
Abstract
Per- and polyfluoroalkyl substances (PFAS) are a class of synthetic chemicals that are highly resistant to degradation because of the strong C-F bond and their unique physico-chemical properties. Several techniques, both destructive and non-destructive, have been explored for removing PFAS from contaminated water. However, the most desirable techniques, ideally capable of effective separation and complete PFAS destruction and mineralization, have not progressed beyond bench-scale testing. This paper provides an overview of the existing treatment techniques demonstrated at laboratory, pilot, and industrial scales, and their associated treatment mechanisms. Insufficient data on pilot-scale and full-scale applications for PFAS remediation has limited the optimization and advancement of these systems at a large scale. Most research related to PFAS-remediation is based on laboratory-scale studies under ideal conditions that do not represent the complexity of PFAS-contaminated media. Factors such as inhibition by competing background compounds and secondary water or air pollution limit the application of some PFAS removal techniques at full-scale. Additionally, high energy intensity, cost, and inappropriate reactor design restrict the scalability of some proposed innovations. Here, we propose integrated systems and treatment trains as potential approaches to effectively remove and destroy PFAS from contaminated waters. This review also offers and contextualizes implementation barriers and scalable approaches for PFAS treatment.
Similar content being viewed by others


Introduction
Per- and polyfluoroalkyl substances (PFAS) are a class of amphipathic molecules with a hydrophilic, charged head group and a hydrophobic, fluorinated tail. Their reactivity and toxicity vary greatly depending on the type and size of these two groups. PFAS are thermally stable and highly resistant to various forms of degradation, including biodegradation, photolysis, and hydrolysis, due to the strong carbon–fluorine (C–F) bond (105 kcal/mol)1,2,3,4. They are commonly referred to as ‘forever chemicals’ because of their persistence in the environment. Other properties of PFAS, such as hydrophilicity and solubility, also pose significant challenges in conventional wastewater treatment processes, as the treatment plants are not equipped to adequately remove PFAS5. There are over 15,000 types of PFAS, but only a select few; such as perfluorooctanoic acid (PFOA) and perfluorooctane sulfonate (PFOS) (both of which contain 8 carbon atoms) have been studied extensively6.
Among several emerging pollutants discovered between 1839 and 2019, PFOA and PFOS garnered a relatively high increase rate of consideration of 18.8% and 13.6% in the years 2018 and 20197. While PFOA and PFOS are the most widely used PFAS in various applications7, shorter chain PFAS can be generated during degradation processes of the long chain PFAS7,8,9,10,11,12. While high PFOA removal rates are standard in many reports, removal of short chain PFAS is much lower. This is because the degradation of long chain PFAS, like PFOA, leads to the generation of shorter chain PFAS, like PFBA, increasing their concentration overtime if they are not immediately degraded13,14,15,16,17,18. Figure 1 illustrates how shorter chain PFAS are generated by the breakdown of longer chain PFAS, here via photocatalytic or photolytic catalysis. In the case of direct photolysis, the C7F15· and CO2 will be generated from perfluorinated carboxyl anions as a result of photoelectrons facilitating the breakage of the C-C bond.

Concluded common pathway for photo-oxidative degradation of PFOA173.
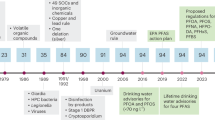
The United States Environmental Protection Agency (USEPA) has set limits for five PFAS, i.e., PFOA, perfluorooctane sulfonic acid (PFOS), perfluorononanoic acid (PFNA), perfluorohexanesulphonic acid (PFHxS), and hexafluoropropylene oxide dimer acid (HFPO-DA—often referred to as GenX Chemicals). The maximum contamination levels for drinking water are 4.0 ppt (ng/L) for PFOA and PFOS, and 10 ppt for PFNA, PFHxS, and HFPO-DA19. In China the drinking water limit is 80 ng/L for PFOA and 40 ng/L for PFOS20. These limits are extremely low and pose a significant challenge for the quantitative detection of these contaminants in aqueous media. Other nations have implemented standards that limit total organofluorine, making complete degradation of PFAS crucial21.
PFAS were developed in the 1940’s as part of the Manhattan project to enrich uranium, and their use grew and diversified rapidly, which resulted in widespread environmental pollution1,22. While high levels are usually found near PFAS manufacturing or discharge sites, some level of PFAS is nearly ubiquitous in water sources worldwide23,24. Even in the least inhabited parts of the world, PFAS contamination is present; for instance, blood tests have revealed PFAS in arctic polar bears25. Other data relating to the bioaccumulation of PFAS and their adverse health effects on humans have been reported4. PFAS exposure has been linked to many negative health effects, including liver and kidney diseases, reproductive issues, impaired immune system function, thyroid dysfunction, pregnancy hypertension, heightened cholesterol levels, and testicular cancer26. In infants, PFAS have been reported to delay mammary gland development, reduce growth, and inhibit response to vaccines27. Two of the commonly used PFAS (PFOA and PFOS) have also been reported as carcinogenic to humans (Group 1) and possibly carcinogenic to humans (Group 2B), for PFOA and PFOS respectively28. Furthermore, PFOS and PFOA concentrations in blood are reported to be much higher than any other PFAS6.
There are several sources from which PFAS may emanate, including manufacturing facilities, industrial complexes, and military fire training sites29,30. However, two of the most dominant sources of PFAS are landfill sites and wastewater treatment plants, both industrial and municipal30. From these sources, PFAS may enter the environment through spills, wastewater discharge, and surface water run-offs31. In order to curb the impacts of PFAS in the environment, there is a need for regulatory interventions from all countries. Past regulatory interventions have halted the production of long-chain PFAS with carbon chain length greater than six32. Short-chain PFAS were developed to replace historical PFAS, but both long-chain and short-chain PFAS persist in the environment and necessitate remediation27. Emerging information on the toxicity of short-chain PFAS has highlighted knowledge gaps surrounding their potential hazardous and bio-accumulative consequences.
Due to PFAS toxicity at extremely low concentrations, many sensitive removal technologies have been developed (Fig. 2). However, most of these technologies have limited efficiency and are not currently feasible for scale up. High cost and energy consumption, new infrastructure requirements, hazardous chemical usage, and the need for pretreatment or preconcentration hinders the use of many technologies in real-world settings33,34. These limitations have warranted a need to identify and develop new technologies at large scale that can eliminate PFAS efficiently to avoid contamination of the environment.

Non-destructive (blue) and destructive (red) techniques to treat PFAS-contaminated water.
This review examines various destructive and non-destructive techniques for the removal of PFAS from water and discusses research needs and opportunities to enhance. We provide an overview of current PFAS treatment techniques by summarizing the progress made to date and discussing the removal or degradation mechanisms underlying each technique and include suggestions for improvements. Additionally, recent progress in integrating non-destructive and destructive techniques to enhance cost-effectiveness in PFAS remediation will be discussed.
Remediation techniques of PFAS: fundamentals, mechanisms, applications, and limitations
Non-destructive separation techniques
Adsorption, ion exchange, and membrane separation are typical examples of non-destructive or phase separation techniques that remove PFAS without transforming them35. The merits and limitations of each technique are discussed, and applications of these techniques at laboratory, pilot and full-scale are compared. Additionally, factors hindering commercialization of these techniques are examined. Overall, non-destructive techniques do not eliminate liability and are hindered by the generation of PFAS-concentrated streams or waste, which require additional treatment before discharge into the environment36. The advantages and disadvantages of non-destructive technologies are presented in Fig. 3 and discussed in detail in the subsequent sections.

Advantages and disadvantages of non-destructive technologies are presented in the figure.
Ion exchange
Ion exchange (IX) is used in the removal of long and short-chain PFAS from water with high removal efficiency and long lifetime because of the ability of the resin to be regenerated37. IX takes advantage of the amphiphilic nature of PFAS through using the electrostatic interactions between the functional group head of PFAS molecules in the appropriate pH range37. The principle of action of these IX resins is as follows: (i) for non-anionic resins, PFAS are removed through the hydrophobic effect38; (ii) for anionic resins, PFAS are adsorbed via electrostatic interactions. Resins are comprised of a hydrophobic polymer backbone and charged functional groups, allowing the polymer to attract PFAS tails via hydrophobic interactions and attract PFAS anionic head groups by exchanging its counterions with the PFAS present in the liquid phase39.
The performance of PFAS-specific resins varies with properties of the resin and matrix composition (presence of other competing species). There are three main characteristics of a resin that influence the removal of PFAS: polymer composition, pore structure, and charged functional group. The polymer composition can be either styrene or acrylic, the pore structure can be gel or microporous, and functional groups are mostly amino complexes, quaternary amines, or tertiary amines39. A comparative study of PFAS removal in the presence of natural organic matter (NOM) used PFAS-specific (A592, polystyrene) and organic scavenger (A860, polyacrylic) resins, and showed high removal efficiency for both short and long-chain PFAS > 90%) while removing 12–15% of NOM38. In contrast, the organic scavenging resin was effective in removing NOM (60–70%) but required 3-fold higher contact time to reach a similar removal efficiency as the PFAS specific resin38. These results corroborate another study showing that polyacrylic-based resins were not as effective for PFAS removal40.
Mechanisms of PFAS removal using IX
Electrostatic and hydrophobic interactions are the fundamental mechanisms of IX41,42. DuPont recently detailed a mechanism of one of their commercial IX resins (IX AMBERLITE™ PSR2 Plus). This resin, like other resins that are used in PFAS applications, is functionalized with tri-N-butylamine, making the exchange sites more hydrophobic and subsequently increasing their affinity for PFAS tails. The hydrophobic effect is the result of PFAS moving towards non-polar hydrophobic surfaces with the help of entropy. It was demonstrated that the hydrophobic effect increases with the C-F chain length43,44. The impact of solution pH on PFAS absorption provided further evidence of the prevalence of electrostatic interactions during the IX process. The uptake of anionic PFAS is directly impacted by the surface charge of IX, which can be changed by protonating the surface functional groups through pH adjustments. Figure 4 depicts PFAS removal through IX principle and mechanism.

(i) A Typical IX resins have charged sites able to attract the oppositely charged ions, here a positively charged resin attracts negatively charged Cl-ions and PFAS. When PFAS and NOM-containing water is passed through the resin, PFAS and Cl-ions are exchanged resulting in the resin absorbing the PFAS, B PFAS is comprised of hydrophobic tail and hydrophilic head- the hydrophobic tail tends to gravitate towards non-polar entities, C the removal of PFAS through IX is impacted by the hydrophobic effect resulting in PFAS associating with NOM, further NOM can compete with PFAS on the resin. (ii) Anion resin comprised of positively charged exchange sites, divinylbenzene cross links, and neutral hydrophobic backbone38,39.
Laboratory-scale and pilot-scale IX applications in the removal of PFAS
IX resins have been extensively used at laboratory-scale, wherein most studies compared the effectiveness of GAC and IX in rapid small-scale column tests (RSSCT) that are considered a model for pilot-scale. Unlike GAC, IX had better PFAS removal efficiency, but at a higher cost. A recent study demonstrated that IX had better adsorption capability than GAC due to both electrostatic and hydrophobic interactions of the IX resin43. Elsewhere, IX resin Amb XAD4 outperformed Dow V493, Dow L493 and removed over 99.99% PFOS at a flow rate of 15 mL/min and was regenerated using a simple organic solvent45. When the performance of two full-scale treatment plant using IX was assessed for PFAS removal. The process was found effective in removing long-chain sulphonated PFAS with an efficiency of more than 92% and less effective in the removal of short-chain PFAS46. At pilot-scale, Woodard et al.38 examined the effectiveness of GAC and IX in the removal of PFAS. The pilot plant treated 422.645 gallons of water and was comprised of 2 parallel pilot test process units (GAC and IX) as depicted in Fig. 5a. For IX, the resin vessels were connected in series; in each vessel were 9 gallons of Sorbix A3F resin providing 2.5 min empty bed contact time (EBCT). The results showed that IX effectively removed PFOA and PFOS, and a mass-to-mass comparison illustrated that IX removed 1.66 mg total PFAS per gram of Sorbix A3F before breakthrough was observed. Key in this study was evaluating Sorbix A3F’s regeneration capability through successive regeneration tests. Using brine and an organic solvent to desorb the PFAS on the resin, the performance of the Sorbix A3F in the lead vessel was restored to near virgin performance. The regenerated resin treated 77.455 gallons of water and the detected PFAS concentration was 4.1 μg/L after 6.824 BVs of treatment. PFOS removal in chromium-plating wastewater was assessed using the pilot plant as shown in (Fig. 5b)47. Interestingly in this work, despite AER being known to effectively adsorb anionic PFAS, it exhibited different adsorption performance for the PFOS and 6:2 FTS. In this study, to estimate the cost to remove PFOS and 6:2 FTS, the costs of virgin adsorbents, disposal costs and power supply were taken into account and linked to the volume treated. Based on the calculations, it would cost $0.24/m3 and $1.99/m3 wastewater to remove PFOS and 6:2 FTS respectively. Elsewhere, anion exchange and GAC were used to treat PFAS concentrate after nanofiltration at pilot scale (Fig. 5c)48. Both raw untreated water and the reject from the membrane were passed through a 400 mL AIX resin A600 and flowrate maintained between 70 and 80 mL min−1. Data showed that the long chain PFAS were better removed than the shorter chain PFAS due to the adsorption of long chain PFAS such as PFOS, PFHxA, PFOA, and PFHxS driven by hydrophobic effects. The data obtained in this work shed light on the practicality of applying AIX to treat membrane reject. Some factors to consider include knowing the quality of the feedwater and the concentrations of PFAS in the water. The AIX noticeably removed 4.1-fold more PFAS mass per AIX volume from the membrane concentrate. Polystyrene and polyacrylic AER resins were applied in a pilot system capable of treating >180.000 BVs (2 min EBCT) and >750.000 BVs (0.5 min EBCT) in the 2 media vessels and lead vessels respectively (Fig. 5d). Single use AERs removed long chain PFCAs and PFSAs more efficiently than regenerable AERs up to 8 times. For the short chain, it was less significant i.e., >50% PFBA breakthrough within 50.000 BVs.

