
Abstract
Fibrosis is involved in 45% of deaths in the United States, and no treatment exists to reverse the progression of lung or kidney fibrosis. Myofibroblasts are key to the progression and maintenance of fibrosis. We investigated features of cell adhesion necessary for monocytes to differentiate into myofibroblasts, seeking to identify pathways key to myofibroblast differentiation. Blocking antibodies against integrins α3, αM, and αMβ2 de-differentiate myofibroblasts in vitro, lower the pro-fibrotic secretome of myofibroblasts, and treat lung fibrosis and inhibit kidney fibrosis in vivo. Decorin’s collagen-binding peptide can be used to direct functionalized blocking antibodies (against integrins-α3, -αM, -αMβ2) to both fibrotic lungs and fibrotic kidneys, reducing the dose of antibody necessary to treat fibrosis. This targeted immunotherapy blocking key integrins may be an effective therapeutic for the treatment of fibrosis.
Similar content being viewed by others


Introduction
Fibrosis is defined by dysregulated extracellular matrix (ECM) deposition leading to scar tissue deposition and increases in tissue stiffness1. Fibrosing diseases—including pulmonary fibrosis, and end-stage kidney disease—are involved in 45% of deaths in the United States1,2. There are only two FDA approved treatments for fibrosis, pirfenidone and nintedanib2,3. Pirfenidone and nintedanib slow, but do not reverse, the progression of fibrosis4, with mechanisms of action that are poorly understood5.
A major goal of research in fibrosis is developing a treatment capable of reversing established fibrosis1. To our knowledge, only one treatment (recombinant pentraxin-2, PRM-151) has thus far shown even a modest ability to reverse even some symptoms of fibrosis in some patients6. To fully reverse fibrosis, it would be beneficial for several cell and ECM-related changes to occur: myofibroblast de-activation would reduce the stiffness of the tissue, a cessation of collagen deposition would stop the progression of fibrosis, and remodeling of existing ECM would allow for a return to healthy tissue function. Interrupting collagen deposition alone can destabilize scar tissue1, and monocyte-derived cells are capable of removing deposited ECM while regenerating tissue7.
Myofibroblasts are cells that adopt a spindle-shaped morphology and actively increase surface stiffness and tension through exertion of force through their cytoskeleton’s attachment to the extracellular matrix. Myofibroblasts also secrete pro-fibrotic factors and collagen1,8,9,10. Myofibroblasts are a heterogenous population that arise from multiple progenitor cells, and the term “myofibroblast” denotes the function of a cell rather than its origin11. Myofibroblasts can arise from several progenitors, including hepatocytes, fibroblasts, epithelial cells, and monocytes1,11,12.
Myofibroblasts can be formed or recruited in response to injury12,13. Persistent myofibroblast activation is associated with tissue damage being resolved by scarring and fibrosis, while myofibroblast de-activation is associated with tissue damage being resolved in regeneration9. Myofibroblasts can de-activate through apoptosis14 or de-differentiation12,15.
Uniquely among myofibroblast precursors, monocytes can be recruited from the bloodstream to different fibrotic tissues in the body16. These monocyte-derived myofibroblasts may provide up to a third of the total number of myofibroblasts in liver17, kidney18, lung19,20, and skin21 fibroses. Whether monocyte-derived cells assume an anti-fibrotic or a pro-fibrotic differentiation state can determine whether tissue damage is resolved with regeneration or progresses to fibrosis22,23. The broad range of signaling that monocyte-derived cells are capable of may explain the importance of monocyte-derived myofibroblasts in the progression of fibrosis22,23,24, despite their smaller proportion of cells25,26,27,28.
Integrins are membrane glycoproteins that are involved in mediating interactions between cells and ECM surfaces29. Integrins comprise 18 α subunits and 8 β subunits, which combine to form 24 αβ heterodimers30. Each αβ heterodimer has a unique binding profile, which allows integrins a broad array of ligands, including ECM proteins, soluble ligands, or other membrane proteins30. Integrins transduce signals from the ECM to the cytoskeleton in what is known as “outside-in” signaling, and transduce force from the cytoskeleton to the ECM in “inside-out” signaling29.
Integrins can be activated by extracellular interactions that induce conformational changes to the integrin’s intracellular domains. Conversely, integrins can be activated by intracellular cytoskeletal interactions that increase the integrin’s ligand binding through conformational changes to its extracellular domains29,31. Among the proteins capable of activating integrins are the intracellular tension-sensing proteins talin1 and talin232. Talins activate integrins through interactions with the β integrin tail29, which induces a conformational change to the αβ integrin’s extracellular domains that increases affinity for ligands.
Focal adhesions (FAs) are subcellular structures rich in integrins that bind to the extracellular ECM, and intracellular talins, which in turn bind to the cytoskeleton33,34,35. FAs are dynamic, deformable, and responsive structures at the cell membrane that integrate signals and transmit/transduce force from the intracellular actin cytoskeleton, transmembrane integrins, and ECM36,37,38,39. Integrins interact with multiple ECM proteins36,37 and are essential to both cellular tension sensing, the formation of FAs, and the maintenance of FAs40. FAs are transient in migrating cells, but can become super-mature in myofibroblasts34. Super-mature FAs are both physically larger34 and have increased concentrations of integrins, talins, and cytoskeletal proteins33,40.
Fibrillar adhesions (FBs) are complexes of cytoskeletal machinery and actin that connect to FAs and localize into long intracellular bundles throughout myofibroblasts41. Many proteins interact in these FAs and FBs, leading to a complex signaling and force-distribution environment33.
The processes by which myofibroblasts respond to—and influence—their mechanical environment are called mechanosensing and mechanotransduction, respectively. Mechanosensing is a dynamic process integrating multiple signals between surface integrins, intracellular cytoskeleton, tension sensing talins, and secreted signals8. Changes in the interactions between integrins, intracellular proteins, and the actin cytoskeleton can lead to signaling changes in what might otherwise appear as a static cellular environment36.
Integrin-αM is found on monocyte-derived cells29. αM forms an αβ heterodimer with integrin-β2, and β2 is also found on monocyte-derived cells29. αM and β2 together form the heterodimer αMβ2 (called MAC1). αMβ2 binds over 30 different ligands, including fibrinogen, and is also a canonical member of the complement pathway (complement receptor 3, CR3)42,43. αMβ2-positive monocyte-derived cells are key to the development of fibrosis44. CRBM1/5 is a mouse anti-human blocking antibody that binds a conformational epitope found only on αMβ2, not on αM alone45. Leukadherin is a small molecule that promotes αMβ2 association46.
Integrin-α3 is an integrin component that binds laminin47. α3 forms an αβ heterodimer with integrin-β1, and is found on both fibroblasts and monocytes.
Decorin’s heparin-binding domain contains a collagen-binding peptide (herein, CBP) that has affinity for fibrotic tissues, and decoration of an antibody with CBP can be used to increase retention in fibrotic lungs48.
Here we show that CBP-functionalized blocking antibodies against αMβ2 (CBP-α-αMβ2), αM (CBP-α-αM), and α3 (CBP-α-α3) de-differentiate myofibroblasts, lower the pro-fibrotic secretome of myofibroblasts, and treat fibrosis in mouse lung and kidney models. These targets were generated by an mRNAseq study comparing monocytes cultured under anti- and pro-fibrotic conditions49. These results raise the possibility of using targeted anti-integrin antibodies as an immunotherapy against fibrosis.
Results
We began this study with the observation that monocyte differentiation into myofibroblasts was dependent on adhesion to a stiff surface49. We sought to identify key pathways of monocyte-to-myofibroblast differentiation by performing an mRNAseq comparison of monocytes cultured on soft (1 kPa; not allowing myofibroblast differentiation) and stiff (12 kPa; allowing myofibroblast differentiation) surfaces (Tables 1–3). We identified a key role for integrins α3, αM, and αMβ2, and blocking antibodies against these integrins de-differentiated existing myofibroblasts and treated fibrosis in murine models of lung fibrosis and kidney fibrosis.
Our mRNAseq study of monocytes cultured on soft (1 kPa) and stiff (12 kPa) surfaces revealed that integrins αM (ITGAM), α3 (ITGA3), and α7 (ITGA7) were directly upregulated by culture on the stiff surface (Table 2). Upstream regulators of β1 (ITGB1), β2 (ITGB2), and αX (ITGAX) were also upregulated (Table 3). Interestingly, no TGFβ-specific integrin or upstream regulator was found to be upregulated, including ITGAV, ITGA11, ITGB3, ITGB5, ITGB6, ITGB78, nor was TGFβ itself. Additionally, several upregulated pathways and functional annotation clustering also indicated the involvement of integrins in the differentiation of myofibroblasts (Table 1).
To test whether the up- and down-regulated sequences identified in our mRNAseq study provided valid protein targets to reverse fibrosis, we assessed the ability of more than 30 anti-integrin blocking antibodies to promote or inhibit myofibroblast differentiation. Myofibroblasts are defined by their morphology (spindle shaped), their protein content (collagen+ and SMA+), and their secretome. In this study we assessed monocyte-to-myofibroblast differentiation by morphology, before protein content and secretome, because a myofibroblast’s spindle shape is easy to differentiate from other monocyte-derived cells.
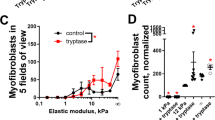
Blocking antibodies against α3 (α-α3), αM (α-αM), and against the heterodimer of αM and β2 (α-αMβ2) consistently de-differentiated monocyte-myofibroblasts (as determined by morphology) when added to culture (Fig. 1A, IC50 in Table 4), while α-α7, α-β1, α-αX inconsistently de-differentiated monocyte-myofibroblasts, and α-β2 caused apoptosis (data not shown50,). Interestingly, transcripts for integrins α3, αM, and β2 are each upregulated in macrophages in idiopathic pulmonary fibrosis (IPF) samples from patients (Supplemental Figs. S1, S2, and S3, data from IPF cell atlas51). Example images of myofibroblast differentiation and de-differentiation can be found in supplemental Fig. S4. An antibody that stabilizes the association of integrin α2β1 (clone Gi14) promoted myofibroblast differentiation (Fig. 1A52,). The small molecule leukadherin, which increases the associated of αM and β2, also promoted myofibroblast differentiation (Fig. 1B46,). Taken together, these results show that our mRNAseq study revealed key integrins that can govern myofibroblast differentiation and de-differentiation when their ligand binding is promoted or inhibited.

