
Abstract
Brown and beige adipocytes express uncoupling protein 1 (UCP1), a mitochondrial protein that dissociates respiration from ATP synthesis and promotes heat production and energy expenditure. However, UCP1−/− mice are not obese1,2,3,4,5, consistent with the existence of alternative mechanisms of thermogenesis6,7,8. Here we describe a UCP1-independent mechanism of thermogenesis involving ATP-consuming metabolism of monomethyl branched-chain fatty acids (mmBCFA) in peroxisomes. These fatty acids are synthesized by fatty acid synthase using precursors derived from catabolism of branched-chain amino acids9 and our results indicate that β-oxidation of mmBCFAs is mediated by the peroxisomal protein acyl-CoA oxidase 2 (ACOX2). Notably, cold exposure upregulated proteins involved in both biosynthesis and β-oxidation of mmBCFA in thermogenic fat. Acute thermogenic stimuli promoted translocation of fatty acid synthase to peroxisomes. Brown-adipose-tissue-specific fatty acid synthase knockout decreased cold tolerance. Adipose-specific ACOX2 knockout also impaired cold tolerance and promoted diet-induced obesity and insulin resistance. Conversely, ACOX2 overexpression in adipose tissue enhanced thermogenesis independently of UCP1 and improved metabolic homeostasis. Using a peroxisome-localized temperature sensor named Pexo-TEMP, we found that ACOX2-mediated fatty acid β-oxidation raised intracellular temperature in brown adipocytes. These results identify a previously unrecognized role for peroxisomes in adipose tissue thermogenesis characterized by an mmBCFA synthesis and catabolism cycle.
Similar content being viewed by others
Main
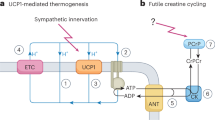
Emerging studies implicate peroxisomes in regulating metabolic homeostasis10. Peroxisomes perform various metabolic functions, including β-oxidation of very long-chain fatty acids and 2-methyl branched-chain fatty acids (BCFA), α-oxidation of phytanic acid and synthesis of ether lipids and bile acids11. These organelles are abundant in thermogenic fat12 and further increase in number with cold exposure in a manner dependent on the thermogenic transcriptional coregulator PRDM16 (ref. 13). Peroxisomes regulate mitochondrial fission, which is required for adipose tissue thermogenesis13,14. However, it is unknown whether peroxisomes directly regulate thermogenesis. Notably, peroxisomes account for up to 20% of total cellular oxygen consumption under basal conditions15. Unlike mitochondrial respiration, peroxisomal oxygen consumption does not lead to ATP synthesis and instead generates heat.
Here, we report that peroxisomes are involved in an alternative mechanism of adipose tissue thermogenesis characterized by synthesis and β-oxidation of monomethyl branched fatty acids (mmBCFAs). These fatty acids are synthesized by fatty acid synthase (FASN) using a short-branched-chain acyl-coenzyme A (CoA) (BrCoA) derived from catabolism of branched-chain amino acids (BCAA) as a precursor9,16. Although mmBCFA are less abundant than conventional straight chain fatty acids (StCFA), brown adipose tissue (BAT) and white adipose tissue (WAT) have the highest mmBCFA concentrations among tissues9. Their relatively low steady-state levels are consistent with a model in which they are rapidly synthesized and degraded to support energy dissipation. The peroxisomal protein acyl-CoA oxidase 2 (ACOX2) is reported to oxidize 2-methyl-branched fatty acids17, and here we show that ACOX2 also mediates β-oxidation of mmBCFAs. Moreover, our studies indicate that FASN-mediated synthesis of mmBCFA also takes place in peroxisomes. Both mmBCFA synthesis and β-oxidation are simultaneously activated by cold exposure. Through use of loss-of-function and gain-of-function mouse models, we demonstrate that this UCP1-independent pathway of thermogenesis is physiologically relevant.
Thermogenic stimuli boost ACOX2 levels
We analysed gene expression in BAT to examine how peroxisomal biogenesis and metabolic pathways are affected by cold exposure in WT and UCP1−/− mice housed at thermoneutrality (30 °C) or adapted to cold (4 °C). Cold exposure increased genes and proteins involved in peroxisomal biogenesis, ether lipid synthesis and β-oxidation (Fig. 1a,b). Among these, the peroxisomal β-oxidation gene ACOX2 showed a marked cold-induced upregulation, particularly in UCP1−/− mice (Fig. 1a,b), suggesting a potential compensatory role in thermogenic adaptation. ACOX2 catalyses the first and rate-limiting step in β-oxidation of 2-mBCFA (Extended Data Fig. 1a) and metabolizes intermediates in bile acid synthesis17,18. Whereas bile acid pathway genes such as AMACR and CYP27A1 decreased in cold-treated UCP1−/− mice, HSD17B4, a downstream enzyme in fatty acid oxidation, increased (Fig. 1a). Cold exposure also promoted ACOX2 expression in inguinal WAT (iWAT) and gonadal WAT but not in the liver (Extended Data Fig. 1b). Acute norepinephrine treatment in UCP1−/− brown adipocytes or subcutaneous WAT from pigs, a species that lacks functional UCP1 protein19, significantly increased ACOX2 expression (Fig. 1c). Similarly, forskolin treatment upregulated ACOX2 in human brown-like adipocytes (Fig. 1d), indicating that ACOX2 is regulated by cyclic AMP (cAMP)-mediated thermogenic signalling.

a, Heatmap of genes involved in various peroxisomal pathways in BAT of mice maintained at thermoneutrality (30 °C) or gradually cold adapted. n = 4. b, Western blot analysis in BAT of warm or cold-treated mice. n = 2. c, ACOX2 gene expression after 2 h of treatment with norepinephrine or vehicle in differentiated mouse UCP1−/− brown adipocytes (norepinephrine 1 μM) or mature adipocytes isolated from fresh pig iWAT (norepinephrine 2 μM). n = 3 biological replicates per group. d, ACOX2 gene expression in cultured human brown-like adipocytes treated with or without forskolin (10 μM, 3 days). n = 3 biological replicates per group. e, Western blot analysis in BAT SVF cells collected on various days during adipogenesis. f, Immunofluorescence of ACOX2 and PMP70 in differentiated WT BAT SVF cells. g, Western blot analysis of ACOX2 knockdown using CRISPR–Cas9 in brown adipocytes. h, Volcano plot depicting log2[fold change] (FC) values of fatty acids. n = 4. i, Quantification of various mmBCFA species. n = 4. j, Atom-transition map illustrating flow of carbons from [U13C6]glucose into newly synthesized mmBCFA. 13C carbons are indicated by closed circles. Created in Adobe Illustrator. k, Measurement of 13C-labelled iso-C15:0 in control and sgACOX2 brown adipocytes after CL316,243 treatment. n = 3. l, OCR in sgACOX2 and control brown adipocytes treated with oligomycin and iso-C17:0, followed by FCCP. n = 7. Data with error bars are reported as mean ± s.e.m. Data in a–d,h,i,k,l are from biologically independent samples. Images in e,f,g are representative of two separate experiments. Two-sided unpaired Student’s t-test in c, d and i. The data in h were adjusted for multiple comparisons using the Benjamini–Hochberg method. Two-way ANOVA with Fisher’s least-significant difference test in k and two-way ANOVA with Sidak’s multiple comparisons test in l. Scale bars, 10 μm.
ACOX2 promotes β-oxidation of mmBCFA
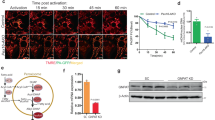
The physiological role of ACOX2 in adipocytes is unknown. Thus, we analysed its expression during adipogenesis. ACOX2 protein levels were low in undifferentiated BAT stromal vascular fraction (SVF) cells and markedly increased during adipogenesis (Fig. 1e). Immunofluorescence analysis showed that ACOX2 is localized in peroxisomes in brown adipocytes (Fig. 1f), consistent with its role in peroxisomal β-oxidation.
ACOX2 mediates β-oxidation of the 2-methyl BCFA acid pristanic acid17, which is obtained either directly from the diet or as the α-oxidation product of the dietary lipid phytanic acid. These fatty acids are not typically present in appreciable amounts in standard cell culture media or rodent diets, suggesting that ACOX2 might be involved in catabolism of other fatty acids in cell types, such as activated brown adipocytes, where it is highly expressed. To identify endogenous fatty acid substrates and intracellular functions of ACOX2, we used CRISPR–Cas9 to delete ACOX2 in brown adipocytes. Western blot analysis of pooled single-guide RNA-treated cells revealed a reduction in ACOX2 expression, although complete ablation was not achieved (Fig. 1g). ACOX2 inactivation had no effect on mitochondrial protein gene expression (Extended Data Fig. 1c) or adipocyte differentiation (Extended Data Fig. 1d) but selectively increased mmBCFA levels on mass spectrometry profiling of fatty acids (Fig. 1h). These fatty acids have a methyl group on the penultimate (iso configuration) or the antepenultimate (ante-iso configuration) carbon (Extended Data Fig. 1e). We validated the structures of major mmBCFA species, including iso-C15:0 and iso-C17:0, in brown adipocytes using tandem mass spectrometry. Other fatty acids with an odd number of carbon atoms, likely mmBCFA species or odd-chain fatty acids (OCFA), both derived from BCAA catabolism20, were also elevated with ACOX2 ablation (Fig. 1h,i). Notably, levels of various StCFA, including very long-chain fatty acids, were not significantly altered or were only minimally affected in ACOX2 knockdown cells (Extended Data Fig. 1f). To determine the cause of mmBCFA accumulation in single guide (sg)ACOX2-treated cells, we traced carbon flux from [U-13C6]-glucose into mmBCFA (Fig. 1j) following β3-adrenergic receptor agonist CL316,243 treatment. In control adipocytes, CL316,243 did not increase 13C incorporation into mmBCFA, indicating balanced synthesis and degradation. However, 13C incorporation into mmBCFA was significantly elevated in sgACOX2 cells (Fig. 1k), suggesting that the absence of ACOX2-mediated β-oxidation allows accumulation of newly synthesized mmBCFA. These results indicate that ACOX2 is essential for maintaining mmBCFA homeostasis by facilitating their β-oxidation.
To further investigate whether ACOX2 is required for mmBCFA β-oxidation and determine its relationship to mitochondrial respiration, we measured oxygen consumption rate (OCR) in control and sgACOX2 adipocytes treated with iso-C17:0 (Fig. 1l). In control cells, iso-C17:0 increased OCR even after oligomycin treatment, indicating that the respiratory induction by the exogenously supplied mmBCFA is independent of mitochondrial ATP synthesis. Subsequent carbonyl cyanide-p-trifluoromethoxyphenylhydrazone (FCCP) treatment did not further increase OCR, suggesting that iso-C17:0 β-oxidation fully engaged cellular respiratory capacity in the presence of ACOX2. By contrast, ACOX2 inactivation significantly decreased both basal and iso-C17:0-induced OCR, confirming that the peroxisomal protein is required for mmBCFA β-oxidation.
We next determined the effect of ACOX2 overexpression on fatty acid levels and β-oxidation. Unlike knockdown, overexpression of ACOX2 selectively decreased mmBCFA levels without significantly affecting other fatty acids (Extended Data Fig. 1g). To determine whether ACOX2 can oxidize mmBCFA and OCFA, we treated brown adipocytes overexpressing ACOX2 or green fluorescent protein (GFP) with iso-C17:0, C15:0 OCFA, C17:0 OCFA or vehicle and measured OCR. ACOX2 overexpression or iso-C17:0 treatment alone promoted oxygen consumption and the combination of these two manipulations markedly increased OCR. Whereas C15:0 OCFA increased OCR, although to a lesser extent than iso-C17:0, treatment with C17:0 OCFA reduced respiration, indicating potential mitochondrial toxicity (Extended Data Fig. 1h). Quantitative real-time PCR analysis showed that ACOX2 overexpression does not alter the expression of mitochondrial protein genes (Extended Data Fig. 1i) or mitochondrial DNA copy number (Extended Data Fig. 1j). Notably, iso-C17:0 treatment increased ACOX2 expression in mouse UCP1−/− brown adipocytes and pig white adipocytes (Extended Data Fig. 1k), indicating that ACOX2 expression is substrate regulated. Together, these findings identify mmBCFAs as endogenous substrates of ACOX2. It is noteworthy that branched short-chain fatty acids, such as isovaleric acid (C5) and isobutyric acid (C4), which serve as precursors for long-chain mmBCFAs (for example, iso-C17:0 and iso-C16:0), have been reported to be catabolized by mitochondrial β-oxidation21,22.
