
Abstract
H9N2 avian influenza virus (AIV) is endemic in poultry worldwide and increasingly zoonotic. Despite the long-term widespread use of inactivated vaccines, H9N2 AIVs remain dominant in chicken flocks. We demonstrated that inactivated vaccines did not prevent the replication of H9N2 AIVs in the upper airway of vaccinated chickens. Viral transmission was enhanced during sequential passage in vaccinated chickens, which was attributed to the restricted production of defective interfering particles and the introduction of stable mutations (NP-N417D, M1-V219I, and NS1-R140W) which enhanced viral replication. Notably, the genetic diversity of H9N2 AIVs was greater and included more potential mammal/human-adapted mutations after passage through vaccinated chickens than through naïve chickens, which might facilitate the emergence of mammal-adapted strains. By contrast, vaccines inducing cellular/mucosal immunity in the upper respiratory tract effectively limit H9N2 AIV. These findings highlight the limitations of inactivated vaccines and the need for revised vaccination strategies to control H9N2 AIV.
Similar content being viewed by others


Introduction
Avian influenza viruses (AIVs) can be divided into two groups according to their virulence, highly pathogenic avian influenza viruses (HPAIVs) and low-pathogenicity avian influenza viruses (LPAIVs). HPAIVs mainly include the H5 and H7 subtype AIVs, whereas H9N2 is the main subtype of the LPAIVs. H9N2 AIV have been endemic in poultry across at least 60 countries globally, with persistent circulation in Asia, the Middle East, and North/West Africa, while also being periodically detected in the Americas and Europe1. Since 2016, H9N2 has been the dominant avian influenza virus subtype in poultry in China, which isolation rate of 9.4–23.3% in live poultry markets2,3,4. Although H9N2 AIV is weakly pathogenic in chickens, it causes significant economic losses from reduced production and coinfections5. Notably, H9N2 AIV is also capable of infecting mammals, including pigs, dogs, cats, foxes, pikas, and humans6,7,8,9,10,11. Increased human receptor-binding property and mammalian adaptation mutations of H9N2 AIVs facilitate the viral fitness in human12,13. The H9N2 serological positivity rate in human populations is 10–37%4,14,15, and the number of human cases of H9N2 AIV infection has shown a dramatic increase in recent years. More than 130 human cases of H9N2 AIV have been reported until November of 2024, including 84 cases after 202016. Furthermore, H9N2 AIVs have been shown to have donated internal genes to multiple emerging human-infecting AIVs, including H5N6, H7N9, H10N8, H3N8, H10N3, and H10N517,18,19,20. Therefore, the control of H9N2 AIVs is important for both the poultry industry and public health.
China has used an inactivated vaccine immunization strategy to control AIVs in poultry for a long time. Although inactivated vaccines efficiently limited outbreaks of H5Ny and H7N9 AIVs in China21,22, H9N2 AIVs are still in national circulation, regardless of the intensive vaccination program undertaken since 19985. H9N2 AIVs with little or no antigenic variation from the vaccine strain are regularly isolated from vaccinated chickens with high antibody levels23. The low efficacy of vaccination against H9N2 viruses has also been frequently reported in other countries24. The reason for the unabated circulation of H9N2 AIV in vaccinated chickens and the impact of its continuous spread in vaccinated poultry on the evolution of the virus have been unclear.
In this study, we identified a unique mechanism by which H9N2 AIV evades the immunity of its hosts that is induced by inactivated vaccines. The replication of the H9N2 virus in the upper airways of chickens was significantly higher than that of other subtypes of AIVs, and the immunoglobulin G (IgG) induced by inactivated vaccines at this site was insufficient to neutralize the H9N2 virus. Furthermore, the replication ability of H9N2 AIV increased during its continuous transmission in vaccinated chickens, with a reduction in defective interfering particles (DIP) production and the coordinated acquisition of mutations across multiple segments of the viral genome. It is noteworthy that the immune pressure induced by the inactivated vaccine also increased the genetic diversity of H9N2, which led to the emergence of more potential mammal- and human-adapted strains. We found that vaccines capable of inducing both cellular and local mucosal immunity are more suitable for controlling H9N2 AIV.
Results
Inactivated vaccine does not prevent H9N2 viral replication or transmission in chickens
We first identified the differences in the effects of inactivated vaccines on different subtypes of AIVs observed clinically. The protective effect of inactivated vaccine against H5Ny, H7N9 and H9N2 AIVs was equally examined. Three strains belonged to different HA lineages from H5Ny, H7N9 or H9N2 subtypes were inactivated and each strain was used to immunize SPF chickens. Chickens with HI antibody titers of 7–8 log2 were intranasally inoculated with 106 EID50 of the homologous virus, and immunologically naïve chickens were infected as controls. Figure 1A shows that neither H5Ny nor H7N9 AIVs were detected in the vaccinated chickens. In contrast, although no H9N2 virus was detected in the lower respiratory tracts of the vaccinated chickens, including the tracheas, bronchi, and lungs, the virus replicated efficiently in the upper respiratory tracts (turbinate and larynx) with mean viral titers of 6.75–7.25 log10 EID50/ml. Consistent with this, in the naïve chickens, the replication levels of all the AIVs were similar in the lower respiratory tracts at 3 dpi, whereas the viral titers of the H9N2 AIVs were higher than those of the H5Ny and H7N9 AIVs in the turbinate and larynx, with an increase of 1.2–2.8 log10 EID50/ml (P < 0.05). This indicates that the particularly effective replication ability of H9N2 AIV in the upper respiratory tract contributed to the shedding of H9N2 AIV by the vaccinated chickens. We also confirmed the failure of the inactivated vaccine to protect chickens from H9N2 infection by observing the concomitant tissue injury. H&E staining of the turbinates, larynges, tracheas, bronchi, and lungs from the naïve group and the group vaccinated with the HB17 virus showed that H9N2 AIV caused the vacuolation of the mucosal cells of the turbinate; the loss of the stratum corneum and the congestion of capillaries in the larynx; and the presence of inflammatory cells around the blood vessels, edema, desquamation, and lesions in the trachea; and bronchial epithelial cells; and bronchopneumonia. However, in the vaccinated group, no lesions were observed in the lower respiratory tract, including the trachea, bronchus and lung, but similar lesions were present in the upper respiratory tracts (turbinate and larynx) (Fig. 1B). In the naïve group, the laryngeal microvilli and a large number of epithelial cells and mucous exudates were shed from the mucosal surface at 3 dpi. The tracheal cilia were shed and the lamina propria cells were exposed. The pathological changes were more serious at 5 dpi. In the vaccinated group, the pathological changes on the laryngeal mucosal surface were similar to those in the naïve group, but there was no loss of tracheal cilia (Fig. 1C). We also demonstrated with reverse genetics that the HA gene of H9N2 AIV is responsible for its replication in vaccinated chickens (Fig. 1D). Because the inactivated vaccine mainly induced humoral immunity, we evaluated the influenza-specific IgG antibodies in the vaccinated chickens. We found that the IgG antibody level in the lavage fluid from the upper respiratory tract was only 3.65% of that detected in blood, which may be insufficient to inhibit the replication of H9N2 AIV at the corresponding upper respiratory sites (Fig. 1E).

A Naïve and inactivated-vaccinated chickens (HI = 7–8 log2) were intranasally inoculated with 106 EID50 of H5Ny, H7N9, or H9N2 AIV. The viral titers in the turbinate, larynx, trachea, bronchus, and lung were measured at 3 dpi. Statistical significance relative to H5Ny or H7N9 AIVs in turbinate and larynx of the naïve group was assessed with two-way ANOVA (*P < 0.05). Horizontal dashed black lines indicate the lower limits of detection. B Representative histopathological changes in H&E-stained turbinate, larynx, trachea, bronchus, and lung sections from HB17-infected groups at 5 dpi. Mucosal cells were swollen and vacuolated (▲); mucosal damage, loss of stratum corneum (△); congestion of capillaries and presence of inflammatory cells around blood vessels (black thick solid arrow); edema, desquamation, and lesion of tracheal epithelial cells (thick open arrow); bronchopneumonia and massive immune cell infiltrates around bronchi and blood vessels (black solid six-pointed star). Scale bar, 50 μm. C Electron microscopy (EM) analysis of larynges and bronchi in HB17-infected groups at 3 and 5 dpi. Scale bar, up, 50 μm; down, 10 μm. D Viral titers of recombinant viruses in the organs of naïve and vaccinated chickens. E IgG antibodies assessed in serum and saliva samples from chickens vaccinated with inactivated-virus vaccine.
We also investigated whether the inactivated vaccine prevented H9N2 viral infection and/or transmission in the chickens with high antibody titers. Three chickens with HI antibody titers of 12 log2 were intranasally inoculated with 106 EID50 of the HB17 virus and after 24 h, were co-housed with another three vaccinated chickens. H9N2 AIV was detected in oropharyngeal swabs from both the inoculated and in-contact chickens (Supplementary Fig. 1), indicating that the H9N2 virus efficiently replicated and was transmitted by chickens immunized with the inactivated vaccine, regardless of their vaccination status.