Antibodies against integrins α3, αM, and αMβ2 de-differentiate human myofibroblasts from monocyte precursors. Freshly isolated human monocytes (A-D) were differentiated into myofibroblasts and subsequently treated with (A) α-αMβ2, α-αM, α-α3, or α-α2β1 antibodies or (B) leukadherin at the indicated concentrations over 1 week, and the number of remaining myofibroblasts was assessed by morphology. Monocyte-derived myofibroblasts were treated with antibodies at 500 ng/ml and leukadherin at 2 ng/ml and analyzed via flow cytometry for (C) αSMA+ collagen I+ double positive cells and (D) talin2+. n ranges from 3 to 15. * = statistical significance of P < 0.05, < 0.01, or < 0.001, significance vs isotype control antibody, (A and B) 2-way ANOVA (with Fisher’s LSD post test), (C and D) Student’s t-test. Myofibroblast images can be found in supplemental Fig. 4, and examples of gating strategy can be found in supplemental Fig. 5.
To determine if we could reduce intracellular markers of myofibroblast differentiation, we added α-α3, α-αM, and α-αMβ2 to monocyte-derived myofibroblasts and measured the number of αSMA+ collagen I+ double positive cells via flow cytometry. The IC50 from Fig. 1A,B was used to generate an effective dose of 500 ng/ml antibody and 2 ng/ml leukadherin. 500 ng/ml α-αM and α-αMβ2 reduced αSMA+ collagen I+ cells from myofibroblast precursors, indicating that de-differentiation of monocyte-myofibroblasts was not limited to morphological changes (Fig. 1C). Similarly, both leukadherin and the integrin α2β1 stabilizing antibody increased the amount of αSMA+ collagen I+ double positive cells, while α-α3 did not reduce the number of αSMA+ collagen I+ double positive cells (Fig. 1C).
In a companion study, we showed that adhesion to a surface of sufficient stiffness (> 12 kPa) is essential for monocyte-myofibroblast differentiation, and that inhibition of the stiffness-sensing spring protein talin2 de-differentiated monocyte-derived myofibroblasts and treated existing lung fibrosis in a mouse model49. Neither integrin blockers (α-α3, α-αM, and α-αMβ2) nor integrin interaction promoters (Gi14, leukadherin) significantly altered talin2 expression in monocyte-derived myofibroblasts (Fig. 1D). Thus de-differentiation of monocyte-derived myofibroblasts by blocking antibodies (α-αMβ2, α-αM, α-α3) does not appear to operate through inhibition of talin2.
Myofibroblasts contribute to the development and maintenance of scar tissue in several distinct ways: by directly secreting ECM components (including collagen I), by using spindle-shaped morphology and cytoskeletal structures (FAs and FBs) to increase tissue rigidity, and by secreting pro-fibrotic cytokines and chemokines.
To determine if de-differentiation is accompanied by a loss of intracellular structures found in myofibroblasts (FAs and FBs), we added 500 ng/ml α-α3, α-αM or α-αMβ2 to cultured monocyte-derived myofibroblasts. α-α3, α-αM or α-αMβ2 eliminated myofibroblasts’ spindle-shaped morphology and caused myofibroblasts to adopt a more macrophage-like morphology (Fig. S4A vs Fig. S4B–D). α-α3, α-αM and α-αMβ2 also induced the complete loss of myofibroblast cytoskeletal structures (FAs and FBs, Fig. S4B–D).
To determine if myofibroblast de-differentiation is accompanied by a change in secretome, we added 500 ng/ml α-α3, α-αM or α-αMβ2 to monocyte-derived-myofibroblasts. α-αM and α-αMβ2 reduced the amount of secreted pro-fibrotic macrophage-chemotactic protein-1 (MCP1) from monocyte-derived myofibroblasts (Fig. S6A). α-αMβ2 also inhibited the amount of secreted IL-6 (Fig. S6C). Antibody clone Gi14 (which stabilizes the association of integrin α2β1) and leukadherin (which stabilizes integrin αMβ2) both promoted the secretion of MCP1 (Fig. S6A) and IL-6 (Fig. S6C) in monocyte-derived myofibroblasts, again showing that modulation of specific integrins can both promote or de-differentiate myofibroblast morphological phenotype and secretome of myofibroblasts.
To determine if modulation of integrins could de-differentiate mouse myofibroblasts, we added α-α3, α-αM, α-αMβ2, α-α2β1 (Gi14) and leukadherin to mouse monocyte-derived myofibroblasts and mouse fibroblast-derived myofibroblasts. Blocking antibodies cross-reactive against mouse integrins α3, αM, αMβ2 (α-α3, α-αM, α-αMβ2) de-differentiated mouse monocyte-derived myofibroblasts (Fig. 2A). Promoting the association of integrin α2β1 (via Gi14) and αMβ2 (via leukadherin) both increased monocyte-derived myofibroblast differentiation (Fig. 2A,B). α-α3, α-αM, α-αMβ2 also reduced the number of αSMA+ and collagen I+ double positive myofibroblasts (Fig. 2C) and did not decrease the amount of talin2+ cells (Fig. 2D). This again suggests that de-differentiating myofibroblasts by blocking α3, αM, αMβ2 operates through a different mechanism than de-differentiating myofibroblasts by inhibition of talin2. Stabilizing integrin α2β1 (through Gi14) and αMβ2 (via leukadherin) increased both the amount of α αSMA+ and collagen I+ double positive monocyte-myofibroblasts, as well as increasing talin2 concentration in these cells (Fig. 2C,D).

Antibodies against integrins α3, αM, and αMβ2 de-differentiate mouse myofibroblasts from monocyte precursors. Freshly isolated mouse monocytes (A-D) were induced to become myofibroblasts with IL-13, MCSF, and β-mercaptoethanol, treated with (A) α-αMβ2, α-αM, α-α3, or α-α2β1 antibodies or (B) leukadherin at the indicated concentrations over 1 week, and the number of myofibroblasts was assessed by morphology. Monocyte-derived myofibroblasts were treated with antibodies at 500 ng/ml and leukadherin at 2 ng/ml, allowed to de-differentiate, and analyzed via flow cytometry for (C) αSMA+ and collagen I+ double positive cells and (D) talin2+. n ranges from 2 to 7. * = statistical significance of P < 0.05, < 0.01, or < 0.001, significance vs isotype control antibody, (A and B) 2-way ANOVA (with Fisher’s LSD post test), (C and D) Student’s t-test.
Antibody CBRM1/5 is raised against the activation epitope of human αMβ253, but shares 2 amino acid overlap with mouse αMβ254. However, CBRM1/5 de-differentiates mouse monocyte-derived myofibroblasts (Fig. 2A), though at a reduced effectiveness vs human monocyte-derived myofibroblasts (Fig. 1A). While it would have been ideal to use an anti-mouse αMβ2 antibody, no such antibody exists raised against the active conformation of mouse αMβ2.
To determine if de-differentiation of mouse myofibroblasts is accompanied by an altered secretome, we assessed the conditioned media from mouse myofibroblasts treated with 500 ng/ml α-α3, α-αM, and α-αMβ2. De-differentiation of myofibroblasts did not alter the amount of secreted anti-fibrotic IL-10, but in some cases significantly reduced (and did not increase in any case) the amount of pro-fibrotic IL-2355, CCL2256, IL-625, CCL1726,56, IL-12 subunit p4057, CXCL127, and TNF-α28 (Figure S7).
While monocytes can become myofibroblasts, the primary cellular component of scar tissue in fibrosis is the fibroblast-derived myofibroblast. To determine if fibroblast-derived myofibroblasts could be de-differentiated, we added 500 ng/ml α-αMβ2, α-αM, α-α3 to fibroblast-derived myofibroblasts and measured the number of αSMA+ and collagen I+ double positive cells, and talin2+cells. α-α3 reduced the number of αSMA+ and collagen I+ fibroblast-derived myofibroblasts, while α-αM and α-αMβ2 did not (Fig. S8A), consistent with the expression of these integrins on monocytes but not fibroblasts. Neither α-αMβ2, α-αM, α-α3 treatment lowered the amount of talin2 (Fig. S8B), again confirming that de-differentiation of myofibroblasts by inhibiting integrin binding and inhibiting tension sensing operate by non-overlapping mechanisms. α-α3 decreased the amount of IL-6 secreted from fibroblast-derived myofibroblasts (Fig. S6D). No antibody treatment lowered the number of mouse αSMA+ and collagen I+ double-positive fibroblast-derived myofibroblasts, or talin2+ cells (Fig. S8C,D).
Treatment with antibodies did not induce cell death from monocyte (Fig. S9A,C) or fibroblast populations (Fig. S9B,D), among both human and mouse cells. This confirms that the de-differentiation of myofibroblasts is not caused by, or accompanied by, an increase in cell death.
Conjugation of decorin’s collagen-binding peptide (CBP) to an antibody increased the proportion of the conjugated CBP-antibody that reached and was retained in fibrotic lungs, compared to non-conjugated antibody48. This finding involved antibodies against soluble factors (α-TNFα and α-TGFβ). To determine if we could deliver an anti-integrin antibody to a fibrotic tissue, we conjugated CY7-CBP to α-αM. We instilled mouse lungs with bleomycin, allowed fibrosis to develop over a week, and injected Cy7-CBP-α-αM and Cy7-α-αM i.v. in mice with healthy and fibrotic lungs. We compared the fluorescence of the harvested organs after 48 h via IVIS (Fig. 3A). Direct comparison of the fluorescence of healthy and fibrotic lungs (Fig. 3B) shows that significantly more fluorescently labeled antibody remained in fibrotic lungs after 48 h. Because αM is present on monocytes, and monocytes are enriched in the spleen, we compared the fluorescence of spleens in healthy mice vs in mice with fibrotic lungs (Fig. 3C). There was significantly less Cy7-CBP-α-αM in the spleen in animals with fibrotic lungs, suggesting that the enrichment of Cy7-CBP-α-αM in the lungs was coming partially at the expense of Cy7-CBP-α-αM in the spleen.

