
Abstract
The eukaryotic circadian clock keeps time by using a transcription–translation feedback loop, which exhibits an architecture that is conserved across a diverse range of organisms, including fungi, plants and animals1. Despite their mechanistic similarity, the molecular components of these clocks indicate a lack of common ancestry2. Our study reveals that RUVBL2, which is a P-loop NTPase enzyme previously shown to affect circadian phase and amplitude as part of mammalian clock super-complexes, influences the circadian period through its remarkably slow ATPase activity, resembling the well-characterized KaiC-based clock in cyanobacteria. A screen of RUVBL2 variants identified arrhythmic, short-period and long-period mutants that altered circadian locomotor activity rhythms following delivery by adeno-associated virus to the murine suprachiasmatic nucleus. Enzymatic assays showed that wild-type RUVBL2 hydrolyses only around 13 ATP molecules a day, a vastly reduced turnover compared with typical ATPases. Notably, physical interactions between RUVBL2 orthologues and core clock proteins in humans, Drosophila and the fungus Neurospora, along with consistent circadian phenotypes of RUVBL2-mutant orthologues across species, reinforce their clock-related function in eukaryotes. Thus, as well as establishing RUVBL2 as a common core component in eukaryotic clocks, our study supports the idea that slow ATPase activity, initially discovered in cyanobacteria, is a shared feature of eukaryotic clocks.
Similar content being viewed by others


Main
The circadian clock operates as a cell-autonomous time-keeping mechanism that orchestrates the rhythmic expression of numerous genes through a complex network of intertwined transcriptional feedback loops to maintain physiological homeostasis3,4. The feedback loop typically involves transcription factors binding to cis elements in DNA promoter regions and activating the expression of target genes, including their own repressors. As repressor proteins accumulate, they translocate from the cytoplasm to the nucleus, interacting with transcription factors and inhibiting their own transcription, generating a roughly 24-h oscillation. The transcription–translation feedback loop (TTFL) model, featuring hierarchical regulation of core and associate loops, has proven effective in various eukaryotic clock systems1.
However, the current TTFL models face several theoretical and experimental challenges. First, there is a question about diversity in clock components. Although eukaryotic clock architectures share a high degree of similarity, the individual core components exhibit notable differences. For instance, plants lack clock components that are structurally and functionally similar to animal period proteins, indicating the lack of a shared ancestor. Current theories propose at least three separate origins for the clocks2,4,5. Second, there is uncertainty about energy consumption. Transcription and translation processes consume approximately 75% of the energy in most cells6. For the presumed TTFL clock to orchestrate these processes into a highly ordered circadian pattern, it must impose a substantial reduction in entropy, thereby creating a high-energy barrier to form oscillations7,8. The third challenge relates to a temporal discrepancy. Experimentally reconstituted transcription feedback cycles exhibit a duration of only 1.5–6.0 h, which is much shorter than the 24-h oscillations observed in natural circadian clocks9,10,11. This discrepancy raises an unresolved ‘slowness’ issue for the TTFL model, because these artificially induced cycles fall far short of the natural circadian duration.
A 2005 study revealed that the KaiABC proteins in cyanobacteria orchestrate a protein-based autonomous phosphorylation–dephosphorylation cycle occurring over a 24-h duration12. It also demonstrated that the extremely weak but stable ATPase activity of KaiC sets the period length of the clock13. Notably, this ATPase activity is highly energy efficient, consuming only 15 molecules of ATP a day, and the slow kinetics of this ATPase activity underline the gradual pace of the 24-h clock14. This KaiC ATPase model elegantly addresses the challenges outlined in the second and third points above. However, it is important to note that the KaiC model is thought to be specific to cyanobacteria and Rhodobacter15, so its applicability regarding both period determination and interactions with clock components may not extend to eukaryotic clocks.
Our study finds that genes of the RuvB-like (RUVBL) AAA+-type ATPase subfamily emerge as compelling candidates to be the common ancestral genes governing eukaryotic clockworks. The evolutionary conservation of these genes (Extended Data Fig. 1a,b), coupled with the localization of RUVBL1/2 in the megadalton super-complex of the mammalian clock machinery (Extended Data Fig. 1c), substantiates this proposition. Notably, like KaiC, RUVBL2 is an extremely weak but stable ATPase, consuming approximately 13 ATP molecules per day (Extended Data Fig. 1d), far lower than typical ATPases, which consume from 103 to 107 ATP molecules per day14. We previously demonstrated that pharmacological perturbation of RUVBL2 resulted in alterations to both the phase and the amplitude of the clock-controlled rhythm in mammals16. Recognizing the circadian period as the most reliable parameter for defining clock components17, we conducted a genetic screen targeting the RUVBL2 gene, revealing multiple mutants with shortened or lengthened periods or displaying arrhythmic behaviour, all of which are tightly correlated to the ATPase activities. Importantly, these findings were corroborated in other eukaryotic clocks. Therefore, as well as confirming that RUVBL2 is a common core component in eukaryotic clocks, our investigation lends support to the notion that slow ATPase activity, originally identified in cyanobacteria, is a shared characteristic of eukaryotic clocks.
A genetic screen of RUVBL2
To explore the potential effects on circadian period changes associated with RUVBL2, we performed a CRISPR–Cas9-based screen using a set of guide RNA molecules (gRNAs) covering the entire coding sequence (CDS) of the RUVBL2 gene in U2OS cells that carry a circadian-regulated reporter18,19 (Fig. 1a,b). We demonstrated that introducing CRISPR–Cas9 alone into U2OS cells did not alter the cellular clock (Extended Data Fig. 2a). However, subsequent administration of individual gRNAs induced random insertion/deletion or point mutations in the approximate sequences of the target region, generating a spectrum of mutant variants. The cellular clock phenotype for each was then detected by longitudinal recording of the circadian reporter in individual clones. Previous studies have revealed that mutants of Per2, a mammalian core clock gene, exhibit bidirectional period changes and arrhythmic behaviour19,20, whereas short interfering RNA (siRNA) knockdown of the ACTB gene does not manifest any discernible circadian phenotype19. Therefore, we selected these two genes, PER2 and ACTB, as the positive and negative controls, respectively (Extended Data Fig. 2b).

a, The genetic screen procedure for circadian phenotypes on a specific gene. b, Schematic representation of single guide RNA (sgRNA) site design on coding sequences of PER2, ACTB and RUVBL2. c, Distribution of circadian parameters in mutants, with upper and lower panels representing period and amplitude distributions, respectively; mutRUVBL2 (n = 822), mutPER2 (n = 210), mutACTB (n = 298). The cut-off for the period is determined using log2(period/mean of mutACTB (25.75 h)). Mutants with a period of less than 24.03 h (log2 value < −0.1) are classified as short hits (green dots), those greater than 27.59 h (log2 value > 0.1) as long hits (orange) and those exceeding 33.98 h (log2 value > 0.4) as arrhythmic (blue). Amplitude is presented as log2(amplitude/mean of mutACTB), and the cut-off was −1.491 and +1.491 for low-amplitude hits (green) and high-amplitude hits (orange). d, Heatmap of Per2-dLuc expression of mutGenes ordered by phase. Each row represents one mutant; data of mutRUVBL2 were normalized between 4 (red) and −4 (blue). For mutRUVBL2, 822 mutants were divided into four groups, with 302 long-period mutants, 443 normal-period mutants, 22 short-period mutants and 55 arrhythmic mutants. e, Luminescence of three representative knock-in Per2-dLuc U2OS mutant cells: wild type (grey, shown in each graph for comparison), short period (green), long period (orange) and arrhythmic (AR, blue); n = 3 biologically independent cells. f, Period-length frequency distribution histogram of mutGenes. g, The proportions of period lengths. h, The distribution of 30 verified in-frame mutations in the RUVBL2 amino acid sequence. Residues for domain I (DI), domain II (DII) and domain III (DIII) are indicated, as well as catalytic motifs. All period data parameters were analysed using MetaCycle. Illustration in a created using BioRender (credit: Juan, C. https://BioRender.com/n10u764; 2024).
In a screen of 822 mutagenized RUVBL2 clones, we identified three distinct clock phenotypes: short period (n = 22), long period (n = 302) and arrhythmic (n = 55). The results resembled those of the positive control (PER2), whereas the negative control (ACTB) displayed very low numbers of outliers (Fig. 1c–g and Extended Data Fig. 2c). Notably, the number of long-period mutants was much greater than that of short-period mutants, aligning with previous findings from genetic and chemical screens for circadian period modifications19,21. A few pioneering genetic screens in circadian research have fortuitously been dominant screens22,23,24,25, in which heterozygous mutants exhibit discernible clock phenotypes. We were in a similarly advantageous position because most of our mutant clones were heterozygous for the RUVBL2 gene. Considering the hexameric nature of this protein26, this occurrence is understandable, because some, although not necessarily all, of the mutant subunits could be sufficient to form a malfunctional protein.
To confirm the bidirectional-period phenotype, we selected all 22 short-period hits and the top 150 long-period hits for expansion of colonies in the follow-up verification. Among them, 75 clones (43.6%) were validated in producing a stable period phenotype, consistent with validation rates observed in cell-based genetic or chemical screens and clonal clock studies19,21,27. Subsequent sequencing of the CDS region of RUVBL2 revealed 68 non-redundant mutations, including 30 in-frame and 38 frame-shift mutants (Extended Data Fig. 2c–e).
The distribution of the 30 verified in-frame mutant locations exhibited non-random patterns, with a notable enrichment in functional motifs and domains. This observation indicates that these motifs and domains may have a role as an extra factor in regulating the circadian period (Fig. 1h). Notably, the region most enriched with mutations is situated around the Walker A motif, also known as the phosphate-binding loop (P-loop), which is known to associate with ATPase activity26. This underscores the correlation between the circadian period and the ATPase function of RUVBL2.
The coexistence of bidirectional period changes and an arrhythmic phenotype in mutants from a specific gene was first seen with the period gene, initially documented in the fruit fly22. This phenomenon is rare and has been observed in only a handful of core clock genes across all model organisms used in circadian research28. Thus, the identification of RUVBL2 mutants with three circadian phenotypes in our study secures its status as a bona fide clock gene.
