplying C1 units to the methionine cycle in Arabidopsis leaves during the photoperiod (Fig. 5). While formate is a recognized C1 donor in yeast and animals, direct evidence in plants has been scarce5. Gashu et al.4 quantified photorespiratory carbon fluxes in Arabidopsis grown under SD using 13CO2 labelling and identified serine as the principal cytosolic C1 source, with formate contributing little and not fitting their flux model. Our results draw a contrasting picture in which serine and formate act complementarily to maintain cytosolic C1 homeostasis. Notwithstanding possible bias from high [13C]formate supplementation, we observed THFS- and MTHFD1-dependent labelling of methionine and 5-methylcytosine, with free methionine labelling conspicuously strong during the photoperiod—consistent with photorespiration driving C1 flux. Together, these observations suggest that the relative contributions of formate and serine to cytosolic FOCM vary with time of day and photoperiod, as reflected by the conditional mthfd1 phenotype.

Formate produced during photorespiration enters C1 metabolism through THFS and MTHFD1, supplying C1 units for thymidylate synthesis, recycling of Hcy to methionine (Met) and SAM-dependent DNA methylation in leaves. Photorespiratory C1 flux (yellow) is modulated by environmental variables. Negative feedback from THFS/MTHFD1 controls serine-derived C1 flux through SHMT4 (blue), stabilizing C1 metabolism under fluctuating conditions. When fluctuations in photorespiratory C1 flux are not compensated by SHMT4, local SAM limitation and SAH build-up drive DNA methylation changes.
Under LD, mthfd1 shows daytime build-up of homocysteine and SAH along with DNA hypomethylation and TE derepression, whereas these signatures are largely attenuated under SD. A longer photoperiod relaxes carbon constraints and increases ATP and NADPH generation by light reactions, which is expected to favour C1 supply via the ATP- and NAD(P)H-dependent steps of the THFS/MTHFD1 branch2,3,12,15. Conversely, a shorter photoperiod probably increases reliance on the serine-derived supply via SHMT4.
This photoperiod dependence intersects with tissue specificity. Earlier studies showed that photorespiratory formate can supply cytosolic C1 for glycine-to-serine conversion in GDC mutants and heterotrophic tissue8,9,44. THFS was recently characterized in Arabidopsis as a cytosolic formate-activating enzyme and appeared to mainly support serine production in roots, where thfs mutants showed reduced growth under SD12. By contrast, under our LD conditions, root growth in thfs was indistinguishable from that in the wild type. Moreover, in leaves, [13C]formate labelling of serine (unlike methionine) was independent of THFS and MTHFD1. Nevertheless, complete suppression of the pleiotropic mthfd1 phenotype—including short roots—by thfs in mt double mutants indicates that the THFS/MTHFD1 branch operates in leaves as well as in roots.
Our genetic analysis of the cytosolic folate network indicates that the THFS/MTHFD1 branch is largely compensable by the SHMT4 branch. The restoration of FOCM in mt double mutants further suggests that THFS negatively regulates SHMT4 activity (Fig. 5), probably via competition for THF8,12,16 and/or accumulation of inhibitory 5-formyl-THF32. This built-in redundancy and feedback regulation underscores the flexibility of the cytosolic folate network across organisms in balancing C1 sources45. By contrast, selectively blocking MTHFD1 while THFS remains intact perturbs this balance, reduces serine- and formate-derived C1 supply for homocysteine-to-methionine recycling, alters diurnal SAM and SAH dynamics, and impairs DNA methylation and TE silencing in Arabidopsis.
Correspondingly, carbon throughput from photorespiration to folate metabolism can directly influence epigenetic regulation. Suppressing photorespiration reduces serine-derived C1 flux by approximately fivefold4. According to our results, formate-derived C1 throughput likewise diminishes, as inferred from the partial restoration of DNA methylation in mthfd1 under hCO2. Together, curbing photorespiratory C1 supply can induce DNA methylation changes, as seen in wild-type plants grown under hCO2, albeit to a lesser extent than in mthfd1, probably because alternative serine production (particularly the PSBP) becomes upregulated, as observed in shmt1 (ref. 18) and in mthfd1.
The C1 metabolic bridge provides a framework for environmentally induced DNA methylation changes24,27,28,46 (Fig. 5). Consistent with this view, a recent study on acclimation to elevated CO2 (1,000 ppm) showed transgenerational DNA methylation changes in Arabidopsis that correlated with enhanced growth47. Persistent DMRs were enriched in gbM and underrepresented in heterochromatic regions47, resembling methylome dynamics in mthfd1. This suggests that gbM may be more vulnerable to limited C1 supply than heterochromatin—potentially because replication-coupled mCG maintenance becomes error-prone when SAM is scarce and the methionine cycle competes with thymidylate synthesis for activated C1 units38. The nuclear localization of SAM synthetase48 further raises the possibility that SAM is preferentially supplied to heterochromatin. In addition, stabilizing mechanisms, including feedback between non-CG methylation and H3 K9 dimethylation49,50, may facilitate the recovery of heterochromatic methylation upon restoration of C1 supply. Our model remains simplified, as illustrated by pericentromeric CG hypomethylation with CHH hypermethylation in the wild type under hCO2.
Because photorespiration is highly sensitive to ambient fluctuations, the formate-based C1 supply described here provides a basis for predicting how climate change may modulate plant methylomes. Although differences in DNA methylation among Arabidopsis accessions are largely explained by genetic variation51, they also correlate with climate of origin25, raising the question of how photorespiration contributes to methylome variation regarding climate change. Canopy-scale models project that photorespiratory losses will decline under future climates: in soybean, the penalty on midday net CO2 assimilation is predicted to drop by ~50% by 2100 under a high-emissions scenario (~1,000 ppm atmospheric CO2; +3.7 °C warming)3—implying lower photorespiratory provision of formate and serine to cytosolic FOCM. That trend may be offset by heat or drought, which increase photorespiration52. However, prior work and our data argue against simple proportionality between photorespiration, C1 flux and DNA methylation, because FOCM is tightly coupled to other pathways—notably the methionine cycle53. Plant FOCM is responsive to cellular demand and can sustain the synthesis of methylated secondary metabolites at levels far exceeding the C1 equivalents in nucleic acids5. Defining the regulatory network that couples environmental fluctuations to the C1 supply–demand balance and DNA methylation warrants further investigation, with implications for predicting adaptation to future climates and for strategies to reduce photorespiratory losses in crops54.
Methods
Plant materials and growth conditions
Arabidopsis thaliana (L.) Heynh. accession Columbia-0 was used throughout. The hypomorphic mthfd1-1 allele, the mthfd1-3 null allele, and wild-type and mthfd1-1 lines carrying SDCpro-GFP have been described previously19. The transfer DNA (T-DNA) insertion mutant thfs (SALK_067510) was obtained from the Arabidopsis Biological Resource Center (Ohio State University), and homozygosity of the T-DNA insertion in THFS (AT1G50480) was confirmed via PCR genotyping (the primer sequences are listed in Supplementary Table 6). The thfs line was crossed with mthfd1-1 SDCpro-GFP and the wild type carrying SDCpro-GFP. Homozygous thfs SDCpro-GFP and mthfd1-1 thfs SDCpro-GFP F2/F3 offspring (mt) were identified via PCR genotyping.
Plants were grown in controlled-environment growth chambers (Weiss Technik & Vötsch) at 23/20 °C (day/night), 65% relative humidity and 100 μmol m−2 s−1 photosynthetically active radiation (400–700 nm) under LD (16 h light/8 h dark) or SD (10 h light/14 h dark) cycles. Unless stated otherwise, the plants were germinated and grown on soil (4:1 (v/v) Floragard multiplication substrate:quartz sand). For isotope labelling and root length measurements, plants were grown in liquid and solid half-strength Murashige and Skoog (MS) medium (Duchefa Biochemie), respectively (see ‘Isotopic labelling’ below and ‘Root length analysis’ in Supplementary Methods).