a A groundwater treatment plant building pilot test equipment installed with 2 parallel pilot test process units IX and GAC, b the system is comprised of parallel GAC adsorption unit and AER adsorption unit, prior to entering the parallel GAC adsorption unit and AER adsorption units the feed was passed through a bag filter. The primary focus of the pilot research took place in column 1 and column 2 was to solely guarantee the effluent's quality, c two nanofiltration membranes were connected in series to treat PFAS contaminated raw water, additionally the raw water and membranes’ reject were subjected to GAC and AIX, d pilot with 24 vessels arranged in eight parallel columns, grey vessels were omitted from the study due to cost constraints38,47,48,174.
Practical considerations and cost estimations of using IX in the removal of PFAS
Despite lab-scale success of IX, there are limitations, such as the cost of secondary waste management. Treating secondary waste uses expensive techniques and requires temperatures of ~1000 °C for incineration. In addition, incineration may emit volatile PFAS into the air, causing environmental concerns. While spent IX resin may be regenerated with brine which presents a safer option, brine still needs further treatment to eliminate toxicity. Other limitations of IX technique include the presence of competing anionic inorganic and organic species which reduces the efficiency of the resins. To select appropriate resins for IX, operators need to know the PFAS concentrations and type (i.e., long or short chained PFAS). Single-use resins have quaternary amines and long hydrocarbon chains which allow hydrophobic attractions and enhances PFAS uptake. Polystyrene resins too, have hydrophobic features allowing PFAS uptake. In locations where long chain PFAS are predominant, high-capacity single-use AERs would be ideal. However, where short chain PFAS are more abundant operators may consider regenerable resins. The configuration of the vessels of the resins in a pilot is something worth considering too. A configuration arranged in series and ensuring the lead vessel is operated to enhance breakthrough prior to media change is ideal. With this setup, operators will be able to swap out the resin with high PFAS loading in the lead vessel while still facilitating adsorption in the lag vessels that would ordinarily be swapped out at a single vessel changeout. Regarding EBCT; ≥2 min was found to optimal unlike shorter EBCTs (i.e., 0.5 min) as rapid detection of PFAS analytes were observed at shorter EBCTs. From an operation and maintenance perspective, the operational costs are often estimated by taking virgin resin costs, used resin disposal, analytical costs, and in other instances power supply costs are also included49. It is worthy of consideration that in full-scale other factors such as labor costs, water quality, depreciating equipment may be considered in estimating operating costs47. Media costs and disposal of IX were estimated at $17.60/kg and $5.300 (per changeout event) respectively50,51. Jiang et al.47 estimated it would cost $7.143–11.429/t (¥50.00080.000/t) to remove PFAS using AER. Operators could consider deploying reverse osmosis (RO) to treat the volume of effluent requiring incineration52. This has been demonstrated to reduce operational costs related to disposal costs and it was shown to reduce ~96.5% of the regeneration volume. In certain instances, cationic polymer-based hydrogels have been proven to be effective in the above.
Adsorption
Heterogeneous adsorption is widely used due to its economic feasibility and ability to adsorb various organic contaminants44. Carbon materials have been used extensively in PFAS removal from water via adsorption45. In PFAS applications, activated carbon is used in several forms: granular activated carbon (GAC) in carbon adsorption columns, commonly found in water and wastewater purification systems; powder-activated carbon (PAC), known as the carbon-activated sludge process for wastewater treatment; and activated carbon filter (ACF)53. GAC, PAC, and ACF differ in their morphological characteristics, such as particle size, surface charge, surface area, and composition. GAC has carbon particles with diameters ranging from 1.2 to 1.6 mm, PAC particles are ~0.1 mm, and ACF ranges from 5 to 30 μm54. The surface charge and carbon composition of AC are mostly determined by the activation process and the type of carbon material used during synthesis55,56. Based on performance, ACF exhibits the best adsorption characteristics for PFAS, followed by PAC, and then GAC57. The high efficiency of ACF is ascribed to its porous structure, large, exposed surface area, concentrated pore size distribution, and smaller fiber diameter57. However, ACF systems are mainly limited by cost of production, as they are synthesized from relatively expensive carbonaceous fibrous precursors such as pitch, phenolic resin, cellulose, and polyacrylonitrile fibers57,58. GAC and PAC are made from less expensive carbonaceous materials such as wood, lignite, coconut, peat, and coal. PAC’s smaller particle size compared to GAC necessitates the incorporation of a primary clarifier to remove PAC, increasing the energy usage and cost of PAC systems. Therefore, the use of GAC is common for PFAS removal. GAC effectively removes long-chain PFAS until breakthrough occurs, but is less effective for shorter-chain PFAS, leading to faster breakthrough and lower GAC loading capacities59.
In addition to carbon-based materials like AC used as adsorbents, metals such as Zn, Fe, and Cu have been used to modulate multi-walled carbon nanotubes for PFOA removal60. Another class of emerging adsorbents include β-Cyclodextrin polymers (CDPs), which was recently used as an alternative to remove 20 target PFAS61. In the study, two CDPs consisting of β‑ cyclodextrin monomers crosslinked with rigid aromatics containing weakly basic or permanently cationic functional groups were found to remove nearly all the PFAS from water after 4-h of contact time61.
Mechanism of PFAS removal with adsorption
The removal of PFAS through adsorption is based on the hydrophobic and electrostatic interactions, ligand exchange, and hydrogen bonding properties of the adsorbent (AC) and adsorbate (PFAS)61. In aqueous solutions of PFAS with pKa values up to 1 form a negatively charged head group61. This negatively charged head is attracted by the positively charged AC surface groups making electrostatic interactions the driving force for the adsorption process61,62. For hydrophobic adsorption mechanisms, an adsorption energy and site preference creates a force-field which displaces water from the hydrophobic surface. This enables short range attractive interactions between hydrophobic PFAS and the AC to take place62, thus facilitating PFAS adsorption. To cater for positively charged and neutral PFAS, AC surface chemistry is important in improving their removal. The significance of AC surface chemistry for PFAS adsorption may be due to the extraordinarily low pKa values (high acid strength) of PFAS, which restrict the charge neutralization during adsorption. In addition, positively charged surface groups in a mostly nonpolar environment are ideal conditions for adsorption of anionic perfluoroalkyl acids (PFAAs) and hydrophobic tail interactions61. Moreover, the presence of NOM molecules which mostly exist in a negatively charged state can lead to repulsive electrostatic interactions toward the anionic PFAS and in some cases hydrophobic interactions. This limits efficiency due to competition for adsorption on limited AC surface area44. Figure 6 depicts various PFAS adsorption removal mechanisms.

a Due to opposing charges, the negatively charged PFAS heads will be attracted to the positively charged surface (electrostatic attraction), b similar charges will result in repulsion, the negatively charged PFAS heads will repel a negatively charged surface (electrostatic repulsion), c when the hydrophobic tails of the PFAS interact with hydrophobic surfaces they tend to cluster together minimizing water interaction (hydrophobic interactions).
Pilot-scale and full-scale application of adsorption for removal of PFAS
The efficiency of a full-scale drinking water treatment plant using GAC was evaluated in removing 15 types of PFAS40. The treatment plant (Fig. 7a) had the following treatment steps: aeration, softening, filtration (dual media) and disinfection. Comparative studies were carried out to assess how a “young” GAC filter (63 operation days) and “old” GAC filter (264 operation days) performed at full-scale. The flowrates of 35 L s−1–45 L s−1 were initially modified to 30 L s−1 and subsequently to 15 L s−1. From the work it was clear that the treatment steps in the treatment plant (i.e., aeration, softening, filtration and disinfection) had no impact on PFAS removal. The GAC filters removed 67–100% of the PFAS, and long chained PFAS were removed more efficiently than short chained PFAS. The removal efficiency of the GAC was influenced by the individual operating times of the filters (young vs old). In addition, lower flow rates enhanced the removal of PFAS. Decreasing the flowrate from 39 to 18 L s−1 after having reached the treatment goal could prolong the lifespan of the GAC by 50%40. It is important when conducting pilot studies to ensure that the results obtained can be utilized and applicable at full-scale. Therefore, hydraulic loading rate is important and enables accurate projections of what would occur at full-scale63. From the study, GACs effectively removed PFAS and exhibited low exhaustion over period, later initial breakthrough, and adsorbed PFSAs better than PFCAs. When comparing F400 GAC to Sorbix A3F resin F400 GAC had lower adsorption capacity; suggesting that the treatment system may require minimal mass of adsorbent/resin if they have high adsorption capacity. The large media volume, high number of pressure vessels needed for GACs made the capital costs higher compared to IX.

a Evaluation of full-scale treatment efficiency (denoted by circles), effect of flowrate (denoted by squares) and long-term efficiency of GAC (denoted by triangles) in the removal of PFAS in a water treatment plant, b depiction of GAC skids equipped with 4 columns and an IX/alternative adsorbent skid equipped with 6 columns, c four GAC columns connected in series in the removal of PFAS, d full-scale plant using columns made up of biological activated carbon (Norit 830 W) and rapid sand filtration at pH 6.840,58,63,64.
Medina et al.63 studied the removal efficiency of PFAS using GAC in a pilot study as shown in Fig. 7b. The pilot-system comprised 3 adsorbent media skids and pre-filtration skid. Of the skids, 2 had 4 columns loaded with GAC. Skid 3 had 6 columns: 2 loaded with AAs and the other 4 loaded with IX. Two 30-µm cartridge filters connected in parallel were found in the prefilter skid. All columns have an effluent sampling port, low totalizer, influent variable area flow meter (5.68–56.8 L/h). All GAC skids had influent pressure gauges while the columns had an effluent sampling port and 1 variable area flow meter (5.68–56.8 L/h).
Elsewhere, in Colorado, to better understand PFAS breakthrough Liu et al.58 evaluated the removal of PFAS in contaminated groundwater. In the system, the feedwater was fed at 2 GPM, passed through a 50 μm cartridge filter, and distributed to separate GAC columns at 0.5 GPM (Fig. 7c). Automated valves (controlled using supervisory control and data acquisition system) were used to regulate the flowrate in the columns. Before use, GACs were immersed for 24 h in deionized water and backwashed to 30% bed expansion. The columns were assembled to 10 min EBCT. The results obtained showed that breakthrough proved to be dependent on chain length, except for PFHpA and PFHpS. For the non-chain length dependent trends of the PFHpA and PFHpS. these were attributed to low influent concentrations and preferential sorption sites. Performance wise, compared to F600 and GCN1240, F400 and GAC400 outperformed them by 40-50% due to the minimal intraparticle diffusion limitations. GAC was assessed in removing PFAS and other organics at full-scale64. The treatment plant is comprised of conventional treatments steps including biological activated carbon filtration, microsieving, and secondary disinfection (Fig. 7d). Based on their findings, the researchers recommended to study novel, cost-efficient treatment techniques for PFAS and desorption trends in GAC filters and through the train of a drinking water treatment plant process.
Comparing the performance of GAC (Filterasorb 400) and anion exchange (EA purolite), short-chain PFAS had short-time breakthrough65. In a separate study38, regenerated GAC (F400) removed PFAS to below the limit of detection and was successfully regenerated to near-virgin conditions. In other 18-month pilot-study comparing GAC, biochar, and anthracite in removal of both short-chain and long-chain PFAS, GAC was found to have high removal efficiency followed by biochar66. Similar behavior for the removal of short-chained PFAS such as PFBA, PFPnA, and PFHxA, were observed, and high concentration of dissolved organic carbon (DOC) reduced the adsorption of PFAS; this study employed an ACF system with adsorption capacity of 108 L/kg at pH 7 for PFOA62. In the study 15 different PFAS were analyzed for the effect of flow rate on the removal efficiency and long-term performance (aging) (Fig. 8). The removal efficiency with the virgin GAC ranged from 92 to 100% whereas the removal efficiency of the spent GAC was between 7% and 100%. The decrease of flowrate by 10 L.s−1, increased the removal efficiency by 14% and 6.5% for used GAC and virgin GAC, respectively. The full-scale results concurred with laboratory and pilot scale studies demonstrating that GAC is less effective in removing short-chain PFAS58. A pertinent challenge in removal efficiency of PFAS was the presence of NOM, which reduces the exposed surface for adsorption and can lead to deactivation. Water samples were collected at different stages of the treatment process except for the disinfection unit. In the post disinfection unit, samples were collected from storage in the underground reservoir which were free of chlorine. The GAC F400 performed better most likely due to the higher surface area of 1050 m2 g−1 for enhanced PFAS removal compared to AquaSorb® 2000 with 950 m2 g−167. Other results also showed that the BET specific surface area was a significant factor affecting PFAS removal55. Strong interactions and sufficient adsorptive surface area accelerate the reaction rate during the PFAS removal process68. Out of several used GAC, one GAC labeled GAC 9, achieved up to 99% removal efficacy for PFAS within 30 days, while another GAC (GAC 8) achieved 67% removal in 360 days. The GAC 8 system exhibited lower adsorption capacity for shorter-chained perfluoroalkyl carboxylic acids (PFCAs) but higher adsorption capacity for branched PFAS. Specifically, GAC 8 removed 66% of linear PFAS, while GAC 9 achieved up to 100% removal of linear PFAS. For branched PFAS, GAC 8 and GAC 9 had a removal efficiency of 37% and 100%, respectively40. Adsorption of 9 short and long-chained PFAS by PC systems was also investigated69. A total of 90% removal efficiency was achieved for long chained PFAS within 120 min (Table 1), whereas for short-chained PFAS only 85% removal was achieved after 240 min. This trend was also observed with RSSCT and batch tests70. Batch tests achieved up to 95% PFAS removal whereas the RSSCT achieved a 50% breakthrough throughput. The positively charged AC surface increased the affinity for PFAS resulting in higher adsorption capacities.

A Filtrasorb® 400 (GAC 1), B Filtrasorb® 400 (GAC 2), C Filtrasorb® 400 (GAC 3), D Filtrasorb® 400 (GAC 4), E 80–90% AquaSorb® 2000 and 10–20% Filtrasorb® 400 (GAC 8) and F Filtrasorb® 400 (GAC 5) (circle denotes PFCAs and triangle denotes PFSAs)40.
Practical considerations and cost estimations of using GAC in the removal of PFAS
The greatest limitation associated with adsorption is its inability to degrade PFAS, resulting in the creation of secondary waste. Further is inefficiency to remove short chain PFAS which is important to some industries like semiconductor manufacturers. The presence of competing species such as NOM and the deactivation of GAC media over time requires constant changeout which is labor intensive and costly. In addition, the spent GAC media requires safe disposal, which if not handled well, may result in secondary contamination55. Although the used GAC can often be regenerated; its efficiency tends to decrease, leading to faster PFAS breakthrough and necessitating the flow rate to be adjusted. Another challenge is that the regenerating spent sorbents typically requires energy-intensive thermal treatment to eliminate PFAS, which can potentially release more toxic byproducts into the atmosphere71. This concern led to a temporary ban on the incineration of PFAS-saturated sorbents, which was lifted in July 202372. Addressing these challenges through the functionalization of GAC-based filters with adsorptive catalytic nanoparticles would offer numerous advantages in the treatment of PFAS73. This functionality would enhance filtering and adsorbing and catalytic properties of GAC, extending its lifetime and reducing replacement costs.
To effectively estimate the operational parameters and costs of using GAC at full-scale depends largely on the treatment goal for the plant40. For a plant with a 25 ng L−1 treatment goal, the cost is estimated at 0.058 euro per m−3. In earlier work, McNamara et al.55 estimated the operating costs (for a plant with a 70 ng L−1 treatment goal) to be 0.038-euro m−3 and 0.025-euro m−3 for regenerated and virgin GAC filters respectively. The costs for GAC regeneration typically account for the majority of the yearly operating costs. Belkouteb et al.40 demonstrated that by adjusting the flowrates to prolong the lifespan of the GAC filters can reduce the costs by 26%. Further, reducing the regeneration costs by 20% also lowers the unit cost by 20%. It must be noted when selecting GAC to opt for a high efficiency and quality as this can lower unit costs by 16%. The costs for media for IX and disposal were $256–$322 per cubic foot and $0.50 per lb respectively. However, the disposal costs are dependent on state regulations which are subject to change. Operators should assess the impact of source water quality and identify the most appropriate IX for individual circumstances at the different sites.
Membrane separation
Membranes used in water treatment are typically ceramic or polymer membranes, and the separation of PFAS is mostly through size exclusion74. RO and nanofiltration (NF) membranes are effective in the removal of PFAS due to their narrow pore sizes which has a diameter that range from 0.0001 to 0.001 μm for RO and 0.001–0.01 μm for NF75. Microfiltration (MF) and ultrafiltration (UF) have a low removal efficiency as their pore sizes are large to reject PFAS. Due to the difficulties of fabricating the ceramic membranes with pore sizes of RO and NF range, there are few studies available on ceramic membranes to remove PFAS from water. Polymeric membranes can be fabricated with RO and NF pores sizes and are the most investigated and applied in the separation of PFAS76.
Mechanisms of PFAS removal with filtration membranes
Membranes can drive separation through adsorption, sieving (size exclusion), and electrostatic interactions (Fig. 9a–c). The separation in membranes is mostly determined by membrane surface properties such as porosity, zeta potential, pore size, and hydrophobicity and/or hydrophilicity. The efficiency of PFAS rejection by a membrane is affected by several parameters such as pH, organic matter concentration, ions, and ionic strength76. Changes in solution pH can affect membrane pore size, flux, and rejection, as well as the surface charge of the membrane depending on the isoelectric point of the membrane material and the associated groups on the membrane surface77. Organic matter present in the solution can also influence PFAS separation by coupling or reacting with the target PFAS compounds, thus changing the membrane surface charge and causing membrane fouling. Electrostatic interactions between PFAS and ions present in water could also cause the ions to bind with the PFAS to form larger clusters, causing partial pore clogging78.