Validation of mouse circadian rhythms
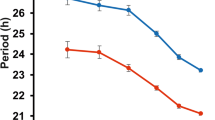
We then tested whether the circadian period phenotype we observed in cell culture could be confirmed in locomotor assays in transgenic animals, based on the dominant circadian phenotypes observed in previous studies20,29. Therefore, we chose nine candidate RUVBL2 variants from the U2OS screen (three short period and six long period) and generated AAV-mediated suprachiasmatic nucleus (SCN)-specific transgenic mice30. Among these candidates obtained from human cells, two short-period and three long-period phenotypes were reproduced behaviourally in mice (Fig. 2a–c and Extended Data Fig. 3a).

a, Luminescence of representative knock-in Per2-dLuc U2OS mutants. The grey line represents RUVBL2WT/WT Per2-dLuc U2OS; n = 3 biologically independent cells. b, Locomotor activity of mice with introduced SCN-specific Ruvbl2 mutations. Top, the actogram of mice; bottom, the corresponding periodogram analysis. The grey line represents mice with GFP introduction in the SCN; n = 7, 8, 9 or 11 biologically independent mice for AAV–WT, K83, 78 K/78E/Δ120–121 and GEP/Δ78–80, respectively. LD indicates 12 h light:12 h dark; DD, always dark; grey shading indicates times of darkness. c, Left and middle: histograms of circadian periods in U2OS cells (left) and SCN-specific transgenic mice (middle). Right: simple linear regression analysis of periods. One-way analysis of variance (ANOVA) differential analysis using Šídák’s multiple comparisons test. d, Locomotor activity of SCN-specific Ruvbl2 knockout mice. Grey shading shows when the lights are off. The control and experimental group had 10 and 12 mice, respectively. e, Luminescence of PER2::LUC SCN with Ruvbl2 knockout in SCN. The right panel indicates amplitude analysis in PER2::LUC SCN recorded ex vivo. Sample sizes: n = 3 biologically independent mice for the control and n = 5 biologically independent mice for the knockout group. All period data parameters were analysed using LumiCycle (c). All bar graphs show mean ± s.e.m.
Knockdown of the RUVBL2 gene in U2OS cells results in an arrhythmic phenotype, indicating its essential role in clock function19. However, conventional knockout (truncation induced by chemical mutagenesis) of this gene leads to embryonic lethality31, preventing further analysis using the ‘gold standard’ mouse locomotor assay. To address this limitation, we generated SCN-specific knockout mice using AAV-mediated delivery32. As anticipated, these knockout mice exhibited arrhythmicity in locomotor activity, owing to the efficient deletion of Ruvbl2 in the SCN (Fig. 2d and Extended Data Fig. 3b–e). Further SCN ex vivo culture results revealed that the clock amplitude was significantly reduced (Fig. 2e), aligning with the observed arrhythmic behaviour in mice. Reintroducing Ruvbl2 into the SCN successfully restored the arrhythmic phenotype of SCN-specific Ruvbl2 knockout mice, highlighting the essential role of Ruvbl2 in maintaining circadian function. Moreover, reintroducing different Ruvbl2 mutants generated the predicted circadian phenotypes, consistent with observations in U2OS cells (Extended Data Fig. 3f).
We have previously demonstrated the relatively high and daily cycling protein expression of RUVBL2 in the SCN16. Similarly, Ruvbl2 gene expression is rhythmic in multiple other tissues, including liver and muscle (Extended Data Fig. 3g). Notably, Ruvbl2 gene expression can be induced by environmental cues, such as food intake in the liver and light in the SCN (Extended Data Fig. 3h,i). Collectively, these findings indicate that the Ruvbl2 gene exhibits intrinsic rhythmicity and is affected by input signals for the circadian clock, further supporting the proposition that it is a core clock gene.
Period determined by ATPase activity
Given that RUVBL proteins are recognized as members of the P-loop NTPase superfamily with extremely low activity26, we evaluated the impact of the ATPase activities of mutants on period regulation. Remarkably, we identified a strong linear correlation between ATPase activity and the corresponding cellular period length, with higher activity leading to shorter period lengths and lower activity resulting in longer period lengths (Fig. 3a–c and Extended Data Fig. 4a), as has been reported for the cyanobacterial clock protein, KaiC13.

a,b, Ultra-high-performance liquid chromatography (UHPLC) determination (a) and relative ATPase activity (b) of RUVBL1WT/RUVBL2WT and RUVBL1WT/RUVBL2MUT proteins. c, Correlation between the ATPase activity of human RUVBL1/2 and the circadian period length of knock-in U2OS cells. d, Primary sequence alignment of human RUVBL1/2 and cyanobacterial KaiC proteins. The sites related to ATPase activity in this study are labelled in blue. The image was created using Snapgene (v.4.3.6). e,f, Effects of the T47E, N86K or A289I mutants of KaiC proteins on fluorescence rhythms in the IVO system. The reaction was at standard condition but 25% of wild-type KaiC was replaced by mutant KaiC. Fluorescence anisotropy (e) is indicated by the colour scale, and period frequency is shown in f. g, Relative ATPase activity of mixing KaiC proteins. h, The position of the water molecule in the ATP-binding pocket in the human RUVBL1/RUVBL2 protein structure (6K0R). i, Relative ATPase activity of the RUVBL1MUT/RUVBL2WT and RUVBL1WT/RUVBL2WT proteins. j,k, Effects of CordyTP on the fluorescence rhythms in the IVO system, showing the fluorescence anisotropy (j) and period frequency (k). l, Relative ATPase activity of KaiC with an additional 0.5 mM ATP or CordyTP. m,n, Effects of cordycepin on the luminescence rhythm in Per2-dLuc U2OS cells, showing normalized luminescence (m) and period frequency (n). o, Relative ATPase activity of RUVBL1/RUVBL2 with an additional 0.5 mM ATP/CordyTP. Sample sizes: n = 3 biologically independent proteins for each group (a, b, c, g, i, l and o); n = 3 biologically independent cells for each group (c, m and n); n = 3 biologically independent samples for each group (e, f, j and k). All period data parameters were analysed using MetaCycle (f and k) and Lumicycle Analysis (n). Statistical tests: simple linear regression analysis (c); one-way ANOVA differential analysis with Šídák’s multiple comparisons test (b, f, g and i); two-tailed t-test differential analysis (k, l, n and o). All graphs show mean ± s.e.m. Credit: h was adapted from ref. 14, American Association for the Advancement of Science.
The RecA-like, P-loop-containing ATPase KaiC, which is a key cyanobacterial clock protein, shares the ATPase domain with the AAA+-type ATPase superfamily that includes RUVBL2 (ref. 33). Moreover, it functions as an extremely low-activity ATPase, consuming only around 13 ATP molecules a day13, which is similar to the number for the RUVBL2 protein34 (Extended Data Fig. 1d). It has been documented that this low activity provides the foundation of the deliberate slowness required for a 24-h cycle oscillation14. Despite the considerable dissimilarity in amino acid sequence (less than 10% similarity) between human RUVBL2 and cyanobacterial KaiC, they exhibit remarkably similar secondary structures in the amino termini of their ATPase domains (approximately 80 amino acids; Extended Data Fig. 4b). This is not particularly surprising, because both KaiC and RUVBL2 belong to a superfamily known as the P-loop NTPases35 (Extended Data Fig. 4c).
In this relatively conserved area of 80 residues, there are two shared outstanding regions: the Walker A motif/P-loop and the helix 3 motif (Fig. 3d). To explore the potential homology between KaiC and RUVBL2, we identified residues in KaiC that correspond to some identified in our U2OS screen to target for mutagenesis in the cyanobacterial system. The RUVBL2Q78E/WT variant (in the P-loop) emerged as a short-period hit, whereas the RUVBL2Δ120-121/WT variant (in helix 3) was identified as a long-period hit. We therefore generated the corresponding mutants in KaiC and assessed their effects on the period. Leveraging a previously developed cell-free reconstituted in vitro oscillator (IVO) system with a fluorescence-based readout36, which is a powerful tool for measuring genetic or pharmacological effects on KaiC oscillation, we observed parallel period changes accompanied by corresponding alterations in ATPase activity (Fig. 3e–g).
Because RUVBL2 shares similarity with KaiC in the origin of slow ATPase activity (Extended Data Fig. 1), we wondered whether they are similar in terms of atomic structure. We proposed that in the active site of the pacemaker CI-ATPase of KaiC, the lytic water molecule is positioned farther away and at an angle that is unfavourable for the nucleophilic attack of ATP relative to other high-activity ATPases, such as myosin and kinesin14. In our previously solved RUVBL1/RUVBL2 crystal structure (PDB: 6K0R), a presumptive lytic water molecule was observed in a similar unfavourable position, near an aspartate residue (D353 of RUVBL1) that presumably activates the water molecule for nucleophilic attack on the γ-phosphate of ATP (Fig. 3h). If this water molecule is the lytic one, a point mutation in D353 would greatly reduce the already low ATPase activity even further. We therefore generated RUVBL1D353Q and found that this single amino acid change did indeed almost completely abolish the ATPase activity of the RUVBL1/RUVBL2 hexameric enzyme, whereas a control mutant, RUVBL1D356Q, left the ATPase activity intact, validating the position of the lytic water molecule and its importance in hydrolysing the ATP (Fig. 3i). Together, these results underscore some intriguing similarities in the functional dynamics of RUVBL2 and cyanobacterial KaiC, despite their disparate primary sequences.
Cordycepin triphosphate (CordyTP) has structural similarity to ATP (Extended Data Fig. 4d) and has been demonstrated to impact the clock function of RUVBL2 (ref. 16). Given that the IVO system provides a straightforward and uncontaminated environment in which to assess KaiC clock activity without interference from other ATP-consuming proteins, we investigated the impact of CordyTP on the cyanobacterial circadian oscillator through KaiC. Remarkably, we observed a period-shortening phenotype, aligning with its biochemical function to stimulate, rather than inhibit, the NTPase (ATPase + CordyTPase) activity of KaiC (Fig. 3j–l). However, cordycepin, which transforms to CordyTP in cells to regulate the clock, exhibited a dose-dependent period-lengthening cellular clock in U2OS cells when pre-incubated with pentostatin to impede its rapid degradation (an EC2h of 500 nM; Fig. 3m,n and Extended Data Fig. 4e). Correspondingly, CordyTP inhibits, rather than stimulates, the NTPase activity of RUVBL2 in a biochemical assay (Fig. 3o). Collectively, these findings highlight that it is the NTPase activity of the respective ATPase (KaiC in cyanobacteria and RUVBL in human cells) that governs the clock period, rather than the compound per se (in this case, a cordycepin derivative).