Replication and transmissibility of H9N2 AIV increased during viral passage in vaccinated chickens
Because the inactivated vaccines did not prevent H9N2 virus shedding by the vaccinated chickens, we also investigated whether H9N2 AIV was continuously transmitted in vaccinated chickens and the impact of the sustained immune pressure induced by the vaccine on H9N2 evolution. A model of influenza transmission under vaccine-induced immune pressure was established, as previously described (Fig. 2A)25,26. H9N2 virus HB17 was passaged among naïve chickens and HB17-immunized chickens during their cohabitation. All the chickens were monitored daily for virus shedding. H9N2 AIV was effectively transmitted through both the naïve and immunized passage lines (Fig. 2A). Initially, the serial interval of P1 (transmission time from P0 to P1 chickens) in the naïve group was 1 day, whereas that in the vaccinated group was 4 days. However, the serial interval in the vaccinated group decreased to 2.5 days by P4 and to 2 days by P5 and P6 (Fig. 2B). The rate of viral replication in the vaccinated group was significantly lower than that in the naïve group between P0 and P3 (P < 0.01), but was similar to that in the naïve group by P4 (Fig. 2C). These results indicate that the transmission of the HB17 virus between vaccinated chickens was initially inhibited or delayed, but viral replication and transmission increased rapidly during its passage.

A Time course of H9N2 virus transmission through naïve and vaccinated passage chains. The schematic diagram was created using Adobe Illustrator and Canva (www.canva.com). B Serial intervals of viral transmission between chickens in the naïve and vaccinated passage chains, which refers to the number of days required for an uninfected animal to become infected after cohabitation with an infected animal. C Growth rate of viral replication in chickens at each passage in the naïve and vaccinated passage chains. D Nucleotide positions and absolute frequencies of mutations. E Numbers of nonsynonymous mutations at different passages. Statistical significance was assessed with two-way ANOVA (*P < 0.05; **P < 0.01).
Genetic diversity of H9N2 AIV was higher in vaccinated chickens than in naïve chickens
The increased transmissibility of H9N2 AIV with passage in vaccinated chickens suggests that the virus had undergone significant genetic changes. Therefore, we used the deep sequencing of oropharyngeal samples taken daily during the transmission study to detect intrahost-acquired SNVs (iSNVs) in the H9N2 AIVs. iSNVs from high-quality datasets containing 44 and 40 samples from the naïve and vaccinated groups, respectively (each with a genome copy number >103/μl and iSNVs detected above a cutoff of 1% of sequence reads), were analyzed according to a previous study27. Notably, although the HA protein is the major target of the antibodies induced by inactivated vaccines, nonsynonymous and synonymous mutations were distributed throughout all the viral gene segments (Fig. 2D). The genetic diversity of the H9N2 AIVs in vaccinated chickens was clearly higher than that in the vaccine-naïve chickens. In total, 107 and 358 amino-acid substitutions were identified in the naïve and vaccinated chains, respectively. More amino-acid substitutions at a frequency >99% were found in the vaccinated chain (Supplementary Fig. 2). However, the genetic diversity in the vaccinated group began to decline in P4 (Fig. 2E).
Transmission bottleneck size was smaller in vaccinated chickens than in naïve chickens
The transmission bottleneck determines how much of the viral diversity generated in one host passes to another during viral transmission, so it plays a vital role in linking within-host processes to larger evolutionary trends28. Therefore, the genetic composition of viral populations immediately before and after viral transmission was compared. The first time point at which infectious virus was detected in the recipients was examined. The vaccinated group had fewer shared polymorphic sites (lie in the middle of the plot) than the naïve group, suggesting a stringent effective bottleneck in the vaccinated groups (Fig. 3A). The maximum-likelihood bottleneck sizes were estimated with the exact beta-binomial method29 to be 3.68 (lambda = 1.15; 95% CI, 2.91–4.14) in the naïve group and 1.25 (lambda = 1.05; 95% CI, 0.49–2.14) in the vaccinated group (Fig. 3B). These results indicate that vaccination narrowed the interhost bottleneck of H9N2 AIVs in chickens.

A Frequency of intrahost-acquired single-nucleotide variants (iSNVs) in both donor and recipient samples. Frequencies <3% and >98% were set to 0% and 100%, respectively. B Estimated bottleneck sizes in 10 transmission events, calculated with the exact beta-binomial method. Statistical significance was assessed with two-way ANOVA (**P < 0.01).
Emergence and transmission of DIPs in H9N2 virus populations were restricted in vaccinated chickens
Previous studies have shown that DIPs significantly inhibit the replication capacity and adaptability of influenza viruses in competition with intact viral particles30. We enriched DIPs through the serial undiluted passage of HB17 virus in CEF, and confirmed a negative correlation between the viral titer and DIP production (Supplementary Fig. 3). The DIP-associated junction formation patterns in the Illumina-based sequencing data derived from oropharyngeal samples were examined, using a previously developed bioinformatics pipeline31. In the naïve group, the manifestation of DIPs within a single host was not homogeneous, but rather showed a fluctuating, wave-like pattern over time, as observed in previous studies32,33,34 (Fig. 4A, B, and Supplementary Fig. 4). In contrast, this wave-like phenomenon was not apparent in the vaccinated group, and the occurrence of DIPs was relatively constant and low. Consistent with previous studies, most DIP-associated deletions were derived from the three polymerase genes, HA and NA, whereas the M and NS segments were almost entirely free of deletions (Fig. 4B). Interestingly, in contrast to the H1N1 and H3N2 strains of human influenza viruses, which showed minimal DIPs associated with NP genes, we found numerous DIPs associated with the NP gene of H9N2 AIVs. DIPs that were transmitted stably in viral populations isolated from the naïve chain were predominantly distributed across the PB2, PB1, and NP genes, but virtually none of these was found in the vaccine line (Fig. 4C). In summary, the production of DIPs was restricted in chickens vaccinated with the inactivated H9N2 virus, which may have promoted viral replication and/or transmission.

A Number of distinct junctions detected in each individual in three passage lineages at the indicated time points. B Number of distinct junctions detected in each viral segment in the naïve and vaccinated lineages. C Plots showing the viral DIP-associated junctions present in the overall viral populations from the naïve group. Each color indicates a particular DIP-associated junction, whose relative proportion corresponds to its abundance in the viral population in the indicated sample.
Mutations that improve viral replication were selected in vaccinated chickens
Antigenic drift induced by the mutation of HA is a major route used by influenza viruses to circumvent vaccine protection26. Therefore, we first hypothesized that the increased transmissibility of the virus in vaccinated chickens after P4 was attributable to antigenic changes in HA. To assess the antigenic variation arising from the passage of the H9N2 virus, 10 plaque clones were purified from P6 of each passage chain and their whole genomes sequenced. Eight monoclonal strains with different HA sequences were identified, but all strains were neutralized with polyclonal hyperimmune serum against the wild-type HB17 virus and showed similar reactions to that of the wild-type HB17 virus, suggesting that the antigenicity of HB17 was not significantly altered during the transmission of the virus in vaccinated chickens (Table S1).
We next hypothesized that the replication ability of the progeny viruses had increased significantly, reducing the neutralization capacity of the antibodies induced by the inactivated vaccines. Ten monoclonal strains with different genotypes were identified among the plaque clones described above, including two from the naïve group and eight from the vaccinated group. The monoclonal strains from the naïve group showed similar replication ability to that of the wild-type virus in CEF (P < 0.05) (Supplementary Fig. 5A). However, five strains of the eight monoclonal strains from the vaccinated group demonstrated significantly higher replication rates than the wild-type strain. We further evaluated the replication and transmission of representative monoclonal strains in chickens. Supplementary Fig. 5B demonstrated that the strains from the vaccinated group possessed enhanced virulence and transmissibility comparing to those form naïve group and the parental strain. This suggests that the increased genetic diversity of H9N2 acquired by vaccinated chickens during serial passages provided a more-diverse quasispecies pool, facilitating the selection of viruses with enhanced replication ability.
We also identified key amino-acid mutations responsible for the increased replication of the progeny viruses in the vaccinated chickens. Three mutations (NP-N417D, M1-V219I, and NS1-R140W) were detected at frequencies of up to 99% after P4, when enhanced viral replication and transmissibility were observed (Fig. 5A). We determined the growth kinetics of the rgHB17(M1-V219I), rgHB17(NS1-R140W), rgHB17(NP-N417D), rgHB17(M1-V219I NS1-R140W), and rgHB17(M1-V219I NS1-R140W NP-N417D) viruses in CEF, and found that the replication of the single and double mutants was significantly higher than that of the wild-type virus (P < 0.05) (Fig. 5B). The replication of the double mutant (M1-V219I and NS1-R140W) was significantly higher than that of the single mutants, and the replication of the triple mutant was still higher (P < 0.05). We confirmed the functional effects of NP-N417D, M1-V219I, and NS1-R140W on the improved replication and transmissibility of H9N2 AIV in chickens (Fig. 5C). Therefore, the fixation of the NP-N417D, M1-V219I, and NS1-R140W mutations may be responsible for enhancing viral replication and reducing the serial interval in the vaccination chain from P4 onwards to levels similar to those in the infected naïve chain.