FASN promotes mmBCFA synthesis
Synthesis of mmBCFA requires FASN, a large multifunctional cytosolic enzyme primarily responsible for palmitate (C16:0) synthesis. Knockout of FASN decreases C16:0 levels but results in accumulation of the substrates acetyl-CoA and malonyl-CoA23. FASN mediates mmBCFA synthesis by using malonyl-CoA from glucose metabolism and BrCoA from BCAA catabolism (Extended Data Fig. 2a)9,20. Deletion of the BCAA catabolic enzyme branched-chain alpha-keto acid dehydrogenase (BCKDHA) in brown adipocytes (Extended Data Fig. 2b) significantly reduced mmBCFA levels (Extended Data Fig. 2c) and increased conventional StCFAs (Extended Data Fig. 2d).
Cold exposure markedly increased the expression of FASN in BAT and iWAT (Fig. 2a and Extended Data Fig. 2e), consistent with previous findings24. Other proteins involved in mmBCFA synthesis, including carnitine acetyltransferase (CRAT), lipogenesis enzymes ATP citrate lyase and acetyl-CoA carboxylase 1 (ACC1), and BCAA catabolism enzymes BCAT2 (branched-chain amino acid transaminase 2) and BCKDH were also significantly increased in BAT and iWAT of cold-treated mice (Fig. 2a and Extended Data Fig. 2e). In addition, treatment with norepinephrine in mouse brown adipocytes or forskolin in human brown adipocytes increased FASN expression (Extended Data Fig. 2f,g), indicating that FASN expression is regulated by cAMP signalling, akin to ACOX2. Thus, thermogenic stimuli not only increase the factors involved in β-oxidation, but also synthesis of mmBCFAs in thermogenic adipocytes.

a, mRNA levels of mmBCFA β-oxidation and synthesis genes in BAT of WT mice maintained at thermoneutrality (30 °C) or 4 °C for 7 days. n = 4. b, Immunofluorescence analysis and quantitative analysis of FASN peroxisomal localization in BAT of WT mice maintained at room temperature (RT) or after cold treatment for 2 h. n = 3. c, Western blot analysis following subcellular fractionation in differentiated BAT SVF cells after norepinephrine (0.2 μM) treatment for 2 h. d, Western blot analysis of FASN in BAT and iWAT of FASN-BKO mice. n = 3. e, Mass spectrometry analysis of a StCFA C16:0 and an mmBCFA iso-C17:0 in BAT of FASN-BKO and control mice housed at 22 °C or 4 °C for 6 h. n = 4. f, Core body temperature of control (n = 13) and FASN-BKO (n = 12) mice after cold exposure (4 °C). g, Energy expenditure of control (n = 4) and FASN-BKO (n = 6) mice after CL316,243 treatment. Data in a,b (right),c–g are from biologically independent samples. Images in b (left) and c are representative of two separate experiments. Data are presented as mean ± s.e.m. Statistical analyses were performed using two-sided unpaired Student’s t-test (a,b right), one-way ANOVA (e) or two-way ANOVA (f–g), followed by Fisher’s least-significant difference post hoc test (e–g). Scale bars, 10 μm.
Previous studies indicate that FASN interacts with peroxisomal fatty acid transporters, including PMP70 (ABCD3)25, which transports BCFAs and C27 bile acids into peroxisomes26, suggesting that the lipogenic enzyme is partially localized at the peroxisomal membrane. To investigate whether the intracellular localization of FASN is influenced by a thermogenic stimulus, we treated cultured brown adipocytes with norepinephrine for 2 h. Immunofluorescence analysis showed that norepinephrine treatment markedly increased the colocalization of FASN with PMP70 (Extended Data Fig. 2h). Similarly, FASN showed peri-lipid droplet localization in BAT of mice maintained at room temperature but had increased colocalization with PMP70 after acute cold exposure (Fig. 2b) or CL316,243 treatment (Extended Data Fig. 2i). Subcellular fractionation of adipocytes confirmed the partial peroxisomal localization of FASN, which was further increased after acute norepinephrine treatment (Fig. 2c). Together, these results indicate that FASN translocates to peroxisomes in response to thermogenic stimuli. In further support of peroxisomal localization, FASN interacts with PEX7 (Extended Data Fig. 2j), an import receptor for a subset of peroxisomal proteins27.
The mmBCFA synthesis protein CRAT is found in peroxisomes and mitochondria across various cell types, including brown adipocytes28,29,30,31. CRAT catalyses the interconversion of acyl-CoA and acylcarnitine and also acts on BrCoA32, a product of BCAA catabolism in mitochondria, to facilitate mmBCFA synthesis9. Norepinephrine treatment increased CRAT localization to peroxisomes in brown adipocytes (Extended Data Fig. 2k). CRISPR-mediated CRAT knockout (Extended Data Fig. 2l) did not affect adipogenesis (Extended Data Fig. 2m) but significantly decreased mmBCFA levels without altering StCFA levels (Extended Data Fig. 2n,o). These findings suggest that thermogenic stimuli promote the translocation of mmBCFA synthesis proteins to peroxisomes, an organelle that houses the mmBCFA β-oxidation protein ACOX2.
To evaluate the impact of inhibiting mmBCFA synthesis on respiration, we measured OCR in CRAT knockout and control brown adipocytes sequentially treated with leucine and norepinephrine. CRAT knockout significantly reduced OCR; notably, this effect was rescued by iso-C17:0 supplementation, indicating that the phenotype results from impaired mmBCFA synthesis (Extended Data Fig. 2p). To test whether ACOX2-mediated respiration involves coordination with CRAT, we overexpressed ACOX2 in CRAT KO cells. Gene expression analysis confirmed these genetic manipulations (Extended Data Fig. 2q). CRAT knockout reduced basal respiration and abolished the effect of ACOX2 overexpression on OCR in FCCP-treated cells (Extended Data Fig. 2r). These results indicate that ACOX2 coordinates with the mmBCFA synthetic machinery to enhance oxygen consumption.
FASN regulates thermogenesis
Previous studies in 3T3-L1 adipocytes indicate that FASN inactivation decreases the levels of StCFA as well as mmBCFA, but the data were confounded by the resulting inhibition of adipogenesis9. Thus, we isolated SVF cells from BAT of FASNLox/Lox neonatal mice, transduced them with retrovirus expressing tamoxifen-inducible Cre or a control virus, differentiated them into adipocytes, and then knocked out FASN using 4-hydroxytamoxifen (4-OHT) (Extended Data Fig. 3a). The inducible knockout of FASN in differentiated adipocytes decreased intracellular lipid content (Extended Data Fig. 3b) and significantly reduced the levels of mmBCFA (Extended Data Fig. 3c), as well as StCFA, such as C16:0 and C16:1 (Extended Data Fig. 3d).
We next investigated the effect of FASN ablation on mmBCFA levels and thermogenesis in mice. Previous studies, including ours, showed that pan-adipose FASN knockout induces iWAT browning33,34. To avoid this confounding effect, we generated BAT-specific FASN knockout (FASN-BKO) mice using constitutive UCP1-Cre. These mice showed selective FASN loss in BAT without affecting FASN or UCP1 expression in iWAT (Fig. 2d). Notably, FASN inactivation did not decrease iso-C17:0 levels in mice housed at room temperature, but significantly decreased the mmBCFA levels in mice subjected to 6 hours of cold exposure. C16:0 was decreased as expected in room temperature or cold-treated FASN-BKO mice (Fig. 2e). Unlike tamoxifen-inducible UCP1-Cre-mediated knockout, which showed no effect35 (probably due to incomplete recombination), constitutive UCP1-Cre knockout of the lipogenesis protein resulted in significantly reduced cold tolerance (Fig. 2f) and impaired CL316,243-induced energy expenditure (Fig. 2g), demonstrating that FASN is required for BAT thermogenesis. FASN-BKO mice showed increased diet-induced obesity (Extended Data Fig. 3e) due to decreased energy expenditure (Extended Data Fig. 3f), with no effect on food intake or locomotor activity (Extended Data Fig. 3g,h).
ACOX2 loss impairs thermogenesis
The cold-induced upregulation of ACOX2 in BAT prompted us to explore its function in thermogenesis and energy metabolism. We analysed ACOX2 knockout (ACOX2−/−) mice36, which were born at expected Mendelian frequencies and appeared normal. Gene expression and western blot confirmed the knockout in adipose tissue (Extended Data Fig. 4a,b). The residual messenger RNA (mRNA) and protein expression in ACOX2−/− mice might reflect mosaicism and/or non-specificity. During a 6-hour cold challenge, ACOX2−/− mice showed significantly impaired cold tolerance (Extended Data Fig. 4c) without notable changes in thermogenesis-related gene expression, except for cell-death-inducing DFFA-like effector A (CIDEA) (Extended Data Fig. 4d).
In BAT, a high-fat diet (HFD) increased ACOX2 expression, but genes involved in BCAA catabolism and mmBCFA synthesis were unchanged (Extended Data Fig. 4e). Thus, we determined the effect of ACOX2 ablation on diet-induced obesity. HFD-fed ACOX2−/− mice gained significantly more weight and fat mass than WT mice (Extended Data Fig. 4f–h), despite similar food intake (Extended Data Fig. 4i) and locomotor activity (Extended Data Fig. 4j). ACOX2−/− mice showed increased adipose depot weights and lipid accumulation (Extended Data Fig. 4k–m). In addition, they showed impaired glucose and insulin tolerance (Extended Data Fig. 4n,o), consistent with defective BAT function.
To determine whether the effects of ACOX2 deficiency on thermogenesis and energy metabolism are intrinsic to adipose tissue, we generated ACOX2LoxP/LoxP mice using CRISPR–Cas9 (Fig. 3a) and crossed them with adiponectin-Cre transgenic mice to create adipose-specific ACOX2 knockout (ACOX2-AKO) mice. Gene expression analysis confirmed successful ACOX2 deletion in adipose tissue (Fig. 3b). ACOX2 deletion did not affect ACOX1 or ACOX3 expression but resulted in reduced FASN levels in BAT (Extended Data Fig. 5a). ACOX2 ablation selectively increased mmBCFA levels in BAT (Extended Data Fig. 5b). Because ACOX2 contributes to bile acid synthesis by degrading C27 intermediates, we measured bile acids in BAT of ACOX2−/− and wild-type (WT) mice. Compared with previously reported hepatic bile acid levels37, bile acids in BAT were virtually undetectable and remained unaffected by ACOX2 ablation (Extended Data Table 1). Furthermore, total serum bile acids were unchanged in ACOX2-AKO mice (Extended Data Fig. 5c).