Conjugation of decorin’s collagen-binding peptide (CBP) to anti-integrin-αM (α-αM) increases antibody concentration in fibrotic lungs. Mice were intranasally instilled with 75 μg of bleomycin sulfate (fibrotic) or PBS (healthy) and injected 1 week later with Cy7-α-αM or Cy7-CBP-α-αM. (A) Heart, lung, spleen, kidneys, and liver were harvested 48 h after injection, and fluorescence intensity was measured via IVIS. (B) The number of photons from lungs or (C) spleen. Fluorescence of the resected organs was pooled and the percentage of fluorescence associated with (D) lungs and (E) spleen. n ranges from 2 to 4. * = statistical significance of P < 0.05, < 0.01, or < 0.001, significance is fluorescence of fibrotic lungs vs fluorescence of healthy lungs, Student’s t-test.
To make certain that our Cy7-CBP-α-αM is not simply remaining in circulation in the mouse for a longer period, we pooled the total fluorescence of all organs for Cy7-CBP-α-αM and CY7-CBP conjugated isotype control antibody, and normalizing the fluorescence for each organ. This analysis showed significantly more Cy7-CBP-α-αM in fibrotic lungs than Cy7-α-αM in fibrotic lungs or Cy7-CBP-α-αM in healthy lungs (Fig. 3D) and less in the spleen (Fig. 3E).
To determine if we could treat existing fibrosis in addition to de-differentiating myofibroblasts, we conjugated non-fluorescent CBP to each of α-α3 (CBP-α-α3), α-αM (CBP-α-αM), and α-αMβ2 (CBP-α-αMβ2). Using our recently published results as a guide48, we used different molar excess of CBP to establish ratios sufficient to attach at least 5 CBP peptides (on average) to each antibody (Fig. S10).
We instilled mouse lungs with bleomycin, allowed fibrosis to develop, and injected α-α3, α-αM, α-αMβ2, CBP-α-α3, CBP-α-αM, and CBP-α-αMβ2 at 7, 9, 11, 14, 16, and 18 days post bleomycin insult. The mice were euthanized 21 days post insult. Only CBP-α-α3 and α-αMβ2 provided weight gain that was statistically higher than the no-treatment control (Fig. 4A–C). CBP-α-α3, α-αM, CBP-α-αM, α-αMβ2, and CBP-α-αMβ2 significantly reduced the total amount of collagen in the right lobe of the lung vs the untreated fibrotic lungs, as assessed by hydroxyproline assay (Fig. 4D). CBP-α-αM and CBP-α-αMβ2 significantly reduced the amount of collagen in the right lungs as a percentage of overall lung weight, compared to the untreated fibrotic lungs (Fig. 4E).

Antibodies against integrins α3, αM, and αMβ2 rescues the fibrotic damage from bleomycin insult to mouse lungs. 50 µg antibody was injected i.v. after 7, 9, 11, 14, 16, and 18 days following insult by 75 μg bleomycin. (A-C) mouse weights after treatment with (A) α-α3 and CBP-α-α3, (B) α-αM and CBP-α-αM, (C) α-αMβ2 and CBP-α-αMβ2. (D) Collagen content from the right, multi-lobed lung assessed by hydroxyproline assay. (E) Data from D divided by dry weight of right lobes of mouse lungs. (F) Blinded Ashcroft scoring. n = 9. * = statistical significance of P < 0.05, < 0.01, or < 0.001, significance vs fibrotic lungs (A-C) 2-way ANOVA (with Fisher’s LSD post-test), (D-F) Student’s t-test.
The left lobes of the bleomycin-insulted lungs were mounted in paraffin, sectioned, stained using Masson’s trichrome, and the histology was blindly scored using the modified Ashcroft method58 by a researcher not involved in administration of the treatment (Fig. 4F). Both α-α3 and CBP-α-α3 reduced the Ashcroft score, as did both α-αMβ2 and CBP-α-αMβ2; with the α-αMβ2 antibody, only the unmodified antibody showed a reduction (Fig. 4F–G). Representative images of the Masson’s trichrome stained left lobes can be seen in Fig. 5A–H.

Representative images of left, single-lobed lungs stained with Massons’ trichrome. (A) Uninsulted lungs instilled with PBS. Lungs insulted with (B-H) 75 μg bleomycin, and instilled with (C) α-α3, (D) CBP-α-α3, (E) α-αM, (F) CBP-α-αM, (G) α-αMβ2, (F) CBP-α-αMβ2. Lungs were harvested 3 weeks after insult and stained via Masson’s trichrome. Scores in Figs. 4, 6 and inset images in supplemental Fig. 11.
To determine if treatment by α-α3, α-αM, and α-αMβ2 and CBP-α-α3, CBP-α-αM, and CBP-α-αMβ2 improved fibrosis in both male and female mice, we analyzed the hydroxyproline-based quantitative collagen data (Fig. 6A–D) and histology-based Ashcroft qualitative data (Fig. 6E–H) based on mouse sex. In general, males develop more fibrosis in the bleomycin-induced lung fibrosis model. Treatment by α-α3, α-αM, and α-αMβ2 and CBP-α-α3, CBP-α-αM, and CBP-α-αMβ2 reduced the absolute amount of collagen in the right lobes of both male and female mice (Fig. 6A,B) though reductions in the amount of collagen as a portion of lung weight were not significant except in the case of CBP-α-αMβ2 in female mice (Fig. 6C,D). Several treatments improved the modified Ashcroft scores for both male and female mice (Fig. 6E,H).

Antibodies against integrins α3, αM, and αMβ2 rescue the fibrotic damage from both male and female mice. Data from Fig. 4, treatment by α-α3, CBP-α-α3, α-αM, CBP-α-αM, α-αMβ2, CBP-α-αMβ2 compared across male (n = 5 or 6, A, C, E) and female (n = 4, B, D, F) mice for amount of collagen in lungs (A, B), collagen as a percentage of dry lung weight (C, D), and blinded Ashcroft scores (E, F). * = statistical significance of P < 0.05, < 0.01, or < 0.001, significance vs fibrotic lungs unless otherwise indicated, Student’s t-test.
Overall, these data indicate that treatment of fibrosis with blocking anti-integrin antibodies directed against α3 and αMβ2 can treat pulmonary fibrosis after a bleomycin insult.
To determine if we could treat existing kidney fibrosis in addition to existing lung fibrosis, we attempted to target CBP-tagged antibodies to fibrotic kidneys, similar to Fig. 3 for lungs. To induce kidney fibrosis, we ligated the descending ureter of the left kidney (unilateral ureteral obstruction, UUO) and allowed the kidneys to become damaged for 1 wk. We then injected Cy7-CBP-α-αM and Cy7-α-αM i.v. and compared the fluorescence of the harvested organs after 48 h via IVIS (Fig. 7A). As in Fig. 3, to make certain that Cy7-CBP-α-αM is not simply remaining in circulation in the mouse for longer periods, we pooled the total fluorescence of all organs for Cy7-CBP-α-αM and CY7-α-αM, and normalizing the fluorescence for each organ. This analysis showed significantly more Cy7-CBP-α-αM in fibrotic kidneys than in healthy kidneys, in comparison to the ratio for Cy7-α-αM (Fig. 7B). We also tested the targeting potential of Cy7-CBP-α-TGFβ, which we had previously shown to be anti-fibrotic when targeted to lungs (Fig. 7B)48. These results indicate we can target the CBP-decorated antibodies to fibrotic kidneys, and in greater quantity than to fibrotic lungs.

Antibodies against TGFβ, integrin α3, and integrin αMβ2 rescue the fibrotic damage from UUO insult to mouse kidneys. The descending ureter of the left kidney was surgically ligated, and 50 µg antibody (either tagged with cy7 or unconjugated) was injected i.v. after 7 days. (A) Heart, lung, spleen, kidneys, and liver were harvested 24 h after injection, and fluorescence intensity was measured via IVIS. (B) Fluorescence of the resected organs was pooled and the percentage of fluorescence associated with healthy and fibrotic kidneys. (C) kidneys were resected 14 post UUO insult (7 days post antibody injection), mounted, and stained via immunohistochemistry for collagen I. The amount of positive IHC staining was compared to the overall amount of kidney tissue, per image. For A and B, n = 3. For C, healthy n = 10, fibrosis n = 10, CBP-antibody treated kidneys n = 8, and unconjugated-antibody treated kidneys n = 6. * = statistical significance of P < 0.05, < 0.01, or < 0.001, (B) significance is fluorescence of fibrotic left kidney vs fluorescence of healthy kidney, Student’s t-test. (C) ANOVA vs fibrosis control, Welch’s correction. Comparison between α-αMβ2 and CBP-α-αMβ2 is Student’s t-test.
To determine if CBP-α-TGFβ, CBP-α-α3, and CBP-α-αMβ2 could treat existing kidney fibrosis, we performed a UUO on the left kidney. At 1 wk post-UUO we injected 50 µg of α-TGFβ, α-α3, α-αMβ2, CBP-α-TGFβ, CBP-α-α3, and CBP-α-αMβ2 i.v. At 2 wk post-UUO, we resected the kidneys, and assessed fibrosis via immunohistochemistry (IHC) for collagen I (Fig. 7, Fig. S12). Because only one kidney per mouse was fibrotic, we confirmed our results by analyzing blood markers of kidney injury, instead of performing a hydroxyproline assay (Fig. S13). CBP-α-TGFβ, CBP-α-α3, and CBP-α-αMβ2 lowered the amount of collagen present in the UUO kidneys (Fig. S12, Fig. 7C). An analysis of blood taken from the kidney UUO mice indicates that each antibody treatment reduced the amount of circulating creatine kinase, but did not change the concentration of blood urea nitrogen (BUN) or uric acid (Fig. S13). This could be because the UUO model leaves mice with one entirely healthy kidney. These results indicate a single 50 µg dose of CBP-α-TGFβ, CBP-α-α3, and CBP-α-αMβ2 is capable of improving kidney fibrosis.
