
Abstract
Breast cancer is a highly heterogeneous disease whose prognosis and treatment as defined by the expression of three receptors—oestrogen receptor (ER), progesterone receptor and human epidermal growth factor receptor 2 (HER2; encoded by ERBB2)—is insufficient to capture the full spectrum of clinical outcomes and therapeutic vulnerabilities. Previously, we demonstrated that transcriptional and genomic profiles define eleven integrative subtypes with distinct clinical outcomes, including four ER+ subtypes with increased risk of relapse decades after diagnosis1,2. Here, to determine whether these subtypes reflect distinct evolutionary histories, interactions with the immune system and pathway dependencies, we established a meta-cohort of 1,828 breast tumours spanning pre-invasive, primary invasive and metastatic disease with whole-genome and transcriptome sequencing. We demonstrate that breast tumours fall along a continuum constrained by three genomic archetypes. The ER+ high-risk integrative subgroup is characterized by complex focal amplifications, similar to HER2+ tumours, including cyclic extrachromosomal DNA amplifications induced by ER through R-loop formation and APOBEC3B-editing, which arise in pre-invasive lesions. By contrast, triple-negative tumours exhibit genome-wide instability and tandem duplications and are enriched for homologous repair deficiency-like signatures, whereas ER+ typical-risk tumours are largely genomically stable. These genomic archetypes, which replicate in an independent cohort of 2,659 primary tumours, are established early during tumorigenesis, sculpt the tumour microenvironment and are conserved in metastatic disease. These complex structural alterations contribute to replication stress and immune evasion, and persist throughout tumour evolution, unveiling potential vulnerabilities.
Similar content being viewed by others

Main
Breast cancer is the most common malignancy in women, accounting for more than 15% of new cancer cases in the USA annually1. Clinically, breast tumours are stratified into three immunohistochemistry subtypes—ER+HER2−, HER2 and triple-negative breast cancer (TNBC)—on the basis of the expression of ER, progesterone receptor and HER2 (ref. 2). Although heterogeneity in gene expression, especially measures of proliferation, within these subtypes correlates with prognosis and patterns of relapse, and is used to guide therapy3, ultimately the paradigm of three major subtypes dictates our understanding of and approach to the disease.
We previously defined eleven subtypes of breast cancer on the basis of integrative clustering (IC) of genomic and transcriptional profiles, and demonstrated their distinct prognosis and relapse trajectories4,5. Among patients with ER+ cancer (80% of cases), one-quarter had a 45% chance of distant recurrence two decades post-diagnosis5. This ER+ ‘high-risk’ subgroup, corresponding to IC1, IC2, IC6 and IC9 subtypes, is enriched for luminal B tumours harbouring focal oncogene amplification and overexpression, similar to ERBB2-amplified tumours (IC5, 10–15%). Moreover, genes within these amplicons mediate resistance to hormonal therapy6,7. TNBC comprises genome-unstable basal-like IC10 and IC4ER− tumours, the latter with relapse risk that persists beyond 5 years.
Although the IC subgroups improve relapse prediction and define new drivers5, their origins, evolution and tumour immune microenvironments (TMEs) remain unknown. To investigate, we assessed the genomic architecture and microenvironmental composition of breast tumours from a meta-cohort of 1,828 tumours spanning pre-invasive ductal carcinoma in situ (DCIS), primary and metastatic lesions, profiled using whole-genome sequencing (WGS) and transcriptome sequencing8,9,10. We further implemented a machine learning framework to determine IC subtypes from DNA-based profiles alone. Our analyses reveal three primary genomic archetypes of breast cancer— (i) TNBC: ICs (IC10 and IC4ER−); (ii) typical-risk ER+HER2− (IC3, IC4ER+, IC7 and IC8); and (iii) high-risk ER+HER2− (IC1, IC2, IC6 and IC9) and HER2+ (IC5) (referred to as ER+ high-risk + HER2+). The last group is characterized by early, recurrent amplifications, including extrachromosomal DNA (ecDNA) owing to APOBEC3B (A3B)-editing at ER-induced R-loops. These genomic patterns, accompanied by variable TMEs, implicate complex rearrangements as a major driver of immune escape and highlight new therapeutic vulnerabilities in aggressive subgroups.
Evolution of the IC subgroups
The mutational processes underlying breast cancer initiation and progression are incompletely understood11,12,13,14. Herein we uniformly processed 1,828 samples from DCIS (n = 406), primary (n = 702) and metastatic (n = 720) lesions using a harmonized, state-of-the-art bioinformatics pipeline to identify single nucleotide variants (SNVs), copy number aberrations (CNAs), structural variants (SVs), ecDNA and mutational signatures (Fig. 1a, Supplementary Fig. 1a–c and Supplementary Table 1). Owing to shallow coverage of the archival DCIS cohort, SNVs and SVs were not called10. Additionally, we used the Molecular Taxonomy of Breast Cancer International Consortium (METABRIC) cohort of primary invasive tumours4 (n = 1,894) with both RNA and DNA profiles and about 20 years of clinical follow-up. To our knowledge, this represents the largest collection of uniformly processed breast tumours spanning all disease stages.

a, Schematic of the study design. BC, breast cancer; HTAN, Human Tumor Atlas Network; sWGS, shallow WGS; TCGA, The Cancer Genome Atlas; PCAWG, Pan-Cancer Analysis of Whole Genomes. b, Schematic of the ENiClust IC classifier. WES, whole-exome sequencing. c, Kaplan–Meier curves of distant relapse-free (DRF) survival of the ER+ typical-risk and ER+ high-risk classes detected by the four IC subtype classifiers. Shaded area represents 95% confidence interval. HR, hazard ratio. d, Difference in distant relapse-free survival probability (top) or delta in Cox proportional hazard ratio (bottom) between ER+ typical-risk and ER+ high-risk classes detected by the four different IC classifiers. Error bars represent the difference in 95% confidence intervals between ER+ typical-risk and ER+ high-risk in each model. e, Differential pattern of relapse across the ICs, illustrated by the cumulative (black) and annual (red) risk of relapse over time. f, IC subgroup (left) and subtype (right) distributions across disease stages. P, primary; M, metastatic. The schematics in a,b were created with BioRender.com.
Although the ICs predict distant relapse and delineate genomic drivers4,5, current methods fail to accurately capture them using DNA profiles alone12. Accordingly, we developed Ensemble Integrative Clustering (ENiClust), which reliably infers IC subtypes from whole-exome sequencing or WGS, across all stages of disease (Fig. 1b and Supplementary Table 2). The final ensemble model yields a nine-class prediction (Fig. 1b and Supplementary Table 2), which is further split into ten on the basis of the ER status of IC4 (that is, IC4ER+ and IC4ER–). These ten classes comprise four clinically distinct IC subgroups—TNBC (IC10 and IC4ER−), HER2+ (IC5), ER+ typical-risk (IC3 + IC7, IC4ER+ and IC8) and ER+ high-risk (IC1, IC2, IC6 and IC9). Throughout we refer to HER2+ tumours as those classified as IC5, enriching for ERBB2 amplification. ENiClust outperformed iC10 DNA alone1,12 (Methods and Supplementary Fig. 1d) and improves patient stratification, with high-risk tumours exhibiting worse distant recurrence-free survival (METABRIC; Fig. 1c–e and Supplementary Fig. 1d–f). Thus, ENiClust identifies clinically meaningful subgroups with distinct biology.
Using ENiClust, we interrogated the distribution of ICs across disease stages. DCIS was enriched for IC5 tumours (Fisher’s exact test P = 2.98 × 10−6; Fig. 1f), corroborating our previous findings10. ER+ high-risk ICs were enriched among metastatic tumours, consistent with their increased relapse risk (Fig. 1f and Extended Data Fig. 1a). IC10 basal-like tumours were depleted in the metastatic cohort, potentially owing to differences in ancestry (Extended Data Fig. 1b–d). The ICs were largely stable from primary to metastasis (concordance = 71.8%; Extended Data Fig. 1e,f).