a, Gene targeting strategy using CRISPR–Cas9 to insert LoxP sites into the ACOX2 locus. The floxed mice were crossed with an adiponectin-Cre mouse to generate ACOX2-AKO mice. b, ACOX2 gene expression analysis in adipose tissue depots of control and ACOX2-AKO mice. n = 4. c, Infrared thermal imaging of mice after a 6 h of cold exposure. Quantification shows average surface temperature of two mice per genotype. d, Core body temperature of control and ACOX2-AKO male mice after cold exposure (4 °C). n = 13. e, VO2 of control (n = 9) and ACOX2-AKO (n = 8) male mice after CL316,243 treatment. f, Western blot analysis in BAT of control (n = 4) and ACOX2-AKO (n = 3) mice. g, Body weight of control and ACOX2-AKO male mice fed HFD. n = 8. h, Body composition analysis of HFD-fed male control and ACOX2-AKO mice. n = 8. i, Regression analysis of energy expenditure with body weight as a covariate in HFD-fed ACOX2-AKO and control male mice. n = 7. j,k, Glucose tolerance testing (GTT) (j) and insulin tolerance testing (ITT) (k) in control and ACOX2-AKO mice after HFD feeding. The mice were dosed with glucose (j) or insulin (k) based on lean body mass; n = 8 for GTT; n = 5 for ITT. Data in b,c (right), d–k are from biologically independent samples. Images in c are representative of two mice per group. Data with error bars are reported as the mean ± s.e.m. Two-sided unpaired Student’s t-test in b,h or two-way ANOVA followed by Fisher’s least-significant difference test in d,e,g,j,k or two-way analysis of covariance (ANCOVA) with Tukey’s test in i. Panel a was created using BioRender (https://biorender.com).
To assess thermogenesis, we used infrared thermal imaging on cold-exposed ACOX2-AKO and control mice. ACOX2-AKO mice showed lower surface temperatures in the cervical and thoracic regions (Fig. 3c), consistent with reduced core body temperature during cold exposure measured using IPTT-300 temperature transponders implanted in the interscapular space (Fig. 3d and Extended Data Fig. 5d). Furthermore, CL316,243-stimulated oxygen consumption (VO2), a marker of thermogenic activation, was significantly lower in ACOX2-AKO mice (Fig. 3e and Extended Data Fig. 5e). Consistent with the gene expression analysis (Extended Data Fig. 5a), western blot analysis showed that ACOX2 deficiency also decreased FASN levels, suggesting coordinated regulation of ACOX2 and FASN expression. However, oxidative phosphorylation protein levels in BAT were unaffected (Fig. 3f).
We next evaluated the role of adipose ACOX2 in diet-induced obesity. HFD-fed ACOX2-AKO male and female mice gained more weight than controls (Fig. 3g and Extended Data Fig. 5f) due to increased fat mass (Fig. 3h). Histological analysis revealed greater lipid accumulation in BAT of ACOX2-AKO mice (Extended Data Fig. 5g). Increased adiposity was not linked to differences in food intake or locomotor activity (Extended Data Fig. 5h,i), but to decreased energy expenditure (Fig. 3i). In addition, HFD-fed ACOX2-AKO mice showed impaired glucose tolerance (Fig. 3j) and reduced insulin sensitivity (Fig. 3k), further evidenced by decreased insulin-stimulated Akt phosphorylation in liver (Extended Data Fig. 5j). Together, these findings demonstrate that ACOX2-mediated β-oxidation is critical for thermogenesis, and its disruption in adipose tissue promotes diet-induced obesity and insulin resistance.
ACOX2 upregulation drives thermogenesis
To assess whether upregulating adipose tissue ACOX2 is sufficient to enhance thermogenesis and energy homeostasis, we generated transgenic mice with adipose-specific ACOX2 overexpression (ACOX2Adipo-OE) (Fig. 4a). Gene expression analysis confirmed ACOX2 overexpression in various adipose depots (Fig. 4b). During cold exposure, ACOX2Adipo-OE mice showed significantly improved cold tolerance (Fig. 4c), indicating that cold-induced ACOX2 upregulation enhances thermogenesis. This was supported by higher VO2 in ACOX2Adipo-OE mice compared with WT controls following CL316,243 treatment (Fig. 4d). ACOX2 overexpression did not affect lipolysis in BAT (Extended Data Fig. 6a) or the levels of various oxidative phosphorylation pathway proteins (Fig. 4e), suggesting that it does not influence substrate liberation or mitochondrial abundance. Notably, the improved cold tolerance associated with adipose-specific ACOX2 overexpression was abolished by treatment of mice with BCAT-IN-2 (Extended Data Fig. 6b), a highly selective inhibitor of BCAT2 (ref. 38). These data demonstrate that a cycle of mmBCFA synthesis through BCAA catabolism, followed by ACOX2-mediated fatty acid oxidation, promotes adipose thermogenesis.

a, Schematic diagram of a transgene construct used to generate adipose-specific ACOX2 overexpression (ACOX2Adipo-OE) mice. b, ACOX2 gene expression in adipose tissue depots of ACOX2Adipo-OE and WT mice. n = 4. c, Core body temperature of ACOX2Adipo-OE and WT mice after cold exposure. n = 10. d, VO2 of ACOX2Adipo-OE and WT mice after CL316,243 treatment. n = 5. e, Western blot analysis of ACOX2 in BAT of ACOX2Adipo-OE and WT mice. n = 2. f, Body weight of HFD-fed ACOX2Adipo-OE and WT female mice. n = 7. g, Body composition of HFD-fed ACOX2Adipo-OE and WT female mice. n = 7. h, Haemotoxylin and eosin staining in adipose depots of HFD-fed ACOX2Adipo-OE and WT female mice. i, GTT analysis in HFD-fed ACOX2Adipo-OE and WT female mice. n = 9. j, ITT analysis in HFD-fed ACOX2Adipo-OE and WT female mice. n = 8. Data in b–g,i,j are from biologically independent samples. Images in h are representative of three mice per group. Data are reported as the mean ± s.e.m. Two-sided unpaired Student’s t-test in b,g or two-way ANOVA followed by Fisher’s least-significant difference test in c,d,f,i,j. Scale bars, 75 μm.
We next examined the impact of ACOX2 overexpression on diet-induced obesity and insulin resistance. HFD-fed female ACOX2Adipo-OE mice showed reduced body weight (Fig. 4f) and fat mass (Fig. 4g), despite no differences in food intake (Extended Data Fig. 6c), fecal lipid content (Extended Data Fig. 6d) or locomotor activity (Extended Data Fig. 6e). Histological analysis revealed protection against obesity-induced lipid accumulation in BAT (Fig. 4h). Similar results were observed in male ACOX2Adipo-OE mice, which were also resistant to diet-induced obesity (Extended Data Fig. 6f,g). Consistent with increased thermogenesis and reduced obesity, ACOX2Adipo-OE mice showed improved glucose tolerance and insulin sensitivity under HFD conditions, in both females (Fig. 4i,j) and males (Extended Data Fig. 6h,i). These findings demonstrate that adipose-specific ACOX2 overexpression promotes thermogenesis and protects against diet-induced obesity and insulin resistance.
UCP1-independent thermogenesis through ACOX2
Next, we investigated whether ACOX2-mediated thermogenesis depends on UCP1 by crossing ACOX2Adipo-OE mice with UCP1−/− mice. Western blot analysis confirmed ACOX2 overexpression and UCP1 knockout in BAT (Fig. 5a). During acute cold exposure, UCP1−/− mice were severely cold intolerant, showing torpor within 3 hours, as previously reported2. However, UCP1−/−/ACOX2Adipo-OE mice showed notable cold tolerance (Fig. 5b), indicating that ACOX2 promotes UCP1-independent thermogenesis, compensating for UCP1 loss.

a, Western blot analysis of ACOX2 and UCP1 in BAT of WT, ACOX2Adipo-OE, UCP1−/− and UCP1−/−/ACOX2Adipo-OE mice. n = 2. b, Body temperature of male mice after cold exposure (4 °C). n = 10. c–g, Body weight (c), body composition (d), adipose tissue weights (e), gross BAT morphology (f) and haemotoxylin and eosin staining of adipose tissue (g) from HFD-fed mice housed at thermoneutrality (30 °C). n = 5 for c–e. Images in f and g are representative of three mice per genotype. h, Western blot analysis of UCP1−/− brown adipocytes transfected with lentivirus overexpressing ACOX2 or LacZ. n = 2. i, OCR measurement using Seahorse in ACOX2 or LacZ-expressing UCP1−/− brown adipocytes. Oligo, oligomycin; AA + R, antimycin A and rotenone. n = 6. j, Measurement of ATP in WT brown adipocytes transfected with LacZ or ACOX2. n = 12. k, Western blot analysis for pAMPK, AMPK, pACC, ACC, ACOX2 and FASN in BAT of ACOX2Adipo-OE and WT mice cold-treated for 6 h. ACC antibodies recognize ACC1 and ACC2. n = 2. l, Schematic of Pexo-TEMP, a peroxisome-localized temperature sensor. m, Fluorescence microscopy analysis of Pexo-TEMP and a peroxisome marker (mCherry-PTS1) in brown adipocytes. Images representative of two separate experiments. n, Fluorescence intensity ratio in response to temperature changes in WT brown adipocytes expressing Pexo-TEMP. n = 16 separate wells. o, Fluorescence intensity ratio in WT brown adipocytes expressing Pexo-TEMP together with LacZ or ACOX2, followed by the indicated treatment. n = 16 separate wells. Data in a–e,h–k,n–o are from biologically independent samples. Data are reported as the mean ± s.e.m. Two-sided unpaired Student’s t-test in d,e,j; two-way ANOVA followed by Fisher’s least-significant difference test in b,c,i; or one-way ANOVA followed by Fisher’s least-significant difference test in o. Scale bars, 75 μm (g), 10 μm (m).
To determine the effect of ACOX2 overexpression on diet-induced obesity, we fed mice a HFD for 18 weeks while they were housed in a thermoneutral room. UCP1−/−/ACOX2Adipo-OE mice had significantly lower body weight (Fig. 5c), fat mass (Fig. 5d) and reduced BAT and iWAT weights compared with UCP1−/− controls (Fig. 5e). BAT in UCP1−/− mice appeared pale with lipid droplet accumulation, whereas BAT from UCP1−/−/ACOX2Adipo-OE mice appeared normal (Fig. 5f,g).
Gene expression analysis revealed that ACOX2 overexpression upregulates FASN in BAT (Extended Data Fig. 7a). Although FASN expression in BAT is lower in mice maintained at thermoneutrality compared with those exposed to cold (Fig. 2a), ACOX2 overexpression drives FASN expression to support the substrate cycle. To determine whether the increased FASN protein levels are associated with peroxisomes, we performed immunofluorescence analysis in HFD-fed UCP1−/− and UCP1−/−/ACOX2Adipo-OE mice housed at thermoneutrality. ACOX2 overexpression increased FASN colocalization with the peroxisome marker PMP70 (Extended Data Fig. 7b). Furthermore, indirect calorimetry of ACOX2Adipo-OE mice housed at thermoneutrality showed that ACOX2 overexpression enhanced energy expenditure during the dark phase (Extended Data Fig. 7c) without significant changes in food intake (Extended Data Fig. 7d) or locomotor activity (Extended Data Fig. 7e). These findings suggest that ACOX2 overexpression promotes metabolic adaptations in BAT by enhancing FASN expression and energy expenditure, even under thermoneutral conditions.
We further examined HFD-fed UCP1−/−/ACOX2Adipo-OE and UCP1−/− control mice housed at room temperature. Similar to thermoneutral conditions, UCP1−/−/ACOX2Adipo-OE mice showed resistance to diet-induced obesity (Extended Data Fig. 7f), reduced fat mass (Extended Data Fig. 7g–i) and preserved BAT morphology, avoiding increased intracellular lipid accumulation seen in UCP1−/− controls (Extended Data Fig. 7j). Furthermore, ACOX2 overexpression improved glucose homeostasis (Extended Data Fig. 7k,l). Together, these findings demonstrate that ACOX2 promotes thermogenesis and metabolic homeostasis, even in the context of UCP1 deficiency.