a Physically sieving solutes that are bigger than the membrane molecular weight cut-off (size exclusion), b elevating pH may deprotonate the carboxyl groups on membrane as a result increasing negative charge which enhances the rejection (electrostatic interaction), c adsorption efficiency can be impacted and influenced by variations in pH as well as ionic strength; mono/divalent cations typically enhance removal efficiency, while increased pH levels tend to decrease adsorption capacity (adsorption) [modified from refs. 74,91,175.
The size exclusion mechanism is mainly applicable to NF and RO membranes that reject PFAS. These membranes have a low molecular weight cut-off (MWCO), which is smaller than the molecular weight of most PFAS78. The molecular weight of PFAS detected in surface water ranges from 213.03 to 613.09 Da, and the MWCO of NF membranes range from 200 to 10,000 Da and for RO is below 200 Da78. By comparing the molecular weight of PFAS and the MWCO of membranes, RO membranes can generally remove all the short-chain and long-chain PFAS, but NF, cannot remove all short-chain PFAS. The size exclusion of PFAS by NF and RO membranes was also confirmed in a study that reported PFAS removal efficiencies of 90–99% for NF and over 99% for RO membranes79.
Electrostatic interactions between charged membrane surfaces and PFAS are another mechanism that influences the removal of PFAS. Most PFAS exist as anions in water, therefore, electrostatic attraction may occur if membrane surface charge is positive79. However, the adsorbed PFAS on the membranes may block the pores of the membranes which in turn reduces water flux and consequently causes membrane fouling80. Electrostatic repulsion between PFAS and the membrane is an ideal interaction to achieve maximum rejection in the removal of short-chain PFAS. This mechanism is highly dependent on the pH.
Generally, when the membrane surface charge is negative, excess protons protonate the negatively charged surface to neutral or positive on the surface, and that could reduce the repulsion forces and allow for PFAS adsorption. The performance of two NF membranes (NF 270 and NTR-7450) were investigated in removal of PFHxA at different pHs (3, 7, and 10). One membrane (NTR-7450) was not affected by the change in pH, but the other membrane exhibited poor performance at pH 3. A general trend of increase in performance with the increase in pH was observed80. The poor performance at pH 3 was attributed to the membrane (NF 270) being negatively charged at low pH and consequently reducing the repulsion forces. It is, therefore, crucial to consider the surface charge of the membranes to achieve maximum rejection through electrostatic repulsion.
Pilot-scale and full-scale application of membranes for removal of PFAS
Coupling membrane technology with adsorption techniques such as activated carbon, and ion exchange resins has been investigated in pilot studies to mitigate the challenge of handling concentrated streams48. NF membranes were used to remove PFAS from water and then the PFAS-concentrated streams were treated with GAC and IX. Membrane technology can also be coupled with catalytic treatment of PFAS, targeting the high concentration brine. Concentration polarization is a common challenge in membrane technology but is of greater concern when the retentate contains PFAS. It is paramount to manage or treat the membrane concentrate to avoid secondary contamination. Regulations state that the total volume of the membrane concentrate waste should always be lower than the daily treatment capacity of the post-treatment systems81. Therefore, it is necessary to optimize water recovery (Y) to reduce the environmental impacts. The water recovery (Y) is defined by Eq. 1:
where qP and qO are the volumetric flow rate of the permeate and the raw-feed water respectively.
The water recovery can be enhanced by shutting down the membrane concentrate’s valve, based on the linear relationship of the flux of the permeate and transmembrane pressure. Another approach is by recycling the concentrate, as shown in Fig. 10a, and through this approach the velocity of the cross is increased which in turn reduces the thickness of the concentration boundary layer81.

a Illustration of single-stage membrane used as single stage and concentrate recycling that can be reduced through reducing thickness of the concentration boundary layer where qO is the volumetric flow rates of the raw-feed water, qF the feed, qP the permeate, qC the retentate and qD the concentrate discharge and CO denotes raw-feed water, CF the feed water, CC, the retentate and finally CP denotes the permeate concentration of the contaminant, b rejection of PFAA using single 4 long high-pressure membranes assembled to hold three 8 × 40 inch spiral membranes equipped with a single membrane spacer81,82.
A pilot-scale membrane filtration system was used to assess the efficiency in rejecting PFAA using high-pressure membranes82. The system operated in a closed-circuit had 4 long pressure vessels housing 8 ×40-inch spiral wound membranes and a membrane spacer. The pilot system was automated with sensors installed in the various probes, the feed flowrate maintained at 10 gpm and flux of 12 gfd (Fig. 10b)82. The membranes used in the pilot were included loose (NF270) and tight (NF90) NF membranes, RO (CR100), and seawater RO (SW30) membranes. A rejection rate higher than 98.3% was observed in the NF90, CR100, and SW30 membranes. The results were correlated with previous studies that indicated that large molecular weights and anionic features facilitated better separation. On the contrary, NF270 had the poorest PFAA rejection.
Hybrid NF and UV-sulfates were coupled for degradation of concentrated streams at pilot scale83. In this specific system, NF rejected 95% of PFAS and the UV-sulfate degraded about 90% PFAS from the concentrated streams after 8 h of treatment. Figure 11 shows results from studies on PFAS removal at laboratory48,84,85,86,87 pilot83, and full scale44,83,88,89.

Summary of membrane separation application of PFAS removal at laboratory, pilot, and full scale.
Practical considerations and cost estimations of using membrane technology in the removal of PFAS
Overall, membrane technology has proven to be a viable approach for removing PFAS from water. However, there are still many challenges that need careful consideration. The major challenges of membrane technology in treatment of PFAS includes generation of concentrated PFAS streams that require further treatment before safe disposal and membrane fouling. To overcome these limitations, researchers have been focusing on the development of hybrid membranes to simultaneously degrade and filter PFAS. Hybrid membranes that have been studied include reactive electrochemical membranes (REM), electromagnetic ceramic membranes, and photocatalytic membranes89,90. These approaches mitigate the challenge of handling concentrated PFAS waste streams. Of the various membranes used in the removing PFAS; NF and RO membrane have recorded significant removal efficiencies. However, the energy consumption requirements increase the costs. The costs to treat wastewater containing PFAS using NF membranes is estimated to be between 0.016 and 0.16 $/m3 and 0.11 $/m3 in a drinking water plant91. Treating landfill leachate containing PFAS using RO was estimated to cost between 1.06 and 2.09 $/m392. As discussed in earlier sections, treatment or management of membrane concentrate is key. As such, integrating other technologies with membrane technology presents a viable solution. However, the cost associated integrated technologies cannot be ignored. In a study where the NF concentrate was treated using GAC and IX, the incurred costs were in the range 0.86 and 3.34 $/m3 respectively51. The estimation when using membrane-adsorption was estimated at 0.28 and 1.31 $/m3. Even higher treatment costs up to 13.1 $/m3 can be expected when integrating electrochemical oxidation and membrane technology as both technologies have high energy requirements91,93. When deciding on which technologies to integrate with membranes, practitioners ought to understand: the composition of the feedwater, PFAS concentrations, and type of PFAS present in the feedwater. Further, special attention needs to be paid when selecting membrane that is fit for purpose, this includes understanding the membrane’s structure, factors affect fouling and cleaning strategies. All these factors combined affect the lifespan and performance of the membranes. Other considerations when handling/treating membrane concentrate include exploring pre-treatment options and developing systems that are energy efficient.
Remediation of PFAS using destructive techniques
Destructive techniques involve the partial or complete breakdown of PFAS bonds, ideally resulting in mineralization, which is often assessed by extent of fluoride release. Supercritical water oxidation, sonochemical oxidation, electrochemical oxidation, and photocatalytic oxidation are examples of such techniques. These techniques are useful in the removal of PFAS at the bench scale. The major limitations to upscaling and commercialization to date include large space requirements, reactor design, high energy consumption, and high capital requirements for new treatment infrastructure. For instance, electrochemical oxidation may need costly electrodes and electrolytes. Table 2 shows the electrical energy per order of reaction for PFAS (PFOA and PFOS) destruction techniques.
On the other hand, destructive techniques namely plasma94, SWO95, and photocatalysis96 revealed higher degradation efficiency for PFOA and PFOS amounting to >99%, 99,9%, and 88% respectively. This review will focus on supercritical water oxidation, sonochemical oxidation, electrochemical oxidation, and photocatalytic oxidation. Further emphasis will be on emerging technologies such as plasma, hydrothermal alkaline treatment (HALT), advanced reduction process (ARP) and electron beam. Figure 12 illustrates an overview of the advantages and disadvantages of several destructive PFAS remediation techniques.

Advantages and disadvantages of various destructive techniques used for removal of PFAS from water.
Supercritical water oxidation (SCWO)
SCWO is considered a clean technology because it results in complete mineralization of PFAS while producing no hazardous materials in the process. SCWO relies on heating water to above 374 °C under a pressure of 221.1 bar in the presence of an oxidant to achieve complete degradation of PFAS to CO2, water, and non-toxic substances97. SCWO has been applied to degrade persistent organics that do not easily undergo natural degradation such as polychlorinated biphenyl and 1,4-dioxane98. SCWO is one of the fastest destruction methods and could rapidly treat a variety of waste (i.e., wastewater, sludge/slurry, and biosolids)88.
The SCWO process consists of pumping the contaminated waste and oxidants into the reaction chamber with controlled temperature and pressure. Under these conditions, water is brought to its supercritical point where it resembles the properties of gas and low-polar organic solvents. Supercritical water dissolves oxidants (oxygen) and organics, creating a homogeneous solution that facilitates the oxidation of organics98. The SWCO technology has been tested98,99. Oxidation of PFOS to HF and CO2 can be achieved (only 70% of PFOS conversion) via SCWO using a stainless-steel batch reactor at temperatures up to 500 °C and 60 min. Autogenic operation of a SCWO reactor can be sustained by a reaction mixture of ethanol and air100,101. The reactor operated in continuous flow up to 650 °C and destroyed 99.999% of PFOS, PFHpS, and PFOA in ~30 s. This is an improvement from earlier results101 obtained from batch reactor, at low temperature 500 °C and longer residence time of 60 min.
Mechanism of PFAS degradation using SCWO
The chief degradation mechanisms of SCWO can occur either via hydrothermal reaction or oxidation95. Typically, the hydrothermal degradation will occur before the supercritical point and is known as sub-critical hydrothermal processes. PFOS degradation and defluorination by hydrothermal reaction has been studies using different acidic, basic, oxidative, and reductive amendments96,102. Alkali amendments such as NaOH and Ca(OH)2 increased degradation efficiency of PFAS at sub-critical water region. The degradation mechanism under basic conditions was catalyzed by OH−. Under hydrothermal conditions PFOA is unstable, and therefore undergoes decarboxylation to form perfluorocarboxylates while releasing 2 F− stepwise until it completely mineralizes to CO2, F−, and HF.
Another study probed the influence of amendments of metals (Fe, Cu, Al, and Zn) at subcritical water oxidation conditions103. Among these metals, Fe was the most influential metal in degradation of PFOS followed by Zn, Cu, and Al with the degradation efficiencies of 99%, 77%, 15.3%, and 6.4%, respectively. On the other hand, HALT is an alternative destructive technique to SCWO and researchers have demonstrated that HALT can effectively degrade PFAS in contaminated groundwater104 and soil104, in aqueous film-forming foam105, in spent GAC96.
Laboratory and pilot-scale application of SCWO in the removal of PFAS
SCWO can achieve PFAS degradation efficiency of over 99% and break the carbon-fluorine bond (C-F) to facilitate PFAS mineralization106. The reaction that takes place is shown in Eq. 2:
Pilot-scale studies have been reported at Battelle (Columbus USA), Aquarden Technologies (Skaeving, Denmark) and 374water (Durhan, USA). These pilot treatment plants operate at maximum temperatures of 590 °C, 590 °C and 595 °C, respectively. For full-scale applications, it is known that some if not most of the companies that commercialized this technique had to shut down the operation due to corrosion and salt precipitation. In applications specific to PFAS, Danish company Aquarden Technologies have successfully scaled up SCWO to full-scale operation. The system includes combination PFAS-selective adsorbents that are then treated with SCWO85. Using a continuous flow SCWO reactor operating in an autogenic regime, the degradation of PFHpS, PFOS, and PFOA were assessed106. The reactor (Fig. 13a) is comprised of 4 influent streams: (i) pilot fuel (EtOH-H2O mixture), (ii) air-H2O mixture, (iii) reagent-H2O mixture and (iv) quench water injection port. A titanium-lined vessel with an approximate volume of 164 mL was used to insulate the reactor. Titanium was used to minimize the likelihood of corrosion failures instead of iron-based alloys as Ti has proven anti-corrosion properties107.

a Illustration of reactor with corresponding sample collection points where TC1 and TC2 are the thermocouples used to track influent temperature, TC3–TC5 track internal reactor fluid temperature, TC6 tracks temperature effluent before the cooler and TC7 temperature after cooler while TC8 and TC9 are used to monitor temperature of the external wall reactor, b continuous mass flow SCWO reactor balance, where AFFF aqueous film-forming foam, SCWO supercritical water oxidation, kg/min kilograms per minute, L liters, NaOH sodium hydroxide, and HP high pressure106,107.
A co-axial nozzle positioned on top of the reactor was used to inject air and fuel. PFAS were injected into near-uniform temperature region using 2 injection ports, and the destruction of the PFAS was evaluated at temperature range of 410–650 °C. For PFOS experiments at low temperatures; a pool of PFCA were observed in the liquid effluents and emissions in gas stream. However, in the PFOA experiments, no PFCAs were detected in the liquid effluent even at the lowest temperature tested. Therefore, the rate-limiting step in PFOS decomposition was the PFOS and PFHpS transition to PFCAs. An intermediate that could only be destroyed at 650 °C trifluoroacetic acid was also detected in the liquid effluents.
Elsewhere, McDonough et al.107 assessed the efficiency of SWCO in destroying 12 PFAAs. The system was equipped with an air-compressor, fuel-fired preheater, titanium-lined insulated reactor, holding tanks and appropriate connections and piping as depicted in Fig. 13b. Results obtained showed a >99.999% destruction and removal efficiency of the12 PFAAs after two successive ~120-min continuous flow. The recorded defluorination % was estimated to be 62.6%. The total PFAS, fluoride concentrations and fluorine mass balance at various temperatures are presented in Fig. 14a. Based on the results obtained and identified possible decomposition pathways PFOS was presented in Fig. 14b96. The ·OH likely attacks the PFOS by cleaving the C-S bond, C8F17· radical, C8F17· radicals are formed which will recombine with ·OH forming C8F17OH through hydroxylation. The weakened C8F17OH will form C7F15COF via F elimination. Through hydrolysis perfluoric acid will be formed from perfluoroketone. Alternatively, PFOS can be destroyed through volatile organic fluorides (VOFs) (depicted by the dashed line in Fig. 14c)96. To understand this pathway a comparison between VOFs yield and composition during PFOS decomposition must be assessed.

