
Abstract
The global demand for fine-flavour cocoa has increased worldwide during the last years. Fine-flavour cocoa offers exceptional quality and unique fruity and floral flavour attributes of high demand by the world's elite chocolatiers. Several studies have highlighted the relevance of cocoa fermentation to produce such attributes. Nevertheless, little is known regarding the microbial interactions and biochemistry that lead to the production of these attributes on farms of industrial relevance, where traditional fermentation methods have been pre-standardized and scaled up. In this study, we have used metagenomic approaches to dissect on-farm industrial fermentations of fine-flavour cocoa. Our results revealed the presence of a shared core of nine dominant microorganisms (i.e. Limosilactobacillus fermentum, Saccharomyces cerevisiae, Pestalotiopsis rhododendri, Acetobacter aceti group, Bacillus subtilis group, Weissella ghanensis group, Lactobacillus_uc, Malassezia restricta and Malassezia globosa) between two farms located at completely different agro-ecological zones. Moreover, a community metabolic model was reconstructed and proposed as a tool to further elucidate the interactions among microorganisms and flavour biochemistry. Our work is the first to reveal a core of microorganisms shared among industrial farms, which is an essential step to process engineering aimed to design starter cultures, reducing fermentation times, and controlling the expression of undesirable phenotypes.
Similar content being viewed by others


Introduction
Cocoa consumption has increased worldwide during the last years in response to its use as a high-quality ingredient in food and nutraceutical products1. Currently, the global market of cocoa is segmented into two different categories: bulk cocoa (95% of the global production) and fine-flavour cocoa (the remaining 5%)2. Fine-flavour cocoa refers to cocoa of exceptional quality and unique flavour attributes (e.g. fruity notes), a highly appreciated attribute by the world's elite chocolatiers. According to International Cocoa Organization (ICCO), 95% of the Colombian cocoa production is recognized as fine-flavour and its production is expected to reach 246,000 t by 2021 in response to the Cocoa development 10-year plan 2012–20213. Moreover, this crop has been in the spotlight for replacing illegal crops (Colombia’s Crop Substitution Program: From Coca to Cocoa) that aims at contributing to the economic development of farmers in high-risk areas, as fine-flavour cocoa offers a new range of unique fruity and floral tastes reaching high-value markets and a premium price4.
High-quality chocolate is traditionally obtained through extensive processing including on-farm spontaneous fermentation, drying, and roasting, which together play an essential role in the production of compounds affecting quality and taste5. More than 600 compounds including alcohols, aldehydes, ketones, esters, pyrazines, acids, peptides, and phenols have been reported to be involved in cocoa flavour as described by Ziegleder6 and reviewed by Aprotosoaie et al.2. Several studies have shown that these compounds can be produced directly during fermentation and kept stable during subsequent roasting stages7,8,9,10, while some of them experienced changes during drying and roasting11. Nevertheless, little is known regarding the microbial interactions and the biochemistry that lead to the production of such compounds during spontaneous cocoa fermentations. It has been described that spontaneous cocoa fermentations involves microorganism such as yeasts, lactic acid bacteria (LAB), acetic acid bacteria (AAB), filamentous fungi and spoilage bacteria12,13,14,15,16,17, which may come from the environment (e.g. air and insects), collecting tools, farmers hands, plant microbiota and the selected fermentation system (e.g. wooden boxes). On the other hand, spontaneous cocoa fermentations also depend on environmental parameters such as humidity, altitude, wind, rain, and temperature, which vary from one region to another and among different fermentation practices16,17.. Despite some traditional fermentation methodologies have been successfully standardized at large scales, cocoa bean fermentation remains a spontaneous/uncontrolled process. Thus, the identification of a core of dominant microorganisms at different locations is an essential step to design started cultures in controlled fermentations.
Up to now, several studies have focused on the identification of microorganisms during bulk cocoa fermentations17,18,19,20,21,22. Comprehensive studies regarding the fermentation of fine-flavour cocoa beans are rare9,23 and this field is still in its infancy. Very recently, the effect of different on-farm traditional fermentation protocols and environmental conditions in the composition of microbial communities have been studied in two different agro-ecological zones in Colombia23.The authors evaluated the effect of dissimilar fermentation protocols, climate, and other parameters on the microbial composition over time. The fermentation protocols at their selected farms were heterogeneous and even changed with the same farm at different seasons. Unfortunately, the authors do not state which clones or clones’ mixtures have been used for each fermentation and what could be their impact on microbial assembly. In this study, we are going one step further by using metagenomic approaches to dissect fermentations of fine-flavour cocoa on farms of industrial relevance (i.e. annual production exceeding 280 tons), where traditional fermentation methods have been scaled up using pre-designed mixtures of clones and reproducible fermentations methodologies. Instead of focusing on spontaneous fermentations on farms with dissimilar protocols, our goal was to identify a core of microorganism between industrial farms, where the same mixture of clones and fermentation methodologies have been used for many years to guarantee reproducibility. Thus, we have used a pre-standardized mixture of fine flavour clones, which is a standard used by the company to produce its origin chocolate. This approach allowed the identification and characterization of a common microorganism’s core shared between two different large-scale producing farms located at two completely different agro-ecological zones in Colombia. Dominant species and microbial groups have been also identified and analyzed regarding the product quality at each farm. Besides, we have reconstructed a genome-scale metabolic model departing from selected species of this core that were integrated to obtain a community metabolic model for the fermentation of fine-flavour cocoa.
Results and discussion
A platform to study on-farm industrial fermentations of fine-flavour cocoa
Cocoa bean fermentation has been extensively reported as a crucial step to produce flavour attributes and bioactive compounds of economical relevance in cocoa products5,6,8,10,11,24. Unlike other fermented foods, cocoa fermentation is an unstandardized/spontaneous process with a large variability all over the world. Currently, there is no universal standard methodology to perform cocoa fermentation and only a few international companies have done a substantial effort to scale up and optimize traditional approaches. Thus, a current challenge in cocoa research is the development of a large-scale standardized methodology that can be transferred to farms of industrial relevance. This is an important step to bring on-farm fermentations to industrial production scales, where controlled and reproducible protocols can be used to guarantee a standard product quality.