There was an increased proportion of luminal B versus luminal A from pre-invasive to primary (Δ(LumB/(LumA + LumB)) = +11%) and primary to metastatic (Δ(LumB/(LumA + LumB)) = +29%; Extended Data Fig. 1g) lesions. Among primary tumours, ER signalling in ER+ high-risk tumours was more akin to that of HER2+ER+ tumours15 and significantly lower than that of ER+ typical-risk tumours (Extended Data Fig. 1h), with no difference between primary and metastatic tumours (Extended Data Fig. 1i). Compared to ER+ typical-risk, ER+ high-risk was enriched among patients with tumours that were resistant to endocrine therapy (odds ratio (OR) ≥ 5.58, P ≤ 0.03; Supplementary Fig. 1g). In a clinical trial (NCT00651976) in early-stage ER+ breast cancer, high-risk tumours had a decreased proliferation score with letrozole treatment but it remained significantly higher than that for typical-risk tumours (P ≤ 0.02; Supplementary Fig. 1h). Thus, ER+ high-risk tumours may experience persistent proliferation despite endocrine treatment. New therapies (selective oestrogen receptor degraders and proteolysis-targeting chimeras) that more fully suppress proliferation might particularly benefit this subgroup.
Early IC-specific SVs fuel progression
The IC subtypes have distinct CNA landscapes (Extended Data Fig. 1j), but their SV landscape and evolution have not been investigated. Leveraging ENiClust, we found that the IC-subgroup-specific genomic landscape of breast cancer is consistent throughout disease progression despite an increased burden of alterations10,13,16,17 (Fig. 2a and Extended Data Fig. 2a,b). Both HER2+ and ER+ high-risk primary and metastatic tumours exhibit characteristic sharp increases in SV burden at their respective recurrently amplified loci (IC5: 17q12; IC6: 8p11; IC2: 11q13; IC1: 17q23). The peak of SV burden at 17q12 (ERBB2) suggests that ERBB2 amplification is fuelled by complex alterations, such as ecDNA18. The mutational burden in primary ER+ typical-risk tumours was minimal (Supplementary Fig. 1i) but increased in metastatic disease (Fig. 2a), in part owing to treatment (Extended Data Fig. 2c). IC10 and IC4ER− tumours exhibit diffuse genome-wide instability with an increased SV burden, although the latter show an attenuated pattern and harbour fewer pathogenic SVs and alterations in DNA repair pathways, confirming previous reports19 (Extended Data Fig. 2d,e). Across metastatic sites, the cumulative burden of alterations was higher in lung and subcutaneous metastases and lower in soft-tissue and in-breast recurrences (Extended Data Fig. 2f). These subgroup-specific alterations were seen in DCIS (Extended Data Fig. 2a), emphasizing early oncogene addiction and mechanisms of malignant transformation.

a, IC group-level CNA profile (shaded area; dark denotes amplification, light denotes deletion) with SV burden (line) as overlay and total alteration burden in primary and metastatic samples. b, Pareto front projection on ternary plot of CNA and SV signature profiles from primary (left) and metastatic (right) tumours independently, resulting in three genomic archetypes. Each plotted circle represents a tumour. c, Lollipop plots illustrating the correlation between mutational features and the distance to each archetype. amp., amplification; BFB, breakage–fusion–bridge; TIC, templated insertion chain; LOH, loss of heterozygosity; WGD, whole-genome doubling; FGA, fraction of genome altered.
Next we characterized CNA and SV signatures in 702 primary breast tumours, replicating the 24 CNA20 and 6 rearrangement8,21 signatures (RSs) previously reported (Supplementary Fig. 2a–c). RS3, RS5 (associated with homologous repair deficiency (HRD); Supplementary Fig. 2d) and CN17 were enriched in IC10 tumours, whereas RS4, RS6 (associated with complex amplifications) and CN7 were enriched in ER+ high-risk and HER2+ tumours (Extended Data Fig. 2g,h and Supplementary Fig. 2e–g). ER+ typical-risk tumours were enriched for CN1 (associated with diploid genomes; Supplementary Fig. 2d,e).
Projected on a two-dimensional plane (Supplementary Fig. 3a,b), the architectural profiles follow a continuum and form a polyhedron reminiscent of Pareto optimum theory, which illustrates trade-offs between biological tasks22. Primary breast cancers map onto three dominant genomic archetypes (Supplementary Fig. 3c–f): TNBC-enriched, ER+ typical-risk-enriched and ER+ high-risk + HER2+-enriched. Tumours dominated by a single mutational process are proximal to a vertex, whereas those characterized by multiple processes cluster at the centre (Fig. 2b and Extended Data Fig. 2i). The TNBC-enriched archetype was positively correlated with genomic instability, HRD and APOBEC-editing SNVs (Fig. 2c and Supplementary Fig. 3g). Compared to ER+ high-risk tumours, HER2+ tumours were enriched for tyfonas (Extended Data Fig. 2j). The ER+ high-risk + HER2+-enriched archetype was positively correlated with complex amplifications, reactive oxygen species and APOBEC-associated SNVs harbouring co-amplification of multiple cytobands (Extended Data Fig. 3a). By contrast, the ER+ typical-enriched archetype negatively correlated with most genomic features.
Tumours predicted to be BRCA-like on the basis of germline or somatic genomic features23 map to the TNBC-enriched archetype (Extended Data Fig. 3b). Indeed, both BRCA1-like and BRCA2-like ER+ and ER− tumours demonstrated significantly higher TNBC-archetype scores than non-HRD tumours, and HRD-like ER+ high-risk tumours were closer to the TNBC-enriched archetype than their non-HRD-like counterparts (OR = 5.09; P = 6.5 × 10−4). Additionally, the mutational patterns of BRCA1-like and BRCA2-like ER− and ER+ tumours were highly concordant (Supplementary Fig. 3h,i). Notably, whereas 43.6% of TNBC tumours were HRD-like, 13.2% of ER+ high-risk tumours were also predicted to be HRD-like, with most being ER+ high-risk IC1 or IC9 (OR = 4.43; P = 0.03; Extended Data Fig. 3c and Supplementary Fig. 3j). Indeed, although foldback inversions and pyrgos were enriched in TNBC (foldback inversion: 17.3%, P = 2.00 × 10−3; pyrgos: 18.8%, P = 9.33 × 10−4), these mutational events were also observed in ER+ tumours (5.1% and 4.1%, respectively; Extended Data Fig. 3d). These data reinforce multiple mechanisms of genome instability in TNBC24 that also affect a subset of ER+ tumours.
The three genomic archetypes replicated in an independent cohort of 2,229 primary tumours from Genomics England21 (Extended Data Fig. 3e). Overall, the genomic landscape of primary breast tumours falls along a continuum with mutational patterns captured by three main genomic archetypes, namely, genome-stable, diploid genomes (ER+ typical-risk-enriched), genome-wide instability (TNBC-enriched) and focal, complex amplifications (ER+ high-risk + HER2+-enriched).
Metastatic lesions exhibit increased SNV and SV burdens compared to unpaired primary tumours, probably owing to therapy, as we and others have shown13,17. Using the above approach, we identified six de novo SV signatures in metastases that correlated with those in primary tumours (Supplementary Fig. 4a,b) and showed similar subgroup-specific enrichment patterns (Extended Data Fig. 3f). Two-dimensional projection again revealed three dominant archetypes (Supplementary Fig. 4c) that overlap with those in primary tumours (Fig. 2b,c, Extended Data Fig. 3g and Supplementary Fig. 4d). Our results were robust to choice of dimensionality reduction algorithm (Supplementary Fig. 4e–g). Thus, the three genomic archetypes of breast cancer are conserved in metastatic disease.
SV signatures were generally conserved, although increased, in metastatic tumours except for RS4 and RS6 in ER+ high-risk and HER2+ tumours, respectively, which were stable (Extended Data Fig. 3h). These data support the early occurrence of complex rearrangements and their persistence through metastasis. Although the distribution of CNA signatures mirrored primary tumours, the Pareto front revealed increased alteration burden and more intermixed profiles in metastasis, consistent with increased whole-genome doubling and genomic instability17 (Extended Data Fig. 3i and Supplementary Fig. 4h,i). Thus, metastatic tumours retain the scars of subgroup-specific mutational processes operative in early-stage disease.