A Linkage analysis of high-frequency (>10%) variants that emerged during homologous vaccinated passage. B Multistep growth curves of the indicated H9N2 AIV mutants in CEF infected at MOI of 0.001. All data are representative of or presented as means ± SD of three independent experiments. C Impact of NP-N417D, M1-V219I, and/or NS1-R140W on the replication and transmission of H9N2 AIV in chickens. Three chickens were intranasally inoculated with 106 EID50 of virus. After 24 h, the inoculated chickens were housed together with three contact chickens. The viral titers in oropharyngeal swabs from inoculated chickens (n = 3) were determined at 3, 5, and 7 dpi and at 3, 5, and 7 dpc for the contact chickens. Statistical significance relative to HB17 was assessed with two-way ANOVA (*P < 0.05; **P < 0.01).
We also investigated the mechanistic effects of the NP-N417D, M1-V219I, and NS1-R140W substitutions on viral replication. Influenza virus vRNPs are transported into the nucleus, where they undergo transcription and replication to generate new viral genomes35. Figure 6A, B show that cells with nuclear localization of NP at 4, 8 and 12 hpi with rgHB17(NP-N417D) virus were higher than those of rgHB17-infected cells, indicating that the NP-N417D facilitated nuclear transport of vRNPs in DF1 cells. The morphology of the rgHB17 and rgHB17(M1-V219I) viruses in MDCK cells was compared with transmission electron microscopy (Fig. 6C). About 66% of the rgHB17(M1-V219I) viral particles were morphologically filamentous, whereas the rgHB17 virions were predominantly spherical or ovoid (85%), with only 15% filamentous particles. M1-V219I increased the proportion of filamentous particles, which is known to increase viral fitness in the host36 (Fig. 6D). The effect of NS1 mutations on the nuclear-cytoplasmic localization of the virus at different stages of infection was examined with immunofluorescence in DF1 cells (Fig. 6E) and quantified (Fig. 6F). The NS1-R140W protein was more strongly localized in the cytoplasm than wild-type NS1. In the leptomycin-treated cells, NS1-R140W protein and wild-type NS1 protein exhibited similar localization patterns, being confined to the nucleus and accumulated near the nuclear membrane (Supplementary Fig. 6). These results indicated that NS1-R140W enhanced the nuclear export of the NS1 protein rather than reducing its nuclear localization function. Previous studies indicate that NS1 protein in the cytoplasm has the ability to inhibit IFN induction and repress the activation of antiviral proteins37. Therefore, the R140W mutation may enhance viral replication by increasing the nuclear export of NS1.

A DF1 cells were infected with rgHB17 or rgHB17(NP-N417D) at MOI of 2. NP was localized with confocal microscopy at the indicated time points (scale bar: 50 μm). B Quantitative analysis of NP localization in virus-infected cells. C H9N2 viral morphology examined with transmission electron microscopy (scale bar: 500 nm). D Quantification of spherical and filamentous virions. E Nuclear-cytoplasmic localization of wild-type HB17 and mutant NS1 protein in DF1 cells during infection. Scale bar, 10 μm. F Quantitative localization of wild-type and mutant NS1 proteins in DF1 cells.
More potential mammal-adapted H9N2 AIVs were generated in the vaccinated group
The increased genetic diversity of H9N2 AIV in the vaccinated group raises concerns about whether vaccination may lead to the emergence of more strains better adapted to mammals. We found 7 and 17 mutations associated with mammal and human adaptation according to previous studies in the naïve and vaccinated chains, respectively (Fig. 7A). Ten plaque clones were purified from each passage (P1–P6) of each passage chain and their whole genomes sequenced. In this way, we identified 14 and 27 genotypes in the naïve and vaccinated groups, respectively. The different genotypes were used to infect mice. We found that 36% and 48% of the monoclonal strains isolated from the naïve and vaccinated groups, respectively, replicated more efficiently in the mouse lungs than did the wild-type virus (Fig. 7B). These results suggest that the increased genetic diversity of H9N2 AIVs in vaccinated chickens provides better opportunity for the emergence of mutants that might cross the host–species barrier.

A Amino-acid substitutions related to mammal-adaptation detected in viral populations isolated from naïve and vaccinated animal chains. B Viral titers of monoclonal strains with significantly higher replication capacity than wild-type HB17 (P < 0.05) in naïve and vaccinated animal. Mice (n = 6) were inoculated with 106 TCID50 of different monoclonal strains. Three mice from each group were euthanized at 3 dpi, and viral titers in the lungs were determined by infection of MDCK cells. Statistical significance relative to wild-type HB17 was assessed with two-way ANOVA (*P < 0.05; **P < 0.01).
Cellular and upper-respiratory-tract mucosal immunity play crucial roles in restricting H9N2 replication in chickens
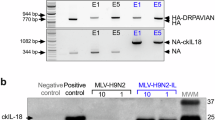
Because the humoral immunity induced by inactivated vaccines is insufficient to prevent the replication and transmission of H9N2 in chickens, we evaluated the protective efficacy of the H9N2 live vector vaccine (rHVT-H9) and the cold-adapted attenuated avian H9N2 influenza vaccine (H9N2-LAIV) which could also induce cellular immunity and/or mucosal immunity in the upper respiratory tract. Our findings revealed that, although low quantities of virus were shed by the chickens in the rHVT-H9 challenge group, the viral load was too low for the effective transmission of the virus to the contact group (Fig. 8A). No virus was detected in either the challenge or contact groups of chickens vaccinated with H9N2-LAIV. Although the humoral immune response induced by rHVT-H9 and H9N2-LAIV was comparable to those elicited by inactivated vaccines, they induced significantly stronger influenza-virus-specific CD4+ and CD8+ T-cell responses than inactivated vaccines (P < 0.01) (Fig. 8B, C). Moreover, the lavage fluid from the upper respiratory tracts of chickens vaccinated with H9N2-LAIV showed significantly elevated IgA levels, and the nasal mucosal cells showed increased IFN-γ expression than in chickens vaccinated with the inactivated vaccine or rHVT-H9 (P < 0.01) (Fig. 8D, E). These results demonstrate that cellular immunity and/or upper-respiratory-tract mucosal immunity are essential for controlling H9N2 AIV with vaccines in chickens.

A Vaccinated chickens were intranasally inoculated with 106 EID50 of HB17 virus. At 24 h after challenge, in-contact vaccinated chickens were placed in physical contact with challenged birds. Viral titers on oropharyngeal swabs were determined at 3, 5, and 7 dpc. Horizontal dashed black line indicates the lower limits of detection. B HI antibody analysis. Sera collected were analyzed with HI assays to test the response to the corresponding vaccine strains. C Cellular immune response analysis. Chickens were vaccinated with different vaccines. Lungs (n = 6) were harvested at HI = 7–8 log2. IFN-γ+CD4+ and IFN-γ+CD8+ T cells were identified in lungs stimulated with corresponding vaccine strains using an intracellular cytokine staining assay. Percentages of IFN-γ+CD4+ or IFN-γ+CD8+ T cells among CD4+ or CD8+ T cells were analyzed. D–E IgG and IgA antibodies measured in saliva samples from chickens vaccinated with either PBS (Mock) or different vaccines. F Numbers of spots of IFNγ-producing cells in turbinates of chickens vaccinated with either PBS or different vaccines. Statistical significance relative to inactivated vaccine group was assessed with two-way ANOVA (**P < 0.01).
Discussion
The control of H9N2 AIVs in chickens is essential to mitigate against disease outbreaks in birds and the emergence of novel zoonotic AIVs. However, the long-term use of H9N2 inactivated vaccines has not reduced the prevalence of H9N2 AIVs in chickens38. On the contrary, the replication of the H9N2 virus has seemingly increased in chickens and mammalian species20,39. In the present study, we found that the use of an inactivated H9N2 vaccine did not prevent or reduce virus shedding, but selected for viral strains with increased replication ability and restricted DIP production, thus facilitating the transmission of H9N2 AIVs in vaccinated chickens. The transmission of H9N2 AIV in vaccinated chickens introduces a further problem in that it increases the diversity of viral genes in the circulating virus, possibly generating more novel strains. Therefore, a policy review is urgently required, together with changes in the current use of inactivated H9N2 viral vaccines in poultry.
Although H9N2 AIVs do not exhibit antigenic drift as significantly as H5 or H7 AIVs in clinic, the emergence of H9N2 variants belonging to novel antigenic group has been shown to reduce the efficacy of inactivated vaccines40,41. More importantly, in this study, we found that the H9 AIV subtype has a greater capacity for replication in the upper respiratory tract than subtype H5 or H7. However, in chickens vaccinated with the H9N2 inactivated vaccine, the IgG antibody levels in the lavage fluid from the upper respiratory tract were only 3.65% of those detected in the blood. Therefore, the humoral immune response induced by the inactivated vaccine in the upper respiratory tract is insufficient to neutralize the H9N2 virus, with its high replication level, at this site. This may explain why H9N2 continues to spread in vaccinated flocks. In contrast, rHVT-H9, which also induces a cellular immune response, and H9N2-LAIV, which induces both a cellular immune response and a local mucosal immune response in the upper respiratory tract, efficiently prevented H9N2 AIV replication in chickens. A similar phenomenon has also been observed for the SARS-CoV-2 virus (responsible for COVID-19). Although vaccines have been effective in reducing the severity and mortality rate of SARS-CoV-2 virus infections, these vaccines do not effectively prevent viral replication and transmission in the upper respiratory tract42,43,44. However, live vector vaccines, which elicit strong cellular immunity, and vaccines that induce local mucosal immunity are effective in blocking the replication and/or transmission of multiple SARS-CoV-2 variants in the respiratory tract45,46. Therefore, inactivated vaccines are insufficient for the effective control of viruses that replicate efficiently in the upper respiratory tract.