We next examined the effect of ACOX2 overexpression on respiration in UCP1−/− brown adipocytes. Consistent with the gene expression analysis, western blot analysis showed that ACOX2 overexpression was associated with FASN upregulation (Fig. 5h). Seahorse analysis showed that overexpression of the peroxisomal β-oxidation enzyme significantly increased basal OCR (Fig. 5i), probably due to increased ATP turnover driven by a substrate cycle. Accordingly, cellular ATP levels were significantly lower in WT brown adipocytes overexpressing ACOX2 (Fig. 5j). This prompted us to determine whether ACOX2 overexpression activates the energy-sensing kinase AMPK, which helps restore cellular energy balance by promoting mitochondrial oxidative phosphorylation and ATP production39. Western blot analysis in BAT of ACOX2Adipo-OE and WT mice revealed that ACOX2 overexpression, which also resulted in upregulation of FASN, increased phosphorylation of AMPK and its downstream target ACC (Fig. 5k), confirming AMPK activation. These findings suggest that the ATP-consuming lipid metabolism mediated by the FASN–ACOX2 pathway activates AMPK as a compensatory mechanism, linking peroxisomal activity to mitochondrial energy production to sustain thermogenesis.
To determine whether ACOX2 overexpression directly affects mitochondrial respiratory function, we measured Complex II-dependent and Complex IV-dependent respiration activities in mitochondria-enriched fraction from BAT of ACOX2Adipo-OE and WT mice. OCR responses were comparable between the transgenic and WT groups, indicating no difference in mitochondrial respiratory capacity (Extended Data Fig. 8a). These results indicate that ACOX2 does not intrinsically enhance mitochondrial respiration.
We next assessed whether ACOX2 functions through futile creatine cycling, an ATP-consuming, UCP1-independent thermogenic mechanism7,40 by evaluating the impact of creatine kinase B (CKB) knockout in ACOX2-overexpressing brown adipocytes (Extended Data Fig. 8b,c). CKB deletion did not affect the ACOX2-induced or iso-C17:0-induced respiration, indicating that ACOX2-mediated thermogenesis operates independently of creatine cycling in mitochondria (Extended Data Fig. 8d).
Next, we examined the role of ACOX2 in non-mitochondrial respiration. WT brown adipocytes expressing either ACOX2 or GFP were treated with antimycin A and rotenone to block mitochondrial electron transfer and subsequently exposed to iso-C17:0 or bovine serum albumin (BSA). ACOX2 overexpression, coupled with mmBCFA treatment, significantly increased OCR (Extended Data Fig. 8e), supporting a UCP1-independent, peroxisome-driven thermogenic mechanism.
ACOX2 induces peroxisomal heat
To confirm peroxisomal origins of ACOX2-mediated thermogenesis, we developed a peroxisome-targeted ratiometric fluorescent temperature sensor (Pexo-TEMP) (Fig. 5l) based on the temperature-sensitive properties of Sirius and mT-Sapphire41 (Extended Data Fig. 9a,b). Confocal microscopy confirmed peroxisomal targeting of Pexo-TEMP in brown adipocytes (Fig. 5m). The fluorescent ratio (509 nm/425 nm) increased with temperature, validating Pexo-TEMP as a functional peroxisomal thermometer (Fig. 5n). Adipocytes expressing ACOX2 and Pexo-TEMP showed heat production in response to iso-C17:0 but not C16:0 treatment (Fig. 5o), with norepinephrine serving as a positive control. These findings indicate that ACOX2-mediated β-oxidation of mmBCFA in peroxisomes generates heat, revealing a previously unrecognized UCP1-independent thermogenic pathway.
Given that peroxisomal β-oxidation is associated with H2O2 production, we next investigated whether ACOX2 overexpression promotes oxidative stress and cytotoxicity using HyPer7-SKL, a peroxisome-targeted H2O2 probe42. ACOX2 overexpression and/or iso-C17:0 treatment increased H2O2 production in brown adipocytes (Extended Data Fig. 9c), supporting the notion that mmBCFAs are metabolized in peroxisomes. Palmitate also elevated H2O2 levels, consistent with previous findings that peroxisomal β-oxidation partially metabolizes palmitate43. This fatty acid metabolism upregulated catalase, a peroxisomal antioxidant enzyme, likely to mitigate potential H2O2-induced toxicity (Extended Data Fig. 9d). To investigate further, we generated catalase KO brown adipocytes using CRISPR–Cas9 (Extended Data Fig. 9e). Treatment with C16:0, iso-C17:0 or H2O2 significantly reduced cell viability in catalase KO adipocytes (Extended Data Fig. 9f), indicating that catalase expression protects against oxidative damage from increased peroxisomal β-oxidation. Consistent with this, BAT from ACOX2Adipo-OE and WT mice showed similar levels of 4-HNE, a marker of lipid peroxidation and oxidative stress44 (Extended Data Fig. 9g). These results demonstrate that ACOX2-mediated β-oxidation enhances thermogenesis without inducing cytotoxicity.
Discussion
These results identify a UCP1-independent pathway of adipose tissue thermogenesis involving a substrate cycle of mmBCFA synthesis and β-oxidation in peroxisomes (Extended Data Fig. 10). Adipose-specific inactivation of ACOX2 impairs thermogenesis, whereas adipose-specific overexpression improves cold tolerance, indicating that the peroxisomal β-oxidation enzyme is necessary and sufficient for adipose tissue thermogenesis. Overexpression of ACOX2 rescues cold intolerance and reduces diet-induced obesity in UCP1−/− mice. ACOX2 overexpression, which also upregulates the expression of FASN, increases basal OCR, reflecting increased ATP turnover due to energy-consuming metabolism of mmBCFA by the FASN–ACOX2 axis. As FASN-mediated lipogenesis is an ATP-consuming process and ACOX2-mediated β-oxidation is not linked to ATP production, this substrate cycle promotes negative energy balance, leading to protection against obesity and insulin resistance. Consistent with recent findings that brown adipocytes rely on mitochondrial ATP production to sustain thermogenesis through the futile creatine cycling pathway45, we observe AMPK activation in ACOX2-overexpressing BAT, suggesting a compensatory increase in mitochondrial ATP synthesis. This supports a self-sustaining mechanism in which ATP-consuming peroxisomal lipid metabolism is balanced by mitochondrial ATP synthesis. Thus, although the FASN–ACOX2 thermogenesis pathway is UCP1-independent and localized to peroxisomes, it remains functionally coupled to mitochondria through its ATP demand. Notably, in contrast to the role of ACOX2-mediated β-oxidation of mmBCFA in metabolic regulation, adipose-specific knockout of ACOX1, which regulates β-oxidation of very long-chain fatty acids, does not affect thermogenesis or diet-induced obesity in mice13.
Several lines of evidence support a functional role for mmBCFAs in BAT: (1) adipose tissue is the primary site of de novo mmBCFA synthesis, with several species of these BCAA-derived fatty acids most abundantly enriched in brown adipocytes9; (2) cold exposure significantly increases mmBCFA synthesis in mouse BAT9 and (3) individuals with obesity show lower mmBCFA levels46,47 and impaired BCAA catabolism21. Increased circulating concentrations of BCAAs are strongly associated with metabolic diseases48. BCAA catabolism has been reported to regulate energy homeostasis due to use of branched amino acids as an alternative fuel source for mitochondrial thermogenesis in BAT49, although recent data show a role for BCAA catabolism in metabolic health independent of a direct effect on thermogenesis38. Our work indicates that BCAA catabolism leads to the synthesis of mmBCFAs, which are then oxidized in peroxisomes to promote thermogenesis. Although mammals, including humans, possess the ability to synthesize mmBCFAs, these fatty acids can also be obtained from dietary sources, including milk and dairy products50. We speculate that dietary interventions implementing use of these branched fatty acids or modulation of proteins involved in their metabolism might be a promising strategy to treat obesity and related metabolic diseases.
Methods
Mouse models and animal experiments
All animal protocols were approved by the Washington University Institutional Animal Care and Use Committee. C57BL/6J WT mice were obtained from the Jackson Laboratory (strain no. 000664). FASLox/Lox mice were previously described in ref. 34. To generate mice with brown-adipose-specific knockout of FAS (FAS-BKO), FASLox/Lox animals were crossed with Ucp1-Cre mice obtained from the Jackson Laboratory (stock no. 024670). FASLox/Lox mice without Cre were used as a control for FAS-BKO mice. ACOX2−/− mice have been previously described in ref. 36 and were generated using two TALEN pairs targeting beginning and end of exon 4 that led to deletion of 176 base pairs covering the entire exon 4, resulting in a frameshift. Founder mice were generated by cytoplasmic injection of TALENs into C57BL/6N mouse zygotes and then backcrossed into the WT C57BL/6J background. ACOX2Lox/Lox mice on the C57BL/6J background were generated using the CRISPR–Cas9 system. CRISPR-mediated mutagenesis was done by the Genome Engineering and IPSC Center at Washington University. To generate mice with adipose-specific knockout of ACOX2 (ACOX2-AKO), ACOX2Lox/Lox animals were crossed with adiponectin-Cre mice obtained from the Jackson Laboratory (stock no. 028020). ACOX2Lox/Lox mice without Cre were used as a control for ACOX2-AKO. To generate mice with adipose-specific overexpression of ACOX2 (ACOX2Adipo-OE), murine ACOX2 was cloned downstream of a previously characterized 5.4-kilobase adiponectin promoter51. The adiponectin promoter plasmid was a generous gift from P. Scherer (University of Texas Southwestern Medical Center). The transgenic construct was microinjected into the pronucleus of a newly fertilized egg from a C57BL/6J × CBA hybrid mouse and implanted into a pseudopregnant female by the Mouse Genetics Core at Washington University. The founders were backcrossed more than seven generations into the WT C57BL/6J genetic background.
UCP1−/− mice were obtained from the Jackson Laboratory (stock no. 003124). To generate mice with adipose-specific overexpression of ACOX2 on the UCP1−/− background, ACOX2Adipo-OE mice were crossed with UCP1−/− mice. An inbred strain of these mice on the C57BL/6J genetic background was developed and used for all experiments. Mice were fed either normal chow diet (Purina 5053) or a HFD (D12492, Research Diets). All animals were randomly allocated into different groups. For metabolic phenotyping studies, both male and female animals were studied, and all data were disaggregated by sex. Mice were maintained under constant temperature (23–25 °C), circulating air and humidity (45–65%) with a 12-h light/dark cycle and provided ad libitum access to food and water. Body composition (fat and lean mass) was measured using an EchoMRI system.
To assess glucose homeostasis, intraperitoneal glucose tolerance tests and intraperitoneal insulin tolerance tests were performed based on body weight or lean body mass, as indicated. Mice were injected intraperitoneally with glucose (2.5 g kg−1 body weight or 2 g kg−1 lean mass) or insulin (0.75 U kg−1 body weight or 1 U kg−1 lean mass) after being fasted for 6 h or 4 h, respectively. Blood glucose levels were measured at 0 min, 15 min, 30 min, 60 min and 120 min after injection by a glucometer.
VO2, VCO2 and respiratory exchange ratio were measured by indirect calorimetry using a PhenoMaster (TSE Systems) metabolic cage system and analysed using CalR web-based software (v.1.3). For measurement of CL316,243-induced energy expenditure, mice were anaesthetized using pentobarbital (90 mg kg−1 intraperitoneally) and acclimated to the environment for 60 min, allowing oxygen consumption to stabilize. The mice were then injected with CL316,243 at a dose of 1 mg kg−1, and data were collected for 2 h in mice housed at 23 °C or 30 °C as previously described52 To assess cold tolerance, body temperature was measured at time 0 and hourly for 6 h during cold exposure using implantable IPTT-300 temperature-sensitive transponders and a DAS-8007 programmable reader from Bio Medic Data Systems as previously described52.