In this work, a platform to study on-farm industrial fermentations of fine-flavour cocoa (i.e. annual production exceeding 280 t) has been established as explained in the experimental section. Thus, two farms of industrial relevance were selected (i.e. Necoclí farm and Arauca farm). The selected farms are located at two completely different agro-ecological zones in Colombia (Fig. 1) and separated by the Andes Mountains (approximately 610 km from each other). The Necoclí farm was founded in 2010 by CASALUKER S.A and is situated on the Caribbean coast (8°28′48.24″N 76°40′59.86″W). It represents the largest fine-flavour cocoa crop in Colombia (550 ha) with an annual production of 280 t. Cocoa liquor from Necoclí farm is known to present an aroma with sour tones, light toasted notes, flavour with pronounced acidity (acetic and citric), a medium balance between cocoa and bitter, medium–low astringency, delicate sweet note (caramel and fruity), and slightly woody tones. In contrast, liquor from Arauca farm (7°01′01″N 71°25′01″W-COMPROCAR collection centre) is recognized for its unique fruity aroma, being the winner of several international chocolate awards during the last decade (e.g. Salon du Chocolat de París 2010–2011). It is known to present an aroma with sour and sweet notes, flavour with delicate sweet tones, and a medium balance between cocoa and bitter, lightly citric and fruity notes, low astringency, mild bitterness, and pronounced acidity. These sensory characteristics are frequently observed on the cocoa liquor produced at both farms where fermentation methodologies and clones’ mixtures are standardized. Sensory evaluations are regularly performed several times a year by the company, always producing the same distinguished profile. In this study, we have obtained the same sensory profiles by using the same methodology and clone’s mixture standardized at each farm described in the experimental section (Fig. 1).

Sensory profiles of cocoa liquor samples produced in Colombia by CASALUKER S.A., from a mixture of commercial clones of fine-flavour cocoa beans from Necoclí (A) and Arauca (B) farms (for more details on selected clones see experimental section). The flavour intensity increases from the centre to the perimeter. Data were obtained through a sensory analysis performed by a trained tasting panel of 6 members. Three replicates were performed. Mean values are shown.
In this study, two wooden large-scale box fermentations were arranged at each farm following the standard methodology used for the company and described in the experimental section. Fermentations at both farms were also studied by recording environmental parameters such as the temperature of the cocoa beans mass and pH (two frequently used parameters to verify proper fermentation at industrial farms). Thus, the temperature profile over fermentation was recorded and showed to be similar between the studied farms (Supplementary Fig. S1). The maximum temperature is always expected in the middle of the fermentation and it is used as a parameter to determine a good quality fermentations25. An increase in temperature during fermentation is frequently associated with ethanol production as an exothermic process25,26,27. The temperature of fermentation mass can vary from 25 to 55 °C depending on several parameters such as aeration, selected clones, fermentation methodology, room temperature, humidity, and other environmental parameters25,26,27. Similarly, the pH of the cocoa beans mass dropped from 6.5 to 6.6 during the first day to 5–5.6 at the end of fermentation at both farms (data not shown) as frequently reported in cocoa fermentation in response to the production of weak acids by acetic and lactic acid bacteria28.
Interesting variations in diversity and abundance of relevant groups of microorganisms over fermentation were found at the studied farms
In this work, we aimed to dissect industrial large-scale fine-flavour cocoa fermentation by using metagenomic approaches. Consequently, the variations of microbial diversity and composition in fermentations at the selected farms were determined as detailed explained in the experimental section.
Initially, the changes in the Shannon Diversity Index for bacteria and fungi during the fermentation of fine flavour cocoa beans at Necoclí and Arauca farms were recorded (Fig. 2). Our data revealed a decrease in bacterial and fungal diversity during fermentation at both farms, which is concomitant with the reported production of typical metabolites affecting microbial growth such as ethanol and weak acids29 and the observed changes in beans mass temperature (Supplementary Fig. S1). This may suggest the creation of a restrictive microenvironment during spontaneous industrial fermentations at both farms. Interestingly, a higher diversity of bacteria was always observed at Necoclí farm as compared to Arauca farms all over the tested replicates (Fig. 2A–C), while fungal diversity was similar between the studied farms (Fig. 2B–D).

Changes in the Shannon Diversity Index during the fermentation of fine flavour cocoa beans at Necoclí (A,B) and Arauca (C,D) farms. Data is shown for bacteria (A,C) and fungi (B,D). Beans samples were collected every 24 h over fermentation as described in the methods section.
To further dissect the microbial composition over fermentation, the relative abundance of selected groups of microorganisms sharing common metabolic characteristics was evaluated every day at the selected farms as mentioned in the experimental section. Selected microorganisms sharing metabolic traits were subsequently clustered in four groups: Lactic Acid Bacteria (LAB), Acetic Acid Bacteria (AAB), Yeasts, and Molds. Their relative abundance was recorded every 24 h until the end of fermentation (Fig. 3). Changes in the relative abundance of LAB and AAB over fine-flavour cocoa fermentations were observed all over the tested replicates at both farms. LABs are recognized as a crucial group of microorganisms in cocoa fermentations30. Our data revealed a higher abundance of these microorganisms at both farms during the first 3 days of fermentation, as reported in the literature28,29. The ability of LAB to transform pulp’s fermentable sugars and citric acid into lactic acid, mannitol, and pyruvate has been previously described31. Such metabolites have been reported to be essential for the release of sour taste during fermentation and spoilage control2,5,30,32,33. Indeed, the lactic acid produced by this group of bacteria is known to be an essential carbon source for the second group of relevant microorganisms during cocoa fermentation: the AAB30. Like LAB, AAB species have been previously reported to play an important role in flavour development5,34 and by affecting the microbial community structure 33,35. Moreover, their ability to convert ethanol into acetic acid is very important to allow the release of enzymes from the dead seeds, which are crucial to producing flavour precursors such as hydrophilic peptides, hydrophobic amino acids, polyphenols, and reducing sugars2,5,8,9. At both farms, the relative abundance of AAB was observed to increase over fermentation (Fig. 3) as previously reported28,29.

Relative abundance of selected microorganism groups sharing common metabolic and physiological characteristics during the fermentation of fine flavour cocoa beans. Schematic representation of the relative abundance of Acetic Acid Bacteria (orange bars) and Lactic Acid Bacteria (blue bars) and yeasts (green bars) and molds (red bars) identified at Necoclí farm (A,B: Necoclí farm, C,D: Arauca farm). Orange and blue lines were used to illustrate the behaviour of AAB and LAB over the fermentation, respectively. Each bar represents a replicate. At least two replicates per day were analysed.
The relative abundance of yeasts, a key group of microorganisms on cocoa fermentation, was also studied at the selected farms (Fig. 3B–D). Yeast species are known to play an important role to transform pulp’s fermentable sugars into ethanol during the first days of fermentation, where an anaerobic environment predominates12,28,33. This group of microorganisms has been also associated with the hydrolytic dissolution of pectin, which is an important step for the release of organic acids, aldehydes, and esters associated with flavour30. Interestingly, yeast flavour-active compounds have been widely associated with aroma in spirits, beers, and other fermented products36. Further metabolomic analysis will be required to highlight their relevance on flavour development during fine-flavour cocoa fermentations. The relative abundance of yeasts over the whole fermentation was similar at both farms (Fig. 3B–D). Our results are different from the observed on bulk cocoa fermentations28,30, where yeasts abundance is reduced during the last days of fermentation possibly due to the accumulation of weak acids 28,30. In this study, yeast abundance was high during the whole fermentation process at both farms. A possible explanation for the observed behaviour in this study relies on the fact that yeast strains display high variability in their response to weak acids, thus their abundance could not be strongly affected by acid concentration37. However, it is important to mention that no formal studies have compared the weak acid concentrations at different industrial and local farms. Moreover, the differences in the response to weak acids between yeast strains from different farms still have to be elucidated and additional studies will be required to further dissect this behaviour.