For experimental suppression of photorespiration, plants were grown in an exposure chamber at the Environmental Simulation phytotron (Helmholtz Munich)55. The growth conditions were 130 µmol m−2 s−1 photosynthetically active radiation at plant height, LD cycle with 23/21 °C and 65% relative humidity. The plants were grown in two Plexiglas cuvettes (55 × 28 × 110 cm3; width × depth × height). One cuvette was flushed with 60 m3 h−1 air at the ambient CO2 level (394 ± 39 ppm, cCO2) and the other with air continuously enriched with CO2 (Air Liquide) to 3,141 ± 53 ppm (hCO2). Airflows were controlled using a 1-l mass flow controller (MKS Instruments GmbH), calibrated with a mass flow meter (ADM 3000, Agilent). CO2 levels were monitored with an infrared gas analyser (BINOS 100, Rosemount Analytical Inc.) calibrated against a certified 2,000-ppm gas standard (Air Liquide) (Supplementary Fig. 1). At ambient CO2, no morphological changes attributable to the phytotron growth systems in either the wild type or mthfd1 were observed via visual inspection. Even so, all analyses were performed within the growth systems to minimize potential system effects.
Unless stated otherwise, samples consisted of rosette leaf tissue harvested 21 days after germination under LD and 28 days after germination under SD. Tissue was harvested 6 h after dawn (ZT6) for single-time-point experiments and at the end of the night (0 h), ZT6, ZT12 and ZT18 for time-course experiments. The samples were flash-frozen in liquid nitrogen and pulverized in a bead mill (Retsch TissueLyser, Qiagen) using two 2-mm steel beads.
Confocal microscopy
SDCpro-GFP expression was quantified via confocal microscopy (Supplementary Methods) of wild-type, mthfd1-1, thfs, mthfd1-1 thfs and CRISPR–Cas9 lines (Supplementary Methods). CTCF was calculated as described in ref. 56 using the formula CTCF = Integrated Density − (Area × Mean Background Fluorescence). For each genotype, data were collected from ten biological replicates (that is, plants) for statistical analysis. Further methodological details are provided in Supplementary Information.
mRNA-seq
The mRNA-seq experiments were performed with three biological replicates. Total RNA was isolated from 100 mg (fresh weight) of leaf tissue pooled from three or four plants using the RNeasy Plant Mini Kit (Qiagen) followed by DNA digestion with amplification-grade DNase I (Thermo Fisher Scientific). Library construction and sequencing were performed by Novogene. For the preparation of non-directional libraries, mRNA was purified from total RNA using oligo-dT-attached magnetic beads. After fragmentation, the first-strand cDNA was synthesized using random hexamer primers, followed by the second-strand cDNA synthesis, end repair, A-tailing, adapter ligation, size selection, amplification and purification. The libraries were sequenced as paired-end reads of 150 bp on NovaSeq Illumina Platforms.
WGBS
The WGBS experiments were performed with two biological replicates. Genomic DNA was isolated from 100 mg (fresh weight) of leaf tissue pooled from three or four plants per sample using the DNeasy Plant Mini Kit (Qiagen), except for the comparison of DNA methylation under SD and LD conditions, where 14-day-old seedlings grown on Phyto agar (RPI Corp.) with half-strength MS basal salt mixture (MP Biomedicals) were used. The isolated DNA was sheared into 200–400-bp fragments via ultrasonication (Covaris S2/S220/E220). WGBS of the wild type, mthfd1-1 and thfs single mutants and mt double mutants was performed by BGI TECH Solutions, proceeding with end repair of the sheared DNA, deoxyadenylation and ligation of methylated sequencing adaptors, followed by bisulfite conversion using the EZ DNA Methylation-Gold kit (ZYMO Research), size selection, PCR amplification and final size selection of libraries. Libraries for WGBS of seedlings grown under SD and LD conditions and CRISPR–Cas9 lines were prepared the same way, except using the KAPA HyperPrep Kit (Roche), TruSeq DNA LT (Illumina)/NEBNext Multiplex Oligos (New England Biolabs) methylated adaptors and the EpiTect Bisulfite Kit (Qiagen), and sequenced at Novogene. WGBS of wild-type and mthfd1-1 plants grown under cCO2 and hCO2 was performed by Novogene using post-bisulfite adaptor tagging with the Accel-NGS Methyl-Seq DNA Library Kit for Illumina (Swift Biosciences). All libraries were sequenced on Illumina platforms (HiSeq 2000/X-Ten, NovaSeq 6000/X Plus) as paired-end reads of 150 bp, except for libraries from seedlings grown under SD and LD, which were sequenced as single-end 50-bp reads.
Isotopic labelling
Seeds of wild-type, mthfd1-1, thfs and mthfd1-1 thfs plants were surface-sterilized with 40% (v/v) commercial bleach at 1,500 rpm and 22 °C for 10 min in a thermoblock, followed by six washes with sterile water. Sterilized seeds were stratified for 2 days at 4 °C and subsequently grown under SD or LD conditions on a shaker at 100 rpm. Approximately 30 seeds were sown per well in six-well plates containing 2.5 ml of half-strength MS medium (Duchefa Biochemie) supplemented with 0.5% sucrose and adjusted to pH 5.7 using KOH.
After 9 days of culture, seedlings had established upright growth, and the medium was replaced according to the labelling treatment. For short-term stable isotope tracing in free amino acids, seedlings were transferred to half-strength MS medium without sucrose. For long-term stable isotope tracing in nucleobases, growth media were supplemented with 1 mM [13C]formate. Control samples received 1 mM unlabelled formate to account for background levels of naturally occurring isotopes. For long-term labelling, both labelled and control media were refreshed after 11 and 13 days of culture. After 15 days of culture under SD conditions, samples were harvested at 6 h after dawn. The seedlings were rinsed thoroughly with deionized water to remove residual medium and carefully patted dry, the roots were excised, and the shoots were harvested and immediately flash-frozen in liquid nitrogen. Long-term labelling was carried out under SD conditions to avoid the observed growth constraints under LD conditions in mthfd1-1.
For short-term isotope tracing in free amino acids, half-strength MS medium was replaced after 13 days of culture under LD with labelled (1 mM [13C]formate or 1 mM [2-13C]glycine) or control (1 mM unlabelled formate or 1 mM unlabelled glycine) media. To assess label incorporation during the day and night, media were exchanged at the onset of the light or dark period, respectively. Samples were collected after 4, 8 and 16 h of incubation as described above. Shoot tissues were ground in liquid nitrogen using a bead mill (Retsch TissueLyser, Qiagen).
We used 100 mg of pulverized plant material per sample for genomic DNA isolation. DNA was dissolved in ultrapure water using ultrasonication for 10 min, transferred to 2-ml amber glass vials with 11-mm crimp tops (Thermo Fisher Scientific) and dried under a constant stream of nitrogen gas for 30 min. For DNA hydrolysis into nucleobases, 100 μl of neat formic acid were added to the sample. The vials were briefly flushed with nitrogen gas and wiped with lint-free Kimwipes (Kimtech) before being sealed with 11-mm crimp caps with PTFE/silicone septa (Agilent). The samples were incubated at 150 °C for 6 h and subsequently dried under a constant stream of nitrogen gas. For nucleobase derivatization, 50 μl of acetonitrile and 50 μl of N,O-bis-(trimethylsilyl)-trifluoracetamid (BSTFA) containing 1% trimethylchlorosilane (TMCS) were added to each sample. The vials were again flushed with nitrogen gas, wiped with Kimwipes and tightly sealed with crimp caps (Agilent). The samples were briefly mixed and incubated at 150 °C for 60 min. The derivatized nucleobases were then transferred to 250-μl glass inserts (Merck) using glass pipettes and placed back into 2-ml amber glass vials.