To determine the effect of CBP-α-α3 or CBP-α-αMβ2 treatment of fibrotic lungs on the cellular level of lung fibrosis, we performed an RNAseq on fibrotic lungs treated with each of CBP-α-α3 and CBP-α-αMβ2. Broadly, treatment of fibrotic lungs with CBP-α-α3 or CBP-α-αMβ2 resulted in upregulation of many pathways associated with healthy, non-fibrotic lungs, and down-regulation of pathways associated with fibrotic lungs.
More specifically, treatment of fibrotic lungs by either CBP-α-α3 or CBP-α-αMβ2 resulted in three distinct clusters of sequences that differ between healthy lungs, fibrotic lungs, and fibrotic lungs treated by either CBP-α-α3 or CBP-α-αMβ2 (Figs. S14, S15). Cluster 1 comprises sequences and pathways that are highly expressed in healthy lungs, have low in expression in fibrotic lungs, and have high expression in fibrotic lungs treated with CBP-α-αMβ2 and moderate expression in fibrotic lungs treated with CBP-α-α3. The most downregulated pathway in cluster 1 is the “dilated cardiomyopathy signaling pathway”, which contains downregulated sequences for each one of the intracellular “mechanical stress” proteins found in pathway (Fig. S16).
Cluster 2 comprises sequences and pathways that have low expression in healthy lungs, are highly expressed in fibrotic lungs, and have moderate expression in fibrotic lungs treated with CBP-α-αMβ2 or with CBP-α-α3. The most upregulated pathway in cluster 2 is the “Pathogen induced cytokine storm signaling pathway.” Individually upregulated sequences associated with this pathway include a large number of chemokines, cytokines, collagens, and TNFα (Supplemental Fig. 17). Other upregulated pathways in cluster 2 are the “Pulmonary fibrosis idiopathic signaling pathway”, the “Hepatic fibrosis / hepatic stellate cell activation pathway,” and “Tumor microenvironment pathway.” (Figs. S18–S20) These pathways include a number of individually upregulated sequences related to fibrosis, including IL-6, TGF-β, chemokines, growth factors, collagens, and integrin α5β3.
Cluster 3 comprises sequences and pathways that are highly expressed in healthy lungs, have low in expression in fibrotic lungs, and have moderate expression in fibrotic lungs treated with CBP-α-αMβ2 and in fibrotic lungs treated with CBP-α-α3. As in cluster 1 the “dilated cardiomyopathy signaling pathway” is the most downregulated pathway in cluster 3. Several other pro-fibrotic pathways are downregulated as well (Supplemental Figure 15).
In summary, integrins α3, αM, and αMβ2 were identified as proteins upregulated during the differentiation of monocytes into myofibroblasts. Blocking antibodies α-α3, α-αM, and α-αMβ2 reverse myofibroblast differentiation and reduce the pro-fibrotic secretome of myofibroblasts. Injected CBP-conjugated antibodies preferentially localize at fibroses in both the lungs and the kidneys, and CBP-conjugated blocking antibodies (CBP-α-TGFβ, CBP-α-α3, CBP-α-αM, and CBP-α-αMβ2) treat lung and kidney fibroses at lower doses than untargeted antibodies. These results raise the possibility of a targeted immunotherapy treatment for fibrosis.
Discussion
Our overall objective in this work is to explore an approach by which to de-differentiate myofibroblasts and treat fibrosis. This study was guided by protein targets identified from an RNAseq comparison of myofibroblasts under anti- and pro-fibrotic culture conditions related to surface stiffness (1 kPa and 12 kPa)49. These targets were subsequently validated and refined by in vitro and in vivo experimentation, and targets that did not consistently inhibit myofibroblast differentiation (as defined by measurements of myofibroblast morphology, SMA+ and collagen content, secretome, fibrotic lung morphology, hydroxyproline content of fibrotic lungs, fibrotic kidney morphology, and kidney function) were discarded. While the anti-fibrotic outcome of individual measurements varied, when all of these tests are examined together each of α-αMβ2, α-αM, and α-α3 blocking antibodies consistently de-differentiated myofibroblasts (Figs. 1, 2, Figs. S4–S8) and treated existing fibrosis in mouse models of lung and kidney fibrosis (Figs. 4–7).
Myofibroblast de-activation has long been a goal of fibrosis research. De-activation can be achieved by myofibroblast apoptosis14 or de-differentiation9. Since no apoptosis was observed by immunofluorescence (Fig. S4B-D) and no increase in live-dead staining was observed after treatment with α-α3, α-αM, or α-αMβ2 (Fig. S9), we conclude we have de-activated myofibroblasts through de-differentiation. We also did not observe a change in Ki-67 staining (a marker of cell proliferation), suggesting that de-differentiated cells did not become senescent. Secretome analysis of the de-differentiated myofibroblasts would also suggest that we have lowered a number of pro-fibrotic and pro-inflammatory secreted signals, suggesting a potentially phenotypic shift to an M2 macrophage phenotype. De-activating monocyte-derived myofibroblasts by de-differentiation seems preferable to apoptosis, since monocyte-derived cells are proficient at removing deposited ECM and regenerating tissue, both of which are beneficial to reversing fibrosis7.
While we showed in this paper that targeting the αMβ2 complex and αM both de-differentiate myofibroblasts and treat fibrosis, targeting β2 (CD18) with blocking antibodies resulted in apoptosis of myofibroblasts50 (data not shown), which rendered this blocking antibody unsuitable for use in this study. However, this reported result may be an example of de-activation of myofibroblasts by apoptosis.
While we have shown that α-αM and α-αMβ2 treat existing fibrosis, some evidence indicates they may have a protective role against fibrosis as well. α-αM is effective in reducing tissue damage in a kidney-ischemia model59, while α-αMβ2 antibody limited both damage from kidney-ischemia and subsequent ischemia-induced fibrosis60.
Only α-α3—and not α-αM or α-αMβ2—de-differentiated fibroblast-derived myofibroblasts based on the secretome of the cells (Fig. S8A). While fibroblasts express integrin α3, the integrins αM and αMβ2 are considered canonical myeloid markers. Thus, it appears that anti-integrin antibodies are sufficiently specific that different lineages of myofibroblasts can be targeted by anti-integrin antibodies to treat fibrosis.
Hinz has proposed the “super-mature” FAs are essential for the development and maintenance of the myofibroblast phenotype9. Super-mature FAs concentrate in one location several different protein components of cell adhesion and tension sensing: integrins, tension-sensing talins, and cytoskeletal machinery36,37. FAs participate in inside-out and outside-in signaling in relation to their size61. Fig. S4A (inset) shows structures remarkably similar to Hinz’s super-mature FAs34, and images in Fig. S4B,C,D show that treatment of myofibroblasts with α-αMβ2, α-αM, and α-α3 both remove both the myofibroblast morphology and the presence of FAs and FBs. That removal of the myofibroblast morphology occurs in parallel with the removal of FAs and FBs perhaps confirms Hinz’s proposal that super-mature FAs are critical structures for the maintenance of myofibroblasts9.
αMβ2 is necessary for maintenance of monocyte binding, allowing the actin reorganization that sustains adhesion62. This suggests that disruption of αMβ2 might remove existing FAs, again recalling Hinz’s hypothesis that FAs are critical to myofibroblast differentiation and maintenance. Treatment of monocyte-derived myofibroblasts with α-αMβ2, α-αM and α-α3 induced a morphology (Fig. S4B–D) that appears similar to fluorescence images from monocytes63 and fibroblasts64,65 that have had talin2 reduced, suggesting that integrin- blocking antibodies and inhibition of talin2 both might de-differentiate myofibroblasts through disruption of existing FAs.
Talins and integrins interact in multiple ways within FAs. Talin’s interaction with the β-tail of integrins29 is essential for “outside-in” signaling of integrins66. Talin2’s affinity for the β-tail of β2 increases the binding of monocytes to cells in two key examples. First, talin2’s interaction with β2 promotes αLβ2 adhesion to ICAM-167. Second, talin2 is also essential for αMβ2-mediated phagocytosis63. The differences in tension-sensing and mechanotransduction between talin1 and talin2 may be primarily due to each isoform’s respective integrin associations30, suggesting the interaction of talins and integrins may play a large role in tension sensing. Intriguingly, integrin-dependent mechanosensing is talin isoform specific65. The difference in talin’s affinity for each integrin30, the subcellular localization of integrins and talins, and interactions with the adhesome are all part of the dynamic mechanosensing and mechanotransduction of monocytes during myofibroblast differentiation8.
While integrin interactions with tension-sensing talins appears to be key to the formation and maintenance of myofibroblasts, integrins themselves may play a role in tension sensing. The speed at which integrin-actin linkages are formed and broken has been suggested to be a cellular tension sensing mechanism in and of itself38,68,69,70. This suggests that integrins may transduce information about specific binding ligands (including ECM components) and surface stiffness. This may explain how several integrins appeared in a screen designed to assess the differences in cultured monocytes between soft (1 kPa) and stiffer (12 kPa) surfaces, on the stiffer surfaces permitting monocyte-to-myofibroblast differentiation.
De-differentiation of mouse and human myofibroblasts resulted in strongly reduced IL-6 and MCP-1 secretion (Figs. S6, S7). Removing the MCP-1 pathway abrogates kidney injury24, again suggesting monocyte-derived cells are quite important in fibrosis. IL-6 is quite pro-fibrotic, in some cases sufficient to induce myofibroblasts71. That MCP-1 and IL-6 were the two consistently inhibited secreted proteins in the cytokine screens suggests that de-differentiation of monocyte-derived myofibroblasts may have additional effects beyond the de-activation of individual myofibroblasts and may contribute to an overall reduction in the pro-fibrotic environment in general.
Several anti-integrin therapies have failed to translate to medicinal use due to side-effects or lack of efficacy29. Our goal was that by targeting antibodies to fibrotic tissue using the decorin-derived CBP (Figs. 3, 7), we could increase the local concentration of anti-integrin antibodies in fibrotic tissues and potentially make anti-integrin antibodies a more useful therapeutic. We further tested a monoclonal antibody that recognizes only the activation epitope of αMβ272, clone CBRM1/5. This antibody binds only to a conformational epitope exposed on activated αMβ2. By using an antibody that recognizes only a conformational epitope, we hope to further reduce the side-effects of anti-integrin treatments.