Cell culture and treatments
Immortalized mouse brown preadipocytes were established and differentiated as previously described13. Immortalized human brown preadipocytes, kindly provided by Y.-H. Tseng (Joslin Diabetes Center), were cultured and differentiated as previously described53,54. For isolation of mature pig adipocytes from subcutaneous WAT, fresh iWAT was digested in a collagenase buffer containing HBSS (GIBCO 14065-45), 12.6 mM CaCl2, 4.9 mM MgCl2, 2% BSA and 800 U g−1 (3 mg g−1 tissue) of type 2 collagenase for roughly 40 min in a 37 °C water bath. The digested material was passed through a 250-μm strainer and washed 3 times with a total of 1 l of KRHB (1× KBH, 25 mM HEPES, 2 mM glucose, 2% BSA) to separate the floating mature adipocytes. The cells were centrifuged at 50g for 3 min. The mature adipocytes (floating cells) were collected and cultured with DMEM/F12 medium.
Mouse brown adipocytes and pig iWAT mature adipocytes were cultured in Dulbecco’s modified eagle medium F12 (DMEM/F12) supplemented with 10% (v/v) fetal bovine serum (FBS), 1% (v/v) penicillin/streptomycin, 1% (v/v) l-glutamine and 1% (v/v) sodium pyruvate. Human embryonic kidney 293T (HEK293T) cells and immortalized human brown adipocytes were cultured in DMEM. All cells were cultured in a humidified incubator at 37 °C with 5% CO2 in air.
Palmitic acid, C15:0 OCFA, C17:0 OCFA and BCFA iso-C17:0 were dissolved in ethanol, then diluted with serum-free medium containing 0.1% fatty acid-free BSA, then added into culture medium at final concentrations of 0.2 μM, 1 μM or 2 μM based on experimental need. Norepinephrine was first dissolved in 1× PBS and diluted into the culture medium at final concentrations of 0.25 μM, 1 μM or 2 μM for different experiments. H2O2 was dissolved in 1× PBS and diluted into the culture medium at a final concentration of 500 μM. Isotope-labelled glucose (U-13C6 glucose) was dissolved in 1× PBS and diluted into the culture medium at a final concentration of 17.5 mM.
Plasmid constructs
Genome-wide guide RNA (gRNA) databases were used to design gRNA oligonucleotides against ACOX2, CRAT, Bckdha, CAT and CKB. The oligonucleotides were ordered from Integrated DNA Technologies and subcloned into lentiCRISPRv2 plasmid. ACOX2 and Hyper7 complementary DNA (cDNA) clones were purchased from transOMIC (BC021339) and Addgene (136466), respectively, and subcloned into pLJM1 lentiviral overexpression plasmid. Sequences of primers used for cloning are listed in Supplementary Table 1. Pexo-TEMP plasmid was generated by inserting PTS2-Sirius-T2A fragment by PCR using primers below. Forward: CGG CGA CCG GTG CCA CCA TGC ACC GGC TGC AGG TGG TGC TGG GCC ACC TGG CCG GCC GGC CCG AGT CCT CCT CCG CCC TGC AGG CCG CCC CCT GCA GCT CGG ATC CCA CCA TG, and reverse: TCA CCA TGA GCT CGG GGC C and inserting mT-Sapphire fragment by PCR using primers below. Forward: CGG CCG AGC TCA TGC ACC GGC TGC AGG TGG TGC TGG GCC ACC TGG CCG GCC GGC CCG AGT CCT CCT CCG CCC TGC AGG CCG CCC CCT GCG TGA GCA AGG GCG AGG AGC T, reverse: TGT GAT GGA TAT CTG CAG AAT TC based on the gTEMP_pcDNA3 plasmid (Addgene no. 89583). To generate recombinant lentiviruses, the overexpression plasmid (pLJM1), gRNA plasmid (lentiCRISPRv2) or short-hairpin RNA plasmid (pLKO.1-puro) together with packaging plasmids (pMD2.G and psPAX2) were cotransfected into HEK293T cells. Lentiviral particles were collected 48 h after transfection and stored at −80 °C until they were used to transduce immortalized BAT SVF cells.
Extraction and mass spectrometric analysis of fatty acids
BAT or brown adipocytes homogenate containing 50 μg of total protein content was hydrolysed in an acid hydrolysis buffer (CH3CN:37% HCl, 4:1) in 90 °C water bath for 2 h. Then, hexane was used to extract the fatty acids. The samples were dried under a stream of nitrogen and redissolved in chloroform, methanol, H2O and 25% NH4OH (50:45:5:0.01). After extraction, the electrospray ionization-mass spectrometry images of the fatty acids in the adipose tissues and adipocytes were obtained by a Thermo Fisher LTQ Orbitrap Velos in the negative-ion mode scanning from 200 m/z to 600 m/z with a resolution of 100,000 (at m/z 400 Da). Data were processed by built-in Xcalibur software as previously described14 and the exogenous docosanoic-22, 22, 22-D3 acid added to samples before extraction was used as an internal standard for quantitation. To verify the iso-form of the fatty acid structures, dried fatty acids were derivatized to the N-(4-aminomethylphenyl) pyridinium derivatives, which were subjected to higher-energy collisional dissociation tandem mass spectrometry for structural identification as described previously55. Data were analysed using R (v.4.2.1).
Carbon flux tracing and lipidomic analysis
To assess incorporation of 13C-label into mmBCFA, BAT SVF cells were differentiated into adipocytes in normal DMEM/F12 medium and then cultured in DMEM/F12 in which glucose was replaced with [U13C6]-glucose, as previously described in ref. 9. Briefly, after differentiation, sgACOX2 or control adipocytes were cultured in the presence of [U13C6]-glucose in DMEM/F12 medium lacking glutamine and pyruvate containing 10% FBS for an extra 3 days, followed by 6 h of treatment with 10 nM CL316,243 or vehicle in a normal DMEM/F12 media and the lipids with labelled mmBCFA were extracted and subjected to high resolution electrospray ionization-mass spectrometry analysis as described above.
Immunofluorescence analysis
Frozen sections or cell samples were fixed with ethanol or 4% paraformaldehyde, followed by primary antibody and the corresponding secondary antibody incubation. Nuclei were counterstained with 4′6-diamidino-2-phenylindole. Samples were subjected to immunofluorescence analysis using rabbit polyclonal anti-ACOX2 antibody (1:100), rabbit polyclonal anti-PMP70 antibody (1:100), rabbit polyclonal anti-FASN antibody (1:100) and rabbit polyclonal anti-CRAT antibody (1:100). Slides were imaged using a Nikon A1Rsi Confocal Microscope. Images were analysed using NIS (v.5.21). Fluorescence intensity and colocalization were calculated using ImageJ (v.1.53) and Colocalization Finder (v.1).
Oil Red O staining
Adipocytes were fixed with 10% formalin overnight and then washed twice with 60% isopropanol. Oil Red O working solution was added, and the cells were incubated for 10 min at room temperature. The Oil Red O solution was removed, and the wells were washed four times with diH2O. The stained cells were photographed under a ×2 objective lens using light microscopy.
Quantitative real-time PCR analysis
Total RNA was isolated using PureLink RNA Mini Kit (Invitrogen, 12183018A) and 2 μg of total RNA was reverse transcribed into cDNA using the iScriptcDNA Synthesis Kit (Bio-Rad) as previously reported23. Quantitative real-time PCR was conducted using PowerUp SYBR Green Master Mix. Relative mRNA expression level was determined using the 2(−ΔΔCT) method and L32 was used as an internal reference. Primers used in PCR analyses are listed in Supplementary Table 1.
Western blot and immunoprecipitation analyses
Cells or mouse tissue samples were homogenized in RIPA buffer (Cell Signaling Technology, 9806S) or homogenization buffer (0.25 M sucrose, 20 mM HEPES in distilled H2O) containing a protease and phosphatase inhibitor cocktail (Sigma, P8465 and P0001). Total protein was extracted and quantitated using a bicinchoninic acid (BCA) kit. Denatured protein was separated by SDS–PAGE electrophoresis and transferred onto polyvinyl difluroride or Nylon membrane, blocked with 5% non-fat milk, incubated overnight with primary antibodies and finally incubated with horse radish peroxidase-conjugated secondary antibodies as reported. Tissue or cell lysates were subjected to immunoblot analysis using rabbit polyclonal anti-ACOX2 antibody (1:1,000), rabbit polyclonal anti-FASN antibody (1:10,000), mouse monoclonal anti-catalase antibody (1:1,000), rabbit polyclonal anti-UCP1 antibody (1:1,000), rabbit polyclonal anti-CRAT antibody (1:500), rabbit polyclonal anti-Ap2 antibody (1:1,000), rabbit polyclonal anti-AKT (1:1,000), rabbit polyclonal anti-PMP70 antibody (1:1,000), mouse monoclonal anti-COX 4 antibody (1:1,000), mouse polyclonal anti-oxidative phosphorylation cocktail (1:1,000), rabbit polyclonal anti-pAMPK (T172) antibody (1:1,000), rabbit polyclonal anti-AMPK antibody (1:1,000), rabbit polyclonal anti-pACC (S79) antibody (1:1,000), rabbit monoclonal anti-ACC antibody (1:1,000), rabbit polyclonal anti-BCKDHA antibody (1:1,000), rabbit polyclonal anti-pAK(T473) antibody (1:1,000), rabbit polyclonal anti-AKT antibody (1:1,000) and mouse monoclonal anti-CKB antibody (1:10,000). Rabbit polyclonal anti-β actin (1:1,000), rabbit polyclonal anti-β Tubulin (1:1,000) and rabbit polyclonal anti-Vinculin (1,000) were used as loading controls. For western blot analysis of ACOX2, initial experiments used either a rabbit polyclonal antibody (Invitrogen; catalogue no. PA5-114814) or a goat polyclonal antibody (Novus; catalogue no. 06011). Subsequently, a rabbit polyclonal anti-ACOX2 antibody from Millipore Sigma (catalogue no. HPA038280) was used. All antibodies used in western blot analyses are listed in the Reporting summary. Proteins were detected with the Odyssey Infrared Imaging System (LI-COR Biosciences). Uncropped raw blots are presented in Supplementary Fig. 1.
Interaction between FASN and PEX7 was assessed by co-immunoprecipitation analysis as described previously56. Briefly, HEK293 cells transiently transfected with Flag-FASN were homogenized using RIPA Lysis Buffer System (Santa Cruz) supplemented with protease and phosphatase inhibitors. Cell lysates were centrifuged at 13,000 rpm in a microcentrifuge for 10 min to remove unlysed cells. Supernatants were collected and subjected to protein quantification using the BCA assay. Cell lysates were then incubated with Anti-Flag M2 Affinity gel (Sigma-Aldrich, A220) overnight. Immunoprecipitates were washed three times with Tris-buffered saline before elution with SDS–PAGE sample buffer and subjected to SDS–PAGE. Cell lysates were subjected to immunoblot analysis using mouse monoclonal anti-HA antibody (1:1,000) and rabbit polyclonal anti-Flag antibody (1:1,000).
Subcellular fractionation of adipocytes
Adipocytes were lysed in Peroxisome Extraction Buffer (Sigma) using a Dounce homogenizer. Lysed cells were centrifuged at 1,000g for 10 min to pellet the nuclear fraction. The supernatant was transferred to a new tube and centrifuged at 12,000g for 15 min to collect the mitochondrial pellet. The supernatant was transferred to a new tube and centrifuged at 21,000g for 40 min to collect the peroxisome pellet. The remaining supernatant was saved as the cytosol fraction.
OCR
OCR in cultured adipocytes was measured using a Seahorse XFe Extracellular Flux Analyzer (Agilent) in a 24-well plate. For mitochondrial stress test, cells were treated with oligomycin (3 μM), FCCP (1.5 μM) and antimycin (2 μM) plus rotenone (1 μM). For measurement of fatty acid-induced respiration, cells were treated with BSA-conjugated 1 mM C15:0, C17:0, iso-C17:0 or BSA alone.