For the isolation of free amino acids, 100 mg of pulverized plant material was incubated in 1 ml of 1 M HCl at 60 °C and 500 rpm for 30 min in a thermoblock. After centrifugation at 10,621 g and 4 °C for 15 min, 800 μl of the supernatant was transferred to a new reaction tube. The centrifugation step was repeated, and 250 μl of the clear supernatant was added to 2-ml amber vials and dried under a constant stream of nitrogen gas for 60 min. Free amino acids were derivatized as described above, except that BSTFA was replaced with N-tert-butyldimethylsilyl-N-methyltrifluoroacetamide containing 1% TMCS. Derivatization was performed for 90 min at 60 °C.
Stable isotope enrichment in derivatized nucleobases and free amino acids was determined using thermal desorption–gas chromatography–mass spectrometry (thermal desorption unit, Gerstel; gas chromatograph: 7890A; mass spectrometer: 5975C, both Agilent Technologies). 1 μl of each sample was injected into the thermal desorption unit and vaporized by increasing the temperature from 40 °C to 280 °C at a rate of 360 °C min−1, with a hold for 2.5 min. Vaporized compounds were cryofocused using a cooled injection system (Gerstel) at −50 °C and subsequently desorbed in splitless mode to 280 °C at 6 °C s−1, holding for 2.5 min. Compounds were separated on a J&W 122-5562G_1- DB-5MS + 10m DG column (Agilent Technologies, 70 m × 250 μm × 0.25 μm) using helium as a carrier gas at a constant flow of 1 ml min−1. Separation of compounds lasted for 31.5 min and started at 90 °C followed by a ramp-up to 150 °C at a rate of 30 °C min−1. Afterwards, the temperature rose to 260 °C at a rate of 5 °C min−1 and subsequently to 310 °C at 100 °C min−1 with a final hold of 7 min. Target compounds were unambiguously identified by mass spectra and retention time comparison with pure standards (Extended Data Fig. 10).
Mass spectral data of tert-butyldimethylsilyl- and trimethylsilyl-derivatized compounds were acquired from total ion chromatogram and selected ion monitoring modes. To obtain label enrichment in target compounds, the selected ion monitoring parameters were set in groups with mass-to-charge (m/z) ratios as follows for amino acids and nucleobases: glycine: start time, 12.1 min; ions, M + 0: m/z 246.0, M + 1: m/z 247.0, M + 2: m/z 248.0; dwell time each, 10 ms, methionine: start time, 19.1 min; ions, M + 0: m/z 320.0, M + 1: m/z 321.0, M + 2: m/z 322.0; dwell time each, 5 ms, serine: start time, 19.9 min; ions, M + 0: m/z 390.0, M + 1: m/z 391.0, M + 2: m/z 392.0; dwell time each, 10 ms; 5-methylcytosine: start time, 11.78 min; ions, M + 0: m/z 254.0, M + 1: m/z 255.0, M + 2: m/z 256.0; dwell time each, 5 ms, adenine: start time, 17.3 min; ions, M + 0: m/z 264.0, M + 1: m/z 265.0, M + 2: m/z 266.0; dwell time each, 10 ms, thymine: start time, 8.5 min; ions, M + 0: m/z 255.0, M + 1: m/z 256.0, M + 2: m/z 257.0; dwell time each, 5 ms. Guanine was not quantified due to weak, tailing peaks indicative of adsorption or degradation under hydrolysis/derivatization conditions.
M + 0, M + 1 and M + 2 represent the ions with zero, one and two heavy isotopes (for example, 13C), respectively, and were used to define the isotopic composition of each target compound. Isotopologue fractions (k = 0, 1, 2) were calculated as percentages of the sum of the different isotopologue abundances; naturally occurring isotope abundances were subsequently removed by subtraction of mean isotopologue fractions obtained from labelling controls supplied with the same concentrations of non-labelled substrates (n, biological replicates):
Enzymatic activity analyses
Spectrophotometric MTHFD and THFS activity measurements were adapted from previous methods57. The liquid chromatography–mass spectrometry-based MTHFR activity assay was adapted from methods previously described58. Details of the protocols are available in Supplementary Information.
Thiol, amino acid and adenosine nucleotide quantification
Amino acids, thiol compounds and adenosines were analysed as described in ref. 59, ref. 60 and ref. 61, respectively. A complete description of the method is included in Supplementary Information.
Folate quantification
Folate quantification was performed as described previously62 and is outlined in detail in Supplementary Information.
mRNA-seq data analysis
mRNA-seq reads were processed using the nf-core/rnaseq pipeline (v.3.10.1)63 in Nextflow (v.22.10.6) executed using singularity containers on the high-performance computing cluster at Helmholtz Munich; the default parameters were used for read trimming/filtering, alignment and quantification with Trim Galore! (v.0.6.7)64, STAR (v.2.6.1d)65 and Salmon (1.9.0)66, respectively, except as stated below. TEs and genes were analysed separately. For TEs, reads were aligned to the TAIR10 reference genome using a transcript-based TE annotation67 and the following extra STAR alignment arguments: outMultimapperOrder, Random; outFilterMultimapNmax, 50; alignIntronMax, 10,000; alignSJoverhangMin, 3. For genes, Araport11 gene models and the following extra STAR alignment arguments were used: outFilterMultimapNmax, 50; alignIntronMax, 10,000; alignSJoverhangMin, 3. The read numbers are summarized in Supplementary Table 1.
Transcript abundance estimates from Salmon were used to generate TE/gene-level count matrices for visualization and differential expression analysis with DESeq2 (v.1.38.3)68 in R (v.4.2.2)69. TEs/genes with less than six counts across all samples were removed. Principal component analysis plots were generated after variance stabilizing transformation using within-group variability. Log fold changes were adjusted using the ashr option for effect size shrinkage. TEs/genes with FDR ≤ 0.05 and absolute fold change value ≥2 were counted as differentially expressed. Araport11 gene annotations including gene names were retrieved from Ensembl Plants with the R package biomaRt (v.2.54.1)70. TE annotations including (sub-)family names and genome positions of start and stop sites were imported from the annotated bed file in ref. 67. Volcano plots were generated with the R package EnhancedVolcano (v.1.16.0)71. Overlaps between differentially expressed TEs/genes from different comparisons were determined on the basis of TE/gene identifier (TAIR ID) and visualized with the R package VennDetail (v.1.14.0)72. Heat maps (Figs. 1e and 4e) were generated using the R package ComplexHeatmap (v.2.14.0)73. Transcripts per million (TPM) values were obtained from the Salmon output files. Heat map rows were hierarchically clustered by Euclidean distance using Ward clustering. For this, mean TPM values across replicates were log2-transformed after adding 1 pseudo count and subsequently standardized (z score) across groups. Groups were clustered by Euclidean distance using complete linkage clustering. For visualization of co-expression, Pearson correlation coefficients were calculated from z scores and Ward clustered by Euclidean distance. Read coverage at SDC was visualized with IGV (v.2.6.1)74 after averaging bigWig coverage files from the nf-core/rnaseq pipeline across replicates at single base bin size using deepTools (v.3.5.1)75.
WGBS data analysis
The raw reads were pre-processed with Trimmomatic (v.0.39)76 using the parameters shown in Supplementary Table 7. Pre-processed reads were mapped to TAIR10 using Bismark (v.0.23.1)77 with parameters N set to 1 (allowing one mismatch in seed alignments) and X (the maximum insert size for valid paired-end alignments) set to 700. Duplicated reads and reads flagged as unconverted due to three or more consecutive methylated CHH sites were discarded. Methylation ratios were calculated separately for CG, CHG and CHH sites as no. of C/(no. of C + no. of T). Non-conversion ratios were calculated from chloroplast cytosine methylation ratios. The read numbers and non-conversion ratios are summarized in Supplementary Table 1. Genome browser tracks showing CG, CHG and CHH methylation ratios at single-cytosine resolution were generated from Bismark genome-wide cytosine methylation output files for cytosine sites with at least four reads.