A CRY-independent mechanism
An important question is how the ATPase activity of RUVBL2 influences the circadian period. Given that RUVBL2 is present in the TTFL supercomplex, we hypothesized that RUVBL2 might regulate the circadian period through the TTFL genes. Two core components of the TTFL, CRY1 and CRY2, have opposing effects on the circadian period: loss of CRY1 shortens the period, whereas loss of CRY2 lengthens it in a dose-dependent manner19,37. To investigate whether the period alterations in the RUVBL2-mutant-U2OS cells are CRY-dependent, we separately knocked down CRY1 and CRY2 using siRNA. Interestingly, we observed that the dose-dependent period-shortening effect of CRY1 knockdown and the period-lengthening effect of CRY2 knockdown were preserved in U2OS cells harbouring either RUVBL2Q78E/WT (high ATPase activity with a short circadian period) or RUVBL2K83I/WT (low ATPase activity with a long circadian period), respectively (Fig. 4a–d). Similar to the wild type, both RUVBL2Q78E/WT and RUVBL2K83I/WT cells exhibited short periods with low-dose CRY1 knockdown but became arrhythmic with high-dose CRY1 knockdown. The effects on the period of the ATPase activity of RUVBL2, in combination with CRY1/CRY2 expression, seem to be additive, rather than synergistic, indicating that the period effects of the ATPase activity of RUVBL2 are independent of the CRY genes.

a,b, Normalized luminescence expression in RUVBL2Q78E/WT (left), RUVBL2WT/WT (middle) and RUVBL2K83I/WT (right) knock-in U2OS cells with CRY1 (a) and CRY2 (b) knockdown from siRNA. The light and dark curves for each colour represent control and high-dose siRNA, respectively. c,d, Circadian periods of knock-in U2OS cells with low (#1) and high (#2) doses of CRY1 (c) and CRY2 (d) siRNA for knockdown; AR, arrhythmic. Periods were calculated by LumiCycle Analysis; n = 3 biologically independent cells. e,f, Knockdown efficiency (left two columns) and effects on clock gene expression (right nine columns) of CRY1 (e) and CRY2 (f) siRNA in unsynchronized U2OS cells; mRNA levels were analysed by quantitative PCR (qPCR), with ACTB as a control; n = 3 technical replicates, representing 3 independent experiments. In parallel experiments, bioluminescence rhythms were recorded and circadian phenotypes were confirmed. Statistical tests: all period data parameters were analysed using LumiCycle Analysis; two-tailed multiple unpaired t-test differential analysis with two-stage step-up (the Benjamini, Krieger and Yekutieli method) (c); two-way ANOVA differential analysis with Dunnett’s multiple comparisons test (d). All bar graphs show mean ± s.e.m.
The TTFL clock genes exhibit network regulatory features37. For example, dose-dependent knockdown of CRY1 proportionally increases the expression of PER1, PER2 and PER3. Furthermore, we know that paralogue compensation occurs in a unidirectional manner because reducing CRY1 levels leads to an increase in CRY2, but not vice versa. We therefore investigated whether these network regulatory features remain in RUVBL2 mutant cells (Fig. 4e,f). We found that these features remain largely intact, further supporting the idea that RUVBL2 regulates the circadian period in a CRY-independent manner.
To exclude the possibility that the aforementioned phenotype is limited to U2OS cells, we introduced the Ruvbl2Q78K mutation into mouse adult fibroblasts (MAFs), resulting in a short-period phenotype similar to that of the RUVBL2Q78K/WT U2OS cells (Extended Data Fig. 5a–c). Similarly, knocking out Cry1 or Cry2 in the Ruvbl2Q78K/WT mutant fibroblasts led to corresponding period shortening or lengthening, respectively, similar to the effects observed in wild-type cells (Extended Data Fig. 5d–g). Together, these results indicate that the ATPase activity of RUVBL2 regulates the circadian period independently of CRY in both human and mouse cells.
Evolutionary conservation
The presence of RUVBL proteins in the mammalian clock supercomplex16 and their high conservation (Extended Data Fig. 1) imply that they have potential interactions with core clock components in other species. To explore this potential connection, we conducted a co-immunoprecipitation assay using well-documented core clock components and corresponding RUVBL subfamily members from various species. Our results demonstrate mutual pull-down interactions between the RUVBL2 homologues and BMAL1, CRY1, CRY2, PER1 and PER2 in humans; Per and Cyc in flies; FRH and FRQ in fungi; and CCA1 and TOC1 in plants, all conducted in co-transfected 293 T cells (Fig. 5a and Extended Data Fig. 6a,b). Interestingly, in a proteomic analysis using GFP-tagged Per (knock-in)38 as a bait from the fruitfly head lysate, both Pontin (the fly’s homologue of RUVBL1) and Reptin (the RUVBL2 homologue) emerged as hits (Fig. 5b and Supplementary Table 1).

a, Western blot analyses of co-immunoprecipitation (co-IP) of clock proteins and RUVBL2 homologues, which represents three independent experiments; antibodies for co-IP and western blot analyses are indicated. For gel data, see Supplementary Fig. 1a. IB, immunoblotting; IP, immunoprecipitation. b, Volcano plot of the proteins enriched by PER::GFP and IGG at zeitgeber time (ZT)16; n = 2 biologically independent experiments; see Methods for more details. c, Effects of CB-6644 in Bmal1-dLuc U2OS cells; n = 3 biologically independent cells for each dose. Left, heatmap of luminescence rhythms that were monitored in the presence of a dose concentration of CB-6644; luminescence intensity is indicated by the colour scale. Right, statistical analysis of the period length change. d, Stacked bar chart showing the rhythmicity of D. melanogaster with ReptinWT and ReptinK79E expression in tim-positive cells. The percentages of rhythmicity (R) and arrhythmicity (AR) are shown on the graph. e, Period length of rhythmic (R) Drosophila; n = 42, 32, 37 and 33 for UAS-WT, UAS-K79E, tim-GAL4;UAS-WT and tim-GAL4;UAS-K79E, respectively. f, Representations of double-plotted averaged actograms of flies measured for 5 days in 12 h:12 h light: dark (LD) and 11 days in constant dark (DD); grey shaded areas represent darkness. The data were analysed using ClockLab Analysis software. g, Relative ATPase activities of PontinWT/ReptinK79E and PontinWT/ReptinWT proteins; n = 3 biologically independent proteins for each group. h, Model of the origins of circadian oscillatory systems. The schematic shows the origins of each organism studied, stemming from the last universal common ancestor (LUCA). The period length is regulated by the activity of KaiC or RUVBL ATPase. Statistical tests: simple linear regression analysis (c); one-way ANOVA differential analysis with Šídák’s multiple comparisons test (e); two-tailed t-test differential analysis (g). All bar graphs show mean ± s.e.m. Illustrations in a and b adapted from BioRender (credit: Juan, C., https://BioRender.com/n10u764; 2024).
A commercially available compound, CB-6644, which was originally designed to treat leukaemia and myeloma in humans, has been identified as a potent inhibitor of the ATPase activities associated with RUVBL proteins34 (Fig. 5c and Extended Data Fig. 7a,b). Consequently, we introduced this compound into U2OS cells and assessed its influence on the cellular period length. We found a dose-dependent effect of CB-6644 on lengthening the cellular clock period, with an effective concentration for at least a 2 h change39 (EC2h) observed at approximately 3 μM (Extended Data Fig. 7c).
Given the highly conserved nature of RUVBL members across eukaryotic species (approximately 85% amino acid similarity), we sought to investigate whether CB-6644, a drug specifically designed for the orthologues of human RUVBL1 and RUVBL2, could also influence the clock. Excitingly, this compound exhibited the capacity to dose-dependently lengthen the periods of clocks in Drosophila, Neurospora and Arabidopsis, albeit with lower potency (an EC2h of approximately 10–20 μM; Extended Data Fig. 7d–f).
To investigate the necessity of Reptin and Pontin in maintaining rhythmic behaviour in Drosophila, we used the GAL4/UAS system to specifically express RNAi in tim-expressing cells40 and in pdf-expressing neurons, which regulate locomotor activity rhythms41. This approach was used to knock down the expression of Reptin and Pontin in clock neurons. Our results revealed that Pontin knockdown led to an almost complete loss of locomotor rhythms in constant darkness. Furthermore, reducing Reptin expression across all clock cells lengthened the circadian period, whereas Reptin knockdown in pdf-expressing neurons not only extended the period under constant darkness, but also resulted in arrhythmic behaviour after five days of darkness (Extended Data Fig. 8). We also generated transgenic flies that express Reptin wild type or mutant in tim-expressing cells using the GAL4/UAS system. Expression of the ReptinK79E variant in all clock cells resulted in a long period or even arrhythmic locomotor activity rhythms, whereas the ReptinWT files were normal (Fig. 5d–f). The ATPase activity of the PontinWT/ReptinK79E mutant variant in Drosophila was lower than that of PontinWT/ReptinWT, which is consistent with our finding of a negative correlation between ATPase activity and circadian period length across mammals and cyanobacteria (Fig. 5g).
In Neurospora, we obtained a rvb2S79I/S79I knock-in mutant strain. This strain lost its growth rhythm in constant darkness, although it maintained a weak growth rhythm in light:dark (12 h:12 h) conditions or temperature cycles (Extended Data Fig. 9a,b). This is reminiscent of the behaviour of the frq mutant, which has been well characterized as a core TTFL component in Neurospora42. The ATPase activity of the RuvB-like protein 1WT/RuvB-like protein 2S79I mutant variant in Neurospora was significantly lower than that of the RuvB-like protein 1WT/RuvB-like protein 2WT (Extended Data Fig. 9c). Together, these findings across different model organisms further confirm the pivotal role of the ATPase activity of RUVBL in regulating circadian clocks in eukaryotes.
Therefore, we propose that with the advent of primordial circadian clocks in ancient organisms, P-loop-containing low-activity ATPases became integral to the circadian circuit as a core component. In cyanobacteria, KaiC, along with its facilitating partners KaiA and KaiB, orchestrates a robust oscillator connecting to a TTFL to govern daily cycling behaviour. RUVBL proteins, the P-loop-containing AAA+ ATPases conserved in various eukaryotic species including fungi, plants, insects and mammals, interact with their respective core TTFL clock proteins. Moreover, as with KaiC, the extremely low activity of RUVBL ATPases determines the circadian period, highlighting the interconnected regulation of slow ATPase and TTFL in both prokaryotic and eukaryotic organisms (Fig. 5h).