a Concentration of PFAS (PFOS, PFHpS and PFCA) intermediates from LC-MS/MS analysis, and the fluoride concentration obtained from ion chromatography, b the quantity of both organic and inorganic fluorine in liquid effluents, c proposed decomposition pathways of PFOS obtained from SWCO using intermediates from GC-MS. Noteworthy: although 1H-perfluorohexane was the only volatile organic fluoride identified other volatile organic fluorides cannot be excluded96.
Practical considerations and energy requirements of using SCWO in the degradation of PFAS
Despite the degradation efficiency of PFAS using SCWO, there are some limitations that hinder upscaling and commercialization efforts, including extensive energy use, salt precipitation, and resilient infrastructure for high temperature and high-pressure operation108. Elevated temperatures are required to achieve the desirable high destruction effectiveness, which unfortunately translates to increased energy requirements. These challenges necessitate reactor designs capable of managing corrosive and exothermic conditions109,110, as well as facilitating effective salt separation. Moreover, it is costly and challenging to produce pressure vessels that can resist the high temperatures and pressures of the SCWO process at industrial scale. The other disadvantage of SCWO is the high energy consumption, however this limitation can be compensated if the energy can be recovered from exothermic reactions as the concentrated waste is treated. In order to determine the financial viability of SWCO, it is essential to determine the energy consumption. In doing these calculations most practitioners may want to analyse per mass of destroyed PFAS. However, these analyses and calculations may not be a true representation as the influent PFAS concentration is directly linked to the required energy needed to destroy PFAS in SCWO. The majority of the energy required in SCWO is used to heat the water to supercritical state. Recently107, electric energy per mass (EEM) was determined to be 1.1 × 105 kW·h/kg PFAS. Even considering the energy required for the air compressor and the transfer pump, it was found that the energy source (diesel) was significant. In addition to the corrosion and salt precipitation, the production of hazardous gases produced during the process are not readily investigated. More research is needed on establishing whether the emitted gases from these full-scale of SWCO are toxic.
Sonochemical oxidation
Sonochemical oxidation uses sound waves to generate large volumes of reactive oxygen species (ROS). It involves the propagation of acoustic waves with a high frequency ranging from 20 to 1000 kHz in a liquid resulting in the formation of acoustic bubbles which are known as acoustic cavitation. Acoustic cavitation produces high temperature, pressure, and free oxidizing radicals70,111. Sonochemical degradation occurs in two ways; via thermal degradation and free radical oxidation112. There have been several studies that have incorporated the sonochemical technique with various catalytic chemicals such as sulfate, persulfate, periodate, and permanganate to improve PFAS degradation efficiency113. The catalyst additives have shown a great improvement in degradation efficiency of PFAS. Among the different oxidizing chemicals that have been incorporated with sonochemical oxidation, persulfate (which generates sulfate radicals) has demonstrated high enhancement in the degradation of PFAS. The performance of sonochemical techniques is directly dependent on the operational parameters such as frequency of ultrasound waves, power density, and solution temperature114. The ultrasonic frequency determines the growth size of the acoustic cavitation bubble. When the cavitation bubbles grow, they absorb the energy of the waves up to the maximum point and then collapse. However, when frequency is increased beyond optimum value, it causes the PFAS degradation rates to decrease due to the rapid collapsing of bubbles115. The cavitation bubble size, duration of the bubble before collapse, transient temperature, and internal pressure of the cavitation bubble are also governed by the power intensity115. Increasing the ultrasound power intensity could raise the maximum collapse temperature as well as the number of collapsed cavities created, and subsequently sonochemical degradation. PFOA and PFOA degradation efficiency has been reported to increase with power density (varied from 30 to 262 W L−1)114. Thus, frequency and power density are crucial parameters for the degradation of PFAS via sonochemical systems.
Mechanism of PFAS degradation using sonochemical technique
The degradation of PFAS with sonochemical technique is via two mechanisms: pyrolysis and oxidation by free radicals. When degradation takes place via pyrolysis it is because of cavitation of the bubbles which releases a very high transient temperature116, while oxidation happens when water undergoes thermal decomposition to form hydroxyl (•OH) radicals as demonstrated in Eq. 3.
In the presence of catalytic additives such as sulfate112, persulfate112, periodate112, sodium hypochlorite (bleach)117, and permanganate112, numerous oxidation species and radicals are formed (IO3•, IO4•, SO4• −, Cl•, MnO2) which increases the degradation rate as illustrated in Eqs. 3–6. Among these radicals, SO4−• has the greatest influence on the degradation of PFAS as it has the highest redox potential (E0 = 2.5–3.1 V). However, not all the radicals formed are able to initiate the degradation of PFAS due to the strength of C–F and C–C bonds118. Pyrolysis initiates the degradation of PFOA via decarboxylation (Eq. 4). Subsequently, the radicals from the catalytic additives react with oxidative radicals to produce perfluorocarbonyl, carbon dioxide and hydrogen fluoride (Eq. 4). The resulting PFAS molecule (C6F13COF) undergoes hydrolysis to produce a shorter chain PFAS (C6F13COOH, PFHpA) (Eq. 5). The C6F13COOH again undergoes the same reactions (Eqs. (4–6)) until it is completely mineralized to CO2 and HF.
Laboratory and pilot-scale apphlication of sonochemical technique for PFAS removal
The defluorination of PFOA and 6:2 fluorotelomer sulfonate (6:2FTS) was reported to increase by over 40% in the presence of persulfate (Fig. 15a–d)70. Similarly, a pilot-scale sonochemical oxidation of PFAS in aqueous film-forming foams reported removal efficiencies from 27% to 91.5% at lower energy costs118. The major challenge with scaling up to full-scale includes the issue of existing infrastructure which represents inertia, people are skeptical about discarding what they have and replacing it with new infrastructure, hence there must be innovation in how to seamlessly integrate new system in existing treatment trains. Fuller et al.119 assessed the extent of degradation of PFAS in resin regeneration stream using the sonochemical technique. In the study the optimal frequency and power were 1000 kHz and 400 W respectively. This yielded the best PFAS removal rates resulting in mineralization forming inorganic fluoride, however it must be noted that small fractions of VOFs were detected. The effects of chlorides, carbonates, organic material on the PFAS degradation were assessed. The efficiency of the PFAS degradation was adversely impacted by TOC and methanol to about 50 g/kg; 5% w:w. However, the addition of chloride enhanced the PFAS degradation, and this was more evident in the longer chained PFAS. The trend indicates that PFAS with higher hydrophobicity were impacted significantly by the enhanced chloride. The improved degradation was also confirmed by high defluorination rates.

a PFOA, b PFOS, c 6:2 FTS and d defluorination. Panels (a–c) display equations and first-order fitting curves70.
Using a 10-litres reactor equipped with 2 transducers operated at 700 and 950 kHz, Kewalramani et al.120 evaluated the scalability of destroying PFAS using sonochemical oxidation (Fig. 16). To fully understand the system the following factors were assessed: frequency, power density, the relative position of the transducer on KI oxidation and calorimetric power. The energy conversion efficiency was recorded at ~50%. Notably, an increase in power density due to reduction in reaction volume enhanced the sonochemical activity. However, as the power density increased, the rate was lowered. Regarding the positioning of the transducer, it can be deduced that the activity was not significantly affected by the transducer’s positioning along the reactor’s side and bottom walls.

The system is used to evaluate the scalability of destroying PFAS using sonochemical oxidation.
Practical considerations and cost estimations of using sonochemical in the degradation of PFAS
The major challenges of implementing sonochemical oxidation on an industrial scale include the extensive energy usage and insufficient comprehensive understanding of the required operating parameters104. Karla et al.121 recently estimated to run a reactor operating at 0.4 kWh in the USA would cost ~$0.12/kWh, $0.18/gal. To reduce the overall energy consumption practitioners ought to decrease the power density, optimize the frequency of the ultrasound, and optimize matrix chemistry to enable efficient transfer from electrical energy to ultrasound. It must be however noted that optimizing frequency in complex water matrices may not be achieved easily as the frequency is pollutant-specific122. For enhanced sonochenical degradation, the integration with other technologies may enhance the degradation123. Additionally, designing large sonochemical reactors and determining the optimal operating frequency of these reactors are significant hurdles. The geometry of the reactors used, the position of transducers, and the total number of transducers used are all crucial in the efficiency of the sonochemical reactor as it impacts on how cavitational occurrences are distributed. It is important to note in sonochemical reactions a low liquid height is not ideal and will not achieve the desired degradation efficiency, therefore altering liquid height significantly impacts on the degradation124,125. The usage of multifrequency transducers unlike the single-frequency transducers also stands to improve the efficiency. More research is needed to design suitable configurations and integrate sonochemical oxidation with other destructive techniques to reduce the energy input and reaction time104.
Electrochemical oxidation
Electrochemical oxidation (EO) requires a direct current power supply, electrolytic cell and electrode array (anode and cathode) as depicted in Fig. 17a, b. When current is applied to an electrolytic cell, the electrons flow from anode to cathode, making the anode oxidative towards any oxidizable solutes present in the solution126. This process involves the transfer of electrons for direct oxidation and production of •OH radicals in situ that can be responsible for the degradation of PFAS. EO processes have demonstrated to be useful techniques in degrading PFAS, with degradation efficiency of over 99%127.

a Typical experimental set-up in EO experiments, b typical degradation pathway using EO where an electron is transferred to the anode or SO4•− resulting in Kolbe decarboxylation reaction, subsequent repeating of a series of CF2-unzipping cycle takes place [adapted from ref. 129], c PFAAs removal using EO in laboratory and semi-pilot scale systems. Inset depicts the pseudo-first-order removal rate for PFAAs for both laboratory and semi-pilot scale systems176.
The efficiency of EO is mostly influenced by anode material, electrolytes, oxidative species generated, and current density127,128. The electrode can either be an active or non-active electrode. The ideal electrodes for degradation of PFAS are those electrodes that have less or no interaction with the generated oxidative species and haves a high oxygen overpotential129. The active electrodes include ruthenium (RuO2), platinum, carbon, graphite and iridium (IrO2). Complete mineralization of PFAS is most likely to occur in non-active electrodes due to high oxygen overpotential and poor interactions with oxidizing species. Non-active electrodes that have been applied in degradation of PFAS include boron-doped diamond (BDD), tin oxide (SnO2), Magneli phase titanium suboxide (TSO), and lead dioxide (PbO2)127. Among these anode electrodes, BDD and TSO are the most investigated electrodes due to their high degradation efficiency towards PFAS129.
Titanium suboxide anode electrodes have also been receiving extensive attention due to their porosity and large surface area which facilitate the electrochemical oxidation of pollutants on the surfaces of the electrodes and are also considered to be inert towards generated ROS. Titanium suboxide can also be applied in fabricating REM130. Other advantages include lower current density and low charge transfer which favors production of OH radicals129. Liang et al.128 reported the degradation of PFOA and PFOS using Magneli phase Ti4O7 in NaSO4 electrolyte solutions. The degradation was found to be 96 and 98.9%, respectively, and the electrolyte was found to enhance the degradation. Electrolytes in electrochemical oxidation serve as charge carriers, however they can also participate in degradation of PFAS through generating oxidative radicals. The common electrolytes are: NaCl, NaClO4, and NaSO4. If chlorinated salts are used, oxidizing chloride species may be formed and lead to the formation of toxic halo-organic compounds. It is important to find ways to inhibit formation of toxic chlorinated byproducts from chlorinated electrolytes. For example, Yang et al.131 probed the formation of these unwanted byproducts by adding hydrogen peroxide (H2O2) to the reaction solutions. The findings showed that ~88% of the chlorinated radicals were suppressed. Improving electrode selectivity is essential to limiting competition between oxidizable species and unwanted side reactions for occurring. One way to do this is concentrating the desired species, PFAS, on the surface of the electrode by using a selective electrode or coating that preferentially adsorbs target compounds. The ‘trapped’ target compounds can then be more efficiently oxidized or “zapped” at the active surface. This strategy has been used before by Yin et al. who coated an anode with a hydrophobic material that adsorbed high levels of PFOA for efficient defluorination once potential was applied132. The disadvantage of EO is the high cost of the electrode, incomplete degradation of PFAS, formation of by-products, and loss of efficiency because of the accumulation of matter on the anode.
Mechanism for PFAS degradation with electrochemical oxidation
The mechanism for degradation of PFAS in EO is either indirect electron transfer (IET) or direct electron transfer (DET) oxidation129 as shown in Fig. 17c. IET is when the anode oxidizes water into OH radicals or electrolytes into oxidative radicals such as Cl•, ClO4•−, or SO4•− which actively degrade PFAS. In DET the pollutant gets oxidized on the surface of the anode by transferring its electrons to the anode. Due to insufficient energy of •OH radical to initiate the degradation of PFAS, degradation is initiated through DET as shown in Eq. 7. The unstable PFAS radicals quickly undergo Kolbe decarboxylation and desulfonation forming perfluoralkyl radicals, shown in Eq. 8. These radicals subsequently react with •OH and form perfluorinated alcohol as shown in Eq. 9. Further reaction with •OH leads to formation of a PFAS radical as shown in Eq. 10. These PFAS radicals can release COF2 to form a shorter chain PFAS as shown in Eq. 11. This shorter chain PFAS repeats Eqs. 9–11 (CF2-unzipping cycle) until it completely mineralizes to CO2 and F−.
Laboratory-scale and pilot-scale electrochemical oxidation in the degradation of PFAS
Recent studies reported 95% degradation efficiency of 17 PFAS using BDD anodes in electrooxidation of municipal landfill leachates133, and 94–99% efficiency for PFAS in IX concentrate133 (Fig. 17c). However, low defluorination was observed, and this was attributed to the foaming of PFAA. There are several modifications that have been done to improve functionality of BDD electrodes such as coupling with other electrodes materials, metals, and non-metals. All electrode modifications exhibited improvement in the degradation of PFAS with efficiency of more than 95%. However, the defluorination remained low. Another studying that the degradation efficiency of 12 PFCAs using BDD doped with silicon (Si/BDD) was dependent on the length of the PFAS chain; the short-chain PFAS were not effectively degraded (removal efficiency of 16–67%) as compared to long-chain PFAS (64–91%), but the with Si/BDD was higher than when a pristine BDD anode was used134. Combining BDD with SiO2, PbO2, and SnO2-F can improve the performance of the BDD anode135. Among these combinations, BDD/SnO2-F had outstanding performance compared to the pure BDD. The extent of defluorination was 62%, compared to 50% using pristine BDD anode electrode135.
Liang et al.14 used an EO reactor to degrade PFAS from spent resin regeneration in still bottoms (Fig. 18) part of the treatment train. The operation of the plant enabled the removal of PFAS from ~260,000 gallons contaminated water. The EO system (utilizing Ti4O7 electrodes) recorded an 80–98% destruction rate for the PFOA and PFOS. The EE/O for removing PFOA and POS was in the range 0.131–0.161 kWh/m3 and 0.071–0.094 kWh/m3 respectively.