Bismark genome-wide cytosine methylation output files were imported into the R package methylKit (v.1.20.0)78 for further analysis. Cytosine sites with less than four reads were excluded. Chromosomal methylation plots were generated by summarizing the numbers of C and T in 10-kb bins with 10 or more covered cytosines in each of the samples across the genome and subsequently calculating CG, CHG and CHH methylation ratios from pooled replicates. Metaplots were generated by dividing each TE/gene from the TAIR10 genome release into 20 bins of equal size between start and stop sites and 100-bp bins in 1-kb flanking regions and averaging weighted methylation levels79 calculated from pooled replicates for each bin across all TEs/genes. Principal component analysis plots were generated from methylation ratios in 100-bp bins with four or more covered cytosines in each sample across the genome (100-bp tiles). DMRs were called from 100-bp tiles with FDR ≤ 0.01 from a logistic regression test with SLIM adjusted P values and absolute methylation differences of at least 0.4, 0.2 and 0.1 for CG, CHG and CHH sequence contexts, respectively. Overlaps between DMRs from different pairwise comparisons were determined and visualized with Intervene (v.0.6.4)80. DMRs from all pairwise comparisons to the control group were combined, and subsequently all DMRs that were separated by 100 bp or less were merged for cluster analysis and heat map visualization using the R package pheatmap (v.1.0.12)81. CG methylation ratios in merged DMRs were calculated from the numbers of C and T after pooling replicates. DMR CG methylation ratios were subsequently standardized (z score) across groups and hierarchically clustered by Euclidean distance using Ward clustering. Groups were clustered by Euclidean distance from CG methylation ratios using complete linkage clustering.
DNA methylation is maintained by different pathways, including the CMT2 pathway, which acts on pericentromeric heterochromatin, and RdDM, which targets repetitive sequences in chromosome arms82. In addition, mCG in coding sequences of expressed genes constitutes gene body methylation82. Overlap percentages of clustered DMRs with defined methylated genomic regions were determined using the function annotateWithFeatures from the R package genomation (v.1.30.0)83. Methylation hypervariable sites (HV) were defined as in ref. 84, except that the HV regions, which were retrieved from Gene Expression Omnibus (GEO) series GSE165095, sample GSM5026060, were merged with 249 spontaneous DMRs identified in ref. 85. gbM regions were defined by the genome coordinates of gbM genes identified in ref. 86. MET1 regions correspond to met1 CG hypo-DMRs identified in ref. 82 that were retrieved from GEO series GSE39901. Regions methylated by the RdDM pathway were defined as RNA polymerase V target sites identified in ref. 87, and the genome coordinates were retrieved from GEO series GSE100010. CMT2 regions representing constitutive heterochromatin were defined as cmt2 CHH hypo-DMRs identified in ref. 82, and the genome coordinates were retrieved from GEO series GSE39901. CG hypo-DMRs from mthfd1-1 mutants, as previously identified19, that did not overlap with HV, gbM, RdDM or CMT2 regions were defined as Other m. Bins separated by 100 bp or less within each of the defined methylated genomic regions were merged. TAIR10 genomic regions that did not overlap with any of the defined methylated genomic regions were called Rest. The precedence of the defined genomic regions in case of multiple overlaps was HV > gbM > RdDM > CMT2 > Other m > Rest. Genome coordinates of Arabidopsis chromatin states (CS1–36) were retrieved from the Plant Chromatin State Database88. Overlaps between clustered DMRs and each of the CS1–36 were quantified using the function countOverlaps from the R package GenomicRanges (v.1.50.2)89 and visualized with pheatmap81.
To plot correlations between DNA methylation and TE expression in different expression clusters, each differentially expressed TE was divided into four bins of equal size between start and stop sites and 250-bp or 500-bp windows in 1-kb flanking regions to generate a matrix of weighted methylation levels79 from pooled replicates for each bin. Subsequently, the methylation levels were rank-compared to log-transformed TPM values (see ‘mRNA-seq data analysis’) to calculate Spearman correlation coefficients in R.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The primary high-throughput sequencing data generated in this study have been deposited in GEO under accession codes GSE292915 and GSE292917. The isotopic labelling data are available via MetaboLights90 under the study MTBLS13444. Source data are provided with this paper. Computational source data for genomic analyses and visualization are also available via Zenodo at https://doi.org/10.5281/zenodo.18135710 (ref. 91).
Code availability
No new algorithms were developed in this study. Reproducible code for genomic analyses and plotting is available via GitHub (release v.1.0.0) and via Zenodo at https://doi.org/10.5281/zenodo.18137716 (ref. 92).
References
Prywes, N., Phillips, N. R., Tuck, O. T., Valentin-Alvarado, L. E. & Savage, D. F. Rubisco function, evolution, and engineering. Annu. Rev. Biochem. 92, 385–410 (2023).
Busch, F. A. Photorespiration in the context of Rubisco biochemistry, CO2 diffusion and metabolism. Plant J. 101, 919–939 (2020).
Walker, B. J., VanLoocke, A., Bernacchi, C. J. & Ort, D. R. The costs of photorespiration to food production now and in the future. Annu. Rev. Plant Biol. 67, 107–129 (2016).
Gashu, K., Kaste, J. A. M., Roje, S. & Walker, B. J. Metabolic flux analysis in leaf metabolism quantifies the link between photorespiration and one carbon metabolism. Nat. Plants 11, 1877–1889 (2025).
Hanson, A. D. & Roje, S. One-carbon metabolism in higher plants. Annu. Rev. Plant Physiol. Plant Mol. Biol. 52, 119–137 (2001).
Oliver, D. J. The glycine decarboxylase complex from plant mitochondria. Annu. Rev. Plant Biol. 45, 323–337 (1994).
Fu, X., Gregory, L. M., Weise, S. E. & Walker, B. J. Integrated flux and pool size analysis in plant central metabolism reveals unique roles of glycine and serine during photorespiration. Nat. Plants 9, 169–178 (2022).
Mouillon, J. M. et al. Glycine and serine catabolism in non-photosynthetic higher plant cells: their role in C1 metabolism. Plant J. 20, 197–205 (1999).
Prabhu, V., Chatson, K. B., Abrams, G. D. & King, J. 13C nuclear magnetic resonance detection of interactions of serine hydroxymethyltransferase with C1-tetrahydrofolate synthase and glycine decarboxylase complex activities in Arabidopsis. Plant Physiol. 112, 207–216 (1996).
Collakova, E. et al. Arabidopsis 10-formyl tetrahydrofolate deformylases are essential for photorespiration. Plant Cell 20, 1818–1832 (2008).
Li, R., Moore, M. & King, J. Investigating the regulation of one-carbon metabolism in Arabidopsis thaliana. Plant Cell Physiol. 44, 233–241 (2003).
Saeheng, S. et al. Formate-tetrahydrofolate ligase: supplying the cytosolic one-carbon network in roots with one-carbon units originating from glycolate. Plant J. 119, 2464–2483 (2024).
Roje, S. et al. Isolation, characterization, and functional expression of cDNAs encoding NADH-dependent methylenetetrahydrofolate reductase from higher plants. J. Biol. Chem. 274, 36089–36096 (1999).
Gorelova, V. et al. Dihydrofolate reductase/thymidylate synthase fine-tunes the folate status and controls redox homeostasis in plants. Plant Cell 29, 2831–2853 (2017).