Discussion
Three key parameters characterize a circadian clock-controlled rhythm: relative phase, amplitude and period43. Our previous findings demonstrated that pharmacological targeting of RUVBL2 led to alterations in the phase and/or amplitude of clock-controlled rhythms16. Here we use a genetic approach to show that RUVBL2 mutants exhibit bidirectional period phenotypes and arrhythmicity (Figs. 1 and 2). This distinctive feature has been reported for only a handful of core clock genes, such as per in fruitflies (and its human orthologue, PER2)20,22 and kaiC in cyanobacteria13. This finding, coupled with the characterization of intrinsic and entrainable rhythmicity44 (Extended Data Fig. 3g–i), establishes RUVBL2 as a core clock gene.
We have previously demonstrated that RUVBL2 interacts with other core clock proteins on chromatin and regulates the circadian phase16. Pharmacological perturbation of RUVBL2 with CordyTP transiently disassembles the circadian supercomplex, leading to reduced occupancy of inhibitory CRY proteins on chromatin, thereby altering the phase of the biological clock. In this study, we show that the ATPase activity of RUVBL2 regulates the circadian period in a CRY-independent manner (Fig. 4 and Extended Data Fig. 5). These findings indicate that the ATPase activity of RUVBL2 in circadian period regulation is not entirely dependent on its role in TTFL, distinguishing it from the mechanism of phase regulation. We interpret these two seemingly distinct mechanisms in a reconciled manner. In our previous study, when cordycepin (becoming the biologically active CordyTP in cells) was suddenly administered in the clock system and quickly degraded by adenosine deaminases, it had an acute effect on the interaction between RUVBL2 and TTFL components such as the BMAL1 and CRY proteins. In the present study, when pre-incubated with pentostatin, which is an adenosine deaminase inhibitor that extends the short half-life of CordyTP, it prolongs the inhibitory effect on RUVBL2 NTPase activity, mimicking the chronic impact of RUVBL2 mutations with low ATPase activity and resulting in a long circadian period in a CRY-independent manner (Extended Data Fig. 4). However, given the broad cellular functions of RUVBL2, further research is needed to explore more specific mechanisms using our screening method, for example, the interaction of RUVBL2 with other core clock components.
Finally, in our proposed model, which is similar to that of KaiC in which the pacemaker signal must ultimately transmit transcriptional outputs to drive clock-controlled behaviour36,45, RUVBL proteins are also required to convey the timing signal produced by ATP hydrolysis to the transcriptional machinery. We have demonstrated that RUVBL proteins interact physically with the core transcription factors of various eukaryotic clocks (Fig. 5a,b), indicating the plausible transduction of the signal. However, unlike the cyanobacterial KaiC model, which encompasses both the ATPase domain for its pacemaker function and the phosphorylation oscillator domain for escapement45, our model currently lacks an identified corresponding escapement component in eukaryotic RUVBL-dependent regulation. Given the classification of RUVBL2 as a DNA helicase and chromatin modifier, it is conceivable that chromatin DNA may store the tension energy generated by ATP hydrolysis, in a similar way to the role played by the phosphorylated oscillator domain in KaiC. Together, these insights collectively imply a close connection between the origins of the primordial prokaryotic and eukaryotic clocks15.
Methods
Plasmid cloning
The plasmids used in this study and the oligonucleotides used for this cloning are listed in Supplementary Table 2.
Cell culture
U2OS cells, mouse adult fibroblast and HEK 293 T cells were usually cultured in Dulbecco’s Modified Eagle’s Medium (Gibco) containing 10% fetal bovine serum (HAKATA), 1% penicillin–streptomycin (10,000 U ml−1, Gibco) at a constant temperature of 37 °C with a 5%-CO2 atmosphere.
RNA extraction and qPCR
Mouse tissue samples were collected after entraining mice to a 12 h:12 h light:dark cycle for two weeks, with lights on at ZT0. Tissues were collected every 4 h over a 24-h period. Mice were anaesthetized and perfused with cold PBS before tissue extraction. All consumables were RNase/DNase free. The tissues were then rapidly frozen in liquid nitrogen and stored at −80 °C. For cell samples, processed cells were washed with cold PBS, treated with Trizol and stored at −80 °C. RNA from all samples was extracted simultaneously using the trizol-phenol-chloroform method16. The RNA was quantified using a Nanodrop, and 500 ng was converted into cDNA using PrimeScript RT Master Mix (Takara). The resulting cDNA was diluted 4-fold for qPCR analysis of gene expression levels following the manufacturer’s instructions for the CFX96 qPCR detection system (Bio-Rad). The sequences of the qPCR primers are listed in Supplementary Table 2.
Screen procedure
For the screen, the Cas9 gene was initially introduced in a U2OS cell line harbouring the Per2-dLuc reporter through a lentiviral approach. The rhythmicity of Per2-dLuc–Cas9 U2OS cells was then assessed using Lumicycle. Subsequently, 51, 30 and 25 sgRNAs (sequences are listed in Supplementary Table 2) with GFP tags targeting the coding regions of PER2, ACTB and RUVBL2 genes, respectively, were designed, packaged and delivered into Per2-dLuc–Cas9 U2OS cells by lentiviral transduction at a controlled multiplicity of infection of around 0.1. Cells expressing both sgRNA and Cas9 were sorted into single clones by flow cytometry and cultured in 96-well plates, and their rhythmicity was recorded using a Tecan luminometer. Based on the recording results, clones exhibiting abnormal rhythms were selected for expansion culture and subjected to secondary inspection with Lumicycle. Finally, clones displaying aberrant biological rhythms were chosen for sequencing to confirm the mutations existing in the coding regions.
Knock-in and knockout
Point mutations or knockout of cells (including Ruvbl2WT/Q78K, Cry1KO/KO, Cry2KO/KO, Cry1KO/KO; Ruvbl2WT/Q78K and Cry2KO/KO; and Ruvbl2WT/Q78K MAFs) were generated using the CRISPR–Cas9 system. Initially, sgRNAs were designed to target the specific gene-editing regions. The guide RNA sequences were then cloned into the pLenti-sgRNA-Lib vector (Addgene, 53121)46. Transient transfection of sgRNA and donor (only sgRNA in the knockout assay) was carried out in Cas9–MAFs using Lipofectamine 3000. Then, 72 h after transfection, fluorescently labelled cells were sorted by flow cytometry and subsequently expanded in culture. These cells were then subjected to single-cell cloning by limiting dilution. Finally, the efficiency and accuracy of gene editing were confirmed by Sanger sequencing. The sgRNA sequence for Q78K is TGCCTGGCTGGCCTGCAATG; the sgRNA sequence for Cry1 knockout is TCGCCGGCTCTTCCAACGT; and the sgRNA sequence for Cry2 knockout is CTGAAAGCGCTTGTAGGTA.
Generation of Neurospora knock-in mutant strains
The Neurospora RUVB2S79I/S79I mutant strains were generated by the homologous recombination method47. The wild type or mutated homologous genome was cloned into the PBS plasmid, which carried hygromycin B (Yeasen Biotechnology). Conidia from Ku70 were transformed by the Gene Pulser Xcell total electroporation system (Bio-Rad) with wild type or mutation cassette and plated on agar plates containing hygromycin B. After an incubation period of 5 days at 30 °C in the dark, colonies were picked onto minimal slants containing hygromycin B for genotyping. Subsequently, the on-target mutant lines were selected and inoculated on race tubes for rhythm detection. The race tubes were then incubated in constant light at 25 °C for 24 h until an even growing front was observed. For constant darkness (DD) conditions, the race tubes were incubated in the dark for several days. For conditions with light and dark (LD), the race tubes were incubated in 12 h:12 h light:dark for several days. For temperature-cycle conditions, the race tubes were incubated at 25 °C for 12 h and 30 °C for 12 h in constant darkness for several days. The period was calculated as described48 using ImageJ (v.1.53c) software.
Circadian-rhythm recording of cells, tissue, fungi and plants
For mammalian cells and the SCN, the culture medium XM used for recording bioluminescence was prepared as described49. The XM medium contained 1× DMEM(Gibco), 1× B27 supplement (Gibco), 4.2 mM NaHCO3 (Gibco), 10 mM HEPES (Gibco), 100 U ml−1 penicillin and streptomycin (Gibco) and luciferin (1 mM for cells and 0.1 mM for tissue; Goldbio). Cells were seeded in a 3.5-cm dish and synchronized by replacing the medium with XM when the confluence rate was 100%. Tissue-preparation buffer was prepared as described16. It contained 1× HBSS, 10 mM HEPES, 4.5 mM NaHCO3, 100 U ml−1 penicillin and 100 U ml−1 streptomycin. SCN samples for recording bioluminescence were sampled at ZT6–10. Mice were euthanized and the brain was cut into slices using a vibrating microtome. The SCN was cut off with a knife, transferred to a 40 μm semi-permeable membrane and then put into a 3.5 cm petri dish containing the recording medium. The dishes were sealed and put into the Lumicycle recorder.
The Neurospora crassa line containing the frq-luc reporter used in the study was donated by X. Liu’s lab. The slants culture medium contained 1× Vogel’s medium, 30 g l−1 sucrose and 15 g l−1 agar50,51. The autoclaved fructose–glucose–sucrose/Vogel’s medium contained 1× Vogel’s medium, 1× fructose–glucose–sucrose, 50 μg l−1 biotin and 50 μM luciferin used for luciferase assay. The conidia suspension was added to a liquid medium without agar and synchronized in constant light conditions. Subsequently, the medium was replaced with a solution containing 1 mM luciferin. The dish was then sealed and placed into the Lumicycle recorder for luminescence recording.
For Arabidopsis thaliana, the plant line with the CCA1-luc reporter was from X. Xu’s lab. The Murashige and Skoog medium (MS medium) used in this experiment contained 4.33 g l−1 MS medium, 30 g l−1 sucrose, 3 g l−1 phytagel at pH 5.8–6.0 (ref. 52). The seeds were sterilized using a sodium hypochlorite solution, followed by rinsing with deionized water. Using a pipette tip, the seeds were planted in MS medium and vernalized at 4 °C for two days. The seedlings were then cultured under a 16 h:18 h light:dark schedule at 23 °C. Seedlings aged 7–14 days were used to record rhythms. The MS medium without phytagel was supplemented with 0.1 mM fluorescein for recording purposes. After adding 500 μl liquid culture medium to a 24-well plate, the seedlings were placed in the culture medium and the roots were kept in contact with the culture medium. The luminescence values were recorded using a Tecan-i-control 2.0.