Although ER+ typical-risk tumours have a favourable prognosis, 29% of patients experience distant relapse4. We investigated whether the genomic archetypes improve risk stratification. Mapping METABRIC onto the Pareto front (Methods, Extended Data Fig. 3j and Supplementary Fig. 4j–l), the position of ER+ typical-risk tumours was predictive of relapse, with recurrent tumours mapping closer to the ER+ high-risk + HER2+ archetype (Extended Data Fig. 3k,l) accompanied by a higher HRD loss-of-heterozygosity score, invasive lobular carcinoma (ILC) histology and increased proliferation.
In METABRIC, ILCs were enriched in ER+ typical-risk tumours (OR = 2.20, P = 2.27 × 10−3, Fisher’s exact test; Supplementary Fig. 4m). Within ER+ high-risk tumours, ILCs exhibited a higher 5-year recurrence risk (39% versus 30%) and cumulative recurrence risk (62% versus 54% at 20 years; Extended Data Fig. 3m). This difference was more marked among ER+ typical-risk tumours (55% versus 37% at 20 years). ILCs were closer to the ER+ typical-risk archetype than their invasive ductal carcinoma (IDC) counterparts (P = 2.10 × 10−5; Extended Data Fig. 3n,o) given their lower levels of whole-genome doubling, ploidy and fraction of genome altered. Thus, given comparable genomic architectures, lobular histology remains a high-risk feature.
ER-induced R-loops fuel ecDNA genesis
ER+ high-risk and HER2+ breast tumours were enriched for complex amplifications in two independent cohorts (OR > 10.1; P < 2.2 × 10−16; Fig. 3a and Extended Data Fig. 4a), motivating further exploration of their origin and nature (Supplementary Fig. 5a,b). There was no difference in cyclic amplifications in HER2+ER− primary tumours compared to HER2+ER+ primary tumours (Extended Data Fig. 4b). Leveraging two independent ecDNA inference methods, 43–67% of primary ER+ high-risk and HER2+ cases were predicted to harbour ecDNA (Extended Data Fig. 4c,d). A proportion of HER2+ primary tumours (25.7%) harboured amplifications in loci specific to the ER+ high-risk subgroup (Extended Data Fig. 4e and Supplementary Fig. 5c), with 8.57% predicted to be on ecDNA. Additionally, we observed a modest enrichment of inversions at the 11q13 locus in primary tumours. HRD and ecDNA were mutually exclusive in primary ER+ high-risk and IC10 tumours (OR = 0.21–0.29; false discovery rate (FDR) < 0.02; Supplementary Fig. 5d). We interrogated complex amplifications in 406 pre-invasive DCIS profiled with shallow WGS (5× median coverage)10. We predicted 35 cyclic and 205 complex non-cyclic amplifications, enriched in ER+ high-risk + HER2+ tumours (OR = 4.21; P = 2.48 × 10−4; Extended Data Fig. 4f and Supplementary Fig. 5e). This pattern replicated in 12 DCIS samples from Genomics England (92.8×)25. Leveraging the clock-like accumulation of mutations, SNV density informs the timing of cyclic amplifications (Methods). Compared to cyclic amplifications in TNBC tumours, cyclic amplifications in ER+ high-risk and HER2+ tumours had a lower SNV density before amplification, suggesting an earlier origin (Fig. 3b and Supplementary Fig. 5f). Median time of cyclic amplification in ER+ high-risk and HER2+ tumours occurs decades earlier than in IC10 tumours, respectively, implicating cyclic amplifications as early events.

a, Proportion (top) and number (bottom) of samples with at least one cyclic or complex non-cyclic amplification in primary or metastatic tumours. b, The density of SNVs occurring before amplification in primary (top) and metastatic (bottom) tumours. Boxplot represents median, 0.25 and 0.75 quantiles with whiskers at 1.5× the interquartile range. c, Illustration showing copy number (CN) and SVs linking together disjoint segments in ecDNA (top), ratio of read depth in the tumor versus normal sample (middle) and location of oncogenes in ecDNA (bottom) in a representative primary IC2 tumour. d, Ratio of sequencing coverage in digested versus parental UCD65 (IC2) cell line in the predicted ecDNA region (dashed red line) compared to 1,000 null regions. e, Proportion of tumours within each IC subtype that harbour cyclic, complex non-cyclic or linear amplification in IC-specific oncogenes. f, Schematic for the genesis of cyclic amplifications. TC-NER, transcription-coupled nucleotide-excision repair. g, The density of ER-induced R-loops in cyclic versus complex non-cyclic amplifications. h, The percentage of breakpoints that overlap ER-induced R-loops with (+) or without (−) E2 treatment. Error bars represent the standard deviation across three replicates. i, The distance of each oncogene to the nearest ER-induced R-loop. The schematic in f was created with BioRender.com.
Most cyclic amplifications in ER+ high-risk (88%) and HER2+ (96%) tumours overlapped at least one COSMIC-defined oncogene (Extended Data Fig. 4g). Of these, 79–92% involved oncogenes in IC-associated cytobands (Extended Data Fig. 1j) and 15% involved two or more cytobands (Extended Data Fig. 4h and Supplementary Table 3). In cell line models of IC2 (UCD65) and IC6 (UCD12) before and after linear DNA digestion, significantly higher sequencing coverage occurred at regions predicted to encode ecDNA, corroborating our computational predictions (Fig. 3c,d and Supplementary Fig. 5g,h). Oncogene incorporation varied across subtypes, with HER2+ tumours harbouring the largest number per megabase (Extended Data Fig. 4i,j). A total of 82% of IC2, 59% of IC5 (HER2+), 48% of IC6 and 32.5% of IC1 tumours had predicted cyclic amplifications at subgroup-defining cytobands, whereas 3% of IC1 and IC9 tumours harboured cyclic amplifications at 20q13, spanning the NCOA3 oncogene (Fig. 3e). Overall 42% of IC9 tumours harbour ecDNA, but these ecDNAs are diffuse along the genome and do not include MYC. In support, focal SV peaks were not observed at 8q24 spanning the MYC oncogene in IC9 primary or metastatic tumours. Instead, a broader region is subject to enhancer hijacking by the long noncoding RNA PVT1, as we previously reported26. PVT1 co-amplifies with MYC in about 90% of tumours (Supplementary Fig. 5i). Frequent enhancer hijacking at MYC may explain the weak correlation between MYC copy number and mRNA abundance (Supplementary Fig. 5j,k).
The subset of ER+ typical-risk tumours harbouring ecDNA fell along the ER+ typical-risk versus high-risk archetype continuum (Extended Data Fig. 4k–l). By contrast, ER− tumours with ecDNA had limited structural conservation (Extended Data Fig. 5a). Across all subgroups, similar patterns were observed in metastatic and pre-invasive tumours (Extended Data Fig. 5b–f).
Increased replication stress has been associated with response to checkpoint27 and DNA repair28 inhibitors, and hence is a therapeutic vulnerability in TNBC8. Assessing replication stress across the IC subgroups, we found increased levels of oncogene-induced replication stress in ER+ high-risk and HER2+ tumours compared to ER typical-risk, IC10 and IC4ER− tumours (FDR < 0.026; Extended Data Fig. 6a,b and Supplementary Table 2). The replication stress signature was positively correlated with TNBC-enriched and ER+ high-risk + HER2+-enriched genomic archetypes (effect size > 0.154, P < 4.98 × 10−15; Extended Data Fig. 6c–e). Within ER+ typical-risk tumours, ILC had a higher replication stress than IDC (FDR = 4.08 × 10−3). Meta-analysis suggests a positive association between ecDNA and replication stress in HER2+, IC1 and IC6 tumours (Extended Data Fig. 6f) and higher levels of type-I interferon signature in ecDNA+ tumours (Supplementary Fig. 6a). Finally, ER+ high-risk and HER2+ tumours demonstrated increased cGAS–STING activity (Extended Data Fig. 6g,h), a possible therapeutic target linked to chromosomal instability and replication stress29.