Our study demonstrates that vaccines capable of inducing cellular immunity and/or local mucosal immunity in the upper respiratory tract, such as rHVT-H9 and live attenuated vaccines, exhibit enhanced protective efficacy against H9N2 infection in chickens. We previously demonstrated that rHVT-H9, which can be used in chicken embryos or in 1-day-old chickens, effectively circumvented interference from maternal antibodies in the field47. Furthermore, the fact that only a single dose is required for rHVT-H9 simplifies the vaccination process and reduces costs. A major challenge in HVT vaccine development is inserting multiple foreign genes without affecting the replication of recombinant HVT. Since the influenza virus genome is segmented, the potential risk of reassortment between live attenuated vaccine and wild-type virus remains a concern. A recent study proposes a strategy for targeted genomic rearrangement to prevent the recombination of specific viral RNA segments48. Although there are currently no commercial vaccines inducing cellular immunity or upper respiratory mucosal immunity against H9N2 AIV, awareness of the limitations of inactivated vaccines could accelerate the development and application of such vaccines.
Understanding the evolution of H9N2 AIVs in vaccinated chickens can shed light on the mechanisms that govern their genetic and biological variation, and thus provide insight into the development of more-effective vaccines or control methods. Our current understanding of the evolution of the influenza viruses is mainly based on studies of strains isolated in epidemiological investigations. Although these analyses are important, they are influenced by a number of factors. Recent studies have focused on the viral diversity present within infected individuals using transmission experiments and the quantitative analysis of host–pathogen interactions, or how transmission bottlenecks mediate the structure and extent of viral genetic diversity in the recipient host25,26,49,50. Such studies are key to understanding how viral and host-associated traits influence the genetic diversity and biological properties of the virus, such as its antigenicity, virulence, and host range, and the epidemiological consequences of within-host evolution. In the present study, H9N2 AIV was passaged by contact transmission in both naïve and vaccinated chickens. We found that the time between transmission events (serial interval) was initially longer in the vaccinated chain than in the naïve chain, but by P4, the serial interval had decreased to that in the infected naïve group. Notably, antigenic drift was not detected in the plaque clones taken from the vaccination groups, but the proportion of quasispecies with increased replication ability was higher in these passages of the vaccinated groups than in the immunologically naïve chickens. In the field, it is common for the antigenicity H5 or H7 AIVs to change significantly through single amino-acid mutations51,52,53, but no similar phenomenon has been detected in H9N2 AIVs. Our results indicate that H9N2 AIVs from vaccinated chickens develop a rapid increase in replication to offset antibody neutralization, unlike H5 and H7 AIVs, which escape vaccine-induced immunity through antigenic variation. The results of our serial passage experiments indicated that the gene diversity in the whole genomes of the vaccination group was significantly higher than that in the naïve group. Previous research has demonstrated a role for glycosylation in facilitating immune evasion by influenza viruses, which compensates for the fitness costs of mutations that allow antigenic escape54. Although influenza viruses primarily evade antibody-mediated inhibition of replication through antigenic drift caused by mutations in HA, these HA mutations may also lead to the emergence of linked or synergistic mutations in other gene segments. Previous studies demonstrated that vaccine-induced immune pressure can also drive mutations in other gene segments26. In the present study, we identified the fitness-enhancing mutation NS1-R140W in P1 of the vaccinated chain with a frequency of 35%, which was fixed in the vaccinated line with 99% frequency. Using reverse genetic manipulation, we found that NS1-R140W significantly increased the replication capacity of the virus. Cooperative mutations were observed in the NP and M genes (M1-V219I and NP-N417D, respectively), suggesting that these sites are under positive selection in this system. This variant combination underwent rapid fixation after transmission, and significantly enhanced the virus’s replication and transmissibility in chickens. These findings extend our understanding of viral adaptive mechanisms in vaccinated hosts.
Virological research is concerned with the production of infectious progeny virus, which represents the primary source of viral pathogenesis and pathogenicity. However, the majority of influenza virus progeny particles are noninfectious55. DIPs carry a large internal deletion in at least one genome segment and are therefore defective in viral replication56. Moreover, they interfere with viral replication57. In the present study, the diversity of DIPs isolated from the vaccinated cohort was lower than that isolated from unvaccinated chickens, suggesting that H9N2 restricts the emergence of DIPs to facilitate viral replication under the immune pressure induced by vaccination. The phenomenon in which viruses enhance their replication by altering their DIP production in a vaccinated host has not been reported previously. Therefore, our results provide new insights into the adaptive evolution of influenza viruses under vaccine-induced immune pressure.
Genetic bottlenecks play a vital role in shaping viral evolution28. Stringent bottlenecks usually limit the diversity of the founding population in the recipient host and alter the mutational composition of the population in the recipient host relative to that in the donor host58. In contrast, if the bottleneck is loose, transmission does not significantly affect the variant frequencies, and the composition of the founding population in the recipient more closely matches that present in the donor host at the time of transmission. We found that the bottleneck size in the vaccinated group was narrower than that in the naïve group, indicating that the selective pressure exerted by vaccine-induced immunity created a narrower bottleneck for the transmission of the H9N2 virus in chickens. The combination of three stable mutations (NP-N417D, M1-V219I, and NS1-R140W) was fixed in the vaccinated chickens, which increased the replication of H9N2 AIVs. Although strong bottlenecks usually lead to a decline in fitness28, we found that mutations steadily increased within the context of pre-existing vaccine-induced immunity, indicating that bottlenecks can also be advantageous for viruses.
In this study, we found that the genetic diversity of H9N2 in the vaccinated group was higher than naïve group and there were more previously reported mammalian-adaptive mutations observed in the viruses in vaccinated group. Meanwhile, there were more monoclonal viruses in vaccinated chickens, which exhibited enhanced replication and pathogenicity in mice. These findings suggest that, when vaccines fail to completely prevent viral shedding and transmission, the diverse progeny viruses might pose a potential risk of cross-species transmission. However, it should be noted that the frequencies of most mammalian adapted mutations in our study are below 10%, and the role of these mutations in mammalian adaptation should be further validated. Moreover, it remains uncertain whether viral adaptation in mice fully reflects the situation in humans. Notably, the long-term and widespread use of inactivated vaccines in China has failed to control H9N2, and H9N2 has even become the predominant subtype in poultry, along with an increased number of human H9N2 cases4,12. Additionally, enhanced mammalian adaptability has been observed in recent strains39. Attention needs to be paid to whether the failure of vaccine immunization against H9N2 AIV might increase the viral threat to public health.
In the present study, viral populations isolated from vaccinated chickens showed greater genetic diversity and the accumulation of mutations that improved their fitness. Because the presently used inactivated H9N2 vaccine is ineffective, and in the face of the continued increase in the replication ability and potential antigenic variation of H9N2 AIVs in chickens39,59, the current vaccination policy for the control of the H9N2 virus requires urgent review. The need to develop vaccines against H9N2 AIV that induce cellular immunity and/or mucosal immunity in the upper respiratory airway cannot be overemphasized. The development of multivalent rHVT vaccines and the improvement of live attenuated vaccine safety represent key directions for future vaccine research. Additionally, the combined use of antiviral drugs could improve the effectiveness of H9N2 prevention and control.
Materials and methods
Ethics statement
All animal research was approved by the Beijing Association for Science and Technology (approval ID SYXK, Beijing, 2007–0023) and was performed in compliance with the Beijing Laboratory Animal Welfare and Ethics guidelines, as issued by the Beijing Administration Committee of Laboratory Animals, and in accordance with the China Agricultural University (CAU) Institutional Animal Care and Use Committee guidelines (ID: AW51706102-2-2) approved by the Animal Welfare Committee of CAU.
Viruses and cells
A/duck/Anhui/01/2006 (AH06, H5N1), A/chicken/Shandong/2019 (SD19, H5N6), A/duck/Jiangxi/A65/2020 (JX20, H5N8), A/pigeon/Shanghai/S1069/2013 (SH13, H7N9), A/chicken/Guangdong/SD098/2017 (GD17, H7N9), A/chicken/Yunnan/SD024/2021 (YN21, H7N9), A/chicken/Beijing/0701/2015 (BJ15, H9N2), A/chicken/Guangdong/f31/2022 (GD22, H9N2), and A/chicken/Hebei/m0530-1/2017 (HB17, H9N2) AIVs are laboratory depository viruses. Each virus was passaged twice after isolation and purified three times with plaque isolation in Madin–Darby canine kidney (MDCK) cells. Stocks were prepared, stored at −80 °C, and titered (50% egg infectious dose [EID50]) in 9–11-day-old specific-pathogen-free (SPF) embryonated chicken eggs, using 10-fold dilutions and 10 eggs per dilution. The infectious dose was calculated according to Reed and Muench. Human embryonic kidney (293 T) cells, MDCK cells, a chicken fibroblast cell line (DF1), and chicken embryonic fibroblasts (CEF) were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Invitrogen, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum, 100 units/ml penicillin, and 100 µg/ml streptomycin. All cell cultures were incubated at 37 °C under 5% CO2.