For the rhythm-detection experiments to investigate the effects of CB-6644 on different species, CB-6644 (MedChemExpress) was dissolved in DMSO and added to the recording medium at the start of the experiments.
For experiments to measure the efficacy of pentostatin and cordycepin, 10 μM pentostatin was dissolved in water and added to XM medium at the start of recording. The cordycepin was then dissolved in DMSO and added to the medium at the first peak in a dose-dependent (200 nM, 500 nM, 1 μM, 2 μM) manner. Data from the lumicycle were analysed using Lumicycle analysis software (v.1.0.0.0). Rhythms were analysed using MetaCycle (v.1.2.0). Bioluminescence curves of all species were drawn using Excel 2021 and the heatmap was drawn using R (v.4.0.2).
Locomotor rhythms of flies
To determine whether ReptinK79E expression altered locomotor activity rhythms, experiments using the attP-PhiC31-mediated transgene of UAS-ReptinWT and UAS-ReptinK79E strains from the attP40 strain were done at Fungene Biotech (http://www.fungene.tech). UAS-ReptinWT and UAS-ReptinK79E strains were crossed to tim-GAL4 to drive expression in all clock cells. To determine the impact of CB-6644 on period, Drosophila melanogaster strains (M(vas-int.Dm)ZH-2A, M(3xP3-RFP.attP) ZH-51C and aM(3xP3-RFP.attP) ZH-86Fb as a wild type were a gift from T.W.’s lab) were raised on a yeast–sucrose–agar food medium. To determine whether knocking down Pontin or Reptin expression altered locomotor activity rhythms, the following strains were obtained from the Vienna Drosophila RNAi Center (VDRC) and the National Institute of Genetics (NIG) in Japan; UAS-Pontin RNAi #1 (VDRC KK105408); UAS-Pontin RNA#2 (NIG 4003R-1); UAS-Reptin RNAi #1 (VDRC KK105408); and UAS-Reptin RNAi #2 (NIG 4003R-1). RNAi strains were crossed to tim-GAL4 to drive expression in all clock cells and pdf-GAL4 to drive expression in pigment dispersing factor expressing brain neurons40,41.
All flies were kept at 25 °C in a 12 h:12 h light:dark cycle. The Drosophila Activity Monitor system (Trikinetics) was used to record locomotor activity as described53. We used 1–5-day fresh male flies to record locomotor activity. They were kept in a light- and temperature-controlled incubator (25 °C), and were monitored in LD cycles for 3–5 days, and then in constant darkness (DD) for a further 6–10 days. Locomotor activity was recorded in 1-min or 5-min bins. The rhythmicity ratio of ReptinWT and ReptinK79E flies was counted, and only the rhythmic flies’ period was calculated. Period length was analysed using ClockLab Analysis (v.1.0.0.272) software. Actograms for each individual fly were generated using the ActogramJ plug-in of the ImageJ program54. The period of RNAi knockdown flies and controls was calculated from the first 6–7 days in DD using Lombe-Scargle periodogram analysis in the ActoJ plug-in of ImageJ software. Visual inspection of actograms in certain RNAi genotypes revealed arrhythmicity in the last 2–3 days of DD. In such cases, period was calculated separately from the first 4 days of DD and the last 2–3 days of DD to report this complex arrhythmic phenotype. Individual values of the period and percentage rhythmicity were plotted using GraphPad Prism (v.9.4.0).
Locomotor activity rhythms of mice
Mice were housed in pathogen-free conditions at a temperature of 23 ± 2 °C and humidity of 40–70%, with food and water provided ad libitum. Male mice aged 2–3 months were randomly assigned to each group, and at least seven mice were included in each group. In the AAV-virus delivery experiment, mice were anaesthetized with avertin and then positioned in a stereotaxic apparatus. To introduce the AAV virus to the bilateral SCN, a craniotomy was done using a microsyringe pump at a speed of 46 nl per min. The coordinates (millimetres from bregma) of the SCN are: AP, +0.1; ML, ±0.25. Ruvbl2 knockout in SCN was done by combining AAV2/9-CMV-spCas9 and AAV2/9-U6-gRNA (Ruvbl2/Gfp) v2.0-CMV-EGFP-WPRE (OBiO) viruses with a titre of 3.0 × 1012 virus genomes per ml, and Ruvbl2 expression was enabled through viruses created at the Genetic Screening Center of the National Institute of Biological Sciences, Beijing (the backbone of AAV expression construct is addgene#37825, pAAV-CAG-GFP), and reintroduction of Ruvbl2 into SCN-specific Ruvbl2-knockout mice was done by mixing these two viruses. After a recovery period of at least eight weeks, the mice were synchronized to a 12 h:12 h light:dark cycle before undergoing continuous darkness conditions for locomotor-activity analysis. Data were collected using ClockLab and analysed using ClockLab Analysis software (v.1.0.0.272). The investigators who did the experiment also analysed related data; it was not a double-blind experiment. All animal experiments were done in accordance with the national guidelines set by the Ministry of Health for the housing and care of laboratory animals. Procedures followed institutional regulations and received prior review and approval from the Institutional Animal Care and Use Committee at the National Institute of Biological Sciences, Beijing. The gRNA sequences for Ruvbl2 knockout and negative control were as follows: gRNA of Ruvbl2, TGCACGGGACTACGACGCCA/TCCGGACACCAATGGAACGG/TGCCCGGCGTGCAGCTGGCG; and gRNA of control, ACGGAGGCTAAGCGTCGCAA.
External cue stimulation assays
For the light-stimulation experiment, mice were housed in constant darkness after being entrained to a 12 h:12 h light:dark cycle. They were exposed to 900-lux white light for 30 min starting at CT15 (75 h after lights off), and samples were taken at CT19. In the fasting–refeeding experiment, mice were housed in the LD cycle and provided with food ad libitum. They were then fasted for 48 h and then refed. Their livers were collected at 0, 3 and 5 h after refeeding. The RNA was extracted and then detected using qPCR. All data analysis was done using GraphPad Prism (v.9.4.0).
Immunofluorescence
After anaesthesia with tribromoethanol, mice were perfused with PBS and their brains were fixed with 4% paraformaldehyde for 4 h. The brains were then dehydrated with 30% sucrose and sliced into 30-μm sections using a cryostat microtome. Brain slices containing the SCN were kept in PBS. The brain slices were treated with PBS containing 0.3% Triton X-100 three times for 6 min each time and then blocked with 5% donkey serum for 2 h at room temperature. Slices were incubated with primary antibody (anti-c-Fos, 1:1,000, Cell Signaling Technology; anti-RUVBL2, 1:1,000, Novus Biologicals) at 4 °C overnight and then washed with PBS containing 0.3% Triton X-100. Slices were then incubated in secondary antibody (Cy3 AffiniPure donkey anti-rabbit IgG, H + L, 1:1,000, Jackson ImmunoResearch) at room temperature for 2 h and then washed three times for 6 min each time. Finally, brain slices were spread on glass and stained with DAPI. Fluorescence signals were detected using a Zeiss LSM 880. Fluorescence-intensity analysis and cell detection were done using ImagJ (v.1.53c).
Transfection, virus packaging and infection
The transfection assay and lentivirus generation were done as previously described16,55. Lenti-vectors pMDL, pREV and pVSVG were transfected to HEK293T using lipo3000 (Thermo Fisher), following the manufacturer’s instructions. After transfection for 48 h, the viruses were collected, filtered and used with a multiplicity of infection of around 0.1.
IP and co-IP
The co-IP procedure of cells was as previously described16. In brief, RUVBL2 with a STREP tag and the circadian clock gene of different species with FLAG-tag plasmids were constructed (the sequences of CCA1 and TOC1 were codon optimized to improve expression level and labelled with OPTI) and overexpressed in pairs in 293T cells (2 μg STREP and 2–4 μg plasmids per well of a 6-well plate), with the groups overexpressing GFP–FLAG and RUVBL2–STREP or overexpressing only RUVBL2–STREP used as controls. Two days after transfection, cells with transfected plasmids were collected on ice and washed with PBS on ice, then the samples were lysed by IP lysis buffer (Beyotime) supplemented with phosphatase inhibitor and protease inhibitors (Mei5bio) for at least 20 min on ice, followed by sonication (30 s on and 30 s off, four cycles) and centrifuged at the maximum speed. The whole cell lysate was incubated with StrepTactin Beads 4FF (Smart-Lifesciences) overnight at 4 °C. Next, the beads were washed with IP lysis buffer and boiled with 1× SDS loading buffer (Beyotime Biotechnology). Boiled samples were analysed by SDS–PAGE, the protein was transferred to a PVDF membrane (ABclonal Biotechnology) and incubated with StrepII antibody (1:1,000, HUABIO) or FLAG antibody (1:15,000, Sigma-Aldrich). The washed membrane was incubated with HRP-linked secondary antibody (1:5,000, Cell Signaling Technology). The washed membrane was incubated with ECL western blotting substrate (EpiZyme or Vazyme) to the dark for exposure.
For IP in fly heads, the procedure follows that of ref. 56. The Drosophila line Per::GFP38 was provided by Y. Zhang’s lab. In brief, the heads of flies entrained for 1–5 days were collected by freezing at ZT16 and total head extracts were prepared by fly lysis buffer. These lysates were then incubated with 8 μl anti-GFP antibody (1:100, Abcam) overnight in 4 °C before incubation with 50 μl protein A + G agarose (Thermo Scientific) for 2 h at room temperature, and the beads were washed with fly lysis buffer or plant lysis buffer and boiled with 1× SDS loading buffer. The boiled samples were analysed by mass spectrometry.