Schematic of the pilot-scale IXR-EO treatment train and sampling locations used in the study14.
Practical considerations and energy requirements of using electrochemical oxidation in the removal of PFAS
Electrochemical oxidation mostly uses BDD anodes, which are relatively expensive and difficult to produce. More research is needed to develop affordable and resilient electrodes to minimize costs. Titanium sub-oxides electrodes, for example, have shown comparable performance with BDD electrodes at a much lower cost136,137. Chaplin et al.138 estimated the cost to produce Ti4O7 electrodes to be ~$0.36 per m2 compared to the estimated $7125 per m2 to produce BDD. Although cost effective unlike BDD, Ti4O7 electrodes do relatively have shorter electrode lifespans compared to BDD139. Regarding energy use, Ti4O7-electrooxidation is reported to be ~80% less energy intensive unlike BDD, the EEO was 3.6 kWh/m3 when using Ti4O7 electrodes and 19.9 kWh/m3 when using BDD139. Furthermore, the excessive amount of perchlorate generated cannot be ignored, as some researchers reported the concentrations to be 65–220 mg/L. To minimize the perchlorate generated, practitioners may consider NF membranes to remove chlorides prior. Additionally, utilizing free chlorine during treatment may reduce the perchlorate generation. Other alternatives of reducing perchlorate include applying H2O2 (50 Mm) and MeOH (100–1000 mM)140. The formation of by-products (i.e., trihalomethanes and haloacetic acids) and incomplete degradation can be minimized through coupling EO with other AOPs preferably photochemical oxidation and photocatalysis. This can enhance the performance and subsequently reduce accumulation of matter on the surface of the electrode which reduces efficiency.
Photocatalytic oxidation
Photocatalytic oxidation uses light as an energy source and catalysts as an active site for PFAS oxidation, which typically occurs through the Decarboxylation, Hydroxylation, Elimination, Hydrolysis (DHEH method). The degradation efficiency of photocatalytic oxidation towards PFAS mostly depends on the activity of the photocatalyst, oxidant and operational parameters such as light sources, pH, dissolved oxygen (DO), and composition of the water141. Increasing the solution temperature improves the photolysis efficiency and increases the photocatalytic rate, but too high a temperature (higher than calcination of photocatalyst, usually 400–500 °C) can destroy the molecular morphology and thus render the catalyst inactive. In addition, pH affects the dissociation of PFAS in solution, which affects photocatalytic performance. Acidic pH favors effective PFAS degradation. Lastly, DO in solution can cause the formation of reactive oxygen species which may participate in unwanted side reactions141. Reactive oxygen species generated from the photocatalytic reactions may participate in the degradation of PFOA, but these ROS species are unable to initiate degradation of PFAS molecules.142,143. Therefore, photocatalysts must provide an active site for PFAS adsorption and direct electron transfer to be effective.
Mechanism of PFAS degradation with photocatalysis
In photocatalytic oxidation PFAS are degraded either via indirect oxidation (radical oxidation) and direct oxidation when the light of suitable energy is radiated to the solution with photocatalyst. Under light irradiation the electrons (e−) in the semiconductor are excited from the valence band to the conduction band leaving positive holes (h+). The excited electrons react with free oxygen to produce ROS and the photogenerated holes can react with PFAS to directly oxidize. In indirect oxidation, the photogenerated hole scavenges water to produce \(\bullet\)OH radicals. However, this is unfavorable because it creates competition between PFAS and water to react with the holes. Photocatalytic degradation of PFOA using titanium dioxide-peroxymonosulfate (TiO2/PMS) as the photocatalyst under visible and UV light can be driven by both sulfate ion radicals and photogenerated holes, indicating both direct and indirect oxidation141. However, this study did not report the repeatability of their experiments and did not offer fluorine balances to assess extent of mineralization. Since other studies have indicated that \(\bullet\)OH radicals are not effective at initiating the decomposition of PFAS, photogenerated holes are essential to achieve enhanced degradation of PFAS. Radical scavenging tests (to discern the importance of hydroxyl radical, superoxide, and photogenerated holes) has also been studied144 have demonstrated the importance of photogenerated holes for initiating PFOA photocatalytic degradation with UV-irradiated hBN144 (Fig. 19).

a Scavenger experiments prove that photogenerated holes are the only species capable of initiating PFOA degradation in a BN-driven photocatalytic system, b in TiO2-driven photocatalytic systems, it was demonstrated that other radical species participate in degradation of PFOA and this was assessed using EDTA (to scavenge holes), SOD (to scavenge superoxide/hydroperoxyl) and TBA (to scavenge hydroxyl radicals)144.
Laboratory-scale and pilot-scale application of photocatalytic oxidation of PFAS
The potential of operating under standard atmospheric conditions and using solar energy has propelled photocatalytic oxidation to be a promising technology for PFAS degradation. Other advantages of photocatalysis include the recyclability and re-use of photocatalyst. At pilot, Liu et al.83 employed an integrated NF/UV-sulfite system. The results showed the membranes achieved rejection ≥95% and the PFAS concentrated streams were effectively destroyed using the UV treatment. The general trend of destruction showed the longer-chain PFAS had preferential degradation compared to the shorter chained PFAS. Elsewhere, the degradation and defluorination utilizing photocatalytic silica-based granular media and NaOH and Na2S2O3 were assessed in a packed column (Fig. 20)145. TiO2 was immobilized on the silica to maximize adsorption of PFAS reduce the electron-hole pair recombination. From the 4-hr long experiments 17 PFAS were assessed; in the experiment where NaOH was added 59% of the detected PFAS were degraded with an efficiency of over 88%.

Schematic representation of the column reactor packed with immobilized TiO2 on the silica145.
To date, direct photocatalysis has been proposed as a cost- and time-effective method for degrading PFAS146. However, compared to other destructive procedures, it has a lower efficiency and might not be the ideal choice for process scale up. Even though photo-enhanced methods for PFAS degradation have produced some interesting results, more research is still needed to determine whether or not highly effective materials could be used in ambient reaction conditions. Table 3 summarizes and compares the degradation efficiency, defluorination rate and the reaction time of some photocatalysts that have been applied in the degradation of PFAS.
Practical considerations, costs and energy requirements of using photocatalysis in PFAS degradation
Some challenges associated with photocatalytic degradation generally include the incomplete degradation of PFAS (defluorination), photocatalyst deactivation, low energy conversions and requires a long reaction time. The low energy conversions are due to absorption, scattering and reflection by other particles present in wastewater147. Finding efficient and effective photocatalysts that are relatively inexpensive for easy commercialization is required. Further, some challenges preventing up-scaling of photocatalysis for the degradation of PFAS is that most of the reported work was done using artificial wastewater or surface water and spiked clean water which are not real representatives of real wastewater.
Considering that wastewater consists of more than one pollutant tends to make photocatalysis less effective due to the presence of competing degradable pollutants. Designing effective photoreactors is also a major limitation for commercialization of photocatalysis. Most laboratory-scale studies are done on semi-batch systems while real life applications require large continuous flow photoreactors. Regarding the reactor design optical thickness is a key scale up parameter, the hydrodynamic flow in the reactor as well as the mass transfer of PFAS reaching the photocatalyst impact the degradation. Estimating the costs of full-scale photocatalytic operations is important in understanding the feasibility. The calculated EEo when using UV activation was found to be in the range >10–100 kWh/m3148. Elsewhere it was found after 4 h operation the degradation of PFCA and PFOS ∼128,000 mJ/cm2 over 2 h and 282,000 mJ/cm2 were needed respectively83.
Emerging technologies
Plasma technology
Plasma technology can be defined as a high-temperature, partially ionized gas which is electrically conductive and can oxidize unwanted pollutants in a water stream149. Plasma treatment is an innovative technology that utilizes plasma to generate highly reactive radicals, such as hydroxyl radicals, hydrogen atoms, and hydrated electrons for the effective degradation of PFAS150,151. Among the various plasma systems, gas discharge reactors with argon bubbling have demonstrated the highest efficiency in treating surfactant-like compounds, including PFAS. In this type of reactor, argon is bubbled through the liquid to create a foam layer at the plasma-liquid interface, where the degradation of PFAS occurs152. This unique configuration enhances the interaction between the reactive species and the PFAS molecules, leading to more efficient breakdown. Figure 21 shows the various reactor geometries that can be employed in instances where the discharge is in either liquid or in contact with liquid153.