Antibodies
Primary antibodies were purchased from the following commercial suppliers: anti-nucleoprotein (NP) monoclonal antibody (mAb) was purchased from GenScript (Nanjing, China); anti-nonstructural protein 1 (NS1) mAb was purchased from Santa Cruz Biotechnology (Dallas, TX, USA). The secondary antibodies used for confocal microscopy were Alexa-Fluor-488-conjugated donkey anti-rabbit IgG antibody and Alexa-Fluor-594-conjugated goat anti-mouse IgG antibody (Life Technologies).
Protective efficacy of vaccines against AIVs in chickens
For the inactivated vaccinated group, 3-week-old SPF white leghorn chickens were obtained from Boehringer Ingelheim Animal Health Care Corporation (Beijing, China). The chickens were vaccinated with inactivated vaccines produced by AIVs of representative lineages of different subtypes. After two rounds of vaccination 2 weeks apart, the chickens with hemagglutination inhibition (HI) antibody titers of 7–8 log2 or high HI titers (12 log2) were intranasally inoculated with 106 EID50 of a homologous strain. The viral titers on oropharyngeal swabs and/or in different tissues were determined at 3 days postinoculation (dpi), and the pathological changes in the chickens were examined with a hematoxylin–eosin (H&E) assay at 5 dpi and with scanning electron microscopy at 3 dpi and 5 dpi. For the group of chickens vaccinated with recombinant turkey herpesviruses containing the hemagglutinin (HA) gene from the H9N2 live vector vaccine (rHVT-H9), 1-day-old SPF chickens were vaccinated with 3000 plaque-forming units of rHVT-H9, which we had developed previously60. At 5 weeks after vaccination, the chickens were challenged intranasally with HB17 virus (106 EID50) in 0.1 mL. Oropharyngeal swabs were collected at 3, 5, and 7 dpi for viral detection and titrated by EID50. To test the cold-adapted attenuated avian H9N2 influenza vaccine (H9N2-LAIV) that we developed previously61, groups of 3-week-old SPF chickens were vaccinated intranasally with 106 EID50 of H9N2-LAIV in 0.4 mL. The chickens were challenged intranasally with 106 EID50 of H9N2 AIV at day 14 post vaccination. Oropharyngeal swabs were collected at 3, 5, and 7 dpi for viral detection and titrated by EID50. In the contact groups, chickens were placed in physical contact with inoculated birds at 24 h postchallenge. Oropharyngeal swabs were collected from all inoculated and contact chickens at 3, 5, and 7 days postcontact (dpc) for viral detection and titration. For the chickens from which tissues were collected, or at the end of the experiment, each chicken was anesthetized by injecting Zoletil® (tiletamine–zolazepam, 20 μg/g; Virbac) into the breast muscle at a dose of 0.18 ml/kg, followed by cervical dislocation once full anesthesia was confirmed by the absence of a palpebral reflex.
Histopathological examination
A portion of turbinate, larynx, trachea, bronchus, and lung from each chicken was immersed in 10% neutral-buffered formalin solution, processed routinely, and embedded in paraffin. The tissues were sectioned to a thickness of 4 µm, and the paraffin sections were stained with H&E for histopathological examination62.
Scanning electron microscopy
Freshly incised samples of the larynx and trachea tissues were washed with saline and fixed in an excess volume of 2% glutaraldehyde in isotonic 50 mM cacodylate buffer containing 150 mM NaCl (pH 7.4). The fixed tissues were washed in the same cacodylate buffer, dehydrated in increasing concentrations of ethanol (30%–100% v/v), dried with hexamethyldisilazane, and sputter-coated with gold-palladium (Polaron™ E5100)63. High-resolution micrographs were obtained from randomly chosen areas in each sample to avoid selection bias.
Viral infection of donor chickens and passage of virus through contact transmission
The transmission study (naïve chain/group) was performed in a group of SPF chickens. Two “seeders” were inoculated intranasally with 106 EID50 of HB17. We used real-time quantitative PCR (qPCR) detection of the virus to confirm of virus excretion by the chickens, and then introduced two immunologically naïve chickens into the same cage. When virus excretion was detected in the introduced naïve chickens, they were moved to a new cage and two further naïve chickens were introduced into the cage. This procedure was repeated six times to establish a transmission chain25. Oropharyngeal swabs were collected every day postinoculation or postcontact, immersed in viral transport medium (VTM; phosphate-buffered saline [PBS] supplemented with 2% tryptose phosphate buffer broth, 2% penicillin/streptomycin, and 2% amphotericin B), vortexed, aliquoted, and stored at −80 °C.
To study viral transmission in vaccinated chickens, a group of chickens was vaccinated with the inactivated H9N2 HB17 virus in an oil-in-water adjuvant; one dose was administered at 3 weeks of age and a second dose 2 weeks later. The chains were initiated with two SPF chickens challenged with 106 EID50 of HB17. When virus excretion was detected with qPCR, two more vaccinated chickens (HI titer, 7–8 log2) were introduced into the same cage, as described above. Once these two vaccinated chickens (P1) were shown to be infected, they were placed in a separate cage and co-housed with the next two recipient vaccinated chickens (P2). The transmission experiment was continued for each immunization group until six passages had been completed.
Detection of virus shedding with reverse transcription (RT)–qPCR
Oropharyngeal sampling was conducted daily in each chicken. The swabs were placed in 1 ml of VTM and vortexed before analysis. The samples were assessed for virus shedding with RT–PCR that detected transcripts of the matrix (M) gene. Transmission was deemed to have occurred when the new recipient chickens returned a positive rapid test, or a positive RT–qPCR result (Ct ≤ 30). A two-step qPCR assay was developed to detect the M gene of HB17 and was optimized for rapid analysis (sample to result in < 4 h), to facilitate the movement of chickens between cages on the same day. Viral RNA was extracted from the oropharyngeal swab with the QIAamp Viral RNA Kit. RNA was reverse transcribed with random hexamer primer and SuperScript™ II Reverse Transcriptase (Invitrogen Life Sciences); the reactions were incubated at room temperature for 10 min, at 42 °C for 30 min, and at 70 °C for 15 min. The qPCR assay was performed with SYBR™ Green mix (Thermo Fisher Scientific) with primers specific for the matrix (M) gene. The samples were incubated at 94 °C for 15 min, and then with 40 cycles of 94 °C for 15 s, 55 °C for 15 s, and 72 °C for 15 s. This was followed by a ramp from 70 to 90 °C in 5 °C increments and a hold time of 10 s. The analysis of serial dilutions of a plasmid containing the HB17 M gene was used to construct a standard curve. The results were analyzed with the QuantaSoft™ software.
Illumina library preparation and sequencing
cDNA corresponding to all eight genomic segments was amplified from 3 μl of viral RNA with SuperScript™ III One-Step RT-PCR with Platinum Taq High Fidelity (Invitrogen). The reactions contained 0.5 μl of SuperScript™ III Platinum Taq Mix, 12.5 μl of 2× reaction buffer, 8 μl of diethyl pyrocarbonate (DEPC)-treated water, and 0.2 μl of 10 μM Uni12/Inf1, 0.3 μl of 10 μM Uni12/Inf3, and 0.5 μl of 10 μM Uni13/Inf1 universal influenza A primers. The thermocycling protocol was: 42 °C for 60 min, 94 °C for 2 min, followed by five cycles of 94 °C for 30 s, 44 °C for 30 s, and 68 °C for 3 min, and then by 28 cycles of 94 °C for 30 s, 57 °C for 30 s, and 68 °C for 3 min. Amplification of all eight segments was confirmed with gel electrophoresis, and 750 ng of each cDNA mixture was sheared to an average size of 300–400 bp with a Covaris S220 focused ultrasonicator. Sequencing libraries were prepared with the NEBNext® Ultra™ DNA Library Prep Kit for Illumina (NEB E7370L), Agencourt AMPure XP beads (Beckman Coulter A63881), and NEBNext® Multiplex Oligos for Illumina (NEB E7600S). The final concentration of each barcoded library was determined with the Quant-iT™ PicoGreen™ dsDNA Assay Kit (Thermo Fisher Scientific), and equal nanomolar concentrations were pooled. Residual primer dimers were removed with the gel isolation of the 300–500 bp bands, which were then purified with the GeneJet Gel Extraction Kit (Thermo Fisher Scientific). The purified pooled libraries were sequenced on the Illumina HiSeq 2500 system with 2 × 125 nucleotide paired-end reads. All raw sequence data have been deposited at the NCBI sequence read archive (BioPro-ject submission ID: PRJNA1117513).