Kirk, C. D., Chen, L., Imeson, H. C. & Cossins, E. A. A 5,10-methylenetetrahydrofolate dehydrogenase: 5,10-methenyltetrahydrofolate cyclohydrolase protein from Pisum sativum. Phytochemistry 39, 1309–1317 (1995).
Nogués, I. et al. Arabidopsis thaliana serine hydroxymethyltransferases: functions, structures, and perspectives. Plant Physiol. Biochem. 187, 37–49 (2022).
Christensen, K. E. & MacKenzie, R. E. Mitochondrial one-carbon metabolism is adapted to the specific needs of yeast, plants and mammals. Bioessays 28, 595–605 (2006).
Rosa-Téllez, S. et al. The serine–glycine–one-carbon metabolic network orchestrates changes in nitrogen and sulfur metabolism and shapes plant development. Plant Cell 36, 404–426 (2024).
Groth, M. et al. MTHFD1 controls DNA methylation in Arabidopsis. Nat. Commun. 7, 11640 (2016).
Xie, G., Du, X., Hu, H. & Du, J. Molecular mechanisms underlying the establishment, maintenance, and removal of DNA methylation in plants. Annu. Rev. Plant Biol. 76, 143–170 (2025).
Rocha, P. S. et al. The Arabidopsis HOMOLOGY-DEPENDENT GENE SILENCING1 gene codes for an S-adenosyl-l-homocysteine hydrolase required for DNA methylation-dependent gene silencing. Plant Cell 17, 404–417 (2005).
Law, J. A. & Jacobsen, S. E. Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat. Rev. Genet. 11, 204–220 (2010).
Yao, N. et al. An evolutionary epigenetic clock in plants. Science 381, 1440–1445 (2023).
Song, X. et al. Inheritance of acquired adaptive cold tolerance in rice through DNA methylation. Cell 188, 4213–4224.e12 (2025).
Kawakatsu, T. et al. Epigenomic diversity in a global collection of Arabidopsis thaliana accessions. Cell 166, 492–505 (2016).
Zheng, X. et al. Transgenerational epimutations induced by multi-generation drought imposition mediate rice plant’s adaptation to drought condition. Sci. Rep. 7, 39843 (2017).
Van Dooren, T. J. M. et al. Mild drought in the vegetative stage induces phenotypic, gene expression, and DNA methylation plasticity in Arabidopsis but no transgenerational effects. J. Exp. Bot. 71, 3588–3602 (2020).
Wibowo, A. et al. Hyperosmotic stress memory in Arabidopsis is mediated by distinct epigenetically labile sites in the genome and is restricted in the male germline by DNA glycosylase activity. eLife 5, e13546 (2016).
Sasaki, T. et al. Arms race between anti-silencing and RdDM in noncoding regions of transposable elements. EMBO Rep. 24, e56678 (2023).
Wang, Q., Liu, W., Leung, C. C., Tarté, D. A. & Gendron, J. M. Plants distinguish different photoperiods to independently control seasonal flowering and growth. Science 383, eadg9196 (2024).
Misselbeck, K., Marchetti, L., Priami, C., Stover, P. J. & Field, M. S. The 5-formyltetrahydrofolate futile cycle reduces pathway stochasticity in an extended hybrid-stochastic model of folate-mediated one-carbon metabolism. Sci. Rep. 9, 4322 (2019).
Goyer, A. et al. 5-Formyltetrahydrofolate is an inhibitory but well tolerated metabolite in Arabidopsis leaves. J. Biol. Chem. 280, 26137–26142 (2005).
Li, W. et al. The 5-formyl-tetrahydrofolate proteome links folates with C/N metabolism and reveals feedback regulation of folate biosynthesis. Plant Cell 33, 3367–3385 (2021).
Okamura, E. & Hirai, M. Y. Novel regulatory mechanism of serine biosynthesis associated with 3-phosphoglycerate dehydrogenase in Arabidopsis thaliana. Sci. Rep. 7, 3533 (2017).
Stover, P. & Schirch, V. Serine hydroxymethyltransferase catalyzes the hydrolysis of 5,10-methenyltetrahydrofolate to 5-formyltetrahydrofolate. J. Biol. Chem. 265, 14227–14233 (1990).
Roje, S., Janave, M. T., Ziemak, M. J. & Hanson, A. D. Cloning and characterization of mitochondrial 5-formyltetrahydrofolate cycloligase from higher plants. J. Biol. Chem. 277, 42748–42754 (2002).
Pawelek, P. D., Allaire, M., Cygler, M. & MacKenzie, R. E. Channeling efficiency in the bifunctional methylenetetrahydrofolate dehydrogenase/cyclohydrolase domain: the effects of site-directed mutagenesis of NADP binding residues. Biochim. Biophys. Acta 1479, 59–68 (2000).
Field, M. S., Kamynina, E., Watkins, D., Rosenblatt, D. S. & Stover, P. J. Human mutations in methylenetetrahydrofolate dehydrogenase 1 impair nuclear de novo thymidylate biosynthesis. Proc. Natl Acad. Sci. USA 112, 400–405 (2015).
Zrenner, R., Stitt, M., Sonnewald, U. & Boldt, R. Pyrimidine and purine biosynthesis and degradation in plants. Annu. Rev. Plant Biol. 57, 805–836 (2006).
Grodzinski, B. A study of formate production and oxidation in leaf peroxisomes during photorespiration. Plant Physiol. 63, 289–293 (1979).
Queval, G. et al. Conditional oxidative stress responses in the Arabidopsis photorespiratory mutant cat2 demonstrate that redox state is a key modulator of daylength-dependent gene expression, and define photoperiod as a crucial factor in the regulation of H2O2-induced cell death. Plant J. 52, 640–657 (2007).
Voll, L. M. et al. The photorespiratory Arabidopsis shm1 mutant is deficient in SHM1. Plant Physiol. 140, 59–66 (2006).
Gout, E. et al. Metabolism of methanol in plant cells: carbon-13 nuclear magnetic resonance studies. Plant Physiol. 123, 287–296 (2000).
Wingler, A., Lea, P. J. & Leegood, R. C. Photorespiratory metabolism of glyoxylate and formate in glycine-accumulating mutants of barley and Amaranthus edulis. Planta 207, 518–526 (1999).
Ducker, G. S. et al. Reversal of cytosolic one-carbon flux compensates for loss of the mitochondrial folate pathway. Cell Metab. 23, 1140–1153 (2016).
Jiang, C. et al. Environmentally responsive genome-wide accumulation of de novo Arabidopsis thaliana mutations and epimutations. Genome Res. 24, 1821–1829 (2014).
Panda, K. et al. The plant response to high CO2 levels is heritable and orchestrated by DNA methylation. New Phytol. 238, 2427–2439 (2023).
Pendle, A. F. et al. Proteomic analysis of the Arabidopsis nucleolus suggests novel nucleolar functions. Mol. Biol. Cell 16, 260–269 (2005).
Hazarika, R. R. et al. Molecular properties of epimutation hotspots. Nat. Plants 8, 146–156 (2022).
Catoni, M. et al. DNA sequence properties that predict susceptibility to epiallelic switching. EMBO J. 36, 617–628 (2017).
Dubin, M. J. et al. DNA methylation in Arabidopsis has a genetic basis and shows evidence of local adaptation. eLife 4, e05255 (2015).
Zhang, Z., Zhu, G. & Peng, X. Photorespiration in plant adaptation to environmental changes. Crop Environ. 3, 203–212 (2024).
Amir, R. Current understanding of the factors regulating methionine content in vegetative tissues of higher plants. Amino Acids 39, 917–931 (2010).
Smith, E. N., van Aalst, M., Weber, A. P. M., Ebenhöh, O. & Heinemann, M. Alternatives to photorespiration: a system-level analysis reveals mechanisms of enhanced plant productivity. Sci. Adv. 11, eadt9287 (2025).