a–d Reactor geometries for the generation of NTP inside the liquid: a point-plate, b point-point, c point-plate with bubbling gas, d wire-to plate. e–h Varying reactor geometries for the generation of NTP above the liquid are shown: e point-to-plate, f DBD with falling liquid film, g gliding arc above liquid, h wire-to-plate153.
Mechanism of PFAS degradation with plasma technology
The mechanism to destroy PFOA and PFOS in plasma treatment is primarily driven by plasma electrons, aqueous electrons, and argon ions. For PFOA, degradation begins with these species attacking the –COOH group, leading to the formation of unstable perfluoroalkyl radicals (e.g., •C7F15). These radicals can recombine with hydroxyl radicals (•OH), forming perfluoro alcohols (C7F15OH) and shortening the carbon chain of PFOA. Similarly, PFOS degradation involves the attack on the C–S bond by electrons or argon ions, cleaving the SO3– group and producing •C8F17 radicals. These radicals propagate and lead to the formation of short-chain PFCAs154.
Advantages of using plasma technology for destruction of PFAS
The plasma technology is considered powerful oxidation processes, it reduces the transportation and disposal costs for industries with challenges effluent, it proves the mineralization or destruction of emerging contaminants including PFAS149. Moreover, for water treatment, the plasma used is typically non-thermal in comparison with that applied in nuclear fusion reactors to generate energy149. The key advantage of this technology is that it only requires energy input to the working gas (such as air, argon, helium, or gas mixtures) to generate plasma, eliminating the need for chemical additives and thereby minimizing secondary pollution150. Additionally, the presence of co-contaminants in environmental samples does not affect the degradation of PFAS150,151. The treatment process is also efficient, with short treatment times of 30, 60, or 90 min.
Limitations for applications plasma technology for PFAS degradation
The challenges include cost-effectiveness, finding the right niches to scale up, and dependence on regulation for market growth149. Plasma treatment achieved high removal efficiency (>90%) for longer-chain PFAS, such as PFOS, PFHxA, and ADONA, within just 10 min, but other compounds demonstrated lower removal rates, indicating incomplete mineralization. Other drawbacks of plasma treatment include the potential formation of short-chain PFAS by-products, high energy consumption, and challenges in scaling up the technology for large-scale applications. Lewis et al.5 used reverse vortex gliding arc plasma to degrade PFCA, PFSA, and FtS. The estimated EEo for PFOS and PFOA were 23.2 kW h m−3 per order and 213.4 kW h m−3 per order respectively. At 150 W (20 min reaction time) a removal rate of 21% and 17% defluorination for PFOA was observed, which is considered the most energy efficient. However, higher removal rates reaching 75% were also attained but to achieve this enhanced removal rate, the system required 918 kJ L−1. PFOS on the other hand, at 180 W (1 min reaction time) the most energy efficient removal only resulted in 25% PFOS destruction. To attain the maximum 90% removal 625 kJ L−1 energy was needed. The high energy demands in the system were attributed to reactor inefficiencies perhaps as a result of the low recycling rate (20 mL min−1).
Hydrothermal alkaline treatment (HALT)
HALT is a subcritical water process that uses an alkaline substance, typically sodium hydroxide (NaOH), to degrade PFAS. Under high-pressure (~25 MPa), high temperature (~350 °C), and highly alkaline conditions (pH > 14) in the compressed liquid phase of water, PFAS can be broken down into inert fluoride salts. This transformation occurs when the reaction conditions are sufficiently harsh, and the treatment allows adequate residence time for the degradation to complete103,155.
Mechanism of PFAS degradation with HALT
The degradation of PFOS is initiated and facilitated by hydroxide ions (OH−) through a series of nucleophilic substitution and decarboxylation reactions. Initially, the OH− ion replaces the sulfonate group, forming unstable alcohols, which is the rate-limiting step of the process. Following this, fluoride ions (F-) are released from the unstable alcohol and ketone intermediates. As ketones undergo hydration, additional F- is released, resulting in the formation of PFOA. This is followed by further decarboxylation of PFOA, gradually producing PFHxA and PFPeA as intermediates, while releasing more F- until the fluorocarbon chain is completely degraded103.
Benefits advantages of using HALT for PFAS degradation
HALT technology offers several benefits including effective degradation and defluorination of a wide range of PFAS species including ultrashort chain compounds such as Trifluoroacetic acid (TFA)105,156. Moreover, the HALT technology can be used to degrade PFAS on spent foam fractionates157 or spent GAC158, offering several advantages, including enhanced degradation of PFAS adsorbed on these materials, reducing the need for disposal or regeneration. This integration also maximizes resource efficiency by treating concentrated PFAS waste streams, minimizes secondary pollution as no additional chemicals are needed, and allows for in situ treatment, simplifying the overall remediation process.
Limitations for HALT application in PFAS destruction
The full-scale application of HALT for PFAS remediation faces several limitations. One significant challenge is the treatment of large water volumes, which makes scaling up the process difficult. Additionally, reactor corrosion due to the harsh conditions, need for pH neutralization after treatment, high temperature, and pressure is a concern, as it can reduce the lifespan of the equipment and increase maintenance costs. The method also requires the development of cost-effective, continuous-flow reactor systems to enable efficient large-scale use. Another major drawback is the high energy consumption needed to maintain the elevated temperatures and pressures required for effective PFAS degradation. Furthermore, managing the disposal of secondary byproducts and the need for specialized materials to withstand extreme operating conditions add to the complexity and cost of the HALT process.
Advanced reduction process (ARP) for PFAS degradation
ARPs utilize highly reactive reducing radicals to degrade PFAS, typically generated by combining ultraviolet (UV) light with water or chemical additives such as sulfite and potassium iodide. The key reactive species in ARPs are hydrated electrons, which are known for their strong reductive power and high efficiency in breaking down and defluorinating PFAS. These processes offer a promising approach for PFAS treatment, as hydrated electrons can attack the carbon-fluorine bonds in PFAS molecules, leading to significant degradation and defluorination159,160.
Mechanism of PFAS degradation with ARP
The proposed degradation pathways for PFOA have been studied by Ren et al.159 who determined that the degradation of PFOA in the UV/sulfite system follows a more complex mechanism. The detection of sulfonated intermediates, unsaturated compounds, and other byproducts indicates that multiple pathways are involved in the breakdown of PFOA. Due to the chemical structure of PFOA, each C–F bond can be attacked by hydrated electrons and participate in the reaction, resulting in the formation of various intermediates from different parent compounds.
Advantages of using ARP for the degradation of PFAS
The generation of hydrated electrons in solution allows for degradation without interacting with a surface, a limitation often seen in electrochemical and nonthermal plasma systems when degrading short and ultrashort PFAS. The UV/sulfite process has demonstrated its effectiveness in degrading PFOA in brine solutions, even in the presence of high pH and chloride concentrations, without being affected by chloride interference, unlike traditional advanced oxidation processes (AOPs).
Limitations for applications ARP in the destruction of PFAS
The presence of background water constituents, such as DO, hydrogen protons (H+), dissolved organic matter, and nitrate, can scavenge reducing radicals, which hinders the degradation of target PFAS159,161. Additionally, the use of UV light results in high energy consumption, posing challenges for scaling up the process. Moreover, PFAS is often found in low concentrations in real water sources, making it difficult to achieve meaningful kinetics in degradation. Furthermore, the reagents employed in ARPs significantly increase the total dissolved solids in water, rendering it impractical for treating water intended for potable reuse or for discharge into freshwater bodies.
Electron beam (e-beam) technology
Electron beam irradiation of water causes the decomposition of water molecules, a process known as water radiolysis, resulting in the formation of reactive species such as protons (H+), hydrated electrons (eaq-), and hydroxyl radicals (•OH), among others. E-beam treatment is regarded as an advanced oxidation/reduction process (AORP) because it simultaneously generates both oxidizing and reducing species162,163.
Mechanism of PFAS degradation with electron beam
Londhe et al.162 performed HRMS suspect screening to observe the degradation of PFOA and PFOS and their intermediates formed. From the intermediates, they found the process involves the stepwise reaction pathway termed decarboxylation—hydroxylation-elimination-hydrolysis (DHEH) process. During the DHEH process, a CO2 moiety is cleaved, leading to the formation of a free radical (CnF2n+1COO- → CnF2n+1•). This step, known as decarboxylation, is followed by hydroxylation, where an OH group is added to the free radical (CnF2n+1–OH). PFAS-alcohols are typically unstable and undergo HF elimination to produce an acyl fluoride, which can then hydrolyze to form a shorter-chain PFCA (Cn−1F2n−1–COO-)
Benefits of using electron beam for PFAS degradation
Electron beam irradiation of PFAS offers effective degradation without the need for chemical additives, resulting in a more environmentally friendly treatment process. Additionally, this method can achieve rapid degradation rates, reduce the formation of toxic byproducts, and treat a wide range of PFAS compounds, making it a versatile solution for addressing PFAS contamination
Limitations for applications of e- beam for the destruction of PFAS
The process requires high energy to produce electron beams. The effectiveness of electron beams decreases in complex matrices and water depth and the inability to treat large flow rates. So far, there have been very few studies examining the effectiveness of this technology for treating PFAS. The results from these limited studies are not directly comparable due to differences in operating conditions and water quality parameters employed across the investigations. Although a study162 an improved understanding of the mechanism of PFAS degradation and revealed that short-chain PFAS are more resistant to defluorination and their levels and regulation in the environment will determine the operating conditions of e-beam and other PFAS treatment technologies162. Another limitation is related to the application of this technology when real-world contaminated groundwater samples were treated. The negative impact is theorized to be due to scavenging of hydrated electrons by matrix components162. However, e-beam was able to simultaneously oxidize PFAS precursors present in these samples, demonstrating its utilization as an oxidative/reductive treatment162.
Overall, the production of short chain PFAS could be regarded as one of the biggest challenges with most PFAS destructive technologies. A further challenge pertains to the confirmation of fluorine mass balance after the degradation of PFAS mainly in real water matrix. For the environmental water sample, tracking targeted PFAS molecule is not limited to the pattern observed with spiked samples prepared in the laboratories. Although this limitation could be attributed to the analytical instrument, the fact that the determination capabilities aren’t always straightforward to measure the fluorine mass balance and confirm the extend of PFAS destruction, this aspect remains an important issue to overcome. Therefore, there is a warrant to consider fluorine mass balance as part of an effective PFAS destructive technologies164.
Proposed remediation of PFAS using two or more coupled/integrated systems
Based on the limitations of current PFAS removal technologies, there is an opportunity to couple non-destructive processes and destructive processes through integrated systems to treat concentrated secondary wastes. An integrated system involves the combination of two or more techniques (sequentially or simultaneously) to overcome the limitations of the individual techniques. Potential integrated systems that have been investigated in degradation of PFAS include the combination of non-destructive (selective adsorption or membrane separation) and destructive technique (AOPs). When these technologies are used in the same system, it is called a parallel treatment train. Integrated parallel treatment trains involve concentrating PFAS via adsorption or membrane separation, then the PFAS concentrates are subsequently subjected to the destructive technique to degrade concentrated PFAS. Since the PFAS concentrates are usually taken off-site for further treatment (safe disposal or incineration)165, parallel treatment trains save transportation cost and reduce risk of secondary contamination. Parallel treatments trains that have been investigated include nanofiltration/electrochemical anodic oxidation (NF-EO), GAC adsorption/activated persulfate (GA- S2O8), ion-exchange resin/electrochemical anodic oxidation (IXR-EO)166,167.
Although treatment train systems can be effective, they may be expensive because each technique operates separately. Coupling of membrane separation and EO, where EO is used to treat the PFAS concentrated brine generated by membrane separation faces the challenge that these technologies have unique capital and operational costs that when combined create an efficient but expensive system165. This has warranted innovation of integrated systems into singular destructive hybrid systems. The innovative integrated systems that have been investigated in degradation of PFAS include REM and photocatalytic membranes. The hybrid systems separate PFAS while degrading them at the same time. Table 4 summarizes the integrated technologies that have been applied in PFAS treatment. For example, Magnéli Phase titanium suboxide was used to remove and degrade both PFOS and PFOA, with a removal efficiency of over 99.9% and greater than 69% defluorination167. The energy consumption to completely degrade PFOA and PFOS were 5.1 and 6.7 kWh m−3 respectively. This energy consumption is one of the lowest compared to other previously reported electrochemical processes88.
The results of one of the largest electrochemical PFAS oxidation tests conducted until today has been reported168, along with details on pilot-scale treatment via sonolysis and low-temperature plasma. A reduction was reported in the electric energy consumed per order of reaction (EEO) for treating PFOA and PFOS in groundwater by the IXR-EO treatment train14. The EEO was lower than that reported for stand-alone electrochemical treatment9. In other work, an integrated system of regenerable IX with plasma treatment result in near zero effluent PFAS94. The integration of adsorption with photocatalysis or EO reduces the need for chemical regeneration of adsorbents169,170,171. Based on the extensive presentation and discussion of the various techniques researched for the removal of PFAS, there is no single technique that can serve as a universally applicable panacea to effectively mineralize PFAS, which suggests that the integration of two or more of scalable technologies into treatment trains may be required for enhanced PFAS removal and destruction efficiency.
Conclusion and perspectives
This review examined the progress made to date in treating PFAS-contaminated water and wastewater, and several remediation techniques were discussed in detail. Despite the demonstrated successes of these techniques at the bench scale, there are still obstacles that limit the full-scale application of existing PFAS removal methods:
-
Presence of competing inorganic and organic species in real water matrices which scavenge adsorption and oxidation capacity, and decrease remediation efficiency
-
Generation of high volumes of PFAS-concentrated streams or solid waste, necessitating costly and energy-intensive treatment for safe disposal
-
High manufacturing costs of adsorbents, electrodes and membranes
-
Incomplete degradation of PFAS and the potential formation of unregulated (and unknown) toxic by-products
-
Lack of large-scale standalone reactors for PFAS degradation or removal
Based on the presented challenges, several recommendations are proposed:
-
Consider integrated systems (including treatment trains) and their optimized integration to enhance removal efficiency and eliminate toxicity. Integrated systems should consist of a non-destructive, adsorptive (or other PFAS separation and concentration) technique, which requires relatively low energy input, and destructive technique that ensures subsequent degradation of PFAS. An example of this would be using a membrane to generate a highly concentrated PFAS waste stream that can be treated with an AOP like EO, resulting in two PFAS-free streams172.
-
Optimize reactor design parameters and conditions for scaling up to full industrial application.
-
The necessity of proving the efficacy of long-term PFAS treatment in the treatments that are discussed.
Additionally, improvements in PFAS analysis should focus on understanding how the presence of co-contaminants influence PFAS behavior during treatment, aiding in predicting their transport and fate in the environment. Cost reductions in manufacturing, chemicals, and energy consumption can be achieved through the integrated treatment-train method for PFAS remediation, employing multiple techniques to treat a broad range of contaminants with minimal treatment chemicals and energy usage. Selective separation approaches may also be needed to concentrate PFAS for more energy-efficient destruction. In conclusion, addressing these challenges through integrated treatment approaches and continuous improvements in technology and cost-effectiveness will be crucial for enhancing water treatment and advancing the full-scale remediation of PFAS-contaminated environments.
Data availability
No datasets were generated or analyzed during the current study.
References