Mass spectrometry and data analyses
Boiled samples were separated by electrophoresis on a 4% tacking gel and 12% resolving protein gel. For protein identification, protein bands on the SDS–PAGE gel were destained and then reduced in 10 mM DTT at 56 °C for 40 min followed by alkylation in 55 mM iodoacetamide in the dark for 1 h. After that, the protein bands were in-gel digested with sequencing grade trypsin (10 ng μl−1 trypsin, 50 mM ammonium bicarbonate, pH 8.0) overnight at 37 °C. Peptides were extracted with 5% formic acid/50% acetonitrile and 0.1% formic acid/75% acetonitrile sequentially and then concentrated to a volume of 20 μl. The extracted peptides were separated by an analytical capillary column (100 μm × 15 cm) packed with 1.9-μm spherical C18 reversed-phase material (Dr. Maisch, Germany) and a pre-column (100 μm x 2 cm) packed with 3-μm spherical C18 reversed-phase material (Dr. Maisch, GmbH, Germany). A Thermo EASY-nLC 1200 ultra-high performance liquid chromatography (UHPLC) system was used to generate the following HPLC gradient: 2–8% B in 10 min, 8–38% B in 30 min, 38–100% B in 15 min, 100–100% B in 15 min (A was 0.1% formic acid in water and B was 0.1% formic acid in 80% acetonitrile). The eluted peptides were sprayed into a Q Exactive mass spectrometer (ThermoFisher Scientific) equipped with a nano-ESI ion source. The mass spectrometer was operated in data-dependent mode with one MS scan followed by ten high-energy collisional dissociation MS/MS scans for each cycle.
For differential analysis of data from D. melanogaster, proteins were searched from raw mass spectrometer files using MaxQuant (v.1.5.1.0)57 with the drosophila fasta database from UniProt. Subsequently, the output data from MaxQuant were imported to ProVision for further analysis. The LFQ values were then filtered for subsequent analysis with a minimum requirement of two unique peptides. After this, log transformation was applied to the LFQ values, and any missing values were imputed from a normal distribution (width, 0.3; down shift, 1.8). The differential analysis was done using the limma package (v.3.54.2)58, and the resulting volcano plots were created in R using ggplot2 (v.3.4.2).
siRNA assay
U2OS cells with the Per2-dLuc reporter were seeded at 8 × 105 cells in 35-mm dishes. Cells were transfected with siRNAs (0.25 pmol or 0.016 pmol) using RNAiMAX transfection reagent (Invitrogen 13778150) following the manufacturer’s instructions. Two days after transfection, the medium was changed to XM medium and the dishes were covered with sterile glass coverslips, sealed with sterile vacuum grease, and placed into the Lumicycle luminometer (Actimetrics) and luminescence levels were measured. RNA was extracted 90 h after transfection for gene expression analysis in parallel experiments. The siRNA sequence of CRY1 was AAGGAATGGAACATTACTAAA and the siRNA sequence of CRY2 was AAGGACGACGGTGGCCAACTA.
Protein expression and purification
The RUVBL1 and RUVBL2 proteins from various species were expressed using Escherichia coli Mach1T1 and BL21(DE3)-competent cells (Beijing Biomed Gene technology) as hosts for plasmid construction and SUMO-6×His-HRV 3C site fusion-protein expression, respectively. The N-terminal SUMO-6×His-HRV 3C site fusion RUVBL2 CDS with a C-terminal Strep tag II was cloned into the pET21 vector, and RUVBL1 CDS with a C-terminal 6×His tag was cloned into the pET28 vector and then co-introduced to E. coli BL21 (DE3). BL21 (DE3) transformed with pET vectors was grown in 1 l of 2 × YT medium containing antibiotics on a rotary shaker (240 rpm) at 37 °C. When the cells reached the mid-exponential phase (an optical density, OD600 nm of around 0.8), induction of protein expression was done with the addition of 0.5 mM IPTG (AMRESCO) and incubated at 24 °C with shaking overnight. Cells were collected by centrifugation at 7,000g. Cell pellets were frozen at −80 °C.
Thawed cell pellets were resuspended in basal buffer (30 mM HEPES buffer containing 150 mM NaCl, 5 mM MgCl2, 1 mM ATP, 5 mM 2-mercaptoethanol, 10 µg ml−1 kanamycin, 10% glycerol, pH 7.5) and lysed by adding 1 mg ml−1 lysozyme (VWR Life Science),1% NP40 and 3 μg ml−1 benzonase nuclease (Smart-Lifesciences). The lysates were centrifuged at 60,000g and filtered through a 0.45-μm PES filter. The supernatant was loaded into prepacked Ni-NTA beads (Smart-Lifesciences) and washed with basal buffer containing 30 mM imidazole. Protein was eluted with Ni-elution buffer (basal buffer with 500 mM imidazole, pH 7.5) and SUMO-6×His tag was cleaved by HRV 3C protease (Sino Biological). Next, the protein was loaded into prepacked StrepTactin beads (Smart-Lifesciences) and washed with basal buffer. RUVBL1/RUVBL2 was eluted with Strep-elution buffer (basal buffer with 30 mM biotin, pH 7.5). The eluted sample was concentrated by an ultrafiltration tube with a 30 kDa aperture at 4 °C and then loaded in a Superdex 200 gel-filtration column (GE Healthcare) with basal buffer; that around a 350 kDa UV peak was collected. After measuring the protein concentration using Bradford reagent (Solarbio), the protein was quickly frozen in liquid nitrogen.
For the KaiA, KaiB and KaiC proteins, overexpression and purification were done as previously described59 using the same 6×His-SUMO-tagged Synechococcus elongatus constructs.
ATPase assay
Human RUVBL1/RUVBL2 protein or KaiC protein (1 μM) was diluted in ATPase buffer (50 mM Tris, 50 mM NaCl, 2 mM MgCl2, 2 mM 2-mercaptoethanol, 1 mM ATP, 10 µg ml−1 kanamycin, pH 7.5) and incubated at 37 °C (RUVBL1/RUVBL2) or 30 °C (KaiC). Samples were collected every 2 h. For each collection, the reaction was stopped by adding five-fold 2 mM EDTA and freezing at −20 °C. The ADP production rate was determined by UHPLC. To test the effect of CordyTP, 0.5 mM of ATP or CordyTP was added to the ATPase buffer. To test the effect of CB-6644, varying concentrations of CB-6644 or DMSO were added in the ATPase buffer. To assess the activity of KaiC ATPase mutants, 25% of KaiC (0.25 μM) in the reaction was replaced with KaiC mutants, the rest being wild-type KaiC (0.75 μM).
LC with a diode array detector
The UHPLC analysis was done using an Agilent 1290 Infinity UHPLC system (Agilent Technologie) equipped with a diode array detector (Agilent G1315D, Agilent Technologies). Separation was achieved using an Agilent Poroshell 120 EC-C18 column (150 mm x 3 mm, 2.7 μm) at a column temperature of 25 °C under isocratic conditions. The mobile phase was potassium phosphate buffer (0.05 M, pH 6.8). The flow rate was maintained at 0.6 ml min−1 and the run time for each injection was 6 min. The injection volume was 5 μl. The detection wavelength was 254 nm. Data processing was done using Agilent Mass Hunter B.07.00 software.
IVO reaction set-up
For KaiA, KaiB and KaiC used in IVO reactions, KaiB was labelled with 5-carboxyfluorescein (5-FAM) as previously described36. The standard IVO reactions contained KaiA (1.2 μM), KaiB (3.45 μM), 5-FAM KaiB (0.05 μM) and KaiC (3.5 μM) in clock reaction buffer (20 mM Tris pH 8.0, 150 mM NaCl, 5 mM MgCl2, 0.5 mM EDTA and 1 mM ATP). To monitor the IVO reactions, 20-μl reactions were dispensed in a 384-well plate before collecting fluorescence anisotropy data at 30 °C, as previously reported36. To test the effect of CordyTP on the IVO reactions, 0.5 mM ATP or CordyTP was added to the clock reaction buffer. To assess the effect of KaiC ATPase mutants on the IVO reactions, 25% of the KaiC (0.88 μM) in the reaction was replaced by KaiC mutants, with the rest being wild-type KaiC (2.62 μM). A standard IVO reaction with 3.5 μM wild-type KaiC was included as a control (to mimic the heterozygous mutant condition in mammalian cells, in which two copies of RUVBL1 and RUVBL2 exist but only one allele of RUVBL2 is the mutant).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Source data are provided with this paper.
Change history
19 June 2025
A Correction to this paper has been published: https://doi.org/10.1038/s41586-025-09237-y
References
Zhang, E. E. & Kay, S. A. Clocks not winding down: unravelling circadian networks. Nat. Rev. Mol. Cell Biol. 11, 764–776 (2010).
Rosbash, M. The implications of multiple circadian clock origins. PLoS Biol. 7, e1000062 (2009).
Hardin, P. E., Hall, J. C. & Rosbash, M. Feedback of the Drosophila period gene product on circadian cycling of its messenger RNA levels. Nature 343, 536–540 (1990).
Takahashi, J. S. Transcriptional architecture of the mammalian circadian clock. Nat. Rev. Genet. 18, 164–179 (2017).
Johnson, C. H., Stewart, P. L. & Egli, M. The cyanobacterial circadian system: from biophysics to bioevolution. Annu. Rev. Biophys. 40, 143–167 (2011).
Lane, N. & Martin, W. The energetics of genome complexity. Nature 467, 929–934 (2010).
Cheng, Y., Chi, Y., Sun, L. & Wang, G.-Z. Dominant constraints on the evolution of rhythmic gene expression. Comput. Struct. Biotechnol. J. 21, 4301–4311 (2023).
Wagner, A. Energy constraints on the evolution of gene expression. Mol. Biol. Evol. 22, 1365–1374 (2005).
Elowitz, M. B. & Leibler, S. A synthetic oscillatory network of transcriptional regulators. Nature 403, 335–338 (2000).
Fung, E. et al. A synthetic gene–metabolic oscillator. Nature 435, 118–122 (2005).
Tigges, M., Marquez-Lago, T. T., Stelling, J. & Fussenegger, M. A tunable synthetic mammalian oscillator. Nature 457, 309–312 (2009).
Nakajima, M. et al. Reconstitution of circadian oscillation of cyanobacterial KaiC phosphorylation in vitro. Science 308, 414–415 (2005).
Terauchi, K. et al. ATPase activity of KaiC determines the basic timing for circadian clock of cyanobacteria. Proc. Natl Acad. Sci. USA 104, 16377–16381 (2007).
Abe, J. et al. Atomic-scale origins of slowness in the cyanobacterial circadian clock. Science 349, 312–316 (2015).
Pitsawong, W. et al. From primordial clocks to circadian oscillators. Nature 616, 183–189 (2023).
Ju, D. et al. Chemical perturbations reveal that RUVBL2 regulates the circadian phase in mammals. Sci. Transl. Med. 12, eaba0769 (2020).
Takahashi, J. S., Shimomura, K. & Kumar, V. Searching for genes underlying behavior: lessons from circadian rhythms. Science 322, 909–912 (2008).