Ghirardo, A. et al. Origin of volatile organic compound emissions from subarctic tundra under global warming. Glob. Change Biol. 26, 1908–1925 (2020).
Régnier, P., Montardi, C., Maciejewski-Duval, A., Marques, C. & Saadoun, D. PUPAID: a R + ImageJ pipeline for thorough and semi-automated processing and analysis of multi-channel immunofluorescence data. PLoS ONE 19, e0308970 (2024).
Vickers, T. J., Murta, S. M. F., Mandell, M. A. & Beverley, S. M. The enzymes of the 10-formyl-tetrahydrofolate synthetic pathway are found exclusively in the cytosol of the trypanosomatid parasite Leishmania major. Mol. Biochem. Parasitol. 166, 142–152 (2009).
Zhang, Y., Sun, K. & Roje, S. An HPLC-based fluorometric assay for serine hydroxymethyltransferase. Anal. Biochem. 375, 367–369 (2008).
Weger, B. D. et al. Extensive regulation of diurnal transcription and metabolism by glucocorticoids. PLoS Genet. 12, e1006512 (2016).
Wirtz, M., Droux, M. & Hell, R. O-Acetylserine (thiol) lyase: an enigmatic enzyme of plant cysteine biosynthesis revisited in Arabidopsis thaliana. J. Exp. Bot. 55, 1785–1798 (2004).
Burstenbinder, K., Rzewuski, G., Wirtz, M., Hell, R. & Sauter, M. The role of methionine recycling for ethylene synthesis in Arabidopsis. Plant J. 49, 238–249 (2007).
Obermaier, L. et al. An improved folate stable isotope dilution assay of unexploited food sources from Brazil. Front. Nutr. 10, 1252497 (2023).
Patel, H. et al. nf-core/rnaseq: nf-core/rnaseq v3.18.0 - Lithium Lynx. Zenodo https://doi.org/10.5281/zenodo.14537300 (2024).
Krueger, F. FelixKrueger/TrimGalore: v0.6.7 - DOI via Zenodo. Zenodo https://doi.org/10.5281/zenodo.5127899 (2021).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Patro, R., Duggal, G., Love, M. I., Irizarry, R. A. & Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 14, 417–419 (2017).
Panda, K. & Slotkin, R. K. Long-read cDNA sequencing enables a ‘gene-like’ transcript annotation of transposable elements. Plant Cell 32, 2687–2698 (2020).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
R Core Team. R: a language and environment for statistical computing (R Foundation for Statistical Computing, 2022).
Durinck, S., Spellman, P. T., Birney, E. & Huber, W. Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat. Protoc. 4, 1184–1191 (2009).
Blighe, K., Rana, S. & Lewis, M. EnhancedVolcano: publication-ready volcano plots with enhanced colouring and labeling (Bioconductor, 2022).
Guo, K. & McGregor, B. VennDetail: a package for visualization and extract details. R package version 1.14.0 (Bioconductor, 2022).
Gu, Z., Eils, R. & Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 32, 2847–2849 (2016).
Robinson, J. T. et al. Integrative Genomics Viewer. Nat. Biotechnol. 29, 24–26 (2011).
Ramírez, F. et al. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 44, W160–W165 (2016).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Krueger, F. & Andrews, S. R. Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 27, 1571–1572 (2011).
Akalin, A. et al. methylKit: a comprehensive R package for the analysis of genome-wide DNA methylation profiles. Genome Biol. 13, R87 (2012).
Schultz, M. D., Schmitz, R. J. & Ecker, J. R. ‘Leveling’ the playing field for analyses of single-base resolution DNA methylomes. Trends Genet. 28, 583–585 (2012).
Khan, A. & Mathelier, A. Intervene: a tool for intersection and visualization of multiple gene or genomic region sets. BMC Bioinform. 18, 287 (2017).
Kolde, R. pheatmap: Pretty Heatmaps. R package version 1.0.12 (CRAN, 2019).
Stroud, H., Greenberg, M. V., Feng, S., Bernatavichute, Y. V. & Jacobsen, S. E. Comprehensive analysis of silencing mutants reveals complex regulation of the Arabidopsis methylome. Cell 152, 352–364 (2013).
Akalin, A., Franke, V., Vlahoviček, K., Mason, C. E. & Schübeler, D. genomation: a toolkit to summarize, annotate and visualize genomic intervals. Bioinformatics 31, 1127–1129 (2015).
Ichino, L. et al. MBD5 and MBD6 couple DNA methylation to gene silencing through the J-domain protein SILENZIO. Science 372, 1434–1439 (2021).
Becker, C. et al. Spontaneous epigenetic variation in the Arabidopsis thaliana methylome. Nature 480, 245–249 (2011).
Williams, C. J., Dai, D., Tran, K. A., Monroe, J. G. & Williams, B. P. Dynamic DNA methylation turnover in gene bodies is associated with enhanced gene expression plasticity in plants. Genome Biol. 24, 227 (2023).
Liu, W. et al. RNA-directed DNA methylation involves co-transcriptional small-RNA-guided slicing of polymerase V transcripts in Arabidopsis. Nat. Plants 4, 181–188 (2018).
Liu, Y. et al. PCSD: a plant chromatin state database. Nucleic Acids Res. 46, D1157–D1167 (2018).
Lawrence, M. et al. Software for computing and annotating genomic ranges. PLoS Comput. Biol. 9, e1003118 (2013).
Yurekten, O. et al. MetaboLights: open data repository for metabolomics. Nucleic Acids Res. 52, D640–D646 (2024).
Groth, M. Computational source data for ‘Photorespiration is linked to DNA methylation by formate as a one-carbon source’. Zenodo https://doi.org/10.5281/zenodo.18135710 (2026).
Groth, M. Photorespiration–DNA methylation: code snapshot (v1.0.0). Zenodo https://doi.org/10.5281/zenodo.18137716 (2026).
Acknowledgements
We thank G. Barthel for plant husbandry and technical support, B. Weber for operating the gas chromatograph–mass spectrometer, F. Antritter for fumigation monitoring, and B. Lange and A. Schäffner for operating the liquid chromatograph–mass spectrometer and for help with the evaluation of mass spectra. We also thank M.-E. Torres-Padilla, R. Schneider and P. Wigge for helpful comments on earlier versions of the paper. This work was supported by the H2020-MSCA individual fellowship no. 798235 to M.G. Work in the labs of M.W. and R.H. at Heidelberg University was funded by the German Research Foundation (project IDs 235736350 and 544882710). Work at the Metabolomics Core Technology Platform was supported by the Excellence Cluster ‘CellNetworks’ (University of Heidelberg, grant no. ZUK 40/2010-3009262).
Funding
Open access funding provided by Helmholtz Zentrum München - Deutsches Forschungszentrum für Gesundheit und Umwelt (GmbH).