Each of α-α3, α-αM, and α-αMβ2 (and their CBP-conjugated variants) de-differentiated mouse myofibroblasts and treated fibrosis in mice at an identical dose to previously published antibody therapeutics for fibrosis48. This indicates that the local concentration of untargeted α-α3, α-αM, and α-αMβ2 is sufficient to treat fibrosis in mice. CBP-conjugated antibodies were twice as concentrated as unconjugated antibodies in fibrotic lungs after 48 h (Fig. 3B) and in fibrotic kidneys after 24 h (Fig. 7B). A single 50 µg dose of CBP-conjugated antibodies was sufficient to treat fibrosis in a kidney model of unreversed UUO kidney fibrosis (Fig. 7 and Fig. S12).
Our results show that α-α3, α-αM, and α-αMβ2 may be potentially useful as translational therapeutics, capable of bringing immunotherapy treatments to fibrosing diseases. Clinical attractiveness may be particularly high for CBP-α-αMβ2 (clone CBRM1/5), which both recognizes only the active ligand-binding configuration for αMβ2, and is also capable of being targeted to sites of fibrosis to increase the local concentration.
Materials and methods
Study design
This study was designed to test the strategy that key pathways of myofibroblast differentiation can be revealed by an RNAseq of myofibroblast precursors (monocytes) cultured on anti-fibrotic soft (1 kPa) and pro-fibrotic stiff (12 kPa) surfaces49. Specifically, this study examines whether antibodies against key integrins upregulated in monocyte-myofibroblasts (α-α3, α-αM, and α-αMβ2) can de-differentiate myofibroblasts and treat fibrosis in a mouse model of lung fibrosis. CBP functionalization was employed to enhance retention in the fibrotic microenvironment.
Group size was selected based on experience with the pulmonary fibrosis model. Mice were randomized into treatment groups within a cage to eliminate cage effects from the experiment. Treatment was performed by multiple researchers over the course of this study, to ensure reproducibility. Lungs were also resected by multiple researchers, and blinded scoring was used on the fibrosis histology images.
Purification of human monocytes
All human blood was acquired through the University of Chicago blood donation center from volunteers who gave informed consent, and the collected PBMC study was performed in accordance with the relevant guidelines and regulations, and approved by the University of Chicago’s institutional review board (IRB). In order to isolate more peripheral blood mononuclear cells (PBMCs) than is possible through a single blood donation, PBMCs were purified from leukocyte reduction filters from de-identified blood donors. Blood was filtered, and leukocytes purified, the same afternoon as a morning blood donation, to reduce the amount of time that PBMCs could adhere to the filter.
Leukocyte reduction filters were sterilized with 70% ethanol, and the blood flow tubes on either end were clamped shut. The tube through which filtered blood had exited the filter was cut below the clamp, and a syringe containing 60 ml phosphate buffered saline (PBS) was inserted into the tube. Following this, the tube through which unfiltered blood had entered the filter was unclamped, and PBS was slowly pushed through the filter in the opposite direction of the original blood flow. Reversing the flow of PBS through the filter resulted in a recovery of approximately 300 million cells per filter.
The collected blood was layered with lymphocyte separation media (LSM), and centrifuged at 1300 × G for 20 min. The PBMC layer was then removed by pipetting.
Monocytes were purified from PBMCs by use of a negative selection kit for human monocytes (Stemcell, Cambridge, MA), per the manufacturer’s instructions. Approximately 20 million monocytes were purified from each filter. Monocytes were then washed by PBS using five successive 300 × G centrifugation steps, in order to remove EDTA from the resulting population. Monocytes were checked for purity using flow cytometry, and average purity was above 95%. Monocytes were cultured immediately following purification, at 100,000 monocytes/cm2.
Purification of mouse monocytes
All the animal experiments performed in this work were approved by the Institutional Animal Care and Use Committee (IACUC) of the University of Chicago, and was reported in this manuscript using ARRIVE guidelines. All mouse experiments were performed under supervision with protocols approved by the University of Chicago IACUC, and the study was performed in accordance with the relevant guidelines and regulations. Spleens were resected from healthy C57BL/6 mice, pooled, and were placed in PBS with 1 mM EDTA, 2% fetal calf serum (FCS) to prevent subsequent clumping of cells. All PBS, plasticware, filters, glassware and magnets were pre-chilled to 4C, and kept cold throughout this procedure, to limit the clumping of cells. Importantly, ACK lysis buffer caused cell death, and so was not used in this procedure.
Spleens were pushed through a 100 μm filter to disassociate the cells. Monocytes were purified from disassociated cells by use of a negative selection kit (Stemcell), following the manufacturer’s instructions. The purified monocytes were then washed by PBS using five successive 300 × G centrifugation steps, in order to remove EDTA from the resulting population. Monocytes were checked for purity using flow cytometry, and average purity was above 95%. The average yield was 1.5 million monocytes per spleen.
Monocytes were cultured immediately following purification, at 250,000 monocytes/cm2.
Culture of human and mouse monocytes
Human and mouse monocytes were cultured as previously described, using serum-free media (SFM)73. Briefly, SFM for human cells is composed of fibrolife (Lifeline, Frederick, MD), with 1 × ITS-3 (Sigma, St. Louis, MO), 1 × HEPES buffer (Sigma), 1 × non-essential amino acids (Sigma), 1 × sodium pyruvate (Sigma), and penicillin–streptomycin with glutamate (Sigma). For mouse monocytes, 2 × concentrations of ITS-3, HEPES buffer, non-essential amino acids, and sodium pyruvate were added, with 50 μM β-mercaptoenthanol (ThermoFisher) and pro-fibrotic supplements M-CSF (25 ng/ml, Peprotech, Rocky Hill, NJ) and IL-13 (50 ng/ml) to induce myofibroblast differentiation. Additionally, M-CSF and IL-13 were refreshed in the media of mouse monocytes after 3 days of culture. Monocytes were allowed to differentiate for 5 days, and counted based on morphology as previously described74.
Myofibroblasts were de-differentiated by addition of antibodies (α-αMβ2, α-αM, α-α3) for 7 days after the 5 day differentiation was completed. Antibodies were free of azide, glycerol, and other preservatives.
Culture of human and mouse fibroblasts
Human fibroblasts (MRC-5, ATCC, Manassas, VA) and mouse fibroblasts (NIH-3T3, ATCC) were cultured in SFM composed for human cells, with 1 × concentrations of additives. 5 ng/ml TGFβ (Peprotech) was added to induce myofibroblast formation75. Cells were cultured at 10,000/cm2, and TGFβ was refreshed in cultures weekly to maintain the myofibroblast phenotype, if necessary. Myofibroblasts were de-differentiated by addition of antibodies for 7 days.
mRNA purification and RNAseq of myofibroblasts
mRNA was purified using the Trizol method76. RNA sequencing was performed by the University of Chicago Center for Research Informatics. Briefly, there were total six samples from three donors, each with paired myofibroblast and control samples. RNA sequencing was run on one flow cell (HFW2JBBXX) using Illumina HiSeq 4000 sequencer. An average of 66.7 million single-end 50 bp sequencing reads were generated for each sample. The following steps and programs were used for data processing and analyzing. All programs were run using default settings except for otherwise specified. The raw read quality was assessed by FastQC v0.11.2, and reads were aligned to the Human reference genome GRCh38 (Gencode v24) using STAR v2.5.2a77. The distribution of reads aligned to genome regions were collected by Picard v2.2.4 (Table 5). Gene expression levels were quantified by featureCount78. Expression data were transformed, normalized and filtered, and differentially expressed genes were detected by79. Track hubs were made to visualize reads aligned to genome in UCSC genome browser. Pathways and GO terms enrichment were explored by Ingenuity Pathway Analyses and DAVID tools v6.8 Beta80.The sequencing quality of raw reads was assessed by FastQC to explore quality distribution per base and other sequence characteristics. It output the overview of sequencing quality for each fasta file to identify possible technical problems. Raw reads were aligned to the Human reference genome GRCh38 (Gencode v24) using the RNA-Seq aligner STAR. Various quality metrics were collected using Picard. Read counts mapped to 58,656 genes were summarized by featureCount, and then processed by edgeR. Counts for each gene were normalized to the effective library size and transformed to CPM (count-per-million). Low expressed genes were filtered out using cutoff CPM > 0:2 (about 10 reads) in at least 2 samples. Likelihood ratio test (LRT) in edgeR was used to detect differentially expressed genes (DEGs) between groups. Donor was used as a factor for the statistical test. Benjamini–Hochberg procedure was used to correct for multiple hypothesis testing error in RNA-Seq analysis. False Discovery Rate (FDR) is the proportion of false positives among all positives. 535 differential expressed genes were detected with False Discovery Rate (FDR) corrected p-value < 0:05 and fold-change > 1:5 comparing myofibroblast to control group. The 535 genes were further explored functional annotation clusters and pathway enrichment analyses. Biological processes related to immune responses are the top terms enriched for this gene list.
mRNA purification and RNAseq of lung tissue
mRNA was purified using the Recoverall isolation kit and following the manufacturers instructions. Briefly, lung tissue was sectioned from paraffin-embedded blocks in four 20 µm sections, for 80 µm total of full lung face tissue. Tissue was processed and digested, and RNA was isolated according to the manufacturer’s instructions using PureLink RNA Mini Columns. mRNA was sequenced by the UIC RNAseq core. There were 5–9 million reads for the gene counts and about 25 k genes detected in each sample. Raw data was processed via FastQC. low-quality bases and adapters were trimmed from reads via cutadapt. Ribosomal RNA content was a assessed against mouse rRNA sequences (BWA MEM), and map reads were to reference genome mm39 in splice-aware manner (STAR). PCR duplicate levels were assessed (Picard MarkDuplicate). Genes were quantified by expression against Ensembl gene IDs (featureCounts), while alternative splicing events were quantified against Ensembl transcript sequences (Kallisto), exon-level expression was quantified against flattened gene exon annotations (featureCounts), and splice junctions quantify with STAR. Gene expression was normalized to counts per million, including trimmed mean of M-values normalization factor (edgeR), assessed with high-level sample clustering using PCA (principle component analysis), and analyzed using clustering and pathway analysis possible.