Michel, B. C. et al. A non-canonical SWI/SNF complex is a synthetic lethal target in cancers driven by BAF complex perturbation. Nat. Cell Biol. 20, 1410–1420 (2018).
Zhang, E. E. et al. A genome-wide RNAi screen for modifiers of the circadian clock in human cells. Cell 139, 199–210 (2009).
Xu, Y. et al. Modeling of a human circadian mutation yields insights into clock regulation by PER2. Cell 128, 59–70 (2007).
Hirota, T. et al. A chemical biology approach reveals period shortening of the mammalian circadian clock by specific inhibition of GSK-3β. Proc. Natl Acad. Sci. USA 105, 20746–20751 (2008).
Konopka, R. J. & Benzer, S. Clock Mutants of Drosophila melanogaster. Proc. Natl Acad. Sci. USA 68, 2112–2116 (1971).
Price, J. L. et al. double-time is a novel Drosophila clock gene that regulates PERIOD protein accumulation. Cell 94, 83–95 (1998).
Sehgal, A., Price, J. L., Man, B. & Young, M. W. Loss of circadian behavioral rhythms and per RNA oscillations in the Drosophila mutant timeless. Science 263, 1603–1606 (1994).
Vitaterna, M. H. et al. Mutagenesis and mapping of a mouse gene, Clock, essential for circadian behavior. Science 264, 719–725 (1994).
Dauden, M. I., López-Perrote, A. & Llorca, O. RUVBL1–RUVBL2 AAA-ATPase: a versatile scaffold for multiple complexes and functions. Curr. Opin. Struct. Biol. 67, 78–85 (2021).
Li, Y. et al. Epigenetic inheritance of circadian period in clonal cells. eLife 9, e54186 (2020).
Cermakian, N. & Sassone-Corsi, P. Multilevel regulation of the circadian clock. Nat. Rev. Mol. Cell Biol. 1, 59–67 (2000).
Yao, Z. & Shafer, O. T. The Drosophila circadian clock is a variably coupled network of multiple peptidergic units. Science 343, 1516–1520 (2014).
Liu, N. et al. A highland-adaptation mutation of the Epas1 protein increases its stability and disrupts the circadian clock in the plateau pika. Cell Rep. 39, 110816 (2022).
Arnold, C. N. et al. A forward genetic screen reveals roles for Nfkbid, Zeb1, and Ruvbl2 in humoral immunity. Proc. Natl Acad. Sci. USA 109, 12286–12293 (2012).
Wang, G. et al. Somatic genetics analysis of sleep in adult mice. J. Neurosci. 42, 5617–5640 (2022).
Swan, J. A. et al. Coupling of distant ATPase domains in the circadian clock protein KaiC. Nat. Struct. Mol. Biol. 29, 759–766 (2022).
Assimon, V. A. et al. CB-6644 is a selective inhibitor of the RUVBL1/2 complex with anticancer activity. ACS Chem. Biol. 14, 236–244 (2019).
Erzberger, J. P. & Berger, J. M. Evolutionary relationships and structural mechanisms of AAA+ proteins. Annu. Rev. Biophys. Biomol. Struct. 35, 93–114 (2006).
Chavan, A. G. et al. Reconstitution of an intact clock reveals mechanisms of circadian timekeeping. Science 374, eabd4453 (2021).
Baggs, J. E. et al. Network features of the mammalian circadian clock. PLoS Biol. 7, e1000052 (2009).
Chen, W., Werdann, M. & Zhang, Y. The auxin-inducible degradation system enables conditional PERIOD protein depletion in the nervous system of Drosophila melanogaster. FEBS J. 285, 4378–4393 (2018).
Hirota, T. et al. Identification of small molecule activators of cryptochrome. Science 337, 1094–1097 (2012).
Emery, P., So, W. V., Kaneko, M., Hall, J. C. & Rosbash, M. CRY, a Drosophila clock and light-regulated cryptochrome, is a major contributor to circadian rhythm resetting and photosensitivity. Cell 95, 669–679 (1998).
Park, J. H. et al. Differential regulation of circadian pacemaker output by separate clock genes in Drosophila. Proc. Natl Acad. Sci. USA 97, 3608–3613 (2000).
Aronson, B. D., Johnson, K. A., Loros, J. J. & Dunlap, J. C. Negative feedback defining a circadian clock: autoregulation of the clock gene frequency. Science 263, 1578–1584 (1994).
Dunlap, J. C. Molecular bases for circadian clocks. Cell 96, 271–290 (1999).
Sehgal, A. et al. Rhythmic expression of timeless: a basis for promoting circadian cycles in period gene autoregulation. Science 270, 808–810 (1995).
Ito-Miwa, K., Terauchi, K. & Kondo, T. in Circadian Rhythms in Bacteria and Microbiomes (eds Johnson, C. H. & Rust, M. J.) 79–91 (Springer, 2021).
Zhou, Y. et al. High-throughput screening of a CRISPR/Cas9 library for functional genomics in human cells. Nature 509, 487–491 (2014).
Colot, H. V. et al. A high-throughput gene knockout procedure for Neurospora reveals functions for multiple transcription factors. Proc. Natl Acad. Sci. USA 103, 10352–10357 (2006).
Schindelin, J. et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682 (2012).
Savelyev, S. A., Larsson, K. C., Johansson, A. S. & Lundkvist, G. B. Slice preparation, organotypic tissue culturing and luciferase recording of clock gene activity in the suprachiasmatic nucleus. J. Vis. Exp. 10.3791/2439 (2011).
Zhou, M. et al. Non-optimal codon usage affects expression, structure and function of clock protein FRQ. Nature 495, 111–115 (2013).
Larrondo, L. F., Olivares-Yañez, C., Baker, C. L., Loros, J. J. & Dunlap, J. C. Circadian rhythms. Decoupling circadian clock protein turnover from circadian period determination. Science 347, 1257277 (2015).
Yuan, L. et al. BBX19 fine-tunes the circadian rhythm by interacting with PSEUDO-RESPONSE REGULATOR proteins to facilitate their repressive effect on morning-phased clock genes. Plant Cell 33, 2602–2617 (2021).
Chiu, J. C., Low, K. H., Pike, D. H., Yildirim, E. & Edery, I. Assaying locomotor activity to study circadian rhythms and sleep parameters in Drosophila. J. Vis. Exp. 10.3791/2157 (2010).
Schmid, B., Helfrich-Förster, C. & Yoshii, T. A new ImageJ plug-in “ActogramJ” for chronobiological analyses. J. Biol. Rhythms 26, 464–467 (2011).
Tiscornia, G., Singer, O. & Verma, I. M. Production and purification of lentiviral vectors. Nat. Protoc. 1, 241–245 (2006).
Fu, J. et al. Codon usage affects the structure and function of the Drosophila circadian clock protein PERIOD. Genes Dev 30, 1761–1775 (2016).
Tyanova, S., Temu, T. & Cox, J. The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat. Protoc. 11, 2301–2319 (2016).
Ritchie, M. E. et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47 (2015).
Fang, M., Chavan, A. G., LiWang, A. & Golden, S. S. Synchronization of the circadian clock to the environment tracked in real time. Proc. Natl Acad. Sci. USA 120, e2221453120 (2023).
Acknowledgements
We thank J. Takahashi, M. Rosbash, S. Kay, J. Hogenesch, Y. Xu, A. Kramer, M. Brunner and members of the Neuroscience Pioneer Club (NPC) for discussions; S. Akiyamay for reanalysis of the RUVBL1/RUVBL2 crystal structure data; G. Mahesh for preliminary analysis of Pontin/Reptin RNAi expression in clock cells; X. Tian for technical support for experiments using Neurospora; and Y. Zhang, L. Zhang, D. Ma and X. Liu for reagents and database information; and J. Chai for providing the original illustration materials drawn using BioRender software. This research was supported by funds from the National Key R&D Program of China (2024YFA1803200), STI2030-Major Project (2021ZD0203400), the Beijing Municipal Government and Tsinghua University to E.E.Z.; and from the US National Institute of General Medical Sciences of the National Institutes of Health to S.S.G. (R35GM118290) and P.E.H. (R01GM124617).
Author information
Authors and Affiliations
Contributions
M. Liao., Y.L., Z.X. and E.E.Z. conceptualized the study. M. Liao., Y.L. and Z.X. did most of the experiments, assisted by Z.Y., Y.C., Z.S., R.H., X.S. and D.J., and received guidance from E.E.Z. M.F. did the IVO experiments under the supervision of S.S.G. N.T. did the fruitfly behavioural analyses using Pontin/Reptin RNAi expression lines under the supervision of P.E.H. J.Y. managed fruitfly crossing under the supervision of T.W. F.H. did the fungi-related work under the guidance of Q.H. M. Liu. did the plant-related work under the supervision of X.X. Q.Z. did the cyanobacteria-related work under the guidance of X.Q. H.H. did the proteomic analysis under the supervision of S.C. X.S. did the metabolomic analysis. M. Liao., Y.L., Z.X. and E.E.Z. wrote the manuscript, with editing support from M.F., S.S.G. and P.E.H.
Corresponding author
Ethics declarations
Competing interests
E.E.Z. and D.J. have applied for a patent associated with this study (WIPO publication WO2018133835A1, Nucleoside analog regulating mammalian circadian rhythm).
Peer review
Peer review information
Nature thanks Joanna Chiu, Carrie Partch and Bin Wang for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 RUVBL2, with low ATPase activity, is conserved in eukaryotes.
a, Left panel: phylogenetic tree schematic illustrating the evolution of RUVBL proteins. Evolutionary analyses were conducted in MEGA11. Right panel: reference name for homologous genes and proteins. b, Protein sequence alignment of RUVBL proteins. c, A list of literature revealing the interactions between core clock proteins with RUVBL proteins. d, UHPLC determination of ATP hydrolysis activities of human RUVBL2 and cyanobacterial KaiC proteins. These activities are corresponding to consuming 12.6 (RUVBL2) and 12.9 (KaiC) ATP molecules per day per ATPase molecule, respectively. Two-tailed t-test differential analysis. n = 3 biologically independent proteins for each group. All bar graphs show mean ± s.e.m. Illustration in a created using BioRender (credit: Juan, C. https://BioRender.com/n10u764; 2024).