Author information
Authors and Affiliations
Contributions
M.G. conceived the study. M.G., M.W., M.R., A.G. and V.H. designed the research. V.H., J.S.K., I.G., L.O., A.G., G.P., M.W. and M.G. performed the experiments and analysed the data. V.H. and A.G. performed the isotopic labelling analysis. A.G. performed the fumigation experiments. L.O. measured folates. G.P. and M.W. measured thiols, amino acids and adenosine nucleotides. V.H. and J.S.K. performed the enzyme activity assays. I.G. cloned the CRISPR–Cas9 plasmids. M.G. performed library preparations for mRNA-seq and WGBS. M.G. performed the bioinformatic analysis. J.-P.S., M.R. and R.H. supervised the mass spectrometry and metabolic analyses. J.D. and M.G. supervised the study. M.G. and V.H. wrote the paper. All authors contributed to editing and revising the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Plants thanks Rocío Díaz de la Garza, Christine Foyer and Stefan Timm for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Mutation of THFS suppresses the mthfd1-1 phenotype.
a, Gene structures of Arabidopsis thaliana THFS and MTHFD1 are indicated along with positions of T-DNA insertions in thfs (SALK_067510) and mthfd1-3 (SALK_015165), the point mutation causing a R175Q substitution in mthfd1-1, and RT-PCR primers flanking the T-DNA insertions in thfs. b, RT-PCR analysis of THFS expression in rosette leaves of wild type and thfs (2 replicates each). Arrow indicates expected amplicon size in wild type; no wild-type transcripts were detected in thfs. L, ladder. c, Representative maximum projections of nuclear SDCpro-GFP expression acquired by confocal laser scanning microscopy (CLSM) z stacks from the ventral side of leaves of 3-week-old mthfd1-1 (m), mthfd1-1 thfs double mutant (mt), thfs (t), and wild type (WT) plants grown under LD. Scale bars, 50 μm. d, Representative images of 5-week-old plants grown under LD. Number of leaves when first flowers emerged. Box plots represent median (centre lines), 25th (bottom) and 75th (top) percentiles, and minimum and maximum points within 1.5 x interquartile range (IQR) (n = 8). Lowercase letters indicate significant differences between genotypes (P < 0.05, one-way ANOVA followed by post-hoc Tukey test). See Supplementary Table 9 for P values. e, Root lengths of 2-week-old plants grown under LD. Box plots indicate medians (centre lines), IQR (boxes), and 1.5×IQR (whiskers). Lowercase letters indicate significant differences between genotypes (n = 15 for m and mt, n = 14 for t and wt, P < 0.05, one-way ANOVA followed by post-hoc Tukey test). See Supplementary Table 10 for P values.
Extended Data Fig. 2 Phenotypes of MTHFD1 mutant alleles are suppressed by T-DNA insertion and CRISPR-Cas9 mutant alleles of THFS.
a, Images of 10-day-old mthfd1-1 CRISPR-Cas9 control (mT-cr23; no deletion in THFS detected by Sanger sequencing) and mthfd1-1 THFS knock out CRISPR-Cas9 (mtΔ-cr20; inversion between sgRNA111 and sgRNA243 detected by Sanger sequencing) T2 seedlings grown under LD. b, Agarose gel images of PCR genotyping for 8 T2 individuals per line (mT-cr23 and mtΔ-cr20). sgRNA target sites, genotyping primers, and expected amplicon sizes are indicated at right. c, Representative maximum projections of nuclear SDCpro-GFP expression acquired by CLSM z stacks from the ventral side of leaves of 3-week-old plants grown under LD. Scale bars, 50 μm. d, Corrected total cell fluorescence (CTCF) of SDCpro-GFP expression quantified from maximum projections. Box plots indicate medians (centre lines), IQR (boxes), and 1.5×IQR (whiskers). Indicated P value is from a two-sided Welch’s t-test (n = 10). e, Mean DNA methylation ratios over genes and TEs and the respective flanking regions in different sequence contexts. Two replicates (#1, #2) per genotype were analysed by whole genome bisulfite sequencing (WGBS). f, Representative pictures of 3-week-old mthfd1-3/mthfd1-3;THFS/thfs (mmTt) and mthfd1-3 thfs (mmtt) F3 plants grown under LD, leaf area quantification from automated phenotyping for mmTt (n = 4) and mmtt (n = 7), and relative DNA methylation levels in rosette leaves from mmTt, mmtt (n = 5) and Col-0 (n = 2) analysed by McrBC-qPCR at locus MG5. Data are presented as mean values +/- s.d.; indicated P value is from a two-sided Welch’s t-test.
Extended Data Fig. 3 Most mthfd1-1 DMRs that are fully suppressed by thfs are located in heterochromatic TE-associated chromatin regions while partially suppressed mthfd1-1 DMRs tend to be located in active gene body-associated chromatin regions.
a, Mean mCG, mCHG, and mCHH ratios in 10 kb windows along chromosomes (Chr) 1 to 5, and signed difference relative to wild type (Δ = mutant - WT). b, Principal component analysis (PCA) of whole-genome bisulfite sequencing (WGBS) data from different genotypes. DNA methylation ratios in CG (left), CHG (middle), and CHH (right panel) were calculated in 100 bp bins across the Arabidopsis genome. Each point represents an individual sample; mthfd1-1 is clearly separated from the other groups along the first principal component. c, Heat maps showing the relative frequency distribution of all merged DMRs from pairwise comparisons of m, t, and mt to wild type over chromatin states (CS) 1-36 (left) and standardized frequencies (z-scores) of DMRs across clusters (A1 to A4, corresponding to Fig. 1d) (middle). Cluster A1 overlaps mostly with CS31-36, whereas cluster A2 overlaps mostly with CS5&6. Chromatin state annotations from the Plant Chromatin State Database (PCSD)88 are shown at right. d, Distribution of mCG ratios in gene body-methylated (gbM; n = 2,448) and CMT2-targeted (n = 2,190) regions in rosette leaves of mthfd1-1 (m), mthfd1-1 thfs double mutant (mt), thfs (t), and wild-type (WT) plants grown under LD. Values were computed from per-site C/T counts and pooled across two biological replicates for each genotype. Box plots indicate medians (centre lines), IQR (boxes), and 1.5×IQR (whiskers); violin width reflects kernel density; n = regions. Different lowercase letters indicate significant differences between genotypes (pairwise Wilcoxon rank-sum tests, two-sided; BH-adjusted P < 0.01). See Supplementary Table 11 for P values. e, Volcano plot of differential TE expression between mthfd1-1 thfs (mt) and wild type (n = 3). Subfamily names of most significant DETEs are shown. f, Principal component analysis (PCA) of mRNA-seq data. Each point represents an individual sample; mthfd1-1 is clearly separated from the other groups along the first principal component. g, Proportions and absolute numbers of DETEs belonging to different TE families for each Cluster A5 to A7, as shown in Fig. 1e.
Extended Data Fig. 4 TE silencing, DNA methylation, enzyme activities and gene expression in mthfd1-1 is sensitive to day length and day time.
a, Distribution of mCHG and mCHH ratios in CMT2-targeted regions (mCHG: n = 1,827; mCHH: n = 1,855) in rosette leaves of mthfd1-1 (m) and wild type (WT) plants grown under SD. Values were computed from per-site C/T counts binned into 100-bp windows and pooled across two biological replicates for each genotype. Box plots indicate medians (centre lines), IQR (boxes), and 1.5×IQR (whiskers); violin width reflects kernel density; n = regions. b, Complete-linkage clustering of samples based on Euclidean distance of mean DNA methylation ratios in 1 kb bins across the Arabidopsis genome for CG, CHG, and CHH sites. c, Steady-state levels of 5-methyl-THF in rosette leaves of mthfd1-1 (m), mthfd1-1 thfs double mutant (mt), thfs (t), and wild type (WT) plants grown under LD. Grey shading depicts dark periods. Data are mean +/- s.d. (n = 3). See Supplementary Table 12 for P values. d, Diurnal enzymatic activities in leaf extracts from wild type and mutants grown under LD. Grey shading depicts dark periods. Data are mean +/- s.d. (n = 3 for MTHFD and MTHFR assays; n = 4 for THFS assays). See Supplementary Table 14 for P values. e, RT-qPCR transcript levels (ΔΔCt) of THFS, MTHFD1, and SHMT4 in wild type and mutants grown under LD. Grey shading depicts dark periods. Data are mean +/- s.d. (n = 3 biological replicates per group; exceptions at 12 h after dawn: t, all genes, n = 2; mt, SHMT4 only, n = 2). See Supplementary Table 15 for P values.