Flow cytometry
Cultured myofibroblasts were lifted with ice cold trypsin-EDTA (Sigma), followed by mechanical agitation by a rubber policeman. Myofibroblasts were fixed and permeabilized using using Cytofix/Cytoperm (BD biosciences, Franklin Lakes, NJ), and live-dead stained (live-dead aqua, ThermoFisher) per manufacturer’s instructions. Antibodies used were α-collagen I (Biolegend, San Diego, CA), anti-α smooth muscle actin (αSMA) (R and D systems, Minneapolis, MN), α-ki-67 (BD biosciences), and α-talin2 (R and D systems). Compensation was performed via UltraComp beads (ThermoFisher) per the manufacturer’s instructions.
Immunofluorescence
Human monocytes from 3 donors were culture in 8-well chamber slides (Millipore-Sigma) in SFM, and allowed to become myofibroblasts over 5 days. Myofibroblasts were then treated with antibodies against integrins α3 (α-α3), -M (α-αM), and αMβ2 (α-αMβ2) at 500 ng/ml for 1 week. After 1 week of de-differentiation, the slides were dried quickly using the airflow from a laminar flow hood, in order to preserve cellular morphology as accurately as possible. Cells were then fixed with ice cold 4% PFA, permeabilized with saponin (Sigma). Primary antibodies (α-talin2, novus) were added at 5 µg/ml overnight. Cells were gently washed 3 times in PBS, and were exposed to DAPI and F-actin-phalloidin-488 (ThermoFisher) for 1 min. Cells were mounted using water-based mowiol mounting media (Southern Biotech, Birmingham, AL) to preserve fluorescence. Slides were imaged immediately using a Confocal microscope (Olympus, Shinjuku City, Tokyo).
Legendplex detection by ELISA
Supernatant from cultured human and mouse myofibroblasts were thawed at 4C, and centrifuged at 4C and 10,000 ×G to pellet cell debris. Supernatant was taken and added to 96 well round bottom plates, in duplicate. Legendplex (Biolegend) beads against general inflammation markers were added, according to the manufacturer’s instructions. Sample readouts were normalized to each individual donor control.
Synthesis of peptide conjugated antibody
Conjugation of decorin’s collagen-binding peptide (CBP) (LRELHLNNNC) was performed as described previously48. Monoclonal antibodies, including mouse α-αM (clone ICRF44 for human experiments, clone M1/70 for mouse experiments72, Biolegend), mouse anti-human/mouse α3 (α-α3, 3F9G4, Proteintech, Rosemont, IL), and mouse anti-human MAC1 (α-αMβ2, CBRM1/5, Biolegend) were incubated with 30 fold molar excess of sulfo-SMCC cross-linker (ThermoFisher) for 30 min at room temperature. The sulfo-SMCC-antibody was then mixed with 20, 30, and 40 fold molar excess CBP peptide (LRELHLNNNC) for 1 h at room temperature, resulting in CBP-conjugated anti-integrin antibodies (CBP-α-αMβ2, CBP-α-αM, CBP-α-α3). The peptide was more than 95% purity, (Genscript, Piscataway, NJ).
Matrix-assisted laser desorption/ionization-time-of-flight (MALDI-TOF) mass spectroscopy
MALDI-TOF was performed as previously described48. Briefly, MALDI-TOF was performed on CBP-conjugated anti-integrin antibodies (CBP-α-αM, CBP-α-α3, CBP-α-αMβ2) using a UltrafleXtreme MALDI TOF/TOF instrument or a Bruker AutoFlex III Smartbeam MALDI TOF. Spectra were collected using Bruker flexControl software and processed with analysis software Bruker flexAnalysis or MATLAB (MathWorks). The matrix used was a saturated solution of α-cyano-4-hydroxycinnamic acid (Sigma-Aldrich) or sinapic acid (Sigma-Aldrich), was prepared in 50:50 (v/v) acetonitrile:(1% trifluoroacetic acid in water) as a solvent. The analyte in phosphate-buffered saline (PBS) (5 μl, 0.1 mg/ml) and the matrix solution (25 μl) were then mixed, and 1 μl of that mixture was deposited on the MTP 384 ground steel target plate. The drop was allowed to dry in a nitrogen gas flow, which resulted in the formation of uniform sample/matrix coprecipitate. All samples were analyzed using the high mass linear positive mode method with 5000 laser shots at a laser intensity of 75%. The measurements were externally calibrated at three points with a mix of carbonic anhydrase, phosphorylase B, and BSA.
In vivo biodistribution study
An in vivo biodistribution study was conducted as previously described48, with minor adjustments.
Fluorescently labeled Cy7-antibodies (CY7-α-αMβ2, CY7-α-αM, CY7-α-α3) were conjugated using sulfo-Cy7 N-hydroxysuccinimide ester (Lumiprobe) according to the manufacturer’s instruction. Unreacted Cy7 was removed by dialysis against PBS.
To make fluorescently labeled Cy7-CBP-antibody, antibodies were incubated with eightfold molar excess of SM(PEG)24 cross-linker (ThermoFisher) for 30 min at room temperature. Unreacted cross-linker was removed using a Zeba spin desalting column (Thermofisher, Waltham, MA), and then 30-fold molar excess of Cy7-labeled CBP ([Cy7]LRELHLNNNC[COOH], Genscript, > 95% purity) was added and reacted for 30 min at room temperature for conjugation to the thiol moiety on the C residue. Unreacted Cy7-CBP was removed by dialysis against PBS, resulting in antibodies labeled with CY7 and CBP: CY7-CBP-α-αMβ2, CY7-CBP-α-αM, and CY7-CBP-α-α3.
7 days following bleomycin insult or UUO surgery, 50 µg Cy7-CBP-antibody or Cy7-antibody were injected via tail vein. 48 h later (in the case of lungs, Fig. 3) or 24 h later (in the case of kidneys, Fig. 7), heart, lungs, spleen, kidneys, and liver were resected and imaged via IVIS (Xenogen) under the following conditions: f/stop, 2; optical filter excitation, 710 nm; emission, 780 nm; exposure time, 5 s; small binning.
Bleomycin induced pulmonary fibrosis model
Male and female mice were acquired at 8 weeks of age (Jackson laboratories, Bar Harbor, ME) with the intent to be used at 12 weeks of age. Due to delays regarding COVID19 lockdown and the allowed resumption of non-COVID19 research, the mice were 32 weeks old when the study began. Mouse lungs were instilled with 0.075 units bleomycin (75 µg, Fresenius Kabi, Switzerland) suspended in endotoxin-free PBS, as previously described48. First, mice were anesthetized via isoflurane inhalation (2%). Mice were then placed upright on an angled surface, their tongue pulled to the side, and a 200 µl narrow pipet was placed at the entrance of their throat. 50 µl of bleomycin/PBS was dispensed to the entrance of the throat, and mice were allowed to inhale. Administration to the lungs was confirmed by listening to the mouse’s breathing for popping noises. Mice were then weighed and placed on a heating pad to recuperate.
Following bleomycin insult, mice were injected i.v. with 50 µg of antibody (α-αMβ2, α-αM, α-α3) or CBP-antibody (CBP-α-αMβ2, CBP-α-αM, CBP-α-α3) via tail-vein injection. The dose schedule was 7, 9, 11, 14, 16, and 18 days following bleomycin insult. Mice were euthanized at 21 days post insult via injecting of euthasol (Covetrus, Portland, ME) instead of CO2 inhalation, which could damage the lungs.
Lung resection and fibrosis scoring
Lungs were harvested, and perfused with 5 ml of PBS via cardiac puncture. After resection, the right and left lobes were separated. The left lobe was fixed in 4% paraformaldehyde overnight, mounted in paraffin, sectioned into 5 μm slices, and stained using Masson’s trichrome. Stained lungs were scanned at high resolution using a CRi Panoramic SCAN 40 × Whole Slide Scanner (Perkin-Elmer, Waltham, MA), and were read for fibrosis using a modified Ashcroft method, as previously described58. Lungs were read unlabeled by a researcher uninvolved with animal treatment.
The right lobe of the lung was frozen, and dehydrated using a tissue lyophilizer (Labconco, Kansas City, MO), weighed, and was assessed for collagen content by hydroxyproline assay81. Briefly, dried lungs were digested in 6N HCl/PBS at 100C for 24 h. Supernatant from this digestion was added to 96 well plates and treated sequentially with chloramine-T solution and Ehrlich’s solution at 65C for 15 min to facilitate the color change reaction. Color was read at 561 nm. Quantification was provided by use of a hydroxyproline (Sigma) dilution series, which was transformed into a standard curve.
Kidney unilateral ureteral obstruction (UUO) fibrosis model
UUO surgery was performed as previously described82, with adjustments. Briefly, Mice were anesthetized via 2% isoflurane inhalation, and injected with meloxicam (1 mg/kg), buprenorphine (0.1 mg/kg) in a saline solution, subcutaneously. Briefly, mice were laid on their right side and an abdominal incision used to visualize the left ureter. The left ureter was ligated in the middle section of the ureter with two ties (2 mm apart) using 7–0 silk sutures. Peritoneum is then closed with 5–0 vicryl and skin is closed with 5–0 nylon.
1 week following UUO ligation, the mice were injected with 50 µg of antibody (α-TGFβ, α-α3, α-αMβ2) or CBP-antibody (CBP-α-TGFβ, CBP-α-α3, CBP-α-αMβ2) via tail-vein injection. 2 weeks post UUO, and 1 week post injection, mice were sacrificed via CO2 inhalation, and their kidneys harvested. At this point, we checked to make sure that the UUO ligation was still in place, and in each case it was.
Assessment of fibrosis in kidneys
Right (healthy) and left (fibrotic) kidneys were placed in 4% PFA for 24 h, mounted in paraffin, sectioned into 5 mm full kidney slices, and stained using immunohistochemistry (IHC) for collagen I (1:4000, polyclonal rabbit, lifespan biosciences, Seattle WA) via a Bond-Max autostaining system (Leica biosystems, Lincolnshire, IL). Stained kidneys were scanned at high resolution using a CRi Panoramic SCAN 40 × Whole Slide Scanner (Perkin-Elmer).