Consistent with the findings of ref. 30, our data showed that cyclic amplifications were significantly enriched for translocations compared to complex non-cyclic amplifications in ER+ primary tumours (Fig. 3f, Extended Data Fig. 7a,b and Supplementary Fig. 6b). These cyclic-amplified ER+ high-risk tumours had a higher ESR1 mRNA abundance (β = 1.27; P = 6.90 × 10−3; Extended Data Fig. 7c) and enriched ER binding within the amplified region (Extended Data Fig. 7d and Supplementary Fig. 6c). Nonetheless, ER signalling was lower in ER+ high-risk compared to typical-risk tumours (Extended Data Fig. 1h). Given the evidence for ecDNA in pre-malignant lesions, we reasoned that ER signalling is increased in DCIS lesions that classify as ER+ high-risk and subsequently decreases in invasive disease. Leveraging 18 paired ER+ DCIS and primary tumours with transcriptome sequencing10, we observed decreased ER signalling in ER+ high-risk tumours (effect size = 0.33; P = 0.03; Extended Data Fig. 7e). These data support the role of ER in ecDNA genesis through translocations and emphasize their early origin.
The mechanism by which ER activation induces translocations remains unknown. ER recruitment of A3B promotes double-stranded breaks (DSBs) at ER binding sites31 (Fig. 3f and Extended Data Fig. 7a). Increased ER-induced transcription leads to the formation of R-loops producing single-stranded DNA, a substrate for A3B-editing32. A3B deaminates cytosine to uracil, which can be repaired by base-excision repair (BER). Single-strand nicks induced by BER coupled with transcription-coupled nucleotide-excision repair processing of the R-loop can result in DSBs31. Together, these findings indicate that A3B can exacerbate chromosomal instability in the pre-invasive setting33. We reasoned that ER-induced R-loops initiate translocation-bridge amplifications through A3B-editing and confirmed that A3B binding in ER+ cell lines was enriched in cyclic versus non-cyclic amplifications (Extended Data Fig. 7d and Supplementary Fig. 6c). Treatment with oestradiol (E2) in MCF7 cell lines induced R-loops (nR-loops = 212) in the same regions where cyclic amplifications were observed in patient tumours (Fig. 3g and Extended Data Fig. 7f,g). This finding was specific to ER-induced R-loops (nR-loops = 13,965; Extended Data Fig. 7h). Unresolved R-loops due to A3B knockout in MCF10A cells were preferentially enriched in regions of cyclic amplifications in primary breast tumours (Extended Data Fig. 7i and Supplementary Fig. 6d). Tumours containing ecDNA were also enriched for transcription-replication collision-associated large tandem duplications (>100 kb), indicative of impaired R-loop resolution28 (Supplementary Fig. 6e). Translocations within cyclic amplifications were significantly closer to ER-induced R-loops than those outside cyclic amplifications (Extended Data Fig. 7j,k). These data support a role for A3B in R-loop resolution, contributing to ecDNA formation.
Accordingly, we reasoned that oestrogen-induced SV breakpoints would be enriched at ER-induced R-loops. Comparing SV patterns in E2-treated MCF7 cells through high-throughput genome-wide translocation sequencing of DSBs forming translocations induced by CRISPR–Cas9 (ref. 30), we confirmed the enrichment for E2-induced breakpoints at E2-induced R-loops (Fig. 3h) compared to all R-loops (Extended Data Fig. 7l). There was no difference in replication timing between cyclic and non-cyclic amplifications (Supplementary Fig. 6f). ER-induced R-loops were enriched closer to the IC-specific oncogenes PAK1 (IC2), ZNF703 (IC6) and MYC (IC9) than to all other COSMIC-defined oncogenes, including ERBB2 (Fig. 3i). There was no enrichment of ER-induced R-loops near IC1 oncogenes.
Germline CNA polymorphisms in A3B have been associated with APOBEC-dependent mutations34 and immune activation in breast cancer35. Despite limited power, our analyses found a modest but nonsignificant decrease in ecDNA prevalence in ER+ high-risk and typical-risk samples with the homozygous deletion allele (n = 5; Extended Data Fig. 7m). Together, these data indicate that ER activity promotes cyclic amplifications through R-loop formation and A3B-editing.
The ICs harbour distinct TMEs
Tumour clonal composition and genomic features are sculpted by immune pressures36, and oncogenic alterations promote both pro-tumour and anti-tumour immune responses37. Using transcriptomic profiles, we characterized the TME in primary (nTCGA = 1,015; nMETABRIC = 1,894) and metastatic (n = 360) tumours focusing on four subtypes defined by immune infiltration and stromal composition: immune-enriched fibrotic, immune-enriched non-fibrotic, fibrotic and depleted38 (Fig. 4a, Extended Data Fig. 8a and Supplementary Table 2). The reproducibility of the TME subtypes is supported by single-cell spatial proteomic profiling (n = 384; Extended Data Fig. 8b) and cell type proportions estimated from bulk transcriptomics (Supplementary Fig. 7a).

a, Schematic of TME subtypes and select immune escape pathways. b, Comparison of TME subtypes by IC subgroup. The number of tumours in each subgroup is indicated on the top of each bar. c, Left: proportion of primary and metastatic samples in each IC subgroup with GIE. Right: proportion of samples with alterations in each pathway stratified by IC subgroup and stage of progression. d, Proportion of alteration types in primary and metastatic samples for each of the immune escape pathways. The schematic in a was adapted from BioRender.com (credit: A. Iwasaki & J.-H. Lee; https://app.biorender.com/biorender-templates/figures/all/t-5f4fb77c3b02b700b74df8c6-mhc-class-i-and-ii-pathways).
We then quantified microenvironmental differences across the IC subgroups. Primary IC10 and IC4ER− were enriched for immune-rich (immune-enriched non-fibrotic and immune-enriched fibrotic) TMEs (OR = 3.004, P = 5.17 × 10−11, Fisher’s exact test; Fig. 4b and Supplementary Fig. 7b), as previously reported39. ER+ high-risk and HER2+ primary tumours harboured immune-depleted TMEs (OR = 3.09, P = 1.06 × 10−15, Fisher’s exact test), whereas genome-stable ER+ typical-risk and IC4ER− primary tumours were enriched for fibrotic signatures (fibrotic and immune-enriched fibrotic subtypes; OR = 5.619, P < 2.2 × 10−16, Fisher’s exact test). These observations replicated using a second transcriptional immune score (Supplementary Fig. 7c,d). Within ER+ high-risk tumours, immune enrichment did not differ across subgroups (Extended Data Fig. 8c). Among ER+ typical-risk tumours, ILCs were enriched for the immune-enriched fibrotic subtype compared with IDCs (OR = 2.18, P = 1.17 × 10−3; Extended Data Fig. 8d).
IC4ER− tumours have a more favourable prognosis but longer-term risk of recurrence than IC10 tumours5 despite similar genomic landscapes (Fig. 2a). To investigate differences in their TME, we leveraged single-cell spatial proteomic data and discovered an increased proportion of fibroblasts and T cells in IC4ER− compared to IC10 tumours (Extended Data Fig. 8e and Supplementary Fig. 7e). In support, previous work has linked increased T cell infiltration with improved overall survival in TNBC39. Compared to primary tumours, ER− metastatic tumours were depleted of immune-enriched non-fibrotic and immune-enriched fibrotic features (OR = 3.01; P = 2 × 10−4; Extended Data Fig. 8f). By contrast, HER2+ and ER+ tumours exhibited stable TMEs through metastasis (Fig. 4b, Extended Data Fig. 8g and Supplementary Fig. 7f), consistent with previous reports that ER promotes immunosuppression and immunoediting in pre-invasive lesions40,41.