Extended Data Fig. 5 Formate incorporation into glycine and serine is daytime dependent.
a, Workflow for isotope label enrichment (M + 1, M + 2) analysis of free amino acids and nucleobases from liquid cultures of Arabidopsis seedlings using GC-MS. b, Schematic model of C1 traces from [2-13C]glycine and [13C]formate in FOCM. Bold arrows illustrate the [13C] trace towards target compounds (bold) in the presumed physiological directions (reaction catalysed by THFS and MTHFD1, and serine-to-glycine conversion are reversible). Mutation of the MTHFD1 and THFS-dependent pathway is highlighted in red. c, Label enrichment (M + 1, M + 2) in glycine and serine after [13C]formate supplementation at the beginning (left) or end (right panel) of the photoperiod. Grey shading depicts dark periods. Data are presented as mean values +/- s.d. (n = 3). See Supplementary Table 16 for P values.
Extended Data Fig. 6 DNA methylation in mthfd1-1 and wild type is sensitive to air CO2 levels.
a, Leaf area quantification of 3-week-old mthfd1-1 (m), wild type (WT), cat2-15, and shmt1 plants grown under LD and control (394 ± 39 ppm, cCO2) or high CO2 (3141 ± 53 ppm, hCO2) conditions. Data are mean +/- s.d. (WT, m hCO2, n = 4; m cCO2, cat2-15 hCO2, shmt1, n = 3; cat2-15 cCO2, n = 2). b,c Mean DNA methylation ratios over genes (b) and TEs (c) and flanking regions in different sequence contexts. Thick lines indicate LOESS-fitted curves. d, Principal component analysis (PCA) of whole-genome bisulfite sequencing (WGBS) data from mthfd1-1 and wild type grown under cCO2 and hCO2. DNA methylation ratios in CG (upper), CHG (middle), and CHH (lower panel) were calculated from per-site C/T counts in 100 bp bins across the Arabidopsis genome. Each point represents an individual sample (colours as in e). e, Mean mCG, mCHG, and mCHH ratios in 10 kb windows along chromosomes (Chr) 1 to 5 in mthfd1-1 and wild type grown under cCO2 and hCO2, and signed difference relative to wild type under cCO2 (Δ). f, Heat maps showing the relative frequency distribution of all merged DMRs from pairwise group comparisons over chromatin states (CS) 1-36 (left) and standardized frequencies (z-scores) of DMRs across clusters (B1 to B4, corresponding to Fig. 4a) (right). Cluster B1, representing CO2-responsive mthfd1 DMRs, overlaps mostly with CS31-36, representing heterochromatic TE-associated chromatin regions, whereas cluster B2 (CO2-insensitive mthfd1-1 DMRs) overlaps mostly with CS3-6 (active/gene body-associated chromatin regions).
Extended Data Fig. 7 Suppression of photorespiration by high CO2 concentration reduces the number of active TEs in mthfd1-1 depending on TE family.
a, Overlaps of DETEs from different comparisons. [CO2]-insensitive (CO2i) and responsive (CO2r) DETEs, as defined hereinafter, are indicated in tan and thistle colours, respectively. b, Principal component analysis (PCA) of mRNA-seq count data. Each point represents a biological replicate; unlike wild type (WT), mthfd1-1 (m) is clearly separated by CO2 condition. c, Total numbers and overlaps of DETEs from all pair-wise group comparisons. d, Distributions of mean (n = 3) expression changes in mthfd1-1 under cCO2 and hCO2 compared to wild type under cCO2 (top left panel), mCG ratios in mthfd1-1 under cCO2 and hCO2 (top right panel), and Spearman correlation between expression and mCG in mthfd1-1 (bottom panel) over TEs and flanking regions for CO2i (n = 569) and CO2r (n = 331) DETEs. mCG ratios for DETEs were computed from per-site C/T counts pooled across two biological replicates for each CO2 condition. Box plots indicate medians (centre lines), IQR (boxes), and 1.5×IQR (whiskers); violin width reflects kernel density; n = DETEs. e, Relative frequencies of TE families among all inducible TEs in Arabidopsis (all), all DETEs between mthfd1-1 and wild type under cCO2 (m cCO2 DETEs), CO2i m cCO2 DETEs, and CO2r mthfd1-1 cCO2 DETEs (left panel), and relative expression changes compared to wild type under cCO2 per TE family (Copia, n = 84; En-Spm, n = 199; Gypsy, n = 297; Harbinger, n = 35; HAT, n = 14; Helitron, n = 52; L1, n = 30; MuDR, n = 223) for CO2i mthfd1-1 cCO2 DETEs and CO2r mthfd1-1 cCO2 DETEs (right panel; box plots indicate median (centre lines), IQR (boxes), and 1.5×IQR (whiskers); violin width reflects kernel density; n = regions). En-Spm elements are strongly upregulated in mthfd1-1 vs. wild type under both cCO2 and hCO2 and therefore enriched in the CO2i subset; by contrast, Gypsy elements are more strongly upregulated in mthfd1-1 under cCO2 vs. hCO2 and therefore enriched in the CO2r subset. f, Overlaps of DEGs from different comparisons. g, Volcano plot of differential gene expression in mthfd1-1 under hCO2 compared to mthfd1-1 under cCO2. Names of DEGs encoding known and putative 5-formyl-THF binding proteins are indicated in blue35.
Extended Data Fig. 8 Suppression of photorespiration by high CO2 concentration suppresses loss of transcriptional silencing of SDC in mthfd1-1.
Genome browser tracks of DNA methylation ratios in CG, CHG, and CHH context for mthfd1-1 (m) and wild type (wt) under cCO2 and hCO2 (2 replicates each) and normalized transcript abundances for mthfd1-1 and wild type under cCO2 and hCO2 (averaged across 3 replicates each) at SDC (AT2G17690).
Extended Data Fig. 9 mthfd1-1 DEGs involved in photorespiration, FOCM, serine biosynthesis, and nitrate assimilation are co-expressed under control and high CO2 levels.
Clustered heatmap showing Pearson correlation coefficients of DEGs from Fig. 4e.
Extended Data Fig. 10 Chromatograms and mass spectra of free amino acids and nucleobases from plant extracts and standards.
Left panels show chromatograms with peaks of amino acids glycine, methionine, serine (a) as well as nucleobases 5-methylcytosine, adenine and thymine (b) and internal standard 4-nitrophenol with retention times from plant extracts and standards. Right panels show mass spectra and structural formulas of tert-butyldimethylsilyl ester derivates of glycine, methionine, and serine (a) and of trimethylsilyl ester derivates of adenine, 5-methylcytosine and thymine (b) acquired from TD-GC-MS analysis of 50 ng μl−1 standards and compared to reference mass spectral data from the library of the National Institute of Standards and Technology (NIST). Ions used to quantify isotopic label enrichment are highlighted with red boxes: glycine M + 0: m/z 246.0, M + 1: m/z 247.0, M + 2: m/z 248.0; methionine M + 0: m/z 320.0, M + 1: m/z 321.0, M + 2: m/z 322.0 and serine M + 0: m/z 390.0, M + 1: m/z 391.0, M + 2: m/z 392.0, adenine M + 0: m/z 264.0, M + 1: m/z 265.0, M + 2: m/z 266.0; 5-methylcytosine M + 0: m/z 254.0, M + 1: m/z 255.0, M + 2: m/z 256.0 and thymine M + 0: m/z 255.0, M + 1: m/z 256.0, M + 2: m/z 257.0.
Supplementary information
Supplementary Information
Supplementary Fig. 1, Tables 1–18, Methods and References.
Source data
Source Data Figs. 1–3 and Extended Data Figs. 1, 2, 4 and 5
Statistical source data.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hankofer, V., Ghirardo, A., Obermaier, L. et al. Photorespiration is linked to DNA methylation by formate as a one-carbon source. Nat. Plants (2026). https://doi.org/10.1038/s41477-026-02222-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41477-026-02222-x