Images were equalized in size, and converted to tif files using CaseViewer. Images were then imported into imageJ, scale set for conversion between microns and pixels, and deconvoluted with the “H DAB” deconvolution option. The resulting blue image was thresholded at 215 to see how many pixels were negative for collagen I, and the brown (IHC positive) image thresholded at 185 to see how many pixels were positive for collagen I. Machine-staining allowed these kidneys to be compared with high reproducibility.
Blood analysis for markers of kidney damage
At the time of euthanasia, blood was collected via submandibular bleed into protein low-bind tubes and allowed to coagulate for 2 h on ice. Coagulated blood was then centrifuged at 10,000 × G for 10 min, and serum collected. Serum was then diluted 4 × in MilliQ water before being placed on deck on an Alfa Wassermann VetAxcel Blood Chemistry Analyzer. All tests requiring calibration were calibrated on the day of analysis and quality controls were run before analyzing samples. Serum tests were run according to kit instructions, and creatine kinase was normalized to calcium ion concentrations where indicated to account for sample hemolysis.
Statistical analysis
Statistical analyses were performed using GraphPad Prism software, and P < 0.05 was considered statistically significant. Either Student’s t-test, 2-way ANOVA (with Fisher’s LSD post test), or 1-way ANOVA with Welch’s correction was used to compare groups.
Declaration of ethics approval
All the animal experiments performed in this work were approved by the Institutional Animal Care and Use Committee (IACUC) of the University of Chicago, and was reported in this manuscript using ARRIVE guidelines. All mouse experiments were performed under supervision with protocols approved by the University of Chicago IACUC, and the study was performed in accordance with the relevant guidelines and regulations.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request. The RNAseq datasets generated and analyzed during the current study are available on the gene expression omnibus (GEO) repository, GSE273713.
Abbreviations
- αSMA:
-
Alpha-smooth muscle actin
- CBP-α-αMβ2:
-
CBP-functionalized blocking antibodies against αMβ2
- CBP-α-αM:
-
CBP-functionalized blocking antibodies against αM
- CBP-α-α3:
-
CBP-functionalized blocking antibodies against α3
- CBP:
-
Collagen-binding peptide (LRELHLNNNC)
- FCS:
-
Fetal calf serum
- FBs:
-
Fibrillar adhesions
- CY7-α-α3:
-
Fluorescently labeled Cy7-blocking antibodies against α3
- CY7-α-αMβ2:
-
Fluorescently labeled Cy7-blocking antibodies against αMβ2
- CY7-α-αM:
-
Fluorescently labeled Cy7-blocking antibodies against αM
- Fas:
-
Focal adhesions
- Gi14:
-
Antibody which stabilizes the association of integrin α2β1
- MRC-5:
-
Human fibroblasts
- Called MAC1:
-
Heterodimer αMβ2
- IPF:
-
Idiopathic pulmonary fibrosis
- IL12p40:
-
IL-12 subunit p40
- α3:
-
Integrin-α3
- αM:
-
Integrin-αM
- CD18:
-
Integrin-β2 (β2)
- MCP1:
-
Macrophage-chemotactic protein-1
- NIH-3T3:
-
Mouse fibroblasts
- α-αMβ2:
-
Blocking antibodies against αMβ2
- α-αM:
-
Blocking antibodies against αM
- α-α3:
-
Blocking antibodies against α3
- PBMC:
-
Peripheral blood mononuclear cells
- PBS:
-
Phosphate buffered saline
- SFM:
-
Serum-free media
- TGFβ:
-
Transforming growth factor β
- TNFα:
-
Tumor necrosis factor α
- UUO:
-
Unilateral ureteral obstruction
References
Wynn, T. A. Cellular and molecular mechanisms of fibrosis. J. Pathol. 214(2), 199–210 (2008).
Wynn, T. A. Fibrotic disease and the T(H)1/T(H)2 paradigm. Nat. Rev. Immunol. 4(8), 583–594 (2004).
Spagnolo, P. Novel treatments for idiopathic pulmonary fibrosis. Am. J. Med. 128, 447–449 (2015).
Xaubet, A., Serrano-Mollar, A. & Ancochea, J. Pirfenidone for the treatment of idiopathic pulmonary fibrosis. Expert Opin. Pharmacother. 15(2), 275–281 (2014).
Knuppel, L. et al. A novel antifibrotic mechanism of nintedanib and pirfenidone. Inhibition of collagen fibril assembly. Am. J. Respir. Cell Mol. Biol. 57(1), 77–90 (2017).
Raghu, G. et al. Long-term treatment with recombinant human pentraxin 2 protein in patients with idiopathic pulmonary fibrosis: An open-label extension study. Lancet Respir. Med. 7(8), 657–664 (2019).
Madsen, D. H. et al. M2-like macrophages are responsible for collagen degradation through a mannose receptor-mediated pathway. J. Cell Biol. 202(6), 951–966 (2013).
Klingberg, F., Hinz, B. & White, E. S. The myofibroblast matrix: Implications for tissue repair and fibrosis. J. Pathol. 229(2), 298–309 (2013).
Hinz, B. Formation and function of the myofibroblast during tissue repair. J. Investig. Dermatol. 127(3), 526–537 (2007).
Liu, F. et al. Feedback amplification of fibrosis through matrix stiffening and COX-2 suppression. J. Cell Biol. 190(4), 693–706 (2010).
Hinz, B. & Gabbiani, G. Fibrosis: Recent advances in myofibroblast biology and new therapeutic perspectives. F1000 Biol. Rep. 2, 78 (2010).
Hinz, B. Mechanical aspects of lung fibrosis: A spotlight on the myofibroblast. Proc. Am. Thorac. Soc. 9(3), 137–147 (2012).
Tomasek, J. J. et al. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat. Rev. Mol. Cell Biol. 3(5), 349–363 (2002).
Lagares, D. et al. Targeted apoptosis of myofibroblasts with the BH3 mimetic ABT-263 reverses established fibrosis. Sci. Transl. Med. 9(420), eaal3765 (2017).
Balestrini, J. L. et al. The mechanical memory of lung myofibroblasts. Integr. Biol. (Camb) 4(4), 410–421 (2012).
Abe, R. et al. Peripheral blood fibrocytes: Differentiation pathway and migration to wound sites. J. Immunol. 166(12), 7556–7562 (2001).
Forbes, S. J. et al. A significant proportion of myofibroblasts are of bone marrow origin in human liver fibrosis. Gastroenterology 126(4), 955–963 (2004).
Direkze, N. C. et al. Multiple organ engraftment by bone-marrow-derived myofibroblasts and fibroblasts in bone-marrow-transplanted mice. Stem Cells 21(5), 514–520 (2003).
Hashimoto, N. et al. Bone marrow-derived progenitor cells in pulmonary fibrosis. J. Clin. Investig. 113(2), 243–252 (2004).
Nakashima, T. et al. Lung bone marrow-derived hematopoietic progenitor cells enhance pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 188(8), 976–984 (2013).
Mori, L. et al. Fibrocytes contribute to the myofibroblast population in wounded skin and originate from the bone marrow. Exp. Cell Res. 304(1), 81–90 (2005).
Murray, P. J. et al. Macrophage activation and polarization: Nomenclature and experimental guidelines. Immunity 41(1), 14–20 (2014).
Wynn, T. A. & Ramalingam, T. R. Mechanisms of fibrosis: Therapeutic translation for fibrotic disease. Nat. Med. 18(7), 1028–1040 (2012).
Furuichi, K. et al. CCR2 signaling contributes to ischemia-reperfusion injury in kidney. J. Am. Soc. Nephrol. 14(10), 2503–2515 (2003).
Le, T. T. et al. Blockade of IL-6 Trans signaling attenuates pulmonary fibrosis. J. Immunol. 193(7), 3755–3768 (2014).
Belperio, J. A. et al. The role of the Th2 CC chemokine ligand CCL17 in pulmonary fibrosis. J. Immunol. 173(7), 4692–4698 (2004).
Sahin, H. & Wasmuth, H. E. Chemokines in tissue fibrosis. Biochim. Biophys. Acta 1832(7), 1041–1048 (2013).
Oikonomou, N. et al. Soluble TNF mediates the transition from pulmonary inflammation to fibrosis. PLoS ONE 1, e108 (2006).
Cox, D., Brennan, M. & Moran, N. Integrins as therapeutic targets: Lessons and opportunities. Nat. Rev. Drug Discov. 9(10), 804–820 (2010).
Anthis, N. J. et al. Structural diversity in integrin/talin interactions. Structure 18(12), 1654–1666 (2010).
Lawson, C. et al. FAK promotes recruitment of talin to nascent adhesions to control cell motility. J. Cell Biol. 196(2), 223–232 (2012).
Anthis, N. J. et al. The structure of an integrin/talin complex reveals the basis of inside-out signal transduction. EMBO J. 28(22), 3623–3632 (2009).
Roca-Cusachs, P. et al. Clustering of alpha(5)beta(1) integrins determines adhesion strength whereas alpha(v)beta(3) and talin enable mechanotransduction. Proc. Natl. Acad. Sci. U. S. A. 106(38), 16245–16250 (2009).
Dugina, V. et al. Focal adhesion features during myofibroblastic differentiation are controlled by intracellular and extracellular factors. J. Cell Sci. 114(Pt 18), 3285–3296 (2001).
Hinz, B. & Gabbiani, G. Cell-matrix and cell-cell contacts of myofibroblasts: Role in connective tissue remodeling. Thromb. Haemost. 90(6), 993–1002 (2003).
Hoffman, B. D., Grashoff, C. & Schwartz, M. A. Dynamic molecular processes mediate cellular mechanotransduction. Nature 475(7356), 316–323 (2011).
Geiger, B., Spatz, J. P. & Bershadsky, A. D. Environmental sensing through focal adhesions. Nat. Rev. Mol. Cell Biol. 10(1), 21–33 (2009).
De, R., Zemel, A. & Safran, S. A. Theoretical concepts and models of cellular mechanosensing. Methods Cell Biol. 98, 143–175 (2010).