Measurement of mitochondrial respiration in BAT
Mitochondrial respiration in mouse BAT was measured using a previously described method57. Briefly, BAT depots from ACOX2Adipo-OE and WT mice were thawed in ice-cold PBS, minced in an Eppendorf tube and mechanically homogenized with 10–20 strokes using Teflon-glass in MAS buffer (70 mM sucrose, 220 mM mannitol, 10 mM KH2PO4, 5 mM MgCl2, 2 mM HEPES, 1.0 mM EGTA and 0.2% (w/v) fatty acid-free BSA). The homogenates were centrifuged at 1,000g for 10 min at 4 °C. The supernatant was collected and loaded into a Seahorse XF96 microplate in 20 µl of MAS (5 µg BAT). The loaded plate was centrifuged at 2,000g for 5 min at 4 °C. An extra 130 µl of MAS buffer was added to each well. Substrate injections were as follows: 5 mM succinate + 2 μM rotenone for port A, 2 μM rotenone + 4 μM antimycin for port B, 0.5 mM TMPD + 1 mM ascorbic acid for port C and 50 mM azide for port D.
Infrared thermal imaging
The surface temperature in the region surrounding the BAT was measured using a high-speed mid-wave infrared camera (Telops FAST M3k) equipped with a 50 mm lens (Telops) and a spacer ring that facilitates a long working distance, resulting in a resolution of 0.25 mm per pixel. The temperature distribution was captured from a top-down perspective at 30 frames per second and a resolution of 320 × 256 pixels. The recorded thermal images were processed using the Reveal-IR software suite v.1.13.0 (Telops), assuming a fur emissivity of 0.75 (refs. 58,59).
MTT assay
To determine whether β-oxidation of mmBCFA results in oxidative stress leading to cytotoxicity, cell viability was assessed using the MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide) assay (Cayman Chemicals). Control and catalase knockout brown adipocytes were treated with iso-C17:0, C16:0, H2O2 or vehicle for 2 h. Cell viability was assessed according to the manufacturer’s instructions.
Lipid peroxidation assay
To determine the effect of ACOX2 overexpression on oxidative stress, BAT samples were collected from ACOX2AdipoOE and WT mice. Lipid peroxidation (4-HNE) protein adducts were quantified in the samples using a 4-HNE assay kit (Abcam) according to the manufacturer’s instructions.
Fecal lipid extraction and measurement
Feces were collected for 24 h from each cage and dried in an incubator for 16 h at 42 °C. One hundred milligrams of feces were homogenized with 1 M NaCl, followed with a chloroform:methanol solution (2:1). Chloroform layers were collected after centrifuge and evaporated under N2 flow until dry. The 2% Triton X-100 in chloroform was added and evaporated again under N2 flow. The samples were dissolved with ddH2O and measured by a commercial kit (FFA, Wako; HR Series NEFA-HA(2)).
Ex vivo lipolysis assay
BAT was collected from WT and ACOX2Adipo OE mice and placed into a well of a 24-well plate containing 1 ml of prewarmed KRBH-BSA with 2% fatty acid-free BSA. The tissues were treated with vehicle or with 10 μM isoproterenol for 2 h at 37 °C. The glycerol levels in the media were measured using a commercial kit (Glycerol Assay Kit; Sigma, MAK117).
ATP assay
The ATP levels in differentiated adipocytes were measured using a commercial kit (ATP Detection Assay Kit; Cayman, 700410). Briefly, adipocytes were homogenized in the ATP detection sample buffer supplied in the kit and cell lysates were transferred to prechilled polypropylene tube. The samples were treated with Reaction Mix at room temperature for 15 min and the luminescence was read in a plate reader.
Bile acid measurement
Bile acids were measured by the Metabolomics Innovation Centre using an LC-multiple-reaction monitoring-MS method, as previously described60, with necessary modifications. Tissue samples were precisely weighed into safe-lock Eppendorf tubes, and water (2 μl per mg of tissue) was added. Samples were homogenized using two metal beads at a shaking frequency of 30 Hz for 2 min on a MM 400 mill mixer. Acetonitrile (8 μl per mg of tissue) was then added, followed by a second homogenization under the same conditions for 3 min. Samples were further subjected to ultrasonication for 2 min in an ice-water bath and centrifuged at 21,000g and 5 °C for 10 min. The clear supernatant (100 μl) was collected and mixed with 75 μl of an internal standard solution containing 14 isotope-labelled bile acids. The mixtures were dried under nitrogen gas, and the residues were reconstituted in 75 μl of 50% methanol. A standard solution containing all targeted bile acids was prepared in the same internal standard solution and serially diluted to generate 10 calibration standards. Aliquots (10 μl) of the calibration and sample solutions were injected into an Agilent 1290 UHPLC system coupled to a 6495B Agilent QQQ mass spectrometer. The mass spectrometer was operated in multiple-reaction monitoring mode with negative-ion detection. Chromatographic separation was performed on a C18 column (150 × 2.1 mm, 1.7 μm) using a binary-solvent gradient with mobile phase A (0.01% formic acid in water) and mobile phase B (0.01% formic acid in acetonitrile). Calibration curves for individual bile acids were generated using linear regression, and bile acid concentrations in the samples were determined by interpolating the calibration curves with the analyte-to-internal standard peak ratios obtained from sample injections, ensuring quantification within the appropriate concentration range.
Statistics and reproducibility
Data are reported as mean ± standard error of the mean (s.e.m.) unless stated otherwise. Statistical comparisons between two groups were performed by using unpaired t-test. Analysis of variance (ANOVA) was used for more than two groups. A P value of less than 0.05 was considered statistically significant. Statistical analysis and graphs were generated using GraphPad Prism software (v.9). All in vitro and in vivo experiments were repeated independently at least twice, and key experiments were performed many times using separate cohorts. All replication attempts yielded consistent results. Representative immunoblot images reflect two independent experiments, and microscopy images are representative of n = 3 per group, unless otherwise specified in the figure legends.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data supporting this study are available in the Article and Supplementary Information. Additional data are available at Figshare (https://figshare.com/s/b8fd1fcaa83a4f3d0ac2)61. Source data are provided with this paper.
References
Dieckmann, S. et al. Susceptibility to diet-induced obesity at thermoneutral conditions is independent of UCP1. Am. J. Physiol. Endocrinol. Metabol. 322, E85–E100 (2022).
Enerbäck, S. et al. Mice lacking mitochondrial uncoupling protein are cold-sensitive but not obese. Nature 387, 90–94 (1997).
Feldmann, H. M., Golozoubova, V., Cannon, B. & Nedergaard, J. UCP1 ablation induces obesity and abolishes diet-induced thermogenesis in mice exempt from thermal stress by living at thermoneutrality. Cell Metab. 9, 203–209 (2009).
Mills, E. L. et al. UCP1 governs liver extracellular succinate and inflammatory pathogenesis. Nat. Metab. 3, 604–617 (2021).
Wang, H. et al. Uncoupling protein-1 expression does not protect mice from diet-induced obesity. Am. J. Physiol. Endocrinol. Metab. 320, E333–E345 (2021).
Ikeda, K. et al. UCP1-independent signaling involving SERCA2b-mediated calcium cycling regulates beige fat thermogenesis and systemic glucose homeostasis. Nat. Med. 23, 1454–1465 (2017).
Kazak, L. et al. A creatine-driven substrate cycle enhances energy expenditure and thermogenesis in beige fat. Cell 163, 643–655 (2015).
Sharma, A. K., Khandelwal, R. & Wolfrum, C. Futile cycles: emerging utility from apparent futility. Cell Metab. 36, 1184–1203 (2024).
Wallace, M. & Green, C. R. Enzyme promiscuity drives branched-chain fatty acid synthesis in adipose tissues. Nat. Chem. Biol. 14, 1021–1031 (2018).
Kleiboeker, B. & Lodhi, I. J. Peroxisomal regulation of energy homeostasis: effect on obesity and related metabolic disorders. Mol. Metab. 65, 101577 (2022).
Wanders, R. J. A., Baes, M., Ribeiro, D., Ferdinandusse, S. & Waterham, H. R. The physiological functions of human peroxisomes. Physiol. Rev. 103, 957–1024 (2023).
Ahlabo, I. & Barnard, T. Observations on peroxisomes in brown adipose tissue of the rat. J. Histochem. Cytochem. 19, 670–675 (1971).
Park, H. et al. Peroxisome-derived lipids regulate adipose thermogenesis by mediating cold-induced mitochondrial fission. J. Clin. Invest. 129, 694–711 (2019).
Hu, D. et al. TMEM135 links peroxisomes to the regulation of brown fat mitochondrial fission and energy homeostasis. Nat. Commun. 14, 6099 (2023).
Boveris, A., Oshino, N. & Chance, B. The cellular production of hydrogen peroxide. Biochem. J. 128, 617–630 (1972).
Green, C. R. et al. Branched-chain amino acid catabolism fuels adipocyte differentiation and lipogenesis. Nat. Chem. Biol. 12, 15–21 (2016).
Vanhove, G. F. et al. The CoA esters of 2-methyl-branched chain fatty acids and of the bile acid intermediates di- and trihydroxycoprostanic acids are oxidized by one single peroxisomal branched chain acyl-CoA oxidase in human liver and kidney. J. Biol. Chem. 268, 10335–10344 (1993).
Van Veldhoven, P. P. Biochemistry and genetics of inherited disorders of peroxisomal fatty acid metabolism. J. Lipid Res. 51, 2863–2895 (2010).
Berg, F., Gustafson, U. & Andersson, L. The uncoupling protein 1 gene (UCP1) is disrupted in the pig lineage: a genetic explanation for poor thermoregulation in piglets. PLoS Genet. 2, e129 (2006).
Crown, S. B., Marze, N. & Antoniewicz, M. R. Catabolism of branched chain amino acids contributes significantly to synthesis of odd-chain and even-chain fatty acids in 3T3-L1 adipocytes. PLoS ONE 10, e0145850 (2015).
Newgard, C. B. et al. A branched-chain amino acid-related metabolic signature that differentiates obese and lean humans and contributes to insulin resistance. Cell Metab. 9, 311–326 (2009).
White, P. J. et al. Branched-chain amino acid restriction in Zucker-fatty rats improves muscle insulin sensitivity by enhancing efficiency of fatty acid oxidation and acyl-glycine export. Mol. Metab. 5, 538–551 (2016).
Guilherme, A. et al. Acetyl-CoA carboxylase 1 is a suppressor of the adipocyte thermogenic program. Cell Rep. 42, 112488 (2023).
Sanchez-Gurmaches, J. et al. Brown fat AKT2 is a cold-induced kinase that stimulates ChREBP-mediated de novo lipogenesis to optimize fuel storage and thermogenesis. Cell Metab. 27, 195–209 e196 (2018).
Hillebrand, M. et al. Identification of a new fatty acid synthesis-transport machinery at the peroxisomal membrane. J. Biol. Chem. 287, 210–221 (2012).
Ferdinandusse, S. et al. A novel bile acid biosynthesis defect due to a deficiency of peroxisomal ABCD3. Hum. Mol. Genet. 24, 361–370 (2015).
Braverman, N. et al. Human PEX7 encodes the peroxisomal PTS2 receptor and is responsible for rhizomelic chondrodysplasia punctata. Nat. Genet. 15, 369–376 (1997).
Elgersma, Y., van Roermund, C. W., Wanders, R. J. & Tabak, H. F. Peroxisomal and mitochondrial carnitine acetyltransferases of Saccharomyces cerevisiae are encoded by a single gene. EMBO J. 14, 3472–3479 (1995).
Houten, S. M., Wanders, R. J. A. & Ranea-Robles, P. Metabolic interactions between peroxisomes and mitochondria with a special focus on acylcarnitine metabolism. Biochim. Biophys. Acta Mol. Basis Dis. 1866, 165720 (2020).
Kramar, R., Huttinger, M., Gmeiner, B. & Goldenberg, H. Beta-oxidation in peroxisomes of brown adipose tissue. Biochim. Biophys. Acta 531, 353–356 (1978).
Ohue, M. & Makita, T. Localization of carnitine acetyltransferase activity in brown adipocytes of the rat. J. Vet. Med. Sci. 56, 329–333 (1994).