We found that 43.86% of primary and 47.67% of metastatic tumours exhibited genetic immune escape (GIE), most of which occurred in a single pathway with varying prevalence across IC subgroups (Fig. 4c and Supplementary Tables 4 and 5). IC2 and IC6 tumours were more immune-depleted than IC1 and IC9 tumours (Extended Data Fig. 8c) but harboured fewer GIE (Extended Data Fig. 9a). Instead, 60% of primary IC6 tumours amplified IDO1, which encodes the heme-containing enzyme indoleamine 2,3-dioxygenase located within 8p11.21 that metabolizes tryptophan involved in immune tolerance42 (Extended Data Fig. 9b,c). ER+ typical-risk ILCs exhibit fewer GIE alterations than ER+ typical-risk IDC tumours (Extended Data Fig. 9d), and GIE was not associated with antigen burden (Supplementary Fig. 7g).
Complex alterations and SVs have been overlooked when evaluating GIE37. We found that about 20% of primary and metastatic tumours with GIE harboured SVs or complex amplifications (Fig. 4d and Extended Data Fig. 9e). HER2+ tumours demonstrated the largest increase in GIE between primary and metastatic disease, potentially owing to greater pressure to evade anti-HER2 therapies (OR = 2.23, FDR = 0.19, Fisher’s exact test; Extended Data Fig. 9f). These data illuminate the role of complex alterations in immune escape and tumour-immune co-evolution during disease progression.
Discussion
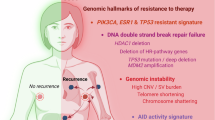
Here we identify three dominant genomic archetypes of breast cancer driven by distinct mutational processes, describing a continuum of genomic profiles and providing a mechanistic basis for these patterns (Fig. 5a). These three archetypes overlap with the main clinical breast cancer subgroups with a notable difference. For a sizeable proportion of ER+ tumours (43.2%), the ER+ high-risk + HER2+ archetype dominates and the mutational processes are indistinguishable from those of HER2+ tumours. Rather than amplifying ERBB2, these ER+ high-risk tumours harbour focal amplifications of other oncogenes (Extended Data Fig. 1j) and have an increased risk of recurrence akin to HER2+ tumours before the introduction of anti-HER2 therapies5. These ER+ high-risk tumours may similarly benefit from agents directed at their amplified oncogenic drivers and/or shared vulnerabilities.

a, Schematic summary of the genomic and microenvironmental characteristics of the three dominant genomic archetypes in breast cancer. b, Temporal changes in genomic stability, ER signalling and immune enrichment from pre-invasive, primary invasive to metastatic disease across subgroups. The schematics in a,b were created with BioRender.com.
A defining feature of the ER+ high-risk + HER2+ archetype is the generation of focally amplified ecDNA through ER-induced R-loops and A3B-editing. ER-induced R-loops create single-stranded DNA, which serves as a substrate for A3B-editing. DSBs arising from BER and nucleotide-excision repair are resolved in the form of interchromosomal translocations. Dicentric chromosomes can form chromosome bridges during mitosis, and breakage of these bridges can generate ecDNA30. ecDNA formation preferentially occurs at loci that define the four ER+ high-risk subgroups and HER2+ disease. Although ecDNA genesis depends on ER, circular amplification may reduce reliance on ER by increasing a particular oncogene’s copy number and rewiring its regulatory network43. This is supported by reduced ER signalling in ER+ high-risk tumours from DCIS to invasive disease. As ER transcriptional activity can contribute to DSBs44, ecDNA formation may balance increased oncogenic signalling with protection against further ER-induced genomic instability (Fig. 5b), and hence reflects an evolutionary trade-off, consistent with mutual exclusivity between complex amplifications and diffuse genome instability.
Beyond tumour subtype, the mutational processes captured by our architectural map may be indicative of distinct therapeutic vulnerabilities. For example, HRD-like tumours are sensitive to PARP inhibition and this has become a mainstay of therapy for TNBC. We find that 44% of TNBC tumours have HRD-like profiles on the basis of WGS, and 13% of ER+ high-risk tumours exhibit BRCA2-like patterns. Although HRD as measured from sequencing data is not confirmed to correlate with PARP inhibitor sensitivity, this result implies that additional patients may benefit from these agents. Further, we find that focally amplified ER+ high-risk tumours exhibit increased replication stress pathway activities, suggesting potential sensitivity to new agents targeting this pathway. Additionally, although APOBEC3 mutagenesis can occur early during tumorigenesis, given its effect on ER activity, A3B represents a potential target in the ER+ high-risk subgroup for which inhibitors are in development45.
The mutational processes that generate and propagate genomic instability both sculpt oncogenic signalling and mediate interactions between tumour cells and the TME. More specifically, SVs contribute to GIE in 9% of breast tumours, but have been overlooked, owing to the need for WGS. Basal-like IC10 tumours, which harbour both high genomic instability and immune infiltrates, probably adapt to this immune pressure through GIE. By contrast, ER+ tumours, both typical- and high-risk, are more immune-depleted at the onset with fewer GIE events, suggesting non-GIE mechanisms37. This is noteworthy given the evolving utility of immunotherapy in breast cancer46. Despite high immune infiltration, up to 62% of TNBC tumours are resistant to current immunotherapies, potentially owing to GIE, whereas 38% of ER+ tumours have immune-enriched TMEs, making them candidates for such agents. Our findings highlight multiple potential strategies for personalizing breast cancer treatment, which will be the focus of ongoing preclinical and translational studies.
Methods
A detailed description of the methods and materials is available in the Supplementary Information.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All cohorts are publicly available. Data for TCGA BRCA samples can be found on the Genomic Data Commons Data Portal (https://portal.gdc.cancer.gov/). DNA-sequencing data for the International Cancer Genome Consortium (ICGC) breast cancer samples can be found on the European Genome-Phenome Archive (accession numbers EGAD00001000141, EGAD00001001322, EGAD00001001334, EGAD00001001335, EGAD00001001336, EGAD00001001337 and EGAD00001001338). In cases in which it was possible, alignments for both TCGA and ICGC samples carried out by the Pancancer Analysis of Whole Genomes were used (https://docs.icgc-argo.org/docs/data-access/icgc-25k-data). RNA-sequencing data for the ICGC breast cancer samples can be found on the European Genome-phenome Archive under accession numbers EGAD00001001323, EGAD00001001339, EGAD00001001340 and EGAD00001001341. Data for metastatic breast cancer samples (Hartwig) are available for academic use under a Data Use 25 Agreement (DR-230) from the Hartwig Medical Foundation (https://www.hartwigmedicalfoundation.nl/en/data/data-acces-request/). Data for HTAN DCIS tumours are available on the database of Genotypes and Phenotypes (accession number phs002371.v4.p1). Data for METABRIC breast cancer samples can be found on the European Genome-Phenome Archive (accession number EGAS00000000083). Data for paired primary and metastatic breast samples are available on the database of Genotypes and Phenotypes under accession numbers phs000730.v1.p1, phs000676.v1.p1 and phs001674.v1.p1, and on the Sequencing Read Archive under accession number SRP055001. Single-cell proteomic data for 384 METABRIC samples can be found at https://idr.openmicroscopy.org/webclient/?experimenter=-1 (idr0076). WGS data from GEL can be accessed through the Genomics England Research Network; the process for joining is described at https://www.genomicsengland.co.uk/research/academic/join-gecip.
Code availability
Code for computational analysis is available via the Curtis Lab GitHub repository at https://github.com/cancersysbio/breast-architecture.
References
Cronin, K. A. et al. Annual report to the nation on the status of cancer, part 1: national cancer statistics. Cancer 128, 4251–4284 (2022).
Gradishar, W. J. et al. NCCN Guidelines® Insights: breast cancer, version 4.2023. J. Natl Compr. Cancer Netw. 21, 594–608 (2023).
Sparano, J. A. et al. Adjuvant chemotherapy guided by a 21-gene expression assay in breast cancer. N. Engl. J. Med. 379, 111–121 (2018).
Curtis, C. et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature 486, 346–352 (2012).
Rueda, O. M. et al. Dynamics of breast-cancer relapse reveal late-recurring ER-positive genomic subgroups. Nature 567, 399–404 (2019).
Turner, N. et al. FGFR1 amplification drives endocrine therapy resistance and is a therapeutic target in breast cancer. Cancer Res. 70, 2085–2094 (2010).