Parsons, J. T., Horwitz, A. R. & Schwartz, M. A. Cell adhesion: Integrating cytoskeletal dynamics and cellular tension. Nat. Rev. Mol. Cell Biol. 11(9), 633–643 (2010).
Katsumi, A. et al. Integrins in mechanotransduction. J. Biol. Chem. 279(13), 12001–12004 (2004).
Praekelt, U. et al. New isoform-specific monoclonal antibodies reveal different sub-cellular localisations for talin1 and talin2. Eur. J. Cell Biol. 91(3), 180–191 (2012).
Hyun, Y. M., Lefort, C. T. & Kim, M. Leukocyte integrins and their ligand interactions. Immunol. Res. 45(2–3), 195–208 (2009).
Podolnikova, N. P. et al. Ligand recognition specificity of leukocyte integrin alphaMbeta2 (Mac-1, CD11b/CD18) and its functional consequences. Biochemistry 54(6), 1408–1420 (2015).
Satoh, T. et al. Identification of an atypical monocyte and committed progenitor involved in fibrosis. Nature 541(7635), 96–101 (2017).
Lomakina, E. et al. Activation of human neutrophil Mac-1 by anion substitution. Blood Cells Mol. Dis. 42(3), 177–184 (2009).
Maiguel, D. et al. Small molecule-mediated activation of the integrin CD11b/CD18 reduces inflammatory disease. Sci. Signal 4(189), ra57 (2011).
Nishiuchi, R. et al. Characterization of the ligand-binding specificities of integrin alpha3beta1 and alpha6beta1 using a panel of purified laminin isoforms containing distinct alpha chains. J. Biochem. 134(4), 497–504 (2003).
Katsumata, K. et al. Targeting inflammatory sites through collagen affinity enhances the therapeutic efficacy of anti-inflammatory antibodies. Sci. Adv. 5(11), eaay1971 (2019).
White, M. J. V., Ozkan, M., Gomez-Medellin, J. E. & Hubbell, J. A. Myofibroblast differentiation is governed by adhesion mechanics, and inhibition of the stress sensor Talin2 reverses lung fibrosis. bioRxiv https://doi.org/10.1101/2021.06.07.447403 (2021).
Walzog, B. et al. Beta2 integrins (CD11/CD18) promote apoptosis of human neutrophils. FASEB J. 11(13), 1177–1186 (1997).
Neumark, N. et al. The idiopathic pulmonary fibrosis cell atlas. Am. J. Physiol. Lung Cell Mol. Physiol. 319(6), L887–L893 (2020).
Camper, L., Heinegard, D. & Lundgren-Akerlund, E. Integrin alpha2beta1 is a receptor for the cartilage matrix protein chondroadherin. J. Cell Biol. 138(5), 1159–1167 (1997).
Diamond, M. S. & Springer, T. A. A subpopulation of Mac-1 (CD11b/CD18) molecules mediates neutrophil adhesion to ICAM-1 and fibrinogen. J. Cell Biol. 120(2), 545–556 (1993).
Oxvig, C., Lu, C. & Springer, T. A. Conformational changes in tertiary structure near the ligand binding site of an integrin I domain. Proc. Natl. Acad. Sci. U. S. A. 96(5), 2215–2220 (1999).
Gasse, P. et al. IL-1 and IL-23 mediate early IL-17A production in pulmonary inflammation leading to late fibrosis. PLoS ONE 6(8), e23185 (2011).
Yogo, Y. et al. Macrophage derived chemokine (CCL22), thymus and activation-regulated chemokine (CCL17), and CCR4 in idiopathic pulmonary fibrosis. Respir. Res. 10, 80 (2009).
Huaux, F. et al. A profibrotic function of IL-12p40 in experimental pulmonary fibrosis. J. Immunol. 169(5), 2653–2661 (2002).
Hubner, R. H. et al. Standardized quantification of pulmonary fibrosis in histological samples. Biotechniques 44(4), 507–11 (2008).
Rabb, H. et al. Role of CD11a and CD11b in ischemic acute renal failure in rats. Am. J. Physiol. 267(6 Pt 2), F1052–F1058 (1994).
Dehnadi, A. et al. Prophylactic orthosteric inhibition of leukocyte integrin CD11b/CD18 prevents long-term fibrotic kidney failure in cynomolgus monkeys. Nat. Commun. 8, 13899 (2017).
Hinz, B. Masters and servants of the force: the role of matrix adhesions in myofibroblast force perception and transmission. Eur. J. Cell Biol. 85(3–4), 175–181 (2006).
Tang, T. et al. A role for Mac-1 (CDIIb/CD18) in immune complex-stimulated neutrophil function in vivo: Mac-1 deficiency abrogates sustained Fcgamma receptor-dependent neutrophil adhesion and complement-dependent proteinuria in acute glomerulonephritis. J. Exp. Med. 186(11), 1853–1863 (1997).
Lim, J. et al. An essential role for talin during alpha(M)beta(2)-mediated phagocytosis. Mol. Biol. Cell 18(3), 976–985 (2007).
Zhang, X. et al. Talin depletion reveals independence of initial cell spreading from integrin activation and traction. Nat. Cell Biol. 10(9), 1062–1068 (2008).
Austen, K. et al. Extracellular rigidity sensing by talin isoform-specific mechanical linkages. Nat. Cell Biol. 17(12), 1597–1606 (2015).
Petrich, B. G. et al. The antithrombotic potential of selective blockade of talin-dependent integrin alpha IIb beta 3 (platelet GPIIb-IIIa) activation. J. Clin. Investig. 117(8), 2250–2259 (2007).
Li, Y. F. et al. The cytosolic protein talin induces an intermediate affinity integrin alphaLbeta2. J. Biol. Chem. 282(33), 24310–24319 (2007).
Bruinsma, R. Theory of force regulation by nascent adhesion sites. Biophys. J. 89(1), 87–94 (2005).
Chan, C. E. & Odde, D. J. Traction dynamics of filopodia on compliant substrates. Science 322(5908), 1687–1691 (2008).
Li, Y., Bhimalapuram, P. & Dinner, A. R. Model for how retrograde actin flow regulates adhesion traction stresses. J. Phys. Condens. Matter 22(19), 194113 (2010).
Gallucci, R. M., Lee, E. G. & Tomasek, J. J. IL-6 modulates alpha-smooth muscle actin expression in dermal fibroblasts from IL-6-deficient mice. J. Investig. Dermatol. 126(3), 561–568 (2006).
Beller, D. I., Springer, T. A. & Schreiber, R. D. Anti-Mac-1 selectively inhibits the mouse and human type three complement receptor. J. Exp. Med. 156(4), 1000–1009 (1982).
Pilling, D., Vakil, V. & Gomer, R. H. Improved serum-free culture conditions for the differentiation of human and murine fibrocytes. J. Immunol. Methods 351(1–2), 62–70 (2009).
Crawford, J. R., Pilling, D. & Gomer, R. H. Improved serum-free culture conditions for spleen-derived murine fibrocytes. J. Immunol. Methods 363(1), 9–20 (2010).
Hinz, B. et al. Myofibroblast development is characterized by specific cell-cell adherens junctions. Mol. Biol. Cell 15(9), 4310–4320 (2004).
Rio, D. C. et al. Purification of RNA using TRIzol (TRI reagent). Cold Spring Harb. Protoc. 2010(6), pdb prot5439 (2010).
Dobin, A. et al. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 29(1), 15–21 (2013).
Liao, Y., Smyth, G. K. & Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30(7), 923–930 (2014).
Robinson, M. D., McCarthy, D. J. & Smyth, G. K. edgeR: A bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26(1), 139–140 (2010).
Huang, D. W. et al. DAVID bioinformatics resources: Expanded annotation database and novel algorithms to better extract biology from large gene lists. Nucleic Acids Res. 35, W169-75 (2007).
Bergheim, I. et al. Critical role of plasminogen activator inhibitor-1 in cholestatic liver injury and fibrosis. J. Pharmacol. Exp. Ther. 316(2), 592–600 (2006).
Cochrane, A. L. et al. Renal structural and functional repair in a mouse model of reversal of ureteral obstruction. J. Am. Soc. Nephrol. 16(12), 3623–3630 (2005).
Subramanian, A. et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U. S. A. 102(43), 15545–15550 (2005).
Acknowledgements
We thank the Human Tissue Resource Center of the University of Chicago for histology analysis. We thank the Integrated Light Microscopy Core of the University of Chicago for Imaging. We thank the Genomics core at the University of Chicago for RNAseq analysis. We also acknowledge blind scoring from Margo MacDonald, and guidance on fibrosis models from Anne Sperling.
Funding
This work was supported in part by the University of Chicago (to JAH) and the rebuilding the kidney consortium (RBK, to JAH). NIH, F30DK123985
Author information
Authors and Affiliations
Contributions
Conceptualization: MJVW, Methodology: MJVW, Surgical techniques: MJVW, KMK, AS, ZJZ, JW, Kidney biology expertise: BN, JW, Investigation: MJVW, MO, JEGM, KK, MMR, ATA, Visualization: MJVW, Funding acquisition: JAH, Project administration: MJVW, JAH, Supervision: MJVW, JW, JAH, Writing – original draft: MJVW, Writing – review & editing: MJVW, JAH.
Corresponding author
Ethics declarations
Competing interests
MJVW and JAH are inventors on U.S. Patent WO2022256824A1. The other authors declare that they have no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
White, M.J.V., Ozkan, M., Gomez-Medellin, J.E. et al. Blocking antibodies against integrin-α3, -αM, and -αMβ2 de-differentiate myofibroblasts, and improve lung fibrosis and kidney fibrosis. Sci Rep 14, 21623 (2024). https://doi.org/10.1038/s41598-024-70737-4
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-024-70737-4
This article is cited by
-
Multidimensional lung imaging: integrated preclinical platforms enabling the identification of translational biomarkers for pulmonary research
Respiratory Research (2026)
-
Engineering IL-10 and rapamycin to bind collagen leads to improved anti fibrotic efficacy in lung and kidney fibrosis
Scientific Reports (2025)
-
Inhibition of Talin2 dedifferentiates myofibroblasts and reverses lung and kidney fibrosis
Scientific Reports (2025)