Sidnell, T., Wood, R. J., Hurst, J., Lee, J. & Bussemaker, M. J. Sonolysis of per-and poly fluoroalkyl substances (PFAS): a meta-analysis. Ultrason. Sonochem. 87, 105944 (2022).
O’Hagan, D. Understanding organofluorine chemistry. an introduction to the C–F bond. Chem. Soc. Rev. 37, 308–319 (2008).
3M. The Science of Organic Fluorochemistry. http://www.fluoridealert.org/wp-content/pesticides/pfos.fr.final.docket.0006.pdf (1999).
Al Amin, M. et al. Recent advances in the analysis of per-and polyfluoroalkyl substances (PFAS)—A review. Environ. Technol. Innov. 19, 100879 (2020).
Lewis, A. J. et al. Rapid degradation of PFAS in aqueous solutions by reverse vortex flow gliding arc plasma. Environ. Sci. Water Res. Technol. 6, 1044–1057 (2020).
https://www.atsdr.cdc.gov/pfas/health-effects/us-population.html (accessed 10 September 2024).
Wee, S. Y. & Aris, A. Z. Revisiting the “forever chemicals”, PFOA and PFOS exposure in drinking water. NPJ Clean. Water 6, 57 (2023).
Deng, Y. et al. The degradation mechanisms of perfluorooctanoic acid (PFOA) and perfluorooctane sulfonic acid (PFOS) by different chemical methods: a critical review. Chemosphere 283, 131168 (2021).
Schaefer, C. E. et al. Electrochemical transformations of perfluoroalkyl acid (PFAA) precursors and PFAAs in groundwater impacted with aqueous film forming foams. Environ. Sci. Technol. 52, 10689–10697 (2018).
Zhuo, Q. et al. Degradation of perfluorinated compounds on a boron-doped diamond electrode. Electrochim. Acta 77, 17–22 (2012).
Carter, K. E. & Farrell, J. Oxidative destruction of perfluorooctane sulfonate using boron-doped diamond film electrodes. Environ. Sci. Technol. 42, 6111–6115 (2008).
Ochiai, T. et al. Efficient electrochemical decomposition of perfluorocarboxylic acids by the use of a boron-doped diamond electrode. Diam. Relat. Mater. 20, 64–67 (2011).
Veciana, M. et al. Electrochemical oxidation processes for PFAS removal from contaminated water and wastewater: fundamentals, gaps and opportunities towards practical implementation. J. Hazard. Mater. 434, 128886 (2022).
Liang, S. et al. Field demonstration of coupling ion-exchange resin with electrochemical oxidation for enhanced treatment of per-and polyfluoroalkyl substances (PFAS) in groundwater. Chem. Eng. J. Adv. 9, 100216 (2022).
Meegoda, J. N., Bezerra de Souza, B., Casarini, M. M. & Kewalramani, J. A. A review of PFAS destruction technologies. Int. J. Environ. Res. Public Health 19, 16397 (2022).
Trautmann, A. M., Schell, H., Schmidt, K. R., Mangold, K. M. & Tiehm, A. Electrochemical degradation of perfluoroalkyl and polyfluoroalkyl substances (PFASs) in groundwater. Water Sci. Technol. 71, 1569–1575 (2015).
Ateia, M., Maroli, A., Tharayil, N. & Karanfil, T. The overlooked short-and ultrashort-chain poly-and perfluorinated substances: a review. Chemosphere 220, 866–882 (2019).
Uwayezu, J. N. et al. Electrochemical degradation of per-and poly-fluoroalkyl substances using boron-doped diamond electrodes. J. Environ. Manag. 290, 112573 (2021).
https://www.epa.gov/sdwa/and-polyfluoroalkyl-substances-pfas (accessed 11 May 2024)
Wang, J. et al. Per-and polyfluoroalkyl substances (PFASs) in Chinese surface water: temporal trends and geographical distribution. Sci. Total Environ. 915, 170127 (2024)
DIRECTIVE (EU) 2020/2184 OF THE EUROPEAN PARLIAMENT AND OF THE COUNCIL of 16 December 2020 on the quality of water intended for human consumption, accessed 21 June 2024. https://eur-lex.europa.eu/eli/dir/2020/2184/oj.
Domingo, J. L. & Nadal, M. Human exposure to per-and polyfluoroalkyl substances (PFAS) through drinking water: a review of the recent scientific literature. Environ. Res. 177, 108648 (2019).
Ackerman Grunfeld, D. et al. Underestimated burden of per-and polyfluoroalkyl substances in global surface waters and groundwaters. Nat. Geosci. 17, 340–346 (2024).
https://www.epa.gov/pfas/our-current-understanding-human-health-and-environmental-risks-pfas (accessed 19 September 2024).
Smithwick, M. et al. Circumpolar study of perfluoroalkyl contaminants in polar bears (Ursus maritimus). Environ. Sci. Technol. 39, 5517–5523 (2005).
Blake, B. E. & Fenton, S. E. Early life exposure to per-and polyfluoroalkyl substances (PFAS) and latent health outcomes: a review including the placenta as a target tissue and possible driver of peri-and postnatal effects. Toxicology 443, 152565 (2020).
Kotlarz, N. et al. Measurement of novel, drinking water-associated PFAS in blood from adults and children in Wilmington, North Carolina. Environ. Health Perspect. 128, 077005 (2020).
https://www.iarc.who.int/news-events/iarc-monographs-evaluate-the-carcinogenicity-of-perfluorooctanoic-acid-pfoa-and-perfluorooctanesulfonic-acid-pfos/ (accessed 24 September 2024).
Anderson, R. H., Long, G. C., Porter, R. C. & Anderson, J. K. Occurrence of select perfluoroalkyl substances at U.S. Air Force aqueous film-forming foam release sites other than fire-training areas: field-validation of critical fate and transport properties. Chemosphere 150, 678–685 (2016).
Bonato, M. et al. PFAS environmental pollution and antioxidant responses: an overview of the impact on human field. Int. J. Environ. Res. Public Health 17, 8020 (2020).
Meegoda, J. N., Kewalramani, J. A., Li, B. & Marsh, R. W. A review of the applications, environmental release, and remediation technologies of per-and polyfluoroalkyl substances. Int. J. Environ. Res. Public Health 17, 8117 (2020).
Brennan, N. M., Evans, A. T., Fritz, M. K., Peak, S. A. & von Holst, H. E. Trends in the regulation of per-and polyfluoroalkyl substances (PFAS): a scoping review. Int. J. Environ. Res. Public Health 18, 10900 (2021).
Interstate Technology and Regulatory Council (ITRC). Interstate technology and regulatory council: Naming conventions and physical and chemical properties of per- and polyfluoroalkyl substances (PFAS). URL: https://pfas-1.itrcweb.org/fact-sheets/ (2020).
Buck, R. C. et al. Perfluoroalkyl and polyfluoroalkyl substances in the environment: terminology, classification, and origins. Integr. Environ. Assess. Manag. 7, 513 (2011).
Li, F. et al. Short-chain per-and polyfluoroalkyl substances in aquatic systems: occurrence, impacts and treatment. Chem. Eng. J. 380, 122506 (2020).
Agenson, K. O. & Urase, T. Change in membrane performance due to organic fouling in nanofiltration (NF)/reverse osmosis (RO) applications. Sep. Purif. Technol. 55, 147 (2007).
Dixit, F., Dutta, R., Barbeau, B., Berube, P. & Mohseni, M. PFAS removal by ion exchange resins: a review. Chemosphere 272, 129777 (2021).
Woodard, S., Berry, J. & Newman, B. Ion exchange resin for PFAS removal and pilot test comparison to GAC. Remediat. J. 27, 19 (2017).
Rahman, M. F. Removal of Perfluorinated Compounds from Ultrapure and Surface Waters by Adsorption and Ion Exchange. University of Waterloo (2014).
Belkouteb, N., Franke, V., McCleaf, P., Köhler, S. & Ahrens, L. Removal of per-and polyfluoroalkyl substances (PFASs) in a full-scale drinking water treatment plant: long-term performance of granular activated carbon (GAC) and influence of flow-rate. Water Res. 182, 115913 (2020).
Du, Z. et al. Adsorption behavior and mechanism of perfluorinated compounds on various adsorbents—a review. J. Hazard. Mater. 274, 443 (2014).
Wei, Z., Xu, T. & Zhao, D. Treatment of per-and polyfluoroalkyl substances in landfill leachate: status, chemistry and prospects. Environ. Sci. Water Res. Technol. 5, 1814 (2019).
Murray, C. C. et al. Removal of per- and polyfluoroalkyl substances using super-fine powder activated carbon and ceramic membrane filtration. J. Hazard. Mater. 366, 160 (2019).
Hung, Y.-T., Lo, H. H., Wang, L. K., Taricska, J. R., & Li, K. H., Granular activated Carbon adsorption. in Physicochemical Treatment Processes (eds Wang, L.K., Hung, Y.-T. & Shammas, N.K.) 573 (Humana Press, 2005).
Senevirathna, S. T. M. L. D. et al. A comparative study of adsorption of perfluorooctane sulfonate (PFOS) onto granular activated carbon, ion-exchange polymers and non-ion-exchange polymers. Chemosphere 80, 647 (2010).
Appleman, T. D. et al. Treatment of poly- and perfluoroalkyl substances in U.S. full-scale water treatment systems. Water Res. 51, 246 (2014).
Jiang, X. et al. Pilot-scale removal of PFAS from chromium-plating wastewater by anion exchange resin and activated carbon: Adsorption difference between PFOS and 6: 2 fluorotelomer sulfonate. Chem. Eng. J. 481, 148569 (2024).
Franke, V., McCleaf, P., Lindegren, K. & Ahrens, L. Efficient removal of per-and polyfluoroalkyl substances (PFASs) in drinking water treatment: nanofiltration combined with active carbon or anion exchange. Environ. Sci. Water Res. Technol. 5, 1836 (2019).
Murray, C. C., Marshall, R. E., Liu, C. J., Vatankhah, H. & Bellona, C. L. PFAS treatment with granular activated carbon and ion exchange resin: comparing chain length, empty bed contact time, and cost. J. Water Process Eng. 44, 102342 (2021).
Marshall, R. Performance and Cost-Effectiveness of Comercially Avaliable Adsorptive Technologies for Treatment of Per- and Polyfluoroalkyl Substances (PFAS) Impacted Groundwater, (Master of Science thesis, ProQuest Number: 27662681, Colorado School of Mines) (2019).
Franke, V. et al. The price of really clean water: Combining nanofiltration with granular activated carbon and anion exchange resins for the removal of per-and polyfluoralkyl substances (PFASs) in drinking water production. ACS ES&T Water 1.4. 782–795 (2021)
Garg, S. et al. Remediation of water from per-/poly-fluoroalkyl substances (PFAS)–challenges and perspectives. J. Environ. Chem. Eng. 9, 105784 (2021).
Najm, I. et al. Per-and polyfluoroalkyl substances removal with granular activated carbon and a specialty adsorbent: a case study. AWWA Water Sci. 3, e1245 (2021).
Wang, Y. et al. Performance and mechanisms for removal of perfluorooctanoate (PFOA) from aqueous solution by activated carbon fiber. RSC Adv. 5, 86927 (2015).
McNamara, J. D., Franco, R., Mimna, R. & Zappa, L. Comparison of activated carbons for removal of perfluorinated compounds from drinking water. J. Am. Water Works Assoc. 110, E2–E14 (2018).
Zaggia, A., Conte, L., Falletti, L., Fant, M. & Chiorboli, A. Use of strong anion exchange resins for the removal of perfluoroalkylated substances from contaminated drinking water in batch and continuous pilot plants. Water Res. 91, 137 (2016).
Lee, T., Ooi, C., Othman, R. & Yeoh, F.-Y. Activated carbon fiber—the hybrid of carbon fiber and activated carbon. Rev. Adv. Mater. Sci. 36, 118 (2014).
Liu, C. J., Werner, D. & Bellona, C. Removal of per-and polyfluoroalkyl substances (PFASs) from contaminated groundwater using granular activated carbon: a pilot-scale study with breakthrough modeling. Environ. Sci. Water Res. Technol. 5, 1844 (2019).
Liu, L., Li, D., Li, C., Ji, R. & Tian, X. Metal nanoparticles by doping carbon nanotubes improved the sorption of perfluorooctanoic acid. J. Hazard. Mater. 351, 206 (2018).
Wu, C., Klemes, M. J., Trang, B., Dichtel, W. R. & Helbling, D. E. Exploring the factors that influence the adsorption of anionic PFAS on conventional and emerging adsorbents in aquatic matrices. Water Res. 182, 115950 (2020).
Wang, M. et al. Removal of PFASs from water by carbon-based composite photocatalysis with adsorption and catalytic properties: a review. Sci. Total Environ. 836, 155652 (2022).
Saeidi, N., Kopinke, F. D. & Georgi, A. What is specific in adsorption of perfluoroalkyl acids on carbon materials? Chemosphere 273, 128520 (2021).
Medina, R. et al. Pilot-scale comparison of granular activated carbons, ion exchange, and alternative adsorbents for per-and polyfluoroalkyl substances removal. AWWA Water Sci. 4, e1308 (2022).
Ullberg, M., Lavonen, E., Köhler, S. J., Golovko, O. & Wiberg, K. Pilot-scale removal of organic micropollutants and natural organic matter from drinking water using ozonation followed by granular activated carbon. Environ. Sci. Water Res. Technol. 7, 535–548 (2021).
McCleaf, P. et al. Removal efficiency of multiple poly- and perfluoroalkyl substances (PFASs) in drinking water using granular activated carbon (GAC) and anion exchange (AE) column tests. Water Res. 120, 77–87 (2017)
Inyang, M. & Dickenson, E. R. The use of carbon adsorbents for the removal of perfluoroalkyl acids from potable reuse systems. Chemosphere 184, 168–175 (2017).
Cantoni, B., Turolla, A., Wellmitz, J., Ruhl, A. S. & Antonelli, M. Perfluoroalkyl substances (PFAS) adsorption in drinking water by granular activated carbon: Influence of activated carbon and PFAS characteristics. Sci. Total Environ. 795, 148821 (2021).
Son, H. & An, B. Investigation of adsorption kinetics for per-and poly-fluoroalkyl substances (PFAS) adsorption onto powder activated carbon (PAC) in the competing systems. Water Air Soil Pollut. 233, 129 (2022).
Son, H., Kim, T., Yoom, H. S., Zhao, D. & An, B. The adsorption selectivity of short and long per-and polyfluoroalkyl substances (PFASs) from surface water using powder-activated carbon. Water 12, 3287 (2020).
Lei, Y. J., Tian, Y., Sobhani, Z., Naidu, R. & Fang, C. Synergistic degradation of PFAS in water and soil by dual-frequency ultrasonic activated persulfate. Chem. Eng. J. 388, 124215 (2020).
Bolan, N. et al. Remediation of poly-and perfluoroalkyl substances (PFAS) contaminated soils–to mobilize or to immobilize or to degrade? J. Hazard. Mater. 401, 123892 (2021).
Huheey, P. Department of Defense United States of America- Incineration Moratorium Report to Congress. https://www.acq.osd.mil/eie/eer/ecc/pfas/docs/reports/Incineration-Mortatorium.pdf accessed in June 2024 (2023).
Li, C., Sun, W., Lu, Z., Ao, X. & Li, S. Ceramic nanocomposite membranes and membrane fouling: a review. Water Res. 175, 115674 (2020).
Liu, C. et al. Evaluating the efficiency of nanofiltration and reverse osmosis membrane processes for the removal of per- and polyfluoroalkyl substances from water: a critical review. Sep. Purif. Technol. 302, 122161 (2022).
Horst, J. et al. Water treatment technologies for PFAS: the next generation. Groundw. Monit. Remediat. 38, 13 (2018).
Crone, B. C. et al. Occurrence of per-and polyfluoroalkyl substances (PFAS) in source water and their treatment in drinking water. Crit. Rev. Environ. Sci. Technol. 49, 2359 (2019).
Johnson, J. K., Hoffman, C. M. Jr., Smith, D. A. & Xia, Z. Advanced filtration membranes for the removal of perfluoroalkyl species from water. ACS Omega 4, 8001 (2019).
Zeng, C., Tanaka, S., Suzuki, Y. & Fujii, S. Impact of feed water pH and membrane material on nanofiltration of perfluorohexanoic acid in aqueous solution. Chemosphere 183, 599 (2017).
Tang, C. Y., Fu, Q. S., Criddle, C. S. & Leckie, J. O. Effect of flux (transmembrane pressure) and membrane properties on fouling and rejection of reverse osmosis and nanofiltration membranes treating perfluorooctane sulfonate containing wastewater. Environ. Sci. Technol. 41, 2008 (2007).
Boo, C. et al. High performance nanofiltration membrane for effective removal of perfluoroalkyl substances at high water recovery. Environ. Sci. Technol. 52, 7279 (2018).
Lee, T., Speth, T. F. & Nadagouda, M. N. High-pressure membrane filtration processes for separation of Per-and polyfluoroalkyl substances (PFAS). Chem. Eng. J. 431, 134023 (2022).
Safulko, A. et al. Rejection of perfluoroalkyl acids by nanofiltration and reverse osmosis in a high-recovery closed-circuit membrane filtration system. Sep. Purif. Technol. 326, 124867 (2023).
Liu, C. J. et al. Pilot-scale field demonstration of a hybrid nanofiltration and UV-sulfite treatment train for groundwater contaminated by per-and polyfluoroalkyl substances (PFASs). Water Res. 205, 117677 (2021).
Tang, C. Y., Fu, Q. S., Robertson, A. P., Criddle, C. S. & Leckie, J. O. Use of reverse osmosis membranes to remove perfluorooctane sulfonate (PFOS) from semiconductor wastewater. Environ. Sci. Technol. 40, 7343 (2006).
Appleman, T. D., Dickenson, E. R., Bellona, C. & Higgins, C. P. Nanofiltration and granular activated carbon treatment of perfluoroalkyl acids. J. Hazard. Mater. 260, 740 (2013).
Zeng, C., Tanaka, S., Suzuki, Y., Yukioka, S. & Fujii, S. Rejection of trace level perfluorohexanoic acid (PFHxA) in pure water by loose nanofiltration membrane. J. Water Environ. Technol. 15, 120 (2017).
Lipp, P., Sacher, F. & Baldauf, G. Removal of organic micro-pollutants during drinking water treatment by nanofiltration and reverse osmosis. Desalin. Water Treat. 13, 226 (2010).
Le, T. X. H., Haflich, H., Shah, A. D. & Chaplin, B. P. Energy-efficient electrochemical oxidation of perfluoroalkyl substances using a Ti4O7 reactive electrochemical membrane anode. Environ. Sci. Technol. Lett. 6, 504 (2019).
Liu, F., Hua, L. & Zhang, W. Influences of microwave irradiation on performances of membrane filtration and catalytic degradation of perfluorooctanoic acid (PFOA). Environ. Int. 143, 105969 (2020).
Eke, J. et al. Dual-functional phosphorene nanocomposite membranes for the treatment of perfluorinated water: an investigation of perfluorooctanoic acid removal via filtration combined with ultraviolet irradiation or oxygenation. Membranes 11, 18 (2021).
Das, S. & Ronen, A. A review on removal and destruction of per-and polyfluoroalkyl substances (PFAS) by novel membranes. Membranes 12, 662 (2022).
Kanchanapiya, P. & Tantisattayakul, T. Analysis of the additional cost of addressing per-and polyfluoroalkyl substance contamination from landfill leachate by reverse osmosis membranes in Thailand. J. Water Process Eng. 45, 102520 (2022).
Soriano, Á., Gorri, D., Biegler, L. T. & Urtiaga, A. An optimization model for the treatment of perfluorocarboxylic acids considering membrane preconcentration and BDD electrooxidation. Water Res. 164, 114954 (2019).
Singh, R. K. et al. Removal of poly-and per-fluorinated compounds from ion exchange regenerant still bottom samples in a plasma reactor. Environ. Sci. Technol. 54, 13973–13980 (2020).
Krause, M. J. et al. Supercritical water oxidation as an innovative technology for PFAS destruction. J. Environ. Eng. 148, 05021006 (2022).
Li, J., Austin, C., Moore, S., Pinkard, B. R. & Novosselov, I. V. PFOS destruction in a continuous supercritical water oxidation reactor. Chem. Eng. J. 451, 139063 (2023).
Xia, C., Lim, X., Yang, H., Goodson, B. M. & Liu, J. Degradation of per-and polyfluoroalkyl substances (PFAS) in wastewater effluents by photocatalysis for water reuse. J. Water Process Eng. 46, 102556 (2022).
Tester, J. W. et al. Supercritical water oxidation technology: process development and fundamental research. Am. Chem. Soc. 35–76 (1993).
Bermejo, M. D. & Cocero, M. J. Supercritical water oxidation: a technical review. AIChE J. 52, 3933 (2006).
Nagar, K. & Deshusses, M. A. Supercritical Water Oxidation (SCWO). [17/08/2022]: from: https://www.enviro.wiki/index.php?title=Supercritical_Water_Oxidation_(SCWO)