Finally, the changes in the relative abundance of mycotoxins-producing molds were evaluated at the selected farms (Fig. 3). Their relative abundance was always low during the whole fermentation at both farms.
Although our data allowed us to identify the variations on key groups of microorganisms during fermentation, we wanted to go one step further by identifying dominant microbial species at the selected farms, which is a crucial step for the design of starter cultures and process engineering. However, the design of starter cultures for cocoa bean fermentation will require to first achieve several challenges as recently reviewed by Figueroa-Hernández et al.38. For instance, it will be first necessary to standardize fermentations according to their location, the mixture of selected clones, and additional omics data.
A core of microorganisms shared between industrial farms at different agro-ecological zones was identified
Bacterial and fungal species were identified every day of fermentation as explained in the experimental section. Our data revealed a core of nine dominant microbial species shared between the studied farms (Fig. 4). Dominant microbial species were defined as those fulfilling the following criteria: (I) species detected all over the tested replicates, and (II) species with relative abundance higher than 1% of the total number of bacterial or fungal species. Thus, the relative abundance of selected dominant bacterial and fungal species was recorded every day of fermentation at each farm (Fig. 5 and Supplementary Fig. S2).

Schematic representation of dominant species observed during the fermentation of fine-flavour cocoa beans at Necoclí and Arauca farms. Dominant species were defined as species detected all over the replicates and showing a relative abundance higher than 1% from the total number of bacterial or fungal species. Data is shown for bacterial (A) and fungal species (B). Intersections on Venn Diagrams represent dominant species identified at both farms (here referred to as microorganism’s core). Bold highlighted names indicate the species selected for genome-scale metabolic model reconstruction that was later integrated to produce a community metabolic model for the fermentation of fine-flavour cocoa.

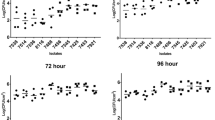
Relative abundance of dominant species of bacteria and fungi from the microorganism’s core during the fermentation of fine flavour cocoa beans at Necoclí (A,C,E and G) and Arauca (B,D,F and H) farms. Data are shown only for species included in the metabolic model (for remaining species included in the core see Supplementary Fig. S3). Dominant species were defined as species detected all over the replicates and showing a relative abundance higher than 1% from the total number of bacterial or fungal species. Each bar represents a replicate. At least two replicates per day have been analysed.
A total of 18 dominant microbial species were found at Necoclí farm, while 10 dominant species were found at Arauca farm (Fig. 4). Interestingly, all dominant bacterial species identified at Arauca farm (i.e. Acetobacter aceti group, Limosilactobacillus fermentum, Bacillus subtilis group, Weissella ghanensis group, and Lactobacillus_uc) were also found to dominate fermentation at Necoclí farm (Fig. 4A). Only four fungal dominant species were shared between the studied farms (i.e. Pestalotiopsis rhododendri, Saccharomyces cerevisiae, Malassezia restricta, and Malassezia globosa) (Fig. 4B).
The Acetobacter aceti group was found to dominate fermentation at both farms as shown in Fig. 5 and Supplementary Fig. S3. The relative abundance of this group of acetic acid bacteria (AAB) increased over fermentation (Fig. 5A,B), which is a frequently reported behaviour2,5,30. Interestingly, the Acetobacter aceti group was also found to be present and dominate the whole fermentation (Fig. 5A,B). This could partially explain the distinctive sour taste of chocolate liquor produced from both farms (Fig. 1). The role of this group of AAB on the release of sour taste during fermentation and spoilage control has been previously described2,5,30,32. In fact, acetic acid is recognized as a crucial driver of flavour cocoa development5,34 and a potential microbial inhibitor33,35. A similar role in the control of microbial populations33 and sour taste5 has been attributed to the lactic acid bacterium Limosilactobacillus fermentum, which is recognized as a representative lactic acid bacterium (LAB) in spontaneous cocoa bean fermentations39,40. Limosilactobacillus fermentum was identified as a dominant species during the first days of fermentation at both farms as shown in Fig. 5C,D. The lactic acid produced by LAB is recognized as an essential carbon source for AAB30. Moreover, it contributes together with AAB to the production of weak acids involved in embryo death, thus allowing the release of enzymes (e.g. proteases, lipases) present in the seeds and several flavour precursors2,5,8,9. In this work, we also identified the lactic acid bacterium Weissella ghanensis recently reported on Ghanaian cocoa fermentations41 and colombian coffee fermentations42. Weissella ghanensis showed similar behaviour to Limosilactobacillus fermentum being dominant only during the first days of fermentation (Supplementary Fig. S2A,B). This species is reported for the first time in Colombian cocoa fermentations.
Interestingly, our data also revealed the relevance of the Bacillus subtilis group during the fermentation of fine flavour cocoa at both farms (Fig. 4). This species group has been previously identified on fermented protein foods such as shoyu and miso43. As shown in Fig. 5 and Supplementary Fig. S3, it was found to dominate all over the fermentation at both farms. In fact, it was the main dominant on day 0 at Arauca farm and its abundance was relatively higher as compared to Necoclí farm (Fig. 5E,F). Although the presence of the Bacillus subtilis group on cocoa fermentations has been early described in1986 by Schwan et al.44, its metabolic contribution to fermentation still has to be elucidated.
In contrast to the Bacillus subtilis group, the role of Saccharomyces cerevisiae on spontaneous fermentation has been extensively described12,28,33. This species has been identified as dominant at both studied farms (Figs. 4 and 5). As expected, its relative abundance was shown to decrease over fermentation at Arauca farm (Fig. 5H). This behaviour has been associated with the production of inhibitory weak acids during the last days of fermentation28,29. Surprisingly, the abundance of Saccharomyces cerevisiae at Necoclí farm was always higher throughout the fermentation step (Fig. 5H). This could be partially explained by the fact that a relatively lower abundance of AAB was observed at this farm as compared to Arauca farm (Fig. 3A–C). However, additional studies will be required to further dissect this particular behaviour, for instance, it is known that the tolerance to acetic acid is a strain-dependent parameter with a large degree of variation among S. cerevisiae strains37.
It is important to mention that other fungal species were also identified as part of the core of the dominant microorganism. For example, our data revealed the presence of Pestalotiopsis rhododendri45 as a dominant species at both farms. This is the first report of this phytopathogen being a dominant relevant species on spontaneous cocoa fermentation. Further studies will be required to elucidate the role of this and other phytopathogens on cocoa fermentation. Similarly, two species of the human-related genus Malassezia46 were identified as dominant at both farms (Fig. 4 and Supplementary Fig. S2). This is also the first report of Malassezia globosa and Malassezia restricta being dominant on cocoa fermentation. Interestingly, these species can utilize fatty acids as a carbon source, which could affect the lipids profile over fermentation. Nevertheless, further research will be required to test this hypothesis and elucidate their role on cocoa fermentations.