Variant detection
The sequencing reads that passed the standard Illumina quality control filters were binned by index and aligned to the reference genome with Bowtie2. Single-nucleotide variants (iSNVs) were identified and analyzed with DeepSNV, which relies on a clonal control to estimate the local error rate within a given sequence context and to identify any strand bias in base calling. The clonal control was a library prepared in an identical fashion from eight plasmids containing the genome of the corresponding circulating reference strain, and was sequenced in the same flow cell to control for batch effects. True positive iSNVs were identified from the raw output tables with the following filtering criteria, in R: (i) Bonferonni corrected P value < 0.01, (ii) average MapQ score on variant reads > 30, (iii) average phred score on variant positions > 35, (iv) average position of variant call on a read > 32 and <94, and (v) variant frequency > 0.01, with iSNVs below 1% frequency considered noise and excluded. To ensure detection accuracy, stringent quality control measures were implemented, excluding samples with viral genome copy numbers below 10³ copies/μL to minimize sequencing errors and false-positive variants, and using synthetic controls to detect contamination or technical artifacts35. We only considered iSNVs identified in a single RT–PCR and sequencing library for samples with copy numbers > 105 genomes/μl of transport medium or in two separate RT–PCRs and sequencing libraries for samples with copy numbers of 103–105 genomes per μl. The strategy used for variant calling is described at https://github.com/lauringlab/variant_pipeline, at which all codes can be found.
Transmission bottleneck analysis
The bottleneck size was calculated with the exact beta-binomial method described in previous research29. Briefly, by deep sequencing viral populations from donor and recipient hosts within the transmission chain, single nucleotide variants (SNVs) were identified to track changes in viral diversity during transmission. To model the probability of SNV passage, binomial and beta-binomial distributions were applied to capture the stochastic effects of genetic drift. A Bayesian framework was then used to infer the most probable transmission bottleneck size representing the number of viral particles that successfully establish infection in the recipient host. Because most samples in the analysis had < 50,000 mapped reads, we considered all sites in the genome, including those in the 3′- and 5′-untranslated regions (UTRs), but excluding the poly-A tail (positions 29,865–29,903), the 18 “highly shared” sites, and those identified in synthetic controls. All sites with > 3% MAF and > 100 read in the assumed source individual were used in the analysis. In the recipients, all reads at these sites were considered, with an error threshold of 0.5% MAF. The 95% confidence intervals (CIs) were calculated with a likelihood ratio test based on previous research29.
Generation of DIPs during high-multiplicity-of-infection (MOI) infections
Confluent MDCK cells were infected in triplicate with HB17 at an MOI of 10 50% tissue culture infectious doses (TCID50)/cell. To harvest the intracellular viral RNA at 3, 6, and 14 h postinfection (hpi), the cells were washed twice with PBS, and RNA was extracted with the Qiagen RNeasy Kit, according to the manufacturer’s instructions. To extract the extracellular RNA from packaged virions, the supernatant was collected from infected cells at 14 and 24 hpi, clarified, and incubated for 30 min with RNase A (0.25 μg). We further confirmed the presence of DIPs. Briefly, viral RNA was extracted by using the QIAamp Viral RNA Minikit (Qiagen, Hilden, Germany); reverse transcriptasePCR (RT-PCR) was then performed by using the PrimeScript One-Step RT-PCR kit (Takara, Beijing, China) with the MBTUni-12 and MBTUni-13 primers. PCR products were purified using the PureLink PCR kit (<300-nt cutoff) and visualized on a 1% agarose gel. The accumulation of DIPs deletion junctions is reflected by the disappearance of the polymerase segments (∼2.3 kb) and the appearance of a smear below the NS segment (∼0.9 kb) ranging from ∼0.3 to 0.8 kb (Supplementary Fig. 3A).
Sequencing analysis of DIP-associated junctions
The raw sequencing reads were quality filtered with Trimmomatic (v0.36) (parameters:ILLUMINACLIP:TruSeq3-PE-2.fa:2:15:10SLIDINGWINDOW:3:20LEADING:28TRA ILING: 28)64, and any reads of < 75 nucleotides were removed from the datasets. The paired reads were concatenated into one file and treated as single-ended when aligned end-to-end against the wild-type reference sequences with Bowtie 2 (v2.3.1) (parameters: –score-min L, 0, −0.3). Bowtie 2 passes these values to a linear function to calculate the total penalty score [f(x) = −0.3 × x], where x is the read length. This option allows Bowtie 2 to align the sequence based on the read length, i.e., the penalty score for a 250-nucleotide read is almost the same as that for a 200-nucleotide read.
The algorithm ViReMa (v0.10) was then used to analyze the remaining unaligned reads (putative junction-spanning reads) (parameters: -DeDup–MicroInDel_Length 20–Defuzz 3–N 1–X 8). The DIP-associated deletion junctions and their read support were extracted from the ViReMa output files and sorted per segment, using an in-house Perl script for data analysis and visualization. To detect any MDCK genome leakage, the datasets were aligned against the dog genome (assembly CanFam3.1)31. All scripts are available at https://github.com/BROOKE LAB/Influenza-virus-DI-identification-pipeline.
Generation of recombinant and mutant H9N2 viruses with reverse genetics
All eight gene segments of the HB17 and GD17 (H7N9) viruses were amplified with RT–PCR, and individually cloned into the dual-promoter plasmid, pHW2000. The recombinant virus was generated with reverse genetics in 293 T cells65. The following reassortant virus were generated with reverse genetics: H9N2HA + NA:H7N9, containing the H9 and N2 surface protein genes and all six H7N9 internal protein genes; the H9N2HA:H7N9 reassortant virus, containing the HA gene from H9N2 and all other genes from H7N9; the H9N2NA:H7N9 reassortant virus, containing the NA gene from H9N2 and all other genes from H7N9; and the reciprocal viruses H7N9HA + NA:H9N2, H7N9HA:H9N2, and H7N9NA:H9N2. The NP-N417D, M1-V219I, or NS1-R140W mutation was introduced into the corresponding gene with the QuikChange Site-Directed Mutagenesis Kit (Agilent, Santa Clara, CA, USA), according to the manufacturer’s instructions. The primer sequences used are available upon request. The rescued viruses containing a single mutation were designated rgHB17(NS1-R140W), rgHB17(M1-V219I), and rgHB17(NP-N417D). The rescued virus containing double mutations was designated rgHB17(NS1-R140W M1-V219I). The rescued virus containing triple mutations was designated rgHB17(NS1-R140W M1-V219I NP-N417D). All viruses were propagated in 9–11-day-old SPF chicken embryos and their sequences verified before use.
Viral titration and growth kinetics in cells
The TCID50 values of viruses were determined in MDCK cells by incubating the cells with 10-fold serially diluted viruses at 37 °C for 72 h. The TCID50 values were calculated with the Reed–Muench method. The multistep replication kinetics were determined in CEF infected with each virus at an MOI of 0.001, overlain with serum-free DMEM containing 1 μg/ml TPCK–trypsin (Sigma-Aldrich, St. Louis, MO, USA), and incubated at 37 °C. The supernatants were sampled at 12, 24, 36, 48, 60, and 72 hpi and titrated in MDCK cells in 96-well plates. Three independent experiments were performed65.
Confocal microscopy
DF1 cells were infected with HB17 or mutant virus at an MOI of 3 for 4, 8, 12, and 16 h, in parallel with uninfected controls. The cells were fixed in 4% paraformaldehyde and permeabilized with 0.2% Triton X-100, then blocked for 1 h at room temperature with 2% bovine serum albumin and 7% fetal bovine serum in PBS. The cells were incubated overnight at 4 °C with primary antibody, and then with Alexa-Fluor-594-conjugated goat anti-mouse IgG or Alexa-Fluor-488-conjugated goat anti-rabbit IgG for 1 h. Finally, coverslips were mounted onto microscope slides with 10–20 μl of 4′,6-diamidino-2-phenylindole (DAPI) for 3 min, and examined with confocal microscopy. Images were produced with a FluoView FV3000 confocal laser scanning microscope (Olympus) and analyzed with the Imaris 9.2 platform66.
Transmission electron micrographs
To visualize the virions, MDCK cells were infected with virus at an MOI of 8. At 18 hpi, the cells were fixed with 2.5% glutaraldehyde in 0.1 M cacodylate buffer for 2–3 h at room temperature or overnight at 4 °C. The cells were then embedded in Eponate 12 resin, sectioned to 80 nm, and stained with 5% uranyl acetate and 2% lead citrate. After sample preparation, the grids were imaged at 75 kV with a JEOL 1200EX transmission electron microscope.
Mouse studies
Groups of 6–8-week-old female BALB/c mice (Charles River Laboratories, Beijing, China) were anesthetized with Zoletil® and inoculated intranasally with 50 μl of serially 10-fold diluted infectious virus in PBS. The test virus clones (106 TCID50) were used for inoculation. Three mice from each group were euthanized at 3 dpi, and the viral titers in their lungs determined by infection of MDCK cells. The remaining three mice were monitored daily for weight loss for 14 days. Euthanasia was performed by trained personnel under deep anesthesia using Zoletil® followed by cervical dislocation.