Lee, K.-M. et al. Proline rich 11 (PRR11) overexpression amplifies PI3K signaling and promotes antiestrogen resistance in breast cancer. Nat. Commun. 11, 5488 (2020).
Nik-Zainal, S. et al. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature 534, 47–54 (2016).
Priestley, P. et al. Pan-cancer whole-genome analyses of metastatic solid tumours. Nature 575, 210–216 (2019).
Strand, S. H. et al. Molecular classification and biomarkers of clinical outcome in breast ductal carcinoma in situ: analysis of TBCRC 038 and RAHBT cohorts. Cancer Cell 40, 1521–1536 (2022).
The ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium. Pan-cancer analysis of whole genomes. Nature 578, 82–93 (2020).
Ali, H. R. et al. Genome-driven integrated classification of breast cancer validated in over 7,500 samples. Genome Biol. 15, 431 (2014).
Hu, Z., Li, Z., Ma, Z. & Curtis, C. Multi-cancer analysis of clonality and the timing of systemic spread in paired primary tumors and metastases. Nat. Genet. 52, 701–708 (2020).
Parker, J. S. et al. Supervised risk predictor of breast cancer based on intrinsic subtypes. J. Clin. Oncol. 27 (2009).
Konecny, G. et al. Quantitative association between HER-2/neu and steroid hormone receptors in hormone receptor-positive primary breast cancer. J. Natl Cancer Inst. 95, 142–153 (2003).
Angus, L. et al. The genomic landscape of metastatic breast cancer highlights changes in mutation and signature frequencies. Nat. Genet. 51, 1450–1458 (2019).
Martínez-Jiménez, F. et al. Pan-cancer whole-genome comparison of primary and metastatic solid tumours. Nature 618, 333–341 (2023).
Vicario, R. et al. Patterns of HER2 gene amplification and response to anti-HER2 therapies. PLoS ONE 10, e0129876 (2015).
Steele, C. D. et al. Signatures of copy number alterations in human cancer. Nature 606, 984–991 (2022).
Everall, A. et al. Comprehensive repertoire of the chromosomal alteration and mutational signatures across 16 cancer types from 10,983 cancer patients. Preprint at medRxiv https://doi.org/10.1101/2023.06.07.23290970 (2023).
Hausser, J. & Alon, U. Tumour heterogeneity and the evolutionary trade-offs of cancer. Nat. Rev. Cancer 20, 247–257 (2020).
Chen, Y. et al. Molecular and epigenetic profiles of BRCA1-like hormone-receptor-positive breast tumors identified with development and application of a copy-number-based classifier. Breast Cancer Res. 21, 14 (2019).
Funnell, T. et al. Single-cell genomic variation induced by mutational processes in cancer. Nature 612, 106–115 (2022).
Caulfield, M. et al. National Genomic Research Library. Figshare https://doi.org/10.6084/M9.FIGSHARE.4530893.V7 (2020).
Cho, S. W. et al. Promoter of lncRNA gene PVT1 is a tumor-suppressor DNA boundary element. Cell 173, 1398–1412 (2018).
Pereira, B. et al. The somatic mutation profiles of 2,433 breast cancers refine their genomic and transcriptomic landscapes. Nat. Commun. 7, 11908 (2016).
McGrail, D. J. et al. Replication stress response defects are associated with response to immune checkpoint blockade in nonhypermutated cancers. Sci. Transl. Med. 13, eabe6201 (2021).
Yang, Y. et al. Transcription and DNA replication collisions lead to large tandem duplications and expose targetable therapeutic vulnerabilities in cancer. Nat. Cancer 5, 1885–1901 (2024).
Li, J. et al. Non-cell-autonomous cancer progression from chromosomal instability. Nature 620, 1080–1088 (2023).
Lee, J. J.-K. et al. ERα-associated translocations underlie oncogene amplifications in breast cancer. Nature 618, 1024–1032 (2023).
Periyasamy, M. et al. APOBEC3B-mediated cytidine deamination is required for estrogen receptor action in breast cancer. Cell Rep. 13, 108–121 (2015).
McCann, J. L. et al. APOBEC3B regulates R-loops and promotes transcription-associated mutagenesis in cancer. Nat. Genet. 55, 1721–1734 (2023).
Venkatesan, S. et al. Induction of APOBEC3 exacerbates DNA replication stress and chromosomal instability in early breast and lung cancer evolution. Cancer Discov. 11, 2456–2473 (2021).
Nik-Zainal, S. et al. Association of a germline copy number polymorphism of APOBEC3A and APOBEC3B with burden of putative APOBEC-dependent mutations in breast cancer. Nat. Genet. 46, 487–491 (2014).
Cescon, D. W., Haibe-Kains, B. & Mak, T. W. APOBEC3B expression in breast cancer reflects cellular proliferation, while a deletion polymorphism is associated with immune activation. Proc. Natl Acad. Sci. USA 112, 2841–2846 (2015).
Martin, T. D. et al. The adaptive immune system is a major driver of selection for tumor suppressor gene inactivation. Science 373, 1327–1335 (2021).
Martínez-Jiménez, F. et al. Genetic immune escape landscape in primary and metastatic cancer. Nat. Genet. 55, 820–831 (2023).
Bagaev, A. et al. Conserved pan-cancer microenvironment subtypes predict response to immunotherapy. Cancer Cell 39, 845–865 (2021).
Denkert, C. et al. Tumour-infiltrating lymphocytes and prognosis in different subtypes of breast cancer: a pooled analysis of 3771 patients treated with neoadjuvant therapy. Lancet Oncol. 19, 40–50 (2018).
Houlahan, K. E. et al. Germline-mediated immunoediting sculpts breast cancer subtypes and metastatic proclivity. Science 384, eadh8697 (2024).
Gil Del Alcazar, C. R. et al. Immune escape in breast cancer during in situ to invasive carcinoma transition. Cancer Discov. 7, 1098–1115 (2017).
Zhai, L. et al. IDO1 in cancer: a Gemini of immune checkpoints. Cell. Mol. Immunol. 15, 447–457 (2018).
Hung, K. L. et al. ecDNA hubs drive cooperative intermolecular oncogene expression. Nature 600, 731–736 (2021).
Stork, C. T. et al. Co-transcriptional R-loops are the main cause of estrogen-induced DNA damage. Elife 5, e17548 (2016).
Roelofs, P. A., Martens, J. W. M., Harris, R. S. & Span, P. N. Clinical implications of APOBEC3-mediated mutagenesis in breast cancer. Clin. Cancer Res. 29, 1658–1669 (2023).
Harris, M. A. et al. Towards targeting the breast cancer immune microenvironment. Nat. Rev. Cancer 24, 554–577 (2024).
Acknowledgements
This research was supported by the National Cancer Institute through the Metastasis Research Network U54 award (CA261719) to C.C. C.C. is also supported by the Breast Cancer Research Foundation. J.L.C.-J. was supported by a National Cancer Institute K08 award (CA252457) and a METAvivor Early Career Research Award. K.E.H. was supported by a CIHR Banting Fellowship. We acknowledge the use of data from the Hartwig Medical Foundation and the TCGA Research Network (https://www.cancer.gov/tcga). This research was made possible through access to data in the National Genomic Research Library, which is managed by GEL (a wholly owned company of the Department of Health and Social Care). The National Genomic Research Library holds data provided by patients and collected by the NHS (National Health Service) as part of their care and data collected as part of their participation in research. The National Genomic Research Library is financed by the National Institute for Health Research and NHS England. The Wellcome Trust, Cancer Research UK and the Medical Research Council have also financed research infrastructure. We thank P. Srinivasan and V. Sankar for bioinformatic support, and M. McClure and other members of the Curtis Lab for feedback on the manuscript.
Author information
Authors and Affiliations
Contributions
Conceptualization: K.E.H., L.M., C.S.-V., A.A. and C.C. Bioinformatic analysis: K.E.H., L.M., C.S.-V., A.A., S.P., A.K. and S.J.P. Statistical analyses: K.E.H., L.M., C.S.-V., A.A., S.P. and S.J.P. Experimental validation: Z.M. Supervision: J.L.C.-J. and C.C. Writing (original draft): K.E.H., L.M., C.S.-V., A.A. and C.C. Writing (review and editing): all authors.