In this study, we have identified a core of nine dominant microorganisms shared between the studied farms. This is a viable tool that can be used to design and develop starter cultures. This is an important step to switch from spontaneous to controlled on-farm fermentations. Nevertheless, additional experiments will be required to increase the number of studied farms and to identify the potential of each nominated species using metabolomics analysis. Besides, we have identified species that are present only on a particular farm (Fig. 4). Such unique species could be used to further explore the microbial differences that could influence variation on the flavour profiles among different agro-ecological zones.
The first community metabolic model for the fermentation of fine-flavour cocoa was reconstructed
A genome-scale metabolic community model was reconstructed to further dissect industrial fermentations of fine flavour cocoa. Hence, four dominant species were selected from the core of microorganisms shared between the studied farms: S. cerevisiae, B. subtilis, L. fermentum and A. aceti (Figs. 4 and 5). Three selection criteria were used to nominate these species: (I) to be present at both evaluated farms as shown in Fig. 4; (II) to represent a functional group of microorganisms involved in the production of typical cocoa fermentation metabolites (i.e. ethanol, acetic acid, and lactic acid) and (III) to dominate at several points through fermentation as shown in Supplementary Fig. S3.
To reconstruct a community metabolic model with these species, two previously reported metabolic models were initially selected: the iMM904 metabolic model for S. cerevisiae47 and the iYO844 metabolic model for B. subtilis48. The S. cerevisiae metabolic model iMM904 was selected as it is recognized as a model of preference to study central carbon metabolism and aminoacids biosynthesis49. It is a compartmentalized model composed of 1226 metabolites, 1577 reactions, and 905 genes.
Regarding the B. subtilis iYO844 metabolic model, it was selected as it represents the unique metabolic reconstruction reported at the BiGG database50 for this microorganism. It is composed of 990 metabolites, 1250 reactions, and 844 genes.
In the case of L. fermentum and A. aceti, de novo metabolic models had to be reconstructed in this study from their reported genomes. The L. fermentum str. IFO 3956 genome (GCF_000010145.1)51 and A. aceti str. TMW2.1153 genome (GCF_002005445.1)52 were then selected to reconstruct de novo metabolic models using the PATRIC database tools53,54. These genomes were selected as they were assembled as one contig, they were annotated into the PATRIC database, and their reconstruction resulted in a large number of reactions related to central carbon metabolism and amino acids biosynthesis. The resulting L. fermentum metabolic model is composed of 1238 metabolites, 1135 reactions, and 1030 genes. For Acetobacter aceti, the reconstructed metabolic model contains 1323 reactions, 1444 metabolites, and 1246 genes. Flux Balance Analysis (FBA)55 was then used to analyze the flow of metabolites into every reconstructed model, which converged to a solution of 29,053 gDW-1 h-1 for L. fermentum and 148,197 gDW-1 h-1 for A. aceti. Complete media was used as a standard to reconstruct each model as it contains all the required metabolites to fill the gaps. Future metabolomics analysis will be required to improve our model in terms of its biochemical composition.
Once these four independent models were obtained, they were integrated into a unique genome-scale community metabolic model (Supplementary File S1) using the SteadyCom optimization framework56. This model contains 5.432 reactions and 4.885 metabolites, thus allowing the identification of reactions of relevance to produce typical cocoa fermentation metabolites (Fig. 6) and flavour precursors (Fig. 7). As shown in Fig. 7, the reported model represents an essential tool for the identification of species-dependent reactions occurring during fermentation. Interestingly, the production of L-leucine is a fundamental step to produce 3-Methyl-2-butanol and 3-Methylbutanal (two important metabolites previously related to fruity and sweet chocolate flavour in fine flavour cocoa products2). While l-phenylalanine and l-tyrosine are related to the production of naringenin, an important precursor of antioxidant compounds of high relevance in the food industry57.

General metabolic network of the central carbon metabolism reconstructed at the community level. The genome-scale metabolic models of four dominant species shared between the studied farms (i.e. S. cerevisiae, B. subtilis, L. fermentum and A. aceti) were used to reconstruct a metabolic community model for fine flavour cocoa fermentation. Production pathways for lactate, ethanol, and acetate are shown (bold-highlighted). Black arrows represent metabolic reactions occurring in all the four species. Colours were used to indicate reactions that took place only on the indicated microorganism.

Network diagram of the amino acid synthesis pathway reconstructed at the community level. Production pathways for l-leucine, l-phenylalanine, l-tyrosine. l-tryptophan, valine and l-threonine are shown (bold-highlighted). These aminoacids have been recognized as crucial precursors of flavour/odour (e.g. 3-Methyl-2-butanol) and bioactive compounds (e.g. naringenin) in fine flavour cocoa. Black arrows represent metabolic reactions occurring in all the species of the community model (i.e. S. cerevisiae, B. subilis, L. fermentum and A. aceti). Colours were used to indicate reactions that took place only on the indicated species.
Our work represents an integrative approximation to understand the metabolic interactions of dominant species during the fermentation of fine-flavour cocoa. Up to now, research has been mainly focused on the reconstruction of individual metabolic models for the typical species of cocoa fermentation58,59. Recently, mathematical models of cocoa bean fermentation have been constructed based on current microbiological and biochemical knowledge28,29. Here, we go one step further by integrating data from genome-scale metabolic reconstructions of several dominant species found at two different farms of industrial relevance. Thus, a genome-scale community metabolic model for industrial fermentations of fine-flavour cocoa has been reconstructed. This model could be used as a platform for the identification of several species-dependent reactions of relevance for process engineering/fine-tuning. Our future work will focus on the construction of a dynamic model by integrating data from metabolomics analysis and including additional microorganisms identified in other industrial farms following similar fermentation protocols.
Conclusion
Our work reveal a core of microorganism shared between farms of industrial relevance. This is a crucial step to rational design starter cultures, reducing the fermentation times, and controlling the expression of undesirable phenotypes. Similar approaches could be applied to other farms of industrial relevance to further dissect fine-flavour cocoa fermentation at different locations. Moreover, our genome-scale community model is the first report of a community metabolic model for industrial fermentations of fine-flavour cocoa. Consequently, it represents a valuable platform to further study species-dependent reactions (Supplementary File S1), which could be further improved by including the reactions of new microorganisms and additional metabolomic/fluxomic data.
Experimental section
Cocoa beans fermentation
Cocoa beans used in this work were originated from a standard mixture of fine-flavour clones (i.e. FTA-2, FSA-13, FSV-41, Fear-5, ICS-39, ICS-95, EET-8, CAUC 37-39, TSH 565 and Luker40) harvested at two different large-scale producing farms in Colombia.