Intracellular staining of T lymphocytes expressing interferon gamma (IFN-γ)
Lung tissue was digested with 50 U/ml DNase (Sigma-Aldrich) and 1 mg/ml collagenase A (Sigma-Aldrich) in RPMI 1640 medium (Invitrogen) containing 5% FBS (Invitrogen) for 1 h at 37 °C. The digested lungs were homogenized and the red blood cells lysed. The cell suspensions from the lungs were then incubated in RPMI 1640 cell-culture medium (10% FBS, 1% penicillin/streptomycin) for 6 h at 37 °C under 5% CO2 in the presence of H9N2 AIV (MOI = 1), 10 U/ml rhIL-2 (PeProTech, Rocky Hill, NJ, USA), and 1 μl/ml brefeldin A (eBioscience, San Diego, CA, USA) for 0 h to test for CD8+ responses or for 2 h to test for CD4+ responses. After 6 h, the cells were washed with PBS containing 2% FBS, then stained with anti-CD8-SPRD and anti-CD4-PE (Southern Biotech, Birmingham, AL, USA) for 30 min on ice. For intracellular IFN-γ staining, the cells were permeabilized with Cytofix/Cytoperm™ (BD Biosciences, San Diego, CA, USA), stained with rabbit anti-chicken IFN-γ antibody (AbD Serotec, Raleigh, NC, USA) for 30 min on ice, and then stained with fluorescein isothiocyanate (FITC)-labeled anti-rabbit antibody (Abcam, Cambridge, UK) for 30 min on ice. The stained cells were evaluated for the expression of CD4, CD8, and IFN-γ with flow cytometry (BD Biosciences). The data were analyzed with the FlowJo software (Tree Star Inc., San Carlos, CA, USA).
Antibody detection with enzyme-linked immunosorbent assays (ELISAs)
Turbinate tissues were collected and repeatedly washed with 0.5 ml of PBS. The lavage liquid was collected and centrifuged at 3000×g at 4 °C for 10 min. Sera were collected with peripheral venous sampling and centrifuged for 15 min at 4000×g. Formalin-inactivated influenza viruses were used as the detection antigens. Briefly, a 96-well Costar medium binding plate (Fisher Scientific Products, Pittsburgh, PA, USA) was coated with 1 μg of influenza virus antigen in 30 mM PBS (pH 7.4) per well overnight at 4 °C. The plate was washed and incubated with the test samples diluted in PBS containing 5% dry milk for 1.5–2 h at room temperature. After three washes, the samples were incubated for 1 h with biotin-labeled goat anti-mouse IgG or IgA antibody. After three additional washes, the plates were incubated with a streptavidin–horseradish peroxidase (HRP) conjugate (Southern Biotechnology) for 1 h at room temperature. Finally, the plates were washed and developed for 15 min with the substrate 3,3′,5,5′-tetramethylbenzidine (TMB; Bio-Rad Laboratories, Melville, NY, USA). The reaction was stopped by the addition of 1 N sulfuric acid. The plates were read at a wavelength of 450 nm with an EMax® Plus Microplate Reader (Molecular Devices, Sunnyvale, CA, USA).
IFN-γ ELISpot analysis
A chicken IFN-γ ELISpot assay was performed with the Chicken IFN-γ ELISpot BASIC Kit (Mabtech), according to the instructions provided. Briefly, 35% ethanol was used to activate the polyvinylidene fluoride membrane for 1 min at room temperature. Next, 96-well ELISpot plates (Millipore) were coated with 3 μl of mouse anti-chicken IFN-γ antibody (Invitrogen) and incubated overnight at 4 °C. After incubation, the plates were washed with PBS and blocked with RPMI 1640 medium containing 10% FBS for 2 h at room temperature. Following the removal of the blocking solution, 106 cells from the turbinates of chickens vaccinated with either PBS or a specific vaccine were added to the wells and incubated for 48 h. The plates were then washed with PBS and incubated with 100 μl of HRP-conjugated streptavidin (BD Biosciences) for 1 h at room temperature. After the plates were washed with PBS, 100 μl of TMB chromogenic solution was added to each well, and the reaction was allowed to develop until visible spots appeared. The reaction was stopped by washing the plates with ultrapure water. The spots were counted with an automatic spot analyzer (Mabtech).
Statistical analysis
Differences between experimental groups were assessed with analysis of variance (ANOVA). Values of P < 0.05 were considered to indicate statistically significant differences. *P < 0.05 and **P < 0.01.
Data availability
Sequence data that support the findings of this study have been deposited in the NCBI sequence read archive (BioPro-ject submission ID: PRJNA1117513, https://dataview.ncbi.nlm.nih.gov/object/PRJNA1117513?reviewer=i5h4a4hbqh31mja4savn05bmqo). All other data supporting the findings of this study are available within the article and its Supporting Information files.
Change history
12 May 2025
In the Acknowledgements section, the funding source Hainan Health Science and Technology Innovation joint project and grant number SQ2023WSJK0575 should have read Joint Program on Health Science & Technology Innovation of Hainan Province and grant number WSJK2024MS235.
References
Zhang, J. et al. Mutational antigenic landscape of prevailing H9N2 influenza virus hemagglutinin spectrum. Cell Rep. 42, 113409 (2023).
Peacock, T. H. P., James, J., Sealy, J. E. & Iqbal, M. A global perspective on H9N2 avian influenza virus. Viruses 11, https://doi.org/10.3390/v11070620 (2019).
Gu, M., Xu, L., Wang, X. & Liu, X. Current situation of H9N2 subtype avian influenza in China. Vet. Res. 48, 49 (2017).
Wang, Z. et al. Mixed selling of different poultry species facilitates emergence of public-health-threating avian influenza viruses. Emerg. Microbes Infect. 12, 2214255 (2023).
Sun, Y. & Liu, J. H9N2 influenza virus in China: a cause of concern. Protein Cell 6, 18–25 (2015).
Wang, J. et al. Infectivity and transmissibility of avian H9N2 influenza viruses in pigs. J. Virol. 90, 3506–3514 (2016).
Sun, X. et al. Evidence of avian-like H9N2 influenza A virus among dogs in Guangxi, China. Infect. Genet. Evol. 20, 471–475 (2013).
Zhou, H. et al. Serological evidence of avian influenza virus and canine influenza virus infections among stray cats in live poultry markets, China. Vet. Microbiol. 175, 369–373 (2015).
Qian, Z. et al. Molecular characteristics of H9N2 influenza viruses isolated from farmed raccoon dogs and arctic foxes in China. Res. Vet. Sci. 135, 542–546 (2021).
Yan, Y. et al. Genetic characterization of H9N2 avian influenza virus in plateau pikas in the Qinghai Lake region of China. Arch. Virol. 162, 1025–1029 (2017).
Peiris, M. et al. Human infection with influenza H9N2. Lancet (Lond., Engl.) 354, 916–917 (1999).
Bi, Y., Li, J. & Shi, W. The time is now: a call to contain H9N2 avian influenza viruses. Lancet Microbe 3, e804–e805 (2022).
Bi, Y. et al. Dominant subtype switch in avian influenza viruses during 2016–2019 in China. Nat. Commun. 11, 5909 (2020).
Song, W. & Qin, K. Human-infecting influenza A (H9N2) virus: a forgotten potential pandemic strain? Zoonoses Public Health 67, 203–212 (2020).
Sun, Y. et al. High genetic compatibility and increased pathogenicity of reassortants derived from avian H9N2 and pandemic H1N1/2009 influenza viruses. Proc. Natl Acad. Sci. USA 108, 4164–4169 (2011).
World Health Organization. Risk Assessments and Summaries of Influenza at the Human–Animal Interface. 2024. Available from: https://www.who.int/teams/global-influenza-programme/avian-influenza/monthly-risk-assessment-summary.
Yang, R. et al. Human infection of avian influenza A H3N8 virus and the viral origins: a descriptive study. Lancet Microbe 3, e824–e834 (2022).
Bi, Y. et al. Genesis, evolution and prevalence of H5N6 avian influenza viruses in China. Cell Host Microbe 20, 810–821 (2016).
Ye, G. et al. Phylogenetic analysis and pathogenicity assessment of two strains of avian Influenza virus subtype H9N2 isolated from migratory birds: high homology of internal genes with human H10N8 virus. Front. Microbiol. 7, 57 (2016).
Pu, J. et al. Evolution of the H9N2 influenza genotype that facilitated the genesis of the novel H7N9 virus. Proc. Natl Acad. Sci. USA 112, 548–553 (2015).
Li, C., Bu, Z. & Chen, H. Avian influenza vaccines against H5N1 ‘bird flu. Trends Biotechnol. 32, 147–156 (2014).
Li, C. & Chen, H. H7N9 Influenza Virus in China. Cold Spring Harbor Perspect. Med. 11, https://doi.org/10.1101/cshperspect.a038349 (2021).
Zhang, P. et al. Characterization of H9N2 influenza viruses isolated from vaccinated flocks in an integrated broiler chicken operation in eastern China during a 5 year period (1998–2002). J. Gen. Virol. 89, 3102–3112 (2008).