Extended Data Fig. 2 A genetic screen of RUVBL2 in U2OS cells.
a, Normalized luminescence expression in Per2-dLuc U2OS cells with or without stable Cas9 expression, n = 3 biologically independent cells. b, The effects of siPER2 and siACTB on Per2-dLuc U2OS cells. Data were reanalyzed from Zhang, E.E. et al.19 Cell. c, Compilation of mutation information for represented RUVBL2 and PER2 mutants in Fig. 1f. d, Compilation of in-frame (left) and frame-shift (right) mutation information for RUVBL2 mutants in secondary validation. e, The procedure of validation across entire screen. All period data parameters were analyzed from LumiCycle Analysis. All graphs show mean ± s.e.m.
Extended Data Fig. 3 RUVBL2 is essential for circadian rhythm of locomotor activity and can be stimulated by external cues.
a, Histograms of circadian periods of SCN-specific transgenic mice. n = 7, 8, 9, 11 biologically independent mice for AAV-WT, Δ151-152, Δ86-87/Δ350-352/Δ83 and GFP. b, Schematic illustration of generating the SCN-specific knockout mice. c, RUVBL2 expression in the SCN. d, Histogram of RUVBL2-positive cells in the SCN. n = 7 and 13 biologically independent mice for control and treat group, position where dense nuclei appeared was defined as 0 μm. e, Periodgram of SCN-specific Ruvbl2 knockout mice. n = 10 and 12 biologically independent mice for control and treat group. f, Left panel: histograms of circadian periods in transgenic mice. Right panel: actograms of mice and the corresponding periodogram. Gray line represents Ruvbl2WT introduction in the SCN. n = 9 mice per group. g, The mRNA expression levels of Ruvbl2 in liver and muscle. Three technical replicates from a mixed sample of 5 mice. h, Statistical analysis of Per2 and Ruvbl2 expression in mouse liver, and blood glucose, G6P as a positive control in fasting-refed experiments. Three technical replicates from a mixed sample of three mice for gene expression experiment. n = 9 (ZT12), 6 (ZT15), 3 (ZT17) biologically independent mice for blood glucose. i, Expression of RUVBL2 and c-Fos in the SCN, light pulse stimulation at CT15, data were analyzed from 5 mice per group. All period data parameters were analyzed from ClockLab Analysis. Two-way ANOVA differential analysis with Dunnett’s multiple comparisons test (d). One-way ANOVA differential analysis with Šídák’s multiple comparisons test (f). Two-tailed multiple unpaired t-test differential analysis with Two-stage step-up (Benjamini, Krieger, and Yekutieli) method (h). Two-tailed t-test differential analysis (i). All graphs show mean ± s.e.m. Illustration in a created using BioRender (credit: Juan, C. https://BioRender.com/n10u764; 2024).
Extended Data Fig. 4 ATP hydrolysis activity of RUVBL and KaiC proteins governs the clock period.
a, SDS-PAGE image of human RUVBL1/2 and cyanobacterial KaiC proteins. For gel source data, see Supplementary Fig. 1b. b, Alignment of the secondary structures of human RUVBL1/2 and cyanobacterial KaiC proteins. c, Eukaryotic RUVBL1/2 (in the HslU-ClpAB/CTD-LonAB-RuvB clade) and cyanobacterial KaiC (RecA homolog) genes belong to the P-loop NTPase superfamily (Figure is adapted from Erzberger et al Ann Rev Biophys Biomol Struct 2006). d, The structures of cordycepin triphosphate and adenosine triphosphate. e, Dose effects of cordycepin on the luminescence rhythms in Per2-dLuc U2OS cells in the presence of pentostatin, which prevents the rapid degradation of cordycepin in the human cells. n = 3 biologically independent cells. One-way ANOVA differential analysis with Šídák’s multiple comparisons test. All period data parameters were analyzed from Lumicycle Analysis. All bar graphs show mean ± s.e.m. Credit: c adapted with permission from ref. 35, Annual Reviews.
Extended Data Fig. 5 The circadian period of MAF cells were regulated by RUVBL2 in a CRY-independent manner.
a, The Ruvbl2Q78K/WT MAF cells were constructed by CRISPR-Cas9 method. b, c, Luminescence rhythm of Ruvbl2WT/WT and Ruvbl2Q78K/WT MAF cells. n = 3 biologically independent cells. d, Validation of CRY1 knock out efficiency and Cry2 knock out efficiency with western blot represents 3 independent experiments and Sanger sequencing results. Gel source data, see Supplementary Fig. 1c. e, Luminescence rhythm and period of WT MAF cells with Cry1 (left panel) or Cry2 (right panel) knock out. n = 3 biologically independent cells. f, Validation of CRY1 knock out efficiency (left panel) and Cry2 knock out efficiency (right panel) of Ruvbl2Q78K/WT MAF cells with western blot represents 3 independent experiments and Sanger sequencing. g, Luminescence rhythm and period of Ruvbl2Q78K/WT MAF cells with Cry1 or Cry2 knock out. n = 3 biologically independent cells. All period parameters were analyzed by LumiCycle Analysis. Two-tailed t-test differential analysis (c). One-way ANOVA differential analysis with Šídák’s multiple comparisons test (e, g). All graphs show mean ± s.e.m.
Extended Data Fig. 6 RUVBL2 interacts core clock proteins in eukaryotes.
Western blot analyses of co-immunoprecipitation (co-IP) of clock proteins from different species which represented 3 independent experiments, antibodies for co-IP and WB analyses are indicated. a, Co-IP by STREP. b, Co-IP by FLAG. For gel source data, see Supplementary Fig. 1d and e. Illustration in a created using BioRender (credit: Juan, C. https://BioRender.com/n10u764; 2024).
Extended Data Fig. 7 Inhibiting the ATPase activity of RUVBL2 with compound lengthens the circadian periods in multiple species.
a, The structure of CB-6644. b, The inhibitory effects of CB-6644 on the ATPase activity of human RUVBL1/2. c, Normalized luminescence rhythm of Bmal1-dLuc U2OS cells shows the period lengthening for 2 h in the presence of CB-6644 (3 μM), n = 3 biologically independent cells. d, Left panel: statistical analysis of the period length changes of Drosophila melanogaster. Right panel: represents of double-plotted averaged actogram of Drosophila melanogaster with CB-6644 or control treatment. n = 24 (DMSO), 22 (10 μM), 21 (25 μM), 23 (50 μM) biologically independent files. e, Left panel: statistical analysis of the period length of Neurospora crassa with CB-6644 treatment. Right panel: normalized luminescence rhythm of Neurospora crassa treated by CB-6644. n = 4 biologically independent fungi. f, Left panel: statistical analysis of the period length of Arabidopsis thaliana with CB-6644 treatment. Right panel: normalized luminescence rhythm of Arabidopsis thaliana treated by CB-6644, cordycepin, and adenosine. n = 3 (100 μM), 4 (DMSO/10/50 μM) biologically independent plants. The Drosophila melanogaster locomotor activity was analyzed by ClockLab Analysis. The period parameter of Neurospora crassa was analyzed from LumiCycle Analysis. The period parameter of Arabidopsis thaliana was analyzed from MetaCycle. One-way ANOVA differential analysis with Šídák’s multiple comparisons test (d, e, f). All bar graphs show mean ± s.e.m.
Extended Data Fig. 8 Reptin and Pontin knockdown in flies lengthens the circadian period or disrupts rhythmicity.
a, Quantification of percent rhythmicity for Pontin and Reptin RNAi knockdown flies. Black bar represents the % of rhythmic individuals and the grey bar represents the % of arrhythmic individuals in each genotype. b, Period of Drosophila lines with Pontin or Reptin RNAi expressed in all clock cells (crossed with tim-GAL4 driver flies), in Pigment Dispersing Factor (PDF) expressing brain neurons (crossed with pdf-GAL4 driver flies), or not expressed (crossed with w1118 flies). w1118 flies as control. n = 8, 16, 31, 14 for w1118, UAS-siRept#1;w1118, UAS-siRept#1;tim-GAL4 and UAS-siRept#1;pdf-GAL4. n = 11, 20, 31, 25 for w1118, UAS-siRept#2;w1118, UAS-siRept#2;tim-GAL4 and UAS-siRept#2;pdf-GAL4. n = 8, 11, 32, 9 for w1118, UAS-siPont#1;w1118, UAS-siPont#1;tim-GAL4 and UAS-siPont#1;pdf-GAL4. n = 11, 15, 15, 14 for w1118, UAS-siPont#2;w1118, UAS-siPont#2;tim-GAL4 and UAS-siPont#2;pdf-GAL4. In certain genotypes, the free-running period was calculated separately for the initial 4 days of DD (1-4 DD) and the last three days (5–7 DD) to detect arrhythmicity that was prominent after day four. The Drosophila melanogaster locomotor activity was analyzed by ClockLab Analysis. The data show mean ± s.e.m.
Extended Data Fig. 9 The rvb2 knock-in mutant Neurospora lost growth rhythm under constant-dark conditions.
a, The schematic illustration of introducing mutation on rvb2 (the RUVBL2 homolog) in Neurospora. b, Growth rhythm detection of rvb2S79I and rvb2WT knock-in mutant strains with race tube assays, and rvb2WT as a control. Two clones of rvb2S79I were presented. There are three conditions (upper: DD conditions, middle: light 12 h: dark 12 h conditions, and lower: 25 °C 12 h: 30 °C 12 h temperature cycles conditions) which were used to evaluate the impact of rvb2K79I on core clock of Neurospora. n is labeled next to it. n = 3 biologically independent fungi. c, The ATPase activity of RUVB1WT/RUVB2WT and RUVB1WT/ RUVB2S79I. n = 3 biologically independent proteins for each group. Two-tailed t-test differential analysis. All bar graphs show mean ± s.e.m.
Supplementary information
Supplementary Fig. 1
Uncropped images for gels and blots. Original scan images are shown for the indicated figure panels, and the red boxes represent the cropped areas. Molecular weight markers and antibodies used are indicated.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Liao, M., Liu, Y., Xu, Z. et al. The P-loop NTPase RUVBL2 is a conserved clock component across eukaryotes. Nature 642, 165–173 (2025). https://doi.org/10.1038/s41586-025-08797-3
Received:
Accepted:
Published:
Issue date:
DOI: https://doi.org/10.1038/s41586-025-08797-3
This article is cited by
-
The cyanobacterial circadian clock
npj Biological Timing and Sleep (2025)
-
Site- and cell-type-specific miRNA and mRNA genes and networks across the cortex, striatum, and hypothalamus
Communications Biology (2025)