For further information regarding the selected clones, please refer to the International Cocoa Germplasm Database (Available at http://www.icgd.reading.ac.uk).
The selected industrial farms: Necoclí farm (8°28′48.24″N 76°40′59.86″W) and Arauca farm (7°01′01″N 71°25′01″W) are located at two different agro-ecological zones, which are separated by the Andes Mountains (approximately 610 km from each other).
During the main harvest season of 2018 (October–November), two industrial large-scale pre-standardized fermentations were performed at the selected farms. Cocoa pods were harvested, piled-up on the ground, and manually opened with a machete. All pods were harvested the same day. The cocoa pulp-bean mass was then scooped out from the pods by hand and collected into plastic barrels (load capacity ~ 50 kg). Once filled, barrels were immediately transferred to a customized fermentation room with pre-arranged wooden boxes. Two wooden boxes for fermentation were arranged at each farm. In Necoclí farm, 400 kg of cocoa pulp-bean mass were weighed and placed into each box. In the case of Arauca farm, a total of 350 kg of cocoa pulp-bean mass were weighed and transferred to fermentation boxes. Cocoa sweatings were drained away over fermentation through holes at the bottom of the boxes. The cocoa pulp-bean mass was mixed every 48 h to allow oxygenation using an iron shovel. All fermentations were completed after six days. Fermentation quality was confirmed by cut test evaluation of cocoa beans (ISO standard 2451:2017)60,61. It is important to mention, that these fermentation conditions are the standardized procedures always conducted at these farms.
Fermentation mass sampling
Samples of 10 beans were collected every 24 h starting at day 0 (i.e. just after placing the cocoa pulp-bean mass into each box) until the end of fermentation (day 6). Beans were collected from different points in the fermentation mass (e.g. surface and bottom) to ensure representative samples. On each farm, a total of six-replicates was collected every day, four of them from fermentation box 1 and the remaining two from fermentation box 2. Each sample was ground using a sanitized coffee grinder and subsequently stored at 4 °C in Nucleic Acid Preservation (NAP) Buffer62 until DNA extraction.
DNA extraction from fermentation samples
The extraction of DNA from fermentation samples was performed according to the protocol described by Ha et al.63. Briefly, DNA was extracted using DNA extraction buffer (Cetyl trimethyl ammonium bromide CTAB/Proteinase K) and precipitated on CTAB/NaCl. Then, ethanol was used to ensure the complete removal of CTAB, and the resulting DNA was finally eluted in Milli-Q water and checked for quality.
Library preparation and Illumina sequencing
Two amplicon libraries were prepared, using V3-V4 hypervariable regions for bacterial 16S rRNA gene, and the internal transcribed spacer ITS1-ITS5 for fungi. PCR reactions were performed in 25 μL containing 5X Hot FirePol Blend Master Mix (SOLIS BIODYNE), primers (Supplementary Table S1) at 0.4 μM, and 2 μL of DNA. PCRs were performed on BIORAD C100 thermal cyclers under the following conditions: initial denaturation at 95 °C for 5 min, followed by 35–40 cycles of 30 s at 95 °C, 30 s at the primer annealing temperature (Supplementary Table S1) and 1 min at 72 °C, and final extension for 10 min. For bacterial 16S amplification, the primers used already contain barcodes and Illumina adapters as previously reported64,65. For the ITS amplicons, Illumina adapters were ligated using the TruSeq Nano HT kit. Samples were purified using AMPure XP beads (BECKMAN COULTER) and normalized with the SequalPrep Normalization Plate Kit (APPLIED BIOSYSTEMS) for a final pooling into two libraries (16S and ITS respectively). DNA sequencing was carried out on Illumina MiSeq sequencer (ILLUMINA, San Diego, CA, USA), PE 2 × 250 using 500 cycle V2 MISEQ Reagent Kits and standard flow cells on an Illumina MiSeq platform located at the laboratory of CorpoGen Corporation (Bogotá, Colombia).
Taxonomic profiling
The paired-end reads generated by Illumina MiSeq were initially merged using the overlapping sequence information. Primer sequences and adapters were subsequently removed to obtain trimmed reads. To filter out those sequences with the low quality a quality filtering was performed using the EzBioCloud66 quality-filtering tool for bacteria, the FASTQC v0.11.9 (http://www.bioinformatics.babraham.ac.uk/projects/fastqc) tool, and ARB-SILVA67 for fungi. Thus, all sequences with lengths of < 100 bp or > 2,000 bp, averaged Q values lower than 30, and not predicted as 16S gene or ITS, were removed. Non-redundant reads were removed, and identical sequences were de-replicated to reduce processing time. These sequences were subsequently used to determine the taxonomic profile for each sample. In all cases, species rarefaction curves were completely saturated. The prokaryotic EzBioCloud 16S database (PKSSU4.0)68 and the UNITE fungal ITS database (https://unite.ut.ee/)69 were used to find and calculate sequence similarities using the VSEARCH program70 for bacteria and fungi, respectively. Different sequence similarity cut-offs were used for taxa identification as reported by Yarza et al.71, where 97% is taken as a cut-off for species-level identification. Chimeric sequences were detected and removed applying UCHIME72 algorithms to trusted non-chimeric reference databases.
Microbial analysis
The Shannon diversity index was calculated per each sample using the previously obtained OTUs (operational taxonomic units) information as reported73. Selected microorganisms sharing metabolic and physiological traits were clustered in four groups: Lactic Acid Bacteria, Acetic Acid Bacteria, Yeasts, and Molds. Clustering was performed based on the taxonomic profiling previously described, where selected genera were clustered according to their current classification: ALB31, AAB74, Yeasts, and Molds75. The relative abundance of these four groups of microorganisms was recorded every 24 h during fermentation starting at day 0 (i.e. just after placing the cocoa pulp-bean mass into each box) until the end of fermentation (day 6). Besides, the relative abundance of selected dominant bacterial and fungal species was also tracked over the fermentation. Dominant species were defined as species detected all over the replicates and showing a relative abundance higher than 1% from the total number of bacterial or fungal species.
Reconstruction of a genome-scale metabolic community model
The genome-scale metabolic models of Saccharomyces cerevisiae, Bacillus subtilis subsp. subtilis str. 168, Limosilactobacillus fermentum IFO 3956 and Acetobacter aceti were used to reconstruct a genome-scale metabolic community model for fine flavour cocoa fermentation. These microorganisms have been selected for the community model as they were identified as dominant species shared between the studied farms. Two of these models (i.e. Saccharomyces cerevisiae model iMM90447 and Bacillus subtilis subsp. subtilis str. 168 iYO844 model48) have been previously reported, while metabolic models for Limosilactobacillus fermentum and Acetobacter aceti were reconstructed from Limosilactobacillus fermentum str. IFO 3956 genome (GCF_000010145.1)51 and Acetobacter aceti str. TMW2.1153 genome (GCF_002005445.1)52, respectively. De novo metabolic models were reconstructed using the PATRIC database53,54 with Reconstruction Metabolic Model Services. A completed media parameter was chosen for metabolic reconstructions. Flux Balance Analysis-FBA55 was performed for each of the four metabolic models. The SteadyCom optimization framework56 was used to reconstruct a community metabolic model. The createCommModel() function was applied to integrate the four independent metabolic models into a unique community model by giving them a different set of identifiers for each organism, which resembles a model with separate compartments. Minor corrections were made to the community model to fix reaction directions. No changes for uptake systems were made. ESCHER tool76 was used to generate metabolic maps and further edited using Inkscape software (0.92.5 version).