Jiang, Z. et al. Review on mechanisms and kinetics for supercritical water oxidation processes. Appl. Sci. 10, 4937 (2020).
Pinkard, B. R., Shetty, S., Stritzinger, D., Bellona, C. & Novosselov, I. V. Destruction of perfluorooctanesulfonate (PFOS) in a batch supercritical water oxidation reactor. Chemosphere 279, 130834 (2021).
Wu, B. et al. Rapid destruction and defluorination of perfluorooctanesulfonate by alkaline hydrothermal reaction. Environ. Sci. Technol. Lett. 6, 630 (2019).
Hori, H. et al. Efficient decomposition of environmentally persistent perfluorooctanesulfonate and related fluorochemicals using zerovalent iron in subcritical water. Environ. Sci. Technol. 40, 1049 (2016).
Hao, S., Choi, Y. J., Deeb, R. A., Strathmann, T. J. & Higgins, C. P. Application of hydrothermal alkaline treatment for destruction of per-and polyfluoroalkyl substances in contaminated groundwater and soil. Environ. Sci. Technol. 56, 6647–6657 (2022).
Tang, X. et al. Corrosion behavior of nickel base alloys, stainless steel and titanium alloy in supercritical water containing chloride, phosphate and oxygen. Chem. Eng. Res. Des. 100, 530–541 (2015).
McDonough, J. T. et al. Validation of supercritical water oxidation to destroy perfluoroalkyl acids. Remediat. J. 32, 75–90 (2022).
Rayaroth, M. P., Aravind, U. K. & Aravindakumar, C. T. Degradation of pharmaceuticals by ultrasound-based advanced oxidation process. Environ. Chem. Lett. 14, 259 (2016).
Cabeza, P. et al. Reactors for supercritical water oxidation processes. in Near-Critical and Supercritical Water and Their Applications for Biorefineries (eds Fang, Z. & Xu, C.) 179 (Springer Netherlands 2014).
Asselin, E., Alfantazi, A. & Rogak, S. Thermodynamics of the corrosion of alloy 625 supercritical water oxidation reactor tubing in ammoniacal sulfate solution. Corrosion 64, 301 (2018).
Peng, K., Qin, F. G., Jiang, R., Qu, W. & Wang, Q. Production and dispersion of free radicals from transient cavitation Bubbles: an integrated numerical scheme and applications. Ultrason. Sonochem. 88, 106067 (2022).
Cao, H., Zhang, W., Wang, C. & Liang, Y. Sonochemical degradation of poly- and perfluoroalkyl substances—a review. Ultrason. Sonochem. 69, 105245 (2020).
Kanthale, P., Ashokkumar, M. & Grieser, F. Sonoluminescence, sonochemistry (H2O2 yield) and bubble dynamics: frequency and power effects. Ultrason. Sonochem. 15, 143 (2008).
Shende, T., Andaluri, G. & Suri, R. Power density modulated ultrasonic degradation of perfluoroalkyl substances with and without sparging Argon. Ultrason. Sonochem. 76, 105639 (2021).
Rodriguez-Freire, L., Balachandran, R., Sierra-Alvarez, R. & Keswani, M. Effect of sound frequency and initial concentration on the sonochemical degradation of perfluorooctane sulfonate (PFOS). J. Hazard. Mater. 300, 662 (2015).
de Souza, B. B., Hewage, S. A., Kewalramani, J. A., van Duin, A. C. & Meegoda, J. N. A ReaxFF-based molecular dynamics study of the destruction of PFAS due to ultrasound. Environ. Pollut. 333, 122026 (2023).
Metz, J., Zuo, P., Wang, B., Wong, M. S. & Alvarez, P. J. Perfluorooctanoic acid degradation by UV/chlorine. Environ. Sci. Technol. Lett. 9, 673–679 (2022).
Gole, V. L. et al. Sono-chemical treatment of per-and poly-fluoroalkyl compounds in aqueous film-forming foams by use of a large-scale multi-transducer dual-frequency based acoustic reactor. Ultrason. Sonochem. 45, 213 (2018).
Fuller, M. E. et al. Sonochemical degradation of PFAS in ion exchange regeneration wastes. J. Hazard. Mater. 471, 134291 (2024).
Kewalramani, J. A., de Souza, B. B., Marsh, R. W. & Meegoda, J. N. Contributions of reactor geometry and ultrasound frequency on the efficiency of sonochemical reactor. Ultrason. Sonochem. 98, 106529 (2023).
Kalra, S. S. et al. Sonolytic destruction of Per-and polyfluoroalkyl substances in groundwater, aqueous Film-Forming Foams, and investigation derived waste. Chem. Eng. J. 425, 131778 (2021).
Campbell, T. Y., Vecitis, C. D., Mader, B. T. & Hoffmann, M. R. Perfluorinated surfactant chain-length effects on sonochemical kinetics. J. Phys. Chem. A 113, 9834–9842 (2009).
Mahamuni, N. N. & Adewuyi, Y. G. Advanced oxidation processes (AOPs) involving ultrasound for waste water treatment: a review with emphasis on cost estimation. Ultrason. Sonochem. 17, 990–1003 (2010).
Son, Y., Lim, M., Song, J. & Khim, J. Liquid height effect on sonochemical reactions in a 35 kHz sonoreactor. Jpn. J. Appl. Phys. 48, 07GM16 (2009).
Lim, M., Ashokkumar, M. & Son, Y. The effects of liquid height/volume, initial concentration of reactant and acoustic power on sonochemical oxidation. Ultrason. Sonochem. 21, 1988–1993 (2014).
Groenen Serrano, K. Chapter 6—Indirect electrochemical oxidation using hydroxyl radical, active chlorine, and peroxodisulfate. in Electrochemical Water and Wastewater Treatment (eds Martínez-Huitle, C.A., Rodrigo, M.A. & Scialdone, O.) 133 (Butterworth-Heinemann, 2018).
Sharma, S., Shetti, N. P., Basu, S., Nadagouda, M. N. & Aminabhavi, T. M. Remediation of per- and polyfluoroalkyls (PFAS) via electrochemical methods. Chem. Eng. J. 430, 132895 (2022).
Liang, S., Pierce, R. D. Jr, Lin, H., Chiang, S. Y. & Huang, Q. J. Electrochemical oxidation of PFOA and PFOS in concentrated waste streams. Remediat. J. 28, 127–134 (2018).
Wang, K., Huang, D., Wang, W., Ji, Y. & Niu, J. Enhanced perfluorooctanoic acid degradation by electrochemical activation of peroxymonosulfate in aqueous solution. Environ. Int. 137, 105562 (2020).
Guo, L., Jing, Y. & Chaplin, B. P. Development and characterization of ultrafiltration TiO2 Magnéli phase reactive electrochemical membranes. Environ. Sci. Technol. 50, 1428–1436 (2016).
Yang, S., Fernando, S., Holsen, T. M. & Yang, Y. Inhibition of perchlorate formation during the electrochemical oxidation of perfluoroalkyl acid in groundwater. Environ. Sci. Technol. Lett. 6, 775 (2019).
Yin, S. et al. Trap-n-zap: Electrocatalytic degradation of perfluorooctanoic acid (PFOA) with UiO-66 modified boron nitride electrodes at environmentally relevant concentrations. Appl. Catal. B Environ. Energy 355, 124136 (2024).
Urtiaga, A., Gomez-Lavin, S. & Soriano, A. Electrochemical treatment of municipal landfill leachates and implications for poly-and perfluoroalkyl substances (PFAS) removal. J. Environ. Chem. Eng. 10, 107900 (2022).
Barisci, S. & Suri, R. Electrooxidation of short-and long-chain perfluoroalkyl substances (PFASs) under different process conditions. J. Environ. Chem. Eng. 9, 105323 (2021).
Zhuo, Q., Wang, J., Niu, J., Yang, B. & Yang, Y. Electrochemical oxidation of perfluorooctane sulfonate (PFOS) substitute by modified boron doped diamond (BDD) anodes. Chem. Eng. J. 379, 122280 (2020).
Liu, G., Zhou, H., Teng, J. & You, S. Electrochemical degradation of perfluorooctanoic acid by macro-porous titanium suboxide anode in the presence of sulfate. Chem. Eng. J. 371, 7–14 (2019).
Guo, S. et al. Progress in preparation and application of titanium sub-oxides electrode in electrocatalytic degradation for wastewater treatment. Catalysts 12, 618 (2022).
Chaplin, B. P. The Prospect of electrochemical technologies advancing worldwide water treatment. Acc. Chem. Res. 52, 596–604 (2019).
Lin, H. et al. Defect engineering on a Ti4O7 electrode by Ce3+ doping for the efficient electrooxidation of perfluorooctanesulfonate. Environ. Sci. Technol. 55, 2597–2607 (2021).
Wang, L., Lu, J., Li, L., Wang, Y. & Huang, Q. Effects of chloride on electrochemical degradation of perfluorooctanesulfonate by Magnéli phase Ti4O7 and boron doped diamond anodes. Water Res. 170, 115254 (2020).
Xu, B., Ahmed, M. B., Zhou, J. L. & Altaee, A. Visible and UV photocatalysis of aqueous perfluorooctanoic acid by TiO2 and peroxymonosulfate: process kinetics and mechanistic insights. Chemosphere 243, 125366 (2020).
Javed, H., Lyu, C., Sun, R., Zhang, D. & Alvarez, P. J. Discerning the inefficacy of hydroxyl radicals during perfluorooctanoic acid degradation. Chemosphere 247, 125883 (2020).
Javed, H. et al. Discerning the relevance of superoxide in PFOA degradation. Environ. Sci. Technol. Lett. 7, 653–658 (2020).
Duan, L. et al. Efficient photocatalytic PFOA degradation over boron nitride. Environ. Sci. Technol. Lett. 7, 613–619 (2020).
McIntyre, H., Minda, V., Hawley, E., Deeb, R. & Hart, M. Coupled photocatalytic alkaline media as a destructive technology for per-and polyfluoroalkyl substances in aqueous film-forming foam impacted stormwater. Chemosphere 291, 132790 (2022).
Olatunde, O. C., Kuvarega, A. T. & Onwudiwe, D. C. Photo enhanced degradation of polyfluoroalkyl and perfluoroalkyl substances. Heliyon 6, e05614 (2020).
Jiang, F., Zhao, H., Chen, H., Xu, C. & Chen, J. Enhancement of photocatalytic decomposition of perfluorooctanoic acid on CeO2/In2O3. RSC Adv. 6, 72015 (2016).
Sadik, W. A., El-Demerdash, A.-G. M., Nashed, A. W., Mostafa, A. A. & Hamad, H. A. Highly efficient photocatalytic performance of Cu2O@TiO2 nanocomposite: Influence of various inorganic oxidants and inorganic anions. J. Mater. Res. Technol. 8, 5405–5414 (2019).
https://www.globalwaterintel.com/articles/can-plasma-based-technologies-strike-success-for-water-treatment (accessed 27 September 2024).
Topolovec, B. et al. Plasma water treatment for PFAS: study of degradation of perfluorinated substances and their byproducts by using cold atmospheric pressure plasma jet. J. Environ. Chem. Eng. 12, 112979–112979 (2024).
Stratton, G. R. et al. Plasma-based water treatment: efficient transformation of perfluoroalkyl substances in prepared solutions and contaminated groundwater. Environ. Sci. Technol. 51, 1643–1648 (2017).
Singh, R. K. et al. Rapid removal of poly- and perfluorinated compounds from investigation-derived waste (IDW) in a pilot-scale plasma reactor. Environ. Sci. Technol. 53, 11375–11382 (2019).
Palma, D., Richard, C. & Minella, M. State of the art and perspectives about non-thermal plasma applications for the removal of PFAS in water. Chem. Eng. J. Adv. 10, 100253 (2022).
Singh, R. K. et al. Breakdown products from perfluorinated alkyl substances (PFAS) degradation in a plasma-based water treatment process. Environ. Sci. Technol. 53, 2731–2738 (2019).
Hao, S., Choi, Y. J., Deeb, R. A., Strathmann, T. J. & Higgins, C. P. Application of hydrothermal alkaline treatment for destruction of per- and polyfluoroalkyl substances in contaminated groundwater and soil. Environ. Sci. Technol. 56, 6647–6657 (2022).
Austin, C. et al. Hydrothermal destruction and defluorination of trifluoroacetic acid (TFA). Environ. Sci. Technol. 58, 8076–8085 (2024).
Hao, S. et al. Hydrothermal alkaline treatment (HALT) of foam fractionation concentrate derived from PFAS-contaminated groundwater. Environ. Sci. Technol. 57, 17154–17165 (2023).
Soker, O., Hao, S., Trewyn, B. G., Higgins, C. P. & Strathmann, T. J. Application of hydrothermal alkaline treatment to spent granular activated carbon: destruction of adsorbed PFASs and adsorbent regeneration. Environ. Sci. Technol. Lett. 10, 425–430 (2023).
Ren, Z., Bergmann, U. & Leiviskä, T. Reductive degradation of perfluorooctanoic acid in complex water matrices by using the UV/sulfite process. Water Res. 205, 117676 (2021).
Houtz, E., Kempisty, D. & Lester, Y. Poly- and perfluoroalkyl substances destruction via advanced reduction processes: assessing scientific and commercial progress and prospects. Curr. Opin. Chem. Eng. 44, 101022 (2024).
Gu, Y., Liu, T., Wang, H., Han, H. & Dong, W. Hydrated electron-based decomposition of perfluorooctane sulfonate (PFOS) in the VUV/sulfite system. Sci. Total Environ. 607-608, 541–548 (2017).
Londhe, K. et al. Application of electron beam technology to decompose per-and polyfluoroalkyl substances in water. Environ. Pollut. 348, 123770 (2024).
Lassalle, J. et al. Degradation of PFOS and PFOA in soil and groundwater samples by high dose Electron Beam Technology. Radiat. Phys. Chem. 189, 109705–109705 (2021).
Smith, S. J. et al. The need to include a fluorine mass balance in the development of effective technologies for PFAS destruction. Environ. Sci. Technol. 58, 2587–2590 (2024).
Soriano, Á., Gorri, D. & Urtiaga, A. Efficient treatment of perfluorohexanoic acid by nanofiltration followed by electrochemical degradation of the NF concentrate. Water Res. 112, 147–156 (2017).
Shi, H. et al. Degradation of perfluorooctanesulfonate by reactive electrochemical membrane composed of magneli phase titanium suboxide. Environ. Sci. Technol. 53, 14528 (2019).
Sharma, S., Shetti, N. P., Basu, S., Nadagouda, M. N. & Aminabhavi, T. M. Remediation of per-and polyfluoroalkyls (PFAS) via electrochemical methods. Chem. Eng. J. 430, 132895 (2022).
Blotevogel, J., Thagard, S. M. & Mahendra, S. Scaling up water treatment technologies for PFAS destruction: current status and potential for fit-for purpose applications. Curr. Opin. Chem. Eng. 41, 100944 (2023).
Mabaso, N. S. N., Tshangana, C. S. & Muleja, A. A. Efficient removal of PFASs using photocatalysis, membrane separation and photocatalytic membrane reactors. Membranes 14, 217 (2024).
Li, F. et al. A concentrate-and-destroy technique for degradation of perfluorooctanoic acid in water using a new adsorptive photocatalyst. Water Res. 185, 116219 (2020).
Zhu, Y. et al. Adsorption and solid-phase photocatalytic degradation of perfluorooctane sulfonate in water using gallium-doped carbon-modified titanate nanotubes. Chem. Eng. J. 421, 129676 (2021).
Wen, Y. et al. Integrated photocatalytic reduction and oxidation of perfluorooctanoic acid by metal–organic frameworks: key insights into the degradation mechanisms. J. Am. Chem. Soc. 144, 11840–11850 (2022).
Wang, S. et al. Photocatalytic degradation of perfluorooctanoic acid and perfluorooctane sulfonate in water: a critical review. Chem. Eng. J. 328, 927–942 (2017).
Ellis, A. C. et al. Pilot study comparison of regenerable and emerging single-use anion exchange resins for treatment of groundwater contaminated by per-and polyfluoroalkyl substances (PFASs). Water Res. 223, 119019 (2022).
Nadagouda, M. N. & Lee, T. Cross-flow treatment of PFAS in water: materials challenges and potential solutions. Acc. Mater. Res. 2, 129 (2021).
Maldonado, V. Y., Becker, M. F., Nickelsen, M. G. & Witt, S. E. Laboratory and semi-pilot scale study on the electrochemical treatment of perfluoroalkyl acids from ion exchange still bottoms. Water 13, 2873 (2021).
Moriwaki, H. et al. Sonochemical decomposition of perfluorooctane sulfonate and perfluorooctanoic acid. Environ. Sci. Technol. 39, 3388–3392 (2005).
Cheng, J., Vecitis, C. D., Park, H., Mader, B. T. & Hoffmann, M. R. Sonochemical degradation of perfluorooctane sulfonate (PFOS) and perfluorooctanoate (PFOA) in groundwater: kinetic effects of matrix inorganics. Environ. Sci. Technol. 44, 445–450 (2010).
Li, Z. et al. Different nanostructured In2O3 for photocatalytic decomposition of perfluorooctanoic acid (PFOA). J. Hazard. Mater. 260, 40 (2013).
Song, C., Chen, P., Wang, C. & Zhu, L. Photodegradation of perfluorooctanoic acid by synthesized TiO2–MWCNT composites under 365nm UV irradiation. Chemosphere 86, 853 (2012).
Zhu, Y. et al. Photocatalytic degradation of GenX in water using a new adsorptive photocatalyst. Water Res. 220, 118650 (2022).
Samuel, M. S., Shang, M. & Niu, J. Photocatalytic degradation of perfluoroalkyl substances in water by using a duo-functional tri-metallic-oxide hybrid catalyst. Chemosphere 293, 133568 (2022).
Shao, T., Zhang, P., Jin, L. & Li, Z. Photocatalytic decomposition of perfluorooctanoic acid in pure water and sewage water by nanostructured gallium oxide. Appl. Catal. B Environ. 142, 654 (2013).
Tan, X. et al. Indium-modified Ga2O3 hierarchical nanosheets as efficient photocatalysts for the degradation of perfluorooctanoic acid. Environ. Sci. Nano 7, 2229 (2020).
Yuan, Y. et al. Efficient removal of PFOA with an In2O3/persulfate system under solar light via the combined process of surface radicals and photogenerated holes. J. Hazard. Mater. 423, 127176 (2022).
Fu, C. et al. Photocatalysis of aqueous PFOA by common catalysts of In2O3, Ga2O3, TiO2, CeO2 and CdS: influence factors and mechanistic insights. Environ. Geochem. Health 44, 2943 (2022).
Lu, D., Sha, S., Luo, J., Huang, Z. & Zhang Jackie, X. Treatment train approaches for the remediation of per- and polyfluoroalkyl substances (PFAS): a critical review. J. Hazard. Mater. 386, 121963 (2020).
Panchangam, S. C., Lin, A. Y.-C., Tsai, J.-H. & Lin, C.-F. Sonication-assisted photocatalytic decomposition of perfluorooctanoic acid. Chemosphere 75, 654 (2009).
Li, Z. et al. Highly efficient degradation of perfluorooctanoic acid: an integrated photo-electrocatalytic ozonation and mechanism study. Chem. Eng. J. 391, 123533 (2020).
Olvera-Vargas, H., Wang, Z., Xu, J. & Lefebvre, O. Synergistic degradation of GenX (hexafluoropropylene oxide dimer acid) by pairing graphene-coated Ni-foam and boron doped diamond electrodes. Chem. Eng. J. 430, 132686 (2022).
Pica, N. E. et al. Electrochemical oxidation of hexafluoropropylene oxide dimer acid (GenX): mechanistic insights and efficient treatment train with nanofiltration. Environ. Sci. Technol. 53, 12602 (2019).
Liu, F. Enhanced removal of perfluorooctanoic acid (PFOA) via microwave-fenton-reactive membrane filtration. Theses. 1774. https://digitalcommons.njit.edu/theses/1774 (2020).
Acknowledgements
Authors C.S.T., S.T.N., A.T.K., T.T.I.N., B.B.M., and A.A.M. gratefully acknowledge funding from the Institute for Nanotechnology and Water Sustainability (iNanoWS) at the College of Science, Engineering and Technology (CSET), University of South Africa. Authors S.G., S.D., P.J.J.A., and A.A.M. acknowledge resources support from Nanotechnology-Enabled Water Treatment (NEWT) at Rice University.
Author information
Authors and Affiliations
Contributions
C.S.T.: Writing original draft, reviewing, and editing. S.T.N.: Writing. S.G.: Reviewing and editing, S.D.: Reviewing and editing. A.T.K.: Editing. T.T.I.N.: Reviewing and funding. B.B.M.: Reviewing and funding. P.J.J.A.: Supervision, resources, reviewing, and editing. A.A.M.: Conceptualization, supervision, funding, writing original draft, reviewing and editing.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Tshangana, C.S., Nhlengethwa, S.T., Glass, S. et al. Technology status to treat PFAS-contaminated water and limiting factors for their effective full-scale application. npj Clean Water 8, 41 (2025). https://doi.org/10.1038/s41545-025-00457-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41545-025-00457-3
This article is cited by
-
PFAS in water environments: recent progress and challenges in monitoring, toxicity, treatment technologies, and post-treatment toxicity
Environmental Systems Research (2025)
-
Bird’s-eye view of per- and polyfluoroalkyl substances pollution research in the African hydrosphere
npj Clean Water (2025)
-
The PFAS treatment evolution
Nature Water (2025)