Corresponding author
Ethics declarations
Competing interests
Unrelated to this work, the following interests are declared: S.P. has equity in Inocras; J.L.C.-J. holds grants from Effector Therapeutics, Novartis and QED Therapeutics; C.C. has advised Bristol Myers Squibb, DeepCell, Genentech, NanoString, Pfizer and 3T Biosciences and has equity in 3T Biosciences, DeepCell and Illumina. All other authors declare no competing interests.
Peer review
Peer review information
Nature thanks Anton Henssen and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 IC subgroup distribution varies across stages of progression, ancestry and histology.
a) IC subgroup (left) and subtype (right) across stages of progression in ER+ samples. b) Inferred ancestry (primary samples, left or metastatic samples, right) across IC subgroups. c) IC subgroup (left) and subtype (right) across inferred ancestry in primary (top) and metastatic (bottom) stages. d) IC subgroup (left) and subtype (right) across inferred ancestry in primary (top) and metastatic (bottom) stages in ER+ samples. e) IC subtypes (left) and subgroups (right) across paired primary and metastatic samples with WES data. f) Graphical network representing primary/primary or primary/metastatic pairs; dots corresponding to a tumour biopsy colored by IC subgroup, the edge between two dots indicates whether the classification is stable (black) or changes (red) through metastasis. The surrounding color represents the PAM50 subtype (gray indicates missing data). g) IC subtype or subgroup consistency with PAM50 in pre-invasive DCIS (left), primary (middle), and metastatic (right) samples. h) ER early transcriptional signature according to subgroup. i) ER early signaling transcriptional signature in primary and metastatic tumours. j) Schematic overview of IC-specific amplification peaks and associated genes. ES, effect size. DCIS, ductal carcinoma in situ; LumA, luminal A; LumB, luminal B. The schematic in j was created with BioRender.com.
Extended Data Fig. 2 Genomic features of primary IC subgroups.
a) IC group-level copy number profile with SV burden overlay in DCIS. b) Fraction of genome altered by subgroup across pre-invasive, primary invasive and metastatic tumours. Boxplot represents median, 0.25 and 0.75 quantiles with whiskers at 1.5x interquartile range. c) Alteration burden in metastatic tumours split based on treatment prior to biopsy. The sample size is indicated at the top of each bar. d) Fraction genome altered, fraction LOH, and number of damaging SVs in IC10 and IC4ER- subtypes. e) Proportion of IC10 and IC4ER- tumours with alterations in genes involved in three key pathways: cell cycle, DNA damage response (DDR), and ubiquitination. f) Alteration burden distribution in metastatic samples across metastatic sites. The sample size for each group is at the top of each bar. g-h) Activity of each of the six rearrangement signatures across the IC subgroups (g) or the ER+ High-risk subtypes (h) in primary tumours. i) Copy number and SV profiles of primary (left) and metastatic (right) samples, each representative of either a TNBC -enriched, ER+ Typical -enriched, ER+ High/HER2 + -enriched, or mixed profile in the center of the Pareto front. j) Proportion that each complex SV event contributes to the total complex SV burden stratified by subgroup. DEL, deletion; LOH, loss-of-heterozygosity; FDR, false discovery rate; BFB, bridge-fusion breakage; CPXDM, complex double minute; DM, double minute; INVDUP, inverted-duplication; TIC, templated insertion chain; TRA, translocation.
Extended Data Fig. 3 Genomic features are conserved though elevated through metastasis.
a) Pareto front projection with tumours colored by presence of co-amplification of two or more amplifications in the following cytobands: 17q23 (IC1), 11q13 (IC2), 17q12 (IC5/HER2 + ), 8p12 (IC6) or 8q24 (IC9). b) Pareto front projection with tumours colored by HRD and ER status. c) Barplot shows the proportion and number of samples predicted to be BRCA1-like or BRCA2-like across the subgroups. d) Proportion of various SV events in BRCA1-like, BRCA2-like or non-HRD tumours across the subgroups. e) Replication of the Pareto front projection using the GEL (primary) cohort. Each dot represents the architecture profile of each tumour colored by IC subgroup. f) Activity of six SV signatures across the IC subgroups in metastatic tumours. g) Distribution of primary and metastatic tumours on Pareto front. h) Comparison of SV signatures in primary and metastatic tumours across the IC subgroups. Barplot shows the log fold change of each rearrangement signature between primary and metastatic tumors across the IC subgroups. i) Transition vector corresponding to the difference in position on the Pareto fronts from (g) between the centroid of primary samples and the centroid of metastatic samples in each IC group. j) Replication of the Pareto front projection using the METABRIC (primary) cohort. Each dot represents the architecture profile of each tumour colored by IC subgroup. k) Forest plot shows the association between the proportion of archetypes and distant relapse free (DRF) survival, correcting for ER and HER2. Dots correspond to estimated hazard ratios and segments to 95% confidence intervals. l) Association between recurrence in the ER+ Typical-risk samples and the distance to each archetype (spanning from 0 to 1, linear regression, top), transcriptomic proliferative and HRD LOH scores (linear regression, bottom) and histological type (IDC or ILC, fisher’s exact test, bottom) in the METABRIC dataset. Significance: P ≤ 0.05 (*), P ≤ 0.01 (**), and P ≤ 0.001 (***). m) Differential pattern of relapse across ER + IC subgroups and by histology (IDC: invasive ductal carcinoma and ILC: invasive lobular carcinoma), illustrated by the cumulative (black) and annual (red) risk of relapse. n) ER+ Typical IDC and ILC distribution on the Primary-Discovery Pareto front. o) ER+ Typical IDC and ILC distribution on the METABRIC Pareto front. SV, structural variant; WGD, whole-genome doubling; HRD, homologous repair deficiency; LOH, loss-of-heterozygosity; IDC, invasive ductal carcinoma; ILC, invasive lobular carcinoma.
Extended Data Fig. 4 Cyclic amplifications preferentially amplify IC-specific oncogenes.
a) Proportion or number of samples with at least one cyclic or complex non-cyclic amplification in primary GEL tumours. b) Proportion or number of samples with at least one cyclic or complex non-cyclic amplification in HER2+ primary tumors stratified by ER positivity. c) Proportion or number of primary samples with at least one cyclic amplification according to JaBbA. d) Proportion of samples where AmpliconArchitect called a cyclic amplification but JaBbA called an alternative type of alteration. Colors indicate which alteration JaBbA called. e) Proportion of HER2+ primary tumours that harbor cyclic or linear amplification in ER+ High-risk-specific oncogenes (left), and the SV types (right). f) Proportion or number of samples with at least one cyclic or complex non-cyclic amplification in DCIS lesions. Left panel: DCIS cohort stratified by subgroup, right panel: DCIS cohort and additional samples from GEL stratified by sequencing method. g) Proportion of cyclic amplifications, stratified by subgroup, that amplify IC-specific or alternative oncogenes in primary tumours, both the discovery and replication (GEL) cohorts. The number of amplifications in each category are included on each bar. h) Number ecDNA involving more than one IC-specific oncogene. i) Number of oncogenes per megabase involved in ecDNA in each subgroup. Boxplot represents median, 0.25 and 0.75 quantiles with whiskers at 1.5x interquartile range. j) Ratio of oncogenes amplified on ecDNA compared vs. oncogenes in the IC-specific cytoband per megabase. k) Proportion of ER+ Typical-risk ecDNA that incorporate each oncogene. l) Proportion of each archetype in ER+ High-risk, ER+ Typical and ER+ Typical containing ecDNA tumours.