Cocoa liquor production and sensory evaluation
The fermented cocoa beans at each farm were sun-dried, roasted, and converted into cocoa liquor by CASALUKER S.A (Bogotá, Colombia) following their standardized protocols. Briefly, cocoa liquor is obtained by subsequent steps of cleaning (i.e. Nibs are separated from shells), roasting, cooling, winnowing, grinding, tempering, molding, and storage for further analysis. Details on liquor production are protected as casaluker´s intellectual property. Sensory evaluation of cocoa liquor was performed by a trained tasting panel of 6 members of CASALUKER S.A. This a regular standardized procedure performed several times a year at the company using liquor produced with material arriving from these farms. Flavour descriptors included acid/sour, astringent, cocoa, bitter, fruity, floral, nutty, moldy, green, malt and others. The analysis was performed in triplicate with a temperature of evaluation between 45–50 °C. The flavour intensity was expressed as numerical values ranging from 0 to 9.
Data availability
All data generated or analyzed during this study are included in this published article (and its Supplementary Information files).
References
Beg, M. S., Ahmad, S., Jan, K. & Bashir, K. Status, supply chain and processing of cocoa—a review. Trends Food Sci. Technol. 66, 108–116 (2017).
Aprotosoaie, A. C., Luca, S. V. & Miron, A. Flavor chemistry of cocoa and cocoa products-an overview. Compr. Rev. Food Sci. Food Saf. 15, 73–91 (2016).
Ortiz-R, O. O., Gallardo, R. A. V. & Rangel, J. M. Applying life cycle management of colombian cocoa production. Food Sci. Technol. 34, 62–68 (2014).
Kadow, D., Bohlmann, J., Phillips, W. & Lieberei, R. Identification of main fine or flavour components in two genotypes of the cocoa tree (Theobroma cacao L.). J. Appl. Bot. Food Qual. 86, 90–98 (2013).
Afoakwa, E. O., Paterson, A., Fowler, M. & Ryan, A. Flavor formation and character in cocoa and chocolate: a critical review. Crit. Rev. Food Sci. Nutr. 48, 840–857 (2008).
Ziegleder, G. Flavour development in cocoa and chocolate. In Industrial Chocolate Manufacture and Use (ed. Beckett, S. T.) 169–191 (Wiley-Blackwell, 2009). https://doi.org/10.1002/9781444301588.ch8.
Voigt, J., Heinrichs, H., Voigt, G. & Biehl, B. Cocoa-specific aroma precursors are generated by proteolytic digestion of the vicilin-like globulin of cocoa seeds. Food Chem. 50, 177–184 (1994).
Toker, O. S., Palabiyik, I., Pirouzian, H. R., Aktar, T. & Konar, N. Chocolate aroma: factors, importance and analysis. Trends Food Sci. Technol. 99, 580–592 (2020).
del Brunetto, M. R. et al. The effect of fermentation and roasting on free amino acids profile in Criollo cocoa (Theobroma cacao L.) grown in Venezuela. Braz. J. Food Technol. 23, 1–12 (2020).
Rohan, T. A. The precursors of chocolate aroma: a comparative study of fermented and unfermented cocoa beans. J. Food Sci. 29, 456–459 (1964).
Rohan, T. A. & Stewart, T. The precursors of chocolate aroma: production of free amino acids during fermentation of cocoa beans. J. Food Sci. 32, 395–398 (1967).
Thi, V., Ho, T., Zhao, J. & Fleet, G. Yeasts are essential for cocoa bean fermentation. Int. J. Food Microbiol. https://doi.org/10.1016/j.ijfoodmicro.2013.12.014 (2013).
Illeghems, K., Vuyst, D., Papalexandratou, L. & Weckx, Z. Phylogenetic analysis of a spontaneous cocoa bean fermentation metagenome reveals new insights into its bacterial and fungal community diversity. PLoS ONE 7, 38040 (2012).
Illeghems, K., Weckx, S. & De Vuyst, L. Applying meta-pathway analyses through metagenomics to identify the functional properties of the major bacterial communities of a single spontaneous cocoa bean fermentation process sample. Food Microbiol. https://doi.org/10.1016/j.fm.2015.03.005 (2015).
Papalexandratou, Z., Vrancken, G., De Bruyne, K., Vandamme, P. & De Vuyst, L. Spontaneous organic cocoa bean box fermentations in Brazil are characterized by a restricted species diversity of lactic acid bacteria and acetic acid bacteria. Food Microbiol. https://doi.org/10.1016/j.fm.2011.06.003 (2011).
Pereira, G. V. M., Miguel, M. G. C. P., Ramos, C. L. & Schwan, R. F. Microbiological and physicochemical characterization of small-scale cocoa fermentations and screening of yeast and bacterial strains to develop a defined starter culture. Appl. Environ. Microbiol. https://doi.org/10.1128/AEM.01144-12 (2012).
Bortolini, C., Patrone, V., Puglisi, E. & Morelli, L. Detailed analyses of the bacterial populations in processed cocoa beans of different geographic origin, subject to varied fermentation conditions. Int. J. Food Microbiol. 236, 98–106 (2016).
Meersman, E. et al. Detailed analysis of the microbial population in Malaysian spontaneous cocoa pulp fermentations reveals a core and variable microbiota. PLoS ONE 8, e81559 (2013).
Ardhana, M. M. & Fleet, G. H. The microbial ecology of cocoa bean fermentations in Indonesia. Int. J. Food Microbiol. 86, 87–99 (2003).
Serra, J. L. et al. Determination of the microbial community in Amazonian cocoa bean fermentation by Illumina-based metagenomic sequencing. LWT 106, 229–239 (2019).
Agyirifo, D. S. et al. Metagenomics analysis of cocoa bean fermentation microbiome identifying species diversity and putative functional capabilities. Heliyon 5, e02170 (2019).
Mota-Gutierrez, J. et al. Dynamics and biodiversity of bacterial and yeast communities during fermentation of cocoa beans. Appl. Environ. Microbiol. 84, 1–17 (2018).
Pacheco-Montealegre, M. E., Dávila-Mora, L. L., Botero-Rute, L. M., Reyes, A. & Caro-Quintero, A. Fine resolution analysis of microbial communities provides insights into the variability of cocoa bean fermentation. Front. Microbiol. 11, 650 (2020).