Choi, J. G. et al. An inactivated vaccine to control the current H9N2 low pathogenic avian influenza in Korea. J. Vet. Sci. 9, 67–74 (2008).
Murcia, P. R. et al. Evolution of an Eurasian avian-like influenza virus in naïve and vaccinated pigs. PLoS Pathog. 8, e1002730 (2012).
Guarnaccia, T. et al. Antigenic drift of the pandemic 2009 A(H1N1) influenza virus in A ferret model. PLoS Pathog. 9, e1003354 (2013).
Debbink, K. et al. Vaccination has minimal impact on the intrahost diversity of H3N2 influenza viruses. PLoS Pathog. 13, e1006194 (2017).
McCrone, J. T. & Lauring, A. S. Genetic bottlenecks in intraspecies virus transmission. Curr. Opin. Virol. 28, 20–25 (2018).
McCrone, J. T. et al. Stochastic processes constrain the within and between host evolution of influenza virus. Elife 7, https://doi.org/10.7554/eLife.35962 (2018)
Frensing, T., Pflugmacher, A., Bachmann, M., Peschel, B. & Reichl, U. Impact of defective interfering particles on virus replication and antiviral host response in cell culture-based influenza vaccine production. Appl. Microbiol. Biotechnol. 98, 8999–9008 (2014).
Alnaji, F. G. et al. Sequencing framework for the sensitive detection and precise mapping of defective interfering particle-associated deletions across Influenza A and B viruses. J. Virol. 93, https://doi.org/10.1128/JVI.00354-19 (2019).
Frensing, T. et al. Continuous influenza virus production in cell culture shows a periodic accumulation of defective interfering particles. PLoS ONE 8, e72288 (2013).
Laske, T., Heldt, F. S., Hoffmann, H., Frensing, T. & Reichl, U. Reprint of “Modeling the intracellular replication of influenza A virus in the presence of defective interfering RNAs. Virus Res. 218, 86–95 (2016).
Kupke, S. Y. et al. Single-cell analysis uncovers a vast diversity in intracellular viral defective interfering RNA content affecting the large cell-to-cell heterogeneity in Influenza A virus replication. Viruses 12, https://doi.org/10.3390/v12010071 (2020).
Eisfeld, A. J., Neumann, G. & Kawaoka, Y. At the centre: influenza A virus ribonucleoproteins. Nat. Rev. Microbiol. 13, 28–41 (2015).
Li, T. et al. The shape of pleomorphic virions determines resistance to cell-entry pressure. Nat. Microbiol. 6, 617–629 (2021).
Hale, B. G., Randall, R. E., Ortín, J. & Jackson, D. The multifunctional NS1 protein of influenza A viruses. J. Gen. Virol. 89, 2359–2376 (2008).
Wang, X. et al. Emergence of a new designated clade 16 with significant antigenic drift in hemagglutinin gene of H9N2 subtype avian influenza virus in eastern China. Emerg. microbes Infect. 12, 2249558 (2023).
Li, X. et al. Genetics, receptor binding property, and transmissibility in mammals of naturally isolated H9N2 Avian Influenza viruses. PLoS Pathog. 10, e1004508 (2014).
Sun, Y. et al. Evaluation of the protective efficacy of a commercial vaccine against different antigenic groups of H9N2 influenza viruses in chickens. Vet. Microbiol. 156, 193–199 (2012).
Zhang, J., Huang, L., Liao, M. & Qi, W. H9N2 avian influenza viruses: challenges and the way forward. Lancet Microbe https://doi.org/10.1016/S2666-5247(22)00305-6 (2022).
Corbett, K. S. et al. Immune correlates of protection by mRNA-1273 vaccine against SARS-CoV-2 in nonhuman primates. Science 373, eabj0299 (2021).
McMahan, K. et al. Correlates of protection against SARS-CoV-2 in rhesus macaques. Nature 590, 630–634 (2021).
Tokunoh, N. et al. A nasal vaccine with inactivated whole-virion elicits protective mucosal immunity against SARS-CoV-2 in mice. Front. Immunol. 14, 1224634 (2023).
Frantz, P. N. et al. A live measles-vectored COVID-19 vaccine induces strong immunity and protection from SARS-CoV-2 challenge in mice and hamsters. Nat. Commun. 12, 6277 (2021).
Deng, S. et al. An intranasal influenza virus-vectored vaccine prevents SARS-CoV-2 replication in respiratory tissues of mice and hamsters. Nat. Commun. 14, 2081 (2023).
Litao, L. et al. Field production efficiency investigation of broilers immunized with a turkey herpesvirus vector vaccine expressing hemagglutinin from H9N2 subtype avian influenza virus. Vaccine 42, 126436 (2024).
Chen, S. et al. A live attenuated H9N2 avian influenza vaccine prevents the viral reassortment by exchanging the HA and NS1 packaging signals. Front. Microbiol. 11, 613437 (2020).
Murcia, P. R. et al. Evolution of equine influenza virus in vaccinated horses. J. Virol. 87, 4768–4771 (2013).
Hoelzer, K. et al. Intrahost evolutionary dynamics of canine influenza virus in naive and partially immune dogs. J. Virol. 84, 5329–5335 (2010).
Li, J. et al. Amino acid substitutions in antigenic region B of hemagglutinin play a critical role in the antigenic drift of subclade 2.3.4.4 highly pathogenic H5NX influenza viruses. Transbound. Emerg. Dis. 67, 263–275 (2020).
Li, X., Gao, Y. & Ye, Z. A single amino acid substitution at residue 218 of hemagglutinin improves the growth of influenza A(H7N9) candidate vaccine viruses. J. Virol. 93, https://doi.org/10.1128/jvi.00570-19 (2019).
Yao, L. et al. Identification of antigenic epitopes in the haemagglutinin protein of H7 avian influenza virus. Avian Pathol.: J. W. V. P. A. 49, 62–73 (2020).
Kosik, I. et al. Influenza A virus hemagglutinin glycosylation compensates for antibody escape fitness costs. PLoS Pathog. 14, e1006796 (2018).
Fonville, J. M., Marshall, N., Tao, H., Steel, J. & Lowen, A. C. Influenza virus reassortment is enhanced by semi-infectious particles but can be suppressed by defective interfering particles. PLoS Pathog. 11, e1005204 (2015).
Laske, T., Heldt, F. S., Hoffmann, H., Frensing, T. & Reichl, U. Modeling the intracellular replication of influenza A virus in the presence of defective interfering RNAs. Virus Res. 213, 90–99 (2016).
Wu, C. A., Harper, L. & Ben-Porat, T. Molecular basis for interference of defective interfering particles of pseudorabies virus with replication of standard virus. J. Virol. 59, 308–317 (1986).
Varble, A. et al. Influenza A virus transmission bottlenecks are defined by infection route and recipient host. Cell Host Microbe 16, 691–700 (2014).
Sun, Y. et al. Genotypic evolution and antigenic drift of H9N2 influenza viruses in China from 1994 to 2008. Vet. Microbiol. 146, 215–225 (2010).
Liu, L. et al. Recombinant Turkey herpesvirus expressing H9 hemagglutinin providing protection against H9N2 avian influenza. Virology 529, 7–15 (2019).
Wei, Y. et al. Generation and protective efficacy of a cold-adapted attenuated avian H9N2 influenza vaccine. Sci. Rep. 6, 30382 (2016).
Liu, L. et al. Truncation of PA-X contributes to virulence and transmission of H3N8 and H3N2 canine influenza viruses in dogs. J. Virol. 94, e00949–20 (2020).
Alnaeem, A. et al. Scanning electron microscopic findings on respiratory organs of some naturally infected dromedary camels with the lineage-B of the middle east respiratory syndrome coronavirus (MERS-CoV) in Saudi Arabia—2018. Pathogens (Basel, Switzerland) 10, https://doi.org/10.3390/pathogens10040420 (2021).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics (Oxf., Engl.) 30, 2114–2120 (2014).
Chen, M. Increased public health threat of avian-origin H3N2 influenza virus caused by its evolution in dogs. Elife 12, e83470 (2023).
Ma, C. et al. p21 restricts influenza A virus by perturbing the viral polymerase complex and upregulating type I interferon signaling. PLoS Pathog. 18, e1010295 (2022).
Acknowledgements
This work was supported by the National Key Research and Development Program of China [Grant Number 2022YFD1801005] and the National Natural Science Foundation of China [Grant Number 32192450], Joint Program on Health Science & Technology Innovation of Hainan Province (Grant Number: WSJK2024MS235).
Author information
Authors and Affiliations
Contributions
YP.S. and JH.L. designed the study. Z.H., H.A., Z.W., SY.H., XX.X., MY.C., JX.W. and W.Y. conducted the experiments; Z.H., H.A., Z.W., HL.S., J.P., JY.S., and CB.B. analyzed the data; Z.H., H.A., Z.W., KC.C., YP.S., and JH.L. wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Hu, Z., Ai, H., Wang, Z. et al. Impact of inactivated vaccine on transmission and evolution of H9N2 avian influenza virus in chickens. npj Vaccines 10, 67 (2025). https://doi.org/10.1038/s41541-025-01115-y
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41541-025-01115-y