Violante, S. et al. Substrate specificity of human carnitine acetyltransferase: implications for fatty acid and branched-chain amino acid metabolism. Biochim. Biophys. Acta 1832, 773–779 (2013).
Guilherme, A. et al. Adipocyte lipid synthesis coupled to neuronal control of thermogenic programming. Mol. Metab. 6, 781–796 (2017).
Lodhi, I. J. et al. Inhibiting adipose tissue lipogenesis reprograms thermogenesis and PPARγ activation to decrease diet-induced obesity. Cell Metab. 16, 189–201 (2012).
Guilherme, A. et al. Neuronal modulation of brown adipose activity through perturbation of white adipocyte lipogenesis. Mol. Metab. 16, 116–125 (2018).
Van Veldhoven, P. Phytol-induced pathology in Acox2−/− mice. In Proc. 59th ICBL, Lipid Fluxes and Metabolism—From Fundamental Mechanisms to Human Disease 129 (International Conference on the Bioscience of Lipids, Helsinki, 2018).
Setchell, K. D. et al. Bile acid concentrations in human and rat liver tissue and in hepatocyte nuclei. Gastroenterology 112, 226–235 (1997).
Verkerke, A. R. P. et al. BCAA-nitrogen flux in brown fat controls metabolic health independent of thermogenesis. Cell 187, 2359–2374 (2024).
Steinberg, G. R. & Hardie, D. G. New insights into activation and function of the AMPK. Nat. Rev. Mol. Cell Biol. 24, 255–272 (2023).
Rahbani, J. F. et al. Creatine kinase B controls futile creatine cycling in thermogenic fat. Nature 590, 480–485 (2021).
Nakano, M. et al. Genetically encoded ratiometric fluorescent thermometer with wide range and rapid response. PLoS ONE 12, e0172344 (2017).
Pak, V. V. et al. Ultrasensitive genetically encoded indicator for hydrogen peroxide identifies roles for the oxidant in cell migration and mitochondrial function. Cell Metab. 31, 642–653 (2020).
Ferdinandusse, S. et al. A novel case of ACOX2 deficiency leads to recognition of a third human peroxisomal acyl-CoA oxidase. Biochim. Biophys. Acta Mol. Basis Dis. 1864, 952–958 (2018).
Shoeb, M., Ansari, N. H., Srivastava, S. K. & Ramana, K. V. 4-Hydroxynonenal in the pathogenesis and progression of human diseases. Curr. Med. Chem. 21, 230–237 (2014).
Bunk, J. et al. The Futile Creatine Cycle powers UCP1-independent thermogenesis in classical BAT. Nat. Commun. 16, 3221 (2025).
Mika, A. et al. A comprehensive study of serum odd- and branched-chain fatty acids in patients with excess weight. Obesity 24, 1669–1676 (2016).
Su, X. et al. Adipose tissue monomethyl branched-chain fatty acids and insulin sensitivity: effects of obesity and weight loss. Obesity 23, 329–334 (2015).
White, P. J. et al. Insulin action, type 2 diabetes, and branched-chain amino acids: a two-way street. Mol. Metab. 52, 101261 (2021).
Yoneshiro, T. et al. BCAA catabolism in brown fat controls energy homeostasis through SLC25A44. Nature 572, 614–619 (2019).
Lu, H. et al. Dietary sources of branched-chain fatty acids and their biosynthesis, distribution, and nutritional properties. Food Chem. 431, 137158 (2024).
Wang, Z. V., Deng, Y., Wang, Q. A., Sun, K. & Scherer, P. E. Identification and characterization of a promoter cassette conferring adipocyte-specific gene expression. Endocrinology 151, 2933–2939 (2010).
Hu, D. & Lodhi, I. J. Thermogenic phenotyping in mice. Methods Mol. Biol. 2662, 117–124 (2023).
Kriszt, R. et al. Optical visualisation of thermogenesis in stimulated single-cell brown adipocytes. Sci. Rep. 7, 1383 (2017).
Xue, R. et al. Clonal analyses and gene profiling identify genetic biomarkers of the thermogenic potential of human brown and white preadipocytes. Nat. Med. 21, 760–768 (2015).
Tatituri, R. V., Wolf, B. J., Brenner, M. B., Turk, J. & Hsu, F. F. Characterization of polar lipids of Listeria monocytogenes by HCD and low-energy CAD linear ion-trap mass spectrometry with electrospray ionization. Anal. Bioanal. Chem. 407, 2519–2528 (2015).
Zoncu, R. et al. mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H(+)-ATPase. Science 334, 678–683 (2011).
Acin-Perez, R. et al. A novel approach to measure mitochondrial respiration in frozen biological samples. EMBO J. 39, e104073 (2020).
McGowan, N. E., Scantlebury, D. M., Maule, A. G. & Marks, N. J. Measuring the emissivity of mammal pelage. Quant. InfraRed Thermogr. J. 15, 214–222 (2018).
McGowan, N. E. et al. Dietary effects on pelage emissivity in mammals: implications for infrared thermography. J. Therm. Biol. 88, 102516 (2020).
Han, J. et al. Metabolic profiling of bile acids in human and mouse blood by LC–MS/MS in combination with phospholipid-depletion solid-phase extraction. Anal. Chem. 87, 1127–1136 (2015).
Liu, X. et al. Peroxisomal metabolism of branched fatty acids regulates energy homeostasis. Figshare https://figshare.com/s/b8fd1fcaa83a4f3d0ac2 (2025).
Acknowledgements
This work was supported by National Institutes of Health grant nos. R01DK133344, R01DK115867, R01DK132239, GM103422, T32DK007120 and S10 OD032315. The core services of the Washington University Diabetes Research Center (grant no. DK020579) and the Nutrition Obesity Research Center (grant no. DK056341) also provided support for this work. The ACOX2−/− mouse model was generated by the FP7-funded European Infrafrontier-I3 project, granted to P.P.V.V.
Author information
Authors and Affiliations
Contributions
X.L. designed and conducted most of the in vitro and in vivo experiments and performed data analysis. A.H. generated Pexo-TEMP and ACOX2Adipo-OE targeting constructs and initially characterized the ACOX2 transgenic mice. D.L., D.H., M.T., A.B., P.G. and BA conducted experiments and/or performed data analysis. B.K. analysed gene expression and mass spectrometry data. B.N.F., M.Z., K.F., J.R.B., A.J., P.W., B.M., P.P.V.V., B.R. and C.F.S. designed experiments and/or provided reagents. I.J.L. conceived the study, secured research funding, designed experiments and performed data analysis. F.-F.H. performed mass spectrometry analysis. X.L. and I.J.L. wrote the paper. All authors read and edited the paper and provided comments.
Corresponding author
Ethics declarations
Competing interests
I.J.L. and X.L. are named on a provisional patent application (serial no. 63/872,889) filed by Washington University related to targeting ACOX2 activation as a treatment for obesity and related metabolic diseases. The other authors declare no competing interests.
Peer review
Peer review information
Nature thanks the anonymous reviewers for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 Characterization of ACOX2 function and regulation in adipocytes.
a), Classical peroxisomal BCFA β-oxidation pathway. For 2-methyl BCFAs, propionyl-CoA is released after the first round of β-oxidation. Created in PowerPoint. b, ACOX2 gene expression in different tissues of wild type mice housed at 4 °C or 22 °C for 7 days. N = 4. c, Gene expression analysis in sgACOX2 and control BAT SVF cells. N = 3. d, Oil Red O staining in ACOX2 KO and control brown adipocytes. Scale bar, 300 mm. e, Structures of C16:0 (palmitate), iso-C17:0, and anteiso-C17:0. f, Mass spectrometry analysis of various straight chain fatty acids in Acox2 KO and control brown adipocytes. n = 4. g, Volcano plot depicting Log2FC values of fatty acids in brown adipocytes overexpressing ACOX2 or GFP. N = 3. h, OCR measurement using Seahorse assay in ACOX2 or GFP-expressing WT brown adipocytes treated with the indicated fatty acid or BSA. N = 7. i, Gene expression analysis in mouse brown adipocytes overexpressing ACOX2 or GFP. N = 3. j, mtDNA copy number normalized to nuclear DNA. N = 6. k, ACOX2 gene expression following treatment with BSA, C16:0 or iso-C17:0 in differentiated mouse UCP1−/− BAT SVF cells (1 mM) or mature adipocytes isolated from fresh pig iWAT (2 mM) for 2 h. N = 3. Data are reported as the mean ± SEM. Data in b, c and f-k are from biologically independent samples. Two-sided unpaired Student’s t test in b, c, f, and i. Comparisons between groups in g were made with a two-tailed unpaired Student’s t-test adjusted for multiple comparisons using the Benjamini-Hochberg method. Two-way ANOVA with Tukey’s multiple comparison’s test in h. One-way ANOVA with Fisher’s LSD in k.
Extended Data Fig. 2 Localization of the mmBCFA synthetic proteins FASN and CRAT to peroxisomes.
a, Schematic of mmBCFA biosynthesis using short branched acylCoA (BrCoA) derived from mitochondrial catabolism of BCAA. b, Western blot analysis of CRISPR/Cas9-mediated KO of BCKDHα in brown adipocytes. N = 2. c-d, Mass spectrometry analysis of mmBCFA (c) and conventional fatty acids (d) in control and BCKDHA KO brown adipocytes. N = 4. e, mRNA levels of mmBCFA β-oxidation and synthesis genes in iWAT of WT mice maintained at thermoneutrality or 4 °C for 7 days. N = 4. f, FASN gene expression in control or norepinephrine (NE)-treated differentiated mouse UCP1−/− brown adipocytes. N = 3. g, FASN gene expression in cultured human brown-like adipocytes treated with or without forskolin. N = 3. h, Immunofluorescence analysis of peroxisomal localization of FASN in wild-type brown adipocytes after NE treatment for 0 or 2 h. N = 3. Scale bar, 10 mm. i, Immunofluorescence analysis of peroxisomal localization of FASN in BAT of WT mice treated with or without CL316,243. N = 3. Scale bar, 10 mm. j, Co-immunoprecipitation of HA-PEX7 with FLAG-FASN in HEK293T cells. k, Immunofluorescence analysis of peroxisomal localization of CRAT in wild-type brown adipocytes after NE treatment for 2 h. Scale bar, 10 mm. l, Western blot analysis of CRISPR/Cas9-mediated KO of CRAT in brown adipocytes. N = 3. m, Oil Red O staining in differentiated CRAT KO and control adipocytes. Scale bar, 300 mm. n,o, Mass spectrometry analysis of mmBCFA (n) and stCFA (o) in control and CRAT KO brown adipocytes. N = 3. p, OCR measurement in sgCRAT and control brown adipocytes at baseline and after sequential treatment with leucine, NE, and iso-C17:0. N = 8 (sgControl); N = 7 (sgCRAT). q, Gene expression analysis in sgCRAT and control brown adipocytes overexpressing ACOX2 or GFP. N = 3. r, OCR measurement in sgCRAT and control brown adipocytes overexpressing ACOX2 or GFP. Oligo, oligomycin; FCCP, carbonyl cyanide-p-trifluoromethoxyphenylhydrazone; AA + R, antimycin A and rotenone. N = 9. Data with error bars are mean ± SEM. Data in b–i, k, l, and n–r are from biologically independent samples. Statistical significance was determined by two-sided unpaired Student’s t test (c–i, k, n–o), two-way ANOVA with Sidak’s (p) or Tukey’s test (r), and one-way ANOVA with Fisher’s LSD (q). Representative images in h–k, and m are from two independent experiments. Panel a was created using BioRender (https://biorender.com).