Jinap, S., Ikrawan, Y., Bakar, J., Saari, N. & Lioe, H. N. Aroma precursors and methylpyrazines in underfermented cocoa beans induced by endogenous carboxypeptidase. J. Food Sci. 73, H141–H147 (2008).
De Vuyst, L. & Weckx, S. The cocoa bean fermentation process: from ecosystem analysis to starter culture development. J. Appl. Microbiol. 121, 5–17 (2016).
Hartuti, S., Karyadi, J. N. W. & Bintoro, N. Effect of aeration on temperature and index fermentation of cocoa beans using a fermentor packed bed. In Proceeding of the 1st International Conference on Tropical Agriculture. 639–646 (Springer International Publishing, 2017). https://doi.org/10.1007/978-3-319-60363-6_64.
Hernández-Hernández, C., López-Andrade, P. A., Ramírez-Guillermo, M. A., Guerra Ramírez, D. & Caballero Pérez, J. F. Evaluation of different fermentation processes for use by small cocoa growers in Mexico. Food Sci. Nutr. 4, 690–695 (2016).
Kresnowati, M. T. A. P., Gunawan, A. Y. & Muliyadini, W. Kinetics model development of cocoa bean fermentation. In AIP Conference Proceedings 1699, 030004 (American Institute of Physics Inc., 2015).
Moreno-Zambrano, M., Grimbs, S., Ullrich, M. S. & Hütt, M. T. A mathematical model of cocoa bean fermentation. R. Soc. Open Sci. 5, 180964 (2018).
De Vuyst, L. & Leroy, F. Functional role of yeasts, lactic acid bacteria, and acetic acid bacteria in cocoa fermentation processes. FEMS Microbiol. Rev. https://doi.org/10.1093/femsre/fuaa014 (2020).
Holzapfel, W. H. & Wood, B. J. B. Lactic Acid Bacteria: Biodiversity and Taxonomy. Lactic Acid Bacteria: Biodiversity and Taxonomy (Wiley Blackwell, 2014).
Schwan, R. F. Cocoa fermentations conducted with a defined microbial cocktail inoculum. Appl. Environ. Microbiol. 64, 1477–1483 (1998).
Schwan, R. F. & Wheals, A. E. The microbiology of cocoa fermentation and its role in chocolate quality. Crit. Rev. Food Sci. Nutr. 44, 205–221 (2004).
Adler, P. et al. The key to acetate: metabolic fluxes of acetic acid bacteria under cocoa pulp fermentation-simulating conditions. Appl. Environ. Microbiol. 80, 4702–4716 (2014).
Camu, N. et al. Influence of turning and environmental contamination on the dynamics of populations of lactic acid and acetic acid bacteria involved in spontaneous cocoa bean heap fermentation in Ghana. Appl. Environ. Microbiol. 74, 86–98 (2008).
Carrau, F., Boido, E. & Dellacassa, E. Yeast diversity and flavor compounds. In Fungal Metabolites (eds Mérillon, J.-M. & Ramawat, K. G.) 569–597 (Springer, 2017).
Swinnen, S., Fernández-Niño, M., González-Ramos, D., van Maris, A. J. A. & Nevoigt, E. The fraction of cells that resume growth after acetic acid addition is a strain-dependent parameter of acetic acid tolerance in Saccharomyces cerevisiae. FEMS Yeast Res. 14, 642–653 (2014).
Figueroa-Hernández, C. et al. The challenges and perspectives of the selection of starter cultures for fermented cocoa beans. Int. J. Food Microbiol. 301, 41–50 (2019).
Papalexandratou, Z. et al. Hanseniaspora opuntiae, Saccharomyces cerevisiae, Lactobacillus fermentum, and Acetobacter pasteurianus predominate during well-performed Malaysian cocoa bean box fermentations, underlining the importance of these microbial species for a successful cocoa. Food Microbiol. 35, 73–85 (2013).
Schwendimann, L., Kauf, P., Fieseler, L., Gantenbein-Demarchi, C. & Miescher Schwenninger, S. Development of a quantitative PCR assay for rapid detection of Lactobacillus plantarum andLactobacillus fermentum in cocoa bean fermentation. J. Microbiol. Methods 115, 94–99 (2015).
De Bruyne, K., Camu, N., Lefebvre, K., De Vuyst, L. & Vandamme, P. Weissella ghanensis sp. Nov., isolated from a Ghanaian cocoa fermentation. Int. J. Syst. Evol. Microbiol. 58, 2721–2725 (2008).
de Oliveira Junqueira, A. C. et al. First description of bacterial and fungal communities in Colombian coffee beans fermentation analysed using Illumina-based amplicon sequencing. Sci. Rep. 9, 1–10 (2019).
Yokotsuka, T. & Sasaki, M. Fermented protein foods in the Orient: shoyu and miso in Japan. In Microbiology of Fermented Foods 351–415 (Springer US, 1998). https://doi.org/10.1007/978-1-4613-0309-1_12.
Schwan, R. F., Vanetti, M. C. D., Silva, D. O., Lopez, A. & Moraes, C. A. Characterization and distribution of aerobic, spore-forming bacteria from cacao fermentations in Bahia. J. Food Sci. 51, 1583–1584 (1986).
Yan-min, Z., Maharachchikumbura, S. S. N., Qing, T. & Hyde, K. D. Pestalotiopsis species on ornamental plants in Yunnan Province, China. (2013).
Amend, A. Pearls from dandruff to deep-sea vents: Malassezia-like fungi are ecologically hyper-diverse. PLoS Pathog. 10, e1004277 (2014).
Mo, M. L., Palsson, B. & Herrgård, M. J. Connecting extracellular metabolomic measurements to intracellular flux states in yeast. BMC Syst. Biol. 3, 37 (2009).
Oh, Y.-K., Palsson, B. O., Park, S. M., Schilling, C. H. & Mahadevan, R. Genome-scale reconstruction of metabolic network in Bacillus subtilis based on high-throughput phenotyping and gene essentiality data. J. Biol. Chem. 282, 28791–28799 (2007).
Lopes, H. & Rocha, I. Genome-scale modeling of yeast: chronology, applications and critical perspectives. FEMS Yeast Res. 17, 1–14 (2017).
Whetten, J. L., Williamson, P. C., Heo, G., Varnhagen, C. & Major, P. W. BiGG models: a platform for integrating, standardizing and sharing genome-scale models. Am. J. Orthodontics Dentofac. Orthop. https://doi.org/10.1016/j.ajodo.2005.02.022 (2015).
Morita, H. et al. Comparative genome analysis of Lactobacillus reuteri and Lactobacillus fermentum reveal a genomic island for reuterin and cobalamin production. DNA Res. 15, 151–161 (2008).
Lu, J. & Salzberg, S. L. Removing contaminants from databases of draft genomes. PLoS Comput. Biol. 14, e1006277 (2018).