Extended Data Fig. 5 Cyclic amplifications are maintained in metastatic tumours.
a-b) Number of recurrent oncogenes per Mbp for each subgroup in primary (a) and metastatic (b) tumors. c-d) Proportion of cyclic amplifications, stratified by subgroup, that amplify IC-specific or alternative oncogenes in metastatic (c) and DCIS (d) lesions. The number of amplifications in each category is included on each bar. e) Proportion of metastatic tumours within each IC subtype that harbor cyclic, complex non-cyclic or linear amplification in the IC-specific oncogenes. The number of tumors within each subtype are indicated at the top of each subpanel. f) Two representative examples of ER+ High-risk DCIS lesions harboring ecDNA containing at 8p11 (IC6). AMP, amplification.
Extended Data Fig. 6 Elevated replication stress in TNBC, ER+ High-risk and HER2+ tumours.
a-b) Replication stress signature stratified by IC subgroup (left) or by histology within ER+ Typical-risk subgroup IDC and ILC (right) in the TCGA (a) and METABRIC (b) datasets. FDR adjusted p-values are reported. c-d) Replication stress signature stratified by IC subtypes in TCGA (c) and METABRIC (d) datasets. e) Pareto projection of METABRIC tumors, colored by replication stress. f) Replication stress signature in HER2+ tumors; IC1, IC6, and IC9 subtypes of ER+ High-risk; and TNBC IC10 stratified by presence of ecDNA. ER+ High IC2 was excluded due to lack of sample size (n = 2 for ecDNA+ IC2). g-h) cGAS/STING signature stratified by IC subgroup (left) or by histology within ER+ Typical-risk subgroup IDC and ILC (right) in the TCGA (g) and METABRIC (h) datasets. FDR adjusted p-values are reported. In a-d, effect sizes (ES) and FDR-adjusted p-values from Mann-Whitney Rank Sum test are shown. In f, ES and p-values from linear regression correcting for cohort are shown. Additionally, the amplicon copy number was corrected for amplicon-driven HER2+ and ER+ Typical tumors.
Extended Data Fig. 7 Model for ER-induced R-loops in ecDNA genesis.
a) Simplified schematic illustrating model. Blue letters correspond to figure panels in Extended Data Fig. 7. Created with BioRender.com. b) Number of translocations in cyclic vs. non-cyclic amplifications across subgroups. Boxplot represents median, 0.25 and 0.75 quantiles with whiskers at 1.5x interquartile range. c) ESR1 mRNA abundance in cyclic amplification-positive vs. -negative (top) and non-cyclic amplification-positive vs. -negative (bottom) primary from Nik-Zainal et al., TCGA or metastatic tumors stratified by the IC subgroups, considering ER+ High-risk and HER2+ subgroups. Odds ratio from logistic regression correcting for tumor purity and error bars represent 95% confidence intervals. d) Density of APOBEC3B and ER ChIP-Seq peaks within cyclic and complex non-cyclic amplifications in primary tumours. e) ER early signaling transcriptional signature in DCIS and primary ER+ Typical vs. ER+ High-risk tumors. f-g) Density of ER-induced R-loops in cyclic and complex non-cyclic amplifications stratified by IC subgroups in primary (f) and metastatic (g) tumours. h) Density of all R-loops in cyclic vs. non-cyclic amplifications in primary and metastatic tumors. i) Difference in number of R-loops between A3B knockout (KO) wildtype (WT) MCF10A cell lines overlapping cyclic or non-cyclic amplifications at baseline or after A3B activation (PMA treatment). j-k) Median distance between a translocation and its closest ER-induced R-loop considering translocations within or outside cyclic amplifications in primary (j) and metastatic (k) tumors. l) Percent of breakpoints that overlap any R-loop with (+) or without (−) E2 treatment. Error bars represent the standard deviation across three replicates. m) Proportion of samples with or without ecDNA stratified by inferred APOBEC3B germline copy number. The total number of samples is included at the top of each bar. In b and j-l fold change (FC) and p-values or false discovery rates (FDR) are from Mann-Whitney Rank Sum test. In d-i, effect sizes (ES) are the difference in medians and p-values are from Mann-Whitney Rank Sum test. BER, base-excision repair; TC-NER, transcription-coupled nucleotide excision repair; E2, estrogen.
Extended Data Fig. 8 IC subgroups harbor distinct TMEs.
a) Schematic illustrating additional transcriptomic profiles and overlap with genomic profiles induced in Fig. 1a. Created with BioRender.com. b) Mean proportion of different cell types from IMC data by TME subtypes. The Wilcoxon test significance was reported above each comparison as follow: ns: not significant, P ≤ 0.05 (*), P ≤ 0.01 (**), P ≤ 0.001 (***), and P ≤ 0.0001 (****). c) Proportion of TME subtypes in primary and metastatic samples for the ER+ High-risk ICs and IC5 (HER2 + ) by ER status. d) Proportion of TME subtypes for primary samples (METABRIC) in ER+ Typical invasive IDC and ER+ Typical ILC. e) Mean proportion of fibroblasts and T cells in TNBC samples with IMC proteomic data obtained from bootstrapping (n = 1000). f) Proportion of TME subtypes for primary and metastatic samples stratified by ER status. g) Proportion of TME subtypes for primary samples and liver metastases by groups. IMC, imaging mass cytometry; SMA, smooth muscle actin.
Extended Data Fig. 9 Genetic mechanisms of immune escape in IC subgroups.
a) Proportion of primary and metastatic samples in each ER+ High-risk subtype with genetic immune escape (GIE) alterations, where values correspond to the number of pathways altered (left). Proportion of samples with alterations in each pathway stratified by IC subtype and disease stage (right). b) Proportion of primary ER+ High-risk samples with co-amplification of IDO1 with FGFR1 or ZNF703 by IC subgroup. c) Proportion of IC6 tumours with immune enriched (IE or IE/F) or immune depleted (D or F) TME subtypes stratified by the co-amplification of IDO1 with FGFR1 or ZNF703 in METABRIC and TCGA. d) Proportion of ER+ Typical IDC and ILC with GIE (left). Odds ratio and p-value from Fisher’s exact test. Proportion of pathways altered in IDC and ILC with GIE (right). e) Number of alteration in immune escape pathways for primary and metastatic samples, normalized by number of samples with alterations. f) Odds ratio for the frequency of GIE pathway alterations, comparing metastatic to primary samples. Background shading indicates FDR adjusted p-values (Fisher’s exact test). The color of the dot represents the direction and magnitude of the odds ratio while the dot size indicates the number of samples with a GIE in each pathway (y-axis). LOH, loss-of-heterozygosity.
Supplementary information
Supplementary Information
Supplementary Figs. 1–7, Materials and Methods, and References.
Supplementary Table 1
Clinical and genomic features of primary and metastatic breast tumours. Patient-level clinical and genomic features for primary (n = 702) and metastatic (n = 720) tumours. Table includes ENiClust subtype, CN and SV signatures, complex amplifications, SV and SNV burden, number of neoantigens. Table also includes a list of genes included in the ENiClust model.
Supplementary Table 2
Transcriptional features of primary and metastatic breast tumours.
Supplementary Table 3
IC-specific oncogenes recurrently incorporated in ecDNA. List of oncogenes in the IC-specific cytoband (Extended Data Fig. 1j) that are recurrently incorporated in ecDNA.
Supplementary Table 4
Description of pathways and alterations considered as genetic mechanisms of GIE.
Supplementary Table 5
Description of patient-level GIE in primary and metastatic tumours.
Supplementary Table 6
Parameters used to detect CNAs using fraction and allele-specific copy number estimates from tumour sequencing (FACETS). List of three parameters selected as the ‘best-fit’ for each sample when calling CNAs with FACETS in WGS or WES.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Houlahan, K.E., Mangiante, L., Sotomayor-Vivas, C. et al. Complex rearrangements fuel ER+ and HER2+ breast tumours. Nature 638, 510–518 (2025). https://doi.org/10.1038/s41586-024-08377-x
Received:
Accepted:
Published:
Issue date:
DOI: https://doi.org/10.1038/s41586-024-08377-x
This article is cited by
-
Modeling tumor relapse using proliferation tracing and ablation transgenic mouse
npj Breast Cancer (2025)
-
Targeting ATM kinase: a promising strategy to disrupt ecDNA maintenance in cancer
Genome Instability & Disease (2025)