Extended Data Fig. 3 Brown adipocyte-specific FASN knockout reduces mmBCFA levels and increases diet-induced obesity.
a, Western blot analysis of FASN in 4-hydroxytamoxifen (4-OHT)-treated FASNLox/Lox brown adipocytes transduced with retrovirus expressing GFP or Cre-ERT2. N = 3. b, Microscopy images of FASN-iKO and control BAT adipocytes prior to and after 4-OHT treatment. In the bottom panel, the cells were stained with Oil Red O. Scale bar, 75 mm (upper panel), 300 mm (lower panel). c, Mass spectrometry analysis of mmBCFA in control and FASN-iKO brown adipocytes. N = 3. d, Mass spectrometry analysis of various straight chain fatty acids in control and FASN-iKO brown adipocytes. N = 3. e, Body weight of HFD-fed FASN-BKO and control female mice. N = 7. f, Regression analysis of energy expenditure in HFD-fed mice. N = 7. g, Cumulative energy intake in HFD-fed FASN-BKO and control female mice. N = 4. h, Locomotor activity in FASN-BKO and control female mice. N = 4. Data are reported as mean ± SEM. Data in a and c-h are from biologically-independent samples. Images in b are representative of two separate experiments. Two-sided unpaired Student’s t test in c and d; two-way ANOVA with Fisher’s LSD in e; and two-way ANCOVA with Tukey’s test in f.
Extended Data Fig. 4 Global knockout of ACOX2 impairs thermogenesis and exacerbates diet-induced obesity and metabolic dysfunction.
a, ACOX2 gene expression in adipose tissue depots of ACOX2−/− and WT mice. N = 3. b, Western blot analysis of ACOX2 in BAT of ACOX2−/− and WT mice. N = 2. c, Core body temperature of ACOX2−/− and WT male mice after cold exposure (4 °C). N = 10. d, Gene expression analysis of proteins involved in UCP1-dependent and -independent pathways of thermogenesis in BAT of ACOX2−/− and WT mice. N = 5. e, Gene expression analysis of WT mice fed normal chow diet or HFD. N = 3. f, Body weight of HFD-fed ACOX2−/− and WT female mice. N = 7. g, Body weight of ACOX2−/− and WT male mice fed with HFD. N = 9. h, Body composition analysis of ACOX2−/− and WT male mice fed HFD. N = 9. i, Food intake of ACOX2−/− and WT mice. N = 5. j, Locomotor activity of ACOX2−/− and WT mice. N = 5. k, Tissue weights of adipose depots of HFD-fed ACOX2−/− and WT male mice. N = 9. l, Gross images of BAT and iWAT from HFD-fed ACOX2−/− and WT male mice. m, Histologic analysis of H&E-stained adipose tissue sections from HFD-fed ACOX2−/− and WT male mice. Scale bar, 75 mm. n, GTT analysis in HFD-fed ACOX2−/− and WT female mice. N = 7. o, ITT analysis in HFD-fed WT and ACOX2−/− female mice. N = 7. Data are reported as the mean ± SEM. Data in a-k and n-o are from biologically independent samples. Images in l and m are representative of 3 mice/group. Two-sided unpaired Student’s t test in a, d, e, and h-k; two-way ANOVA followed by Fisher’s LSD test in c, f, g and n-o.
Extended Data Fig. 5 Adipose-specific ACOX2 deletion impairs thermogenesis and promotes diet-induced obesity.
a, Gene expression analysis of ACOX1, ACOX3 and FASN in BAT of ACOX2-AKO and control mice. N = 4. b, Volcano plot depicting Log2FC values of fatty acids in BAT of ACOX2-AKO and control mice. N = 5. c, Total serum bile acids in ACOX2-AKO and control mice. N = 9. d, Core body temperature of ACOX2-AKO and control female mice after cold exposure (4 °C). N = 11. e, VO2 of female ACOX2-AKO (N = 6) and control mice (N = 7) after CL316,243 treatment. f, Body weight of HFD-fed ACOX2-AKO and control female mice. N = 8. g, Histologic analysis of H&E-stained adipose tissue sections from HFD-fed ACOX2−/− and WT male mice. Scale bar, 250 mm. h, Cumulative energy intake of ACOX2-AKO (N = 5) and control mice (N = 4). i, Locomotor activity of ACOX2-AKO (N = 5) and control mice (N = 4). j, Western blot analysis of insulin-stimulated AKT phosphorylation in BAT of liver of HFD-fed ACOX2-AKO and control mice. The bar graph shows quantification of AKT phosphorylation (N = 2). Data with error bars are reported as the mean ± SEM. Data in a-f and h-j are from biologically independent samples. Images in g are representative of 3 mice/genotype. Two-sided unpaired Student’s t test in a and c; two-sided unpaired Student’s t-test adjusted for multiple comparisons using the Benjamini-Hochberg method in b; two-way ANOVA with Fisher’s LSD in d-f.
Extended Data Fig. 6 Adipose-specific ACOX2 overexpression enhances thermogenesis and metabolic health.
a, Media glycerol levels of fresh BAT explants from ACOX2Adipo-OE (N = 5) or WT (N = 4) mice incubated in media containing vehicle or isoproterenol for 2 h. b, Core body temperature prior to and after cold exposure in ACOX2Adipo-OE and WT mice treated with 25 mg/kg BCAT-IN-2 or vehicle for 1 week. N = 7. c, Food intake of ACOX2Adipo-OE and WT mice. N = 7. d, Fecal FFA content of ACOX2Adipo-OE and WT mice. N = 6. e, Locomotor activity of ACOX2Adipo-OE and WT mice. N = 6. f, Body weight of HFD-fed ACOX2Adipo-OE and WT male mice. N = 8. g, Body composition analysis in HFD-fed ACOX2Adipo-OE and WT male mice. N = 8. h, GTT analysis in HFD-fed ACOX2Adipo-OE and WT male mice. N = 8. i, ITT analysis in HFD-fed ACOX2Adipo-OE and WT male mice. N = 8. Data are reported as mean ± SEM and are from biologically-independent samples. One-way ANOVA with Fisher’s LSD in a and b and two-way ANOVA with Fisher’s LSD in f, h, and i; Two-sided unpaired Student’s t test in g.
Extended Data Fig. 7 ACOX2 overexpression increases energy expenditure and improves metabolic health in UCP1−/− mice.
a, Relative FASN expression in BAT of UCP1−/− (N = 8) and UCP1−/−/ACOX2Adipo-OE (N = 7) mice. b, Immunofluorescence analysis and quantitative analysis of FASN peroxisomal localization in BAT of UCP1−/− and UCP1−/−/ACOX2Adipo-OE mice housed at thermoneutrality. N = 3. Scale bar, 10 mm. c-e, Indirect calorimetry analysis of (c) energy expenditure, (d) cumulative food intake, and (e) locomotor activity in ACOX2Adipo-OE (N = 8) and WT (N = 7) mice housed at thermoneutrality. f, Body weight of HFD-fed male UCP1−/− and UCP1−/−/ACOX2Adipo-OE mice fed mice housed at normal room temperature (22 °C). N = 7. g, Body composition analysis of HFD-fed UCP1−/− and UCP1−/−/ACOX2Adipo-OE mice housed at 22 °C. N = 7. h, Adipose tissue weights of HFD-fed UCP1−/− and UCP1−/−/ACOX2Adipo-OE mice housed at 22 °C. N = 7. i, Gross images of BAT from HFD-fed UCP1−/− and UCP1−/−/ACOX2Adipo-OE mice fed housed at 22 °C. j, H&E staining in adipose depots of HFD-fed UCP1−/− and UCP1−/−/ACOX2Adipo-OE mice housed at 22 °C. Scale bar, 75 mm. k, GTT analysis in HFD-fed UCP1−/− and UCP1−/−/ACOX2Adipo-OE female mice housed at 22 °C. N = 8. l, ITT analysis in HFD-fed UCP1−/− (n = 7) and UCP1−/−/ACOX2Adipo-OE (N = 8) female mice fed housed at 22 °C. Data are presented as mean ± SEM. Data in b-h, k, and l are from biologically independent samples. Images in i and j are representative of three mice per genotype. Statistical analysis was performed using two-sided unpaired Student’s t-test (a, b, g, h), two-way ANOVA with Tukey’s test (c), or Fisher’s LSD test (f, k, l).
Extended Data Fig. 8 Creatine cycling is dispensable for ACOX2-induced enhancement of oxygen consumption.
a, Activities of complex II and complex IV in isolated mitochondria from BAT of ACOX2Adipo-OE and WT mice measured using Seahorse (N = 9). b,c, Gene expression (b) and Western blot (c) analysis in sgCKB and control brown adipocytes overexpressing ACOX2 or GFP. N = 3 for panel b and N = 2 for panel c. d, OCR measurement in sgCKB and control brown adipocytes overexpressing ACOX2 or GFP and treated with iso-C17:0 or BSA (N = 12). e, OCR measurement using Seahorse in ACOX2 or GFP-expressing WT brown adipocytes treated with AA + R to block mitochondrial respiration, followed by treatment with C17:0-iso or BSA. N = 4. Data are from biologically-independent samples and are reported as mean ± SEM. One-way ANOVA with Fisher’s LSD in b and two-way ANOVA with Tukey’s test in d and e.
Extended Data Fig. 9 ACOX2 overexpression induces catalase to attenuate oxidative stress.
a, Temperature-dependent fluorescence spectra of Sirius fluorescent protein. b, Temperature-dependent fluorescence spectra of mTSapphire fluorescent protein. c, Fluorescence microscopy analysis and quantification of peroxisome-localized H2O2 sensor HyPer7-SKL in brown adipocytes transfected with lentivirus expressing ACOX2 or LacZ and treated with BSA, C17:0-iso, C16:0 or H2O2. N-3. Scale bar, 20 mm. d, Catalase gene expression in GFP or ACOX2-overexpressing brown adipocytes treated with BSA, C16:0 or iso-C17:0. N = 3. e, Western blot analysis of catalase knockout using CRISPR/Cas9 in brown adipocytes. N = 2. f, MTT assay in control and CAT KO brown adipocytes after treatment with BSA, iso-C17:0, C16:0 or H2O2. N = 12. g, 4-HNE levels in BAT of ACOX2Adipo-OE and WT mice. N = 7. Data are reported as the mean ± SEM. Data in c-g are from biologically independent samples. Images in c are representative of three separate experiments. Two-sided unpaired Student’s t test in c (quantification) and f. One-way ANOVA followed by Fisher’s LSD test in d. Panels a,b adapted from ref. 41, PLOS, under a Creative Commons licence CC BY 4.0.
Extended Data Fig. 10 UCP1-independent thermogenesis through an ATP-consuming cycle of monomethyl branched chain fatty acid (mmBCFA) synthesis and β-oxidation.
Mitochondrial catabolism of branched chain amino acids (BCAA) generates a short-branched acylCoA (BrCoA), such as isovaleryl-CoA. Carnitine acetyltransferase (CRAT), which is localized in mitochondria and peroxisomes, catalyses interconversion of branched-chain acylcarnitine and BrCoA, which is exported to the cytosol. The de novo lipogenesis enzyme FASN translocates to peroxisomes in response to acute cold exposure or β-adrenergic receptor stimulation and mediates synthesis of mmBCFA using BrCoA as a precursor. ACOX2 promotes β-oxidation of mmBCFA, generating heat in the process. Activation of the FASN–ACOX2 thermogenic pathway depletes ATP, triggering AMPK activation to promote compensatory ATP synthesis and sustain thermogenesis. Graphic created using BioRender (https://biorender.com).
Supplementary information
Supplementary Fig. 1
Uncropped raw western blots for main figures and extended data figures.
Supplementary Table 1
Oligonucleotide sequences used in this study, including primers for genotyping, cloning and quantitative real-time PCR.
Source data
Supplementary Data
Source Data Figs. 1–5, Extended Data Figs. 1–9.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Liu, X., He, A., Lu, D. et al. Peroxisomal metabolism of branched fatty acids regulates energy homeostasis. Nature (2025). https://doi.org/10.1038/s41586-025-09517-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41586-025-09517-7
This article is cited by
-
Novel UCP1-independent thermogenic mechanism identified
Nature Reviews Endocrinology (2025)