Gillespie, J. J. et al. PATRIC: the comprehensive bacterial bioinformatics resource with a focus on human pathogenic species. Infect. Immun. 79, 4286–4298 (2011).
Wattam, A. R. et al. Improvements to PATRIC, the all-bacterial bioinformatics database and analysis resource center. Nucleic Acids Res. 45, D535–D542 (2017).
Orth, J. D., Thiele, I. & Palsson, B. O. What is flux balance analysis?. Nat. Biotechnol. 28, 245–248 (2010).
Chan, S. H. J., Simons, M. N. & Maranas, C. D. SteadyCom: predicting microbial abundances while ensuring community stability. PLoS Comput. Biol. 13, e1005539 (2017).
Spinnler, H. E. Flavors from amino acids. In Food Flavors: Chemical, Sensory and Technological Properties 121–136 (2011). https://doi.org/10.1201/b11187-7.
Illeghems, K., De Vuyst, L. & Weckx, S. Complete genome sequence and comparative analysis of Acetobacter pasteurianus 386B, a strain well-adapted to the cocoa bean fermentation ecosystem. BMC Genom. 14, 47–76 (2013).
Pelicaen, R., Gonze, D., Teusink, B., De Vuyst, L. & Weckx, S. Genome-scale metabolic reconstruction of Acetobacter pasteurianus 386B, a candidate functional starter culture for cocoa bean fermentation. Front. Microbiol. 10, 2801 (2019).
Schwan, R. F., Fleet, G. H. & Fleet, G. H. Methods of Cocoa Fermentation and Drying. 90–147 (2014). https://doi.org/10.1201/B17536-8.
Santos, F. A., Palmeira, E. S. & Jesus, G. J. An image dataset of cut-test-classified cocoa beans. Data Br. 24, 103916 (2019).
Camacho-Sanchez, M., Burraco, P., Gomez-Mestre, I. & Leonard, J. A. Preservation of RNA and DNA from mammal samples under field conditions. Mol. Ecol. Resour. 13, 663–673 (2013).
Ha, L. T. V., Vanlerberghe, L., Toan, H. T., Dewettinck, K. & Messens, K. Comparative evaluation of six extraction methods for DNA quantification and PCR detection in cocoa and cocoa-derived products. Food Biotechnol. 29, 1–19 (2015).
Kozich, J. J., Westcott, S. L., Baxter, N. T., Highlander, S. K. & Schloss, P. D. Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the miseq illumina sequencing platform. Appl. Environ. Microbiol. 79, 5112–5120 (2013).
Epp, L. S. et al. New environmental metabarcodes for analysing soil DNA: potential for studying past and present ecosystems. Mol. Ecol. 21, 1821–1833 (2012).
Yoon, S. H. et al. Introducing EzBioCloud: a taxonomically united database of 16S rRNA gene sequences and whole-genome assemblies. Int. J. Syst. Evol. Microbiol. 67, 1613–1617 (2017).
Pruesse, E. et al. SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res. 35, 7188–7196 (2007).
Park, S.-C. & Won, S. Evaluation of 16S rRNA databases for taxonomic assignments using a mock community. Genom. Inform. 16, e24 (2018).
Nilsson, R. H. et al. The UNITE database for molecular identification of fungi: handling dark taxa and parallel taxonomic classifications. Nucleic Acids Res. 47, D259–D264 (2019).
Rognes, T., Flouri, T., Nichols, B., Quince, C. & Mahé, F. VSEARCH: A versatile open source tool for metagenomics. PeerJ 2016, e2584 (2016).
Yarza, P. et al. Uniting the classification of cultured and uncultured bacteria and archaea using 16S rRNA gene sequences. Nat. Rev. Microbiol. 12, 635–645 (2014).
Edgar, R. C. Accuracy of taxonomy prediction for 16S rRNA and fungal ITS sequences. PeerJ 2018, e4652 (2018).
Morris, E. K. et al. Choosing and using diversity indices: insights for ecological applications from the German biodiversity exploratories. Ecol. Evol. 4, 3514–3524 (2014).
Gomes, R. J., de Borges, M. F., de Rosa, M. F., Castro-Gómez, R. J. H. & Spinosa, W. A. Acetic acid bacteria in the food industry: systematics, characteristics and applications. Food Technol. Biotechnol. 56, 139–151 (2018).
Pleadin, J., Frece, J. & Markov, K. Mycotoxins in food and feed. In Advances in Food and Nutrition Research 89, 297–345 (Academic Press Inc., 2019).
King, Z. A. et al. Escher: a web application for building, sharing, and embedding data-rich visualizations of biological pathways. PLoS Comput. Biol. 11, e1004321 (2015).
Acknowledgements
We thank the researchers at Grupo de Diseño de Productos y Procesos (GDPP) for their constant contributions, correct observations, and great knowledge.
Author information
Authors and Affiliations
Contributions
Conceptualization: M.F.N., M.J.C., P.D.P., S.R. and A.F.G.B. Data curation: M.F.N., M.J.R.C., F.H.R. and J.M.A. Formal analysis: M.F.N., M.J.R.C., F.H.R. and J.M.A.. Funding acquisition: M.F.N., S.R., M.J.C., H.H.O. and A.F.G.B. Investigation: M.F.N., M.J.R.C., F.H.R., J.M.A., M.L.C.H., J.L.A.M., M.J.C., H.H.O., C.R.L., D.C., A.R., P.D.P., S.R. and A.F.G.B. Methodology: M.F.N., M.J.R.C., F.H.R., M.L.C.H., J.L.A.M., P.D.P., D.C., J.M.A., C.R.L. and A.F.G.B. Project administration: M.J.C., M.F.N. and A.F.G.B. Supervision & Validation: M.F.N. and A.F.G.B. Visualization: M.F.N., M.J.R.C., F.H.R. and M.J.C. Writing ± original draft: M.F.N., M.J.R.C., F.H.R., J.M.A., M.J.C. and A.F.G.B. Writing ± review & editing: M.F.N., M.J.R.C., F.H.R., J.M.A., M.L.C.H., J.L.A.M., M.J.C., H.H.O., C.R.L., D.C., A.R., P.D.P., S.R. and A.F.G.B. M.F.N. and M.J.R.C. contributed in the same way. All authors have approved the final version.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Fernández-Niño, M., Rodríguez-Cubillos, M.J., Herrera-Rocha, F. et al. Dissecting industrial fermentations of fine flavour cocoa through metagenomic analysis. Sci Rep 11, 8638 (2021). https://doi.org/10.1038/s41598-021-88048-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-88048-3
This article is cited by
-
A defined microbial community reproduces attributes of fine flavour chocolate fermentation
Nature Microbiology (2025)
-
Dissecting fine-flavor cocoa bean fermentation through metabolomics analysis to break down the current metabolic paradigm
Scientific Reports (2021)
