
Abstract
Compared to traditional intermediate concepts, specific bioinformatics entities are more informative and higher directional. This study is based on the BITOLA system and combines bioinformatics methods to determine the intermediate concept which is key to improve efficiency of Literature-based Knowledge Discovery, proposes the concept of “Swanson framework + Bioinformatics”, and conducts practice of Literature-based Knowledge Discovery to improve the scientificity and efficiency of research and development. Firstly, detected the disease related genes (i.e. differentially expressed genes) according to the results of gene functional analysis as intermediate concepts to carry out Literature-based Knowledge Discovery. Taking the disease “Autism Spectrum Disorder (ASD)” as an example, the potential “disease-drug” association was predicted, and the predicted drugs were verified from the perspective of bioinformatics. Two drugs potentially associated with ASD were found: Fish oil and Forskolin, which were closely related to ASD in bioinformatics analysis results and literature verification. The two “disease-drug” association results showed better scientificity. The BIOINF-ABC+ model improves the accuracy of calculations by 76% compared to using the BITOLA system alone. In addition, it also shows high accuracy and credibility in literature verification. The BIOINF-ABC+ model based on the “Swanson framework + Bioinformatics” has good practicality, applicability, and accuracy in conducting “disease-drug” association prediction in the biomedical field, and can be used for mining “disease-drug” relationships.
Similar content being viewed by others


Telephone number: +8,613,753,162,525.
The origin, present situation and development of literature-based knowledge discovery
Data mining appeared in the late 1980s and was first developed in the field of databases, which is called Knowledge Discovery in Databases (KDD)1. The concept of knowledge discovery was first formally proposed at the 11th International Joint Artificial Intelligence Conference held in the United States in 1989. Since then, knowledge discovery has begun to flourish.
The so-called Literature-based Knowledge Discovery is a classical information science method that identifies effective, novel, potentially useful and ultimately understandable knowledge from the content of unrelated literature through literature mining to discover the cross domain knowledge transfer and implicit correlation2. Professor Swanson2,3,4 proposed the concept of “undiscovered public knowledge” in 1985, and developed a knowledge discovery research method based on unrelated literature in 1986. It describes how to obtain the undiscovered implied association from two types of unrelated literature. The general idea is: if one published article reports the meaningful association between A and B, and the other reports the association between B and C, but there is no literature about the association between A and C, The new relationship between A and C can be obtained by considering the two literatures together. Professor Swanson2,4 developed a knowledge discovery tool based on the principle of this method and put forward two hypotheses. One is that eating Fish oil may change some blood parameters to treat Raynaud’s syndrome, and the other is that magnesium deficiency can lead to migraine. These two hypotheses were later verified by clinical experiments5,6.
On this basis, many scientists continue to put forward new ideas. Gordon, a professor at the University of Michigan in the United States, and his collaborators successfully reproduced the scientific hypothesis of “the relationship between edible Fish oil and Raynaud’s disease” and “the relationship between magnesium and migraine”7,8, and developed a set of methods for knowledge discovery based on computer retrieval. According to the model from source literature to intermediary literature and then to target literature, they were used to assist in knowledge discovery of unrelated literature. Weeber9 proposed a “two-step discovery model”, which successfully reproduced the relationship between Raynaud’s disease and Fish oil, magnesium and migraine, and formally defined the two steps of the process of knowledge discovery as “open discovery” and “closed verification”, that is, the process of open knowledge discovery is to find the intermediate word B through A, and then find C; The process of closed knowledge discovery is a process of testing hypotheses, starting from A and C to find a common intermediate concept B. Stegmann and Grohmann10 verified the process of Swanson’s knowledge discovery by using co-occurrence word clustering analysis, found eigenvalues based on the ratio of centripetality and density, and quickly determined the clustering of possible intermediate words and unrelated literature words. Hristovski, et al.11 proposed a literature-based interactive biomedical discovery support system BITOLA, which aims to discover the potential relationship between biomedical concepts (including MeSH (medical subject title) and human genes from HUGO) by mining MEDLINE database, so as to help biomedical researchers propose or verify new knowledge discoveries.
In the above studies, Arrowsmith12, a knowledge discovery tool, mainly selects intermediate concepts based on semantics and co-occurrence frequency. Gordon believes that the intermediary literature is best identified by absolute word frequency, and the target literature is best generated from the intermediary literature by using relative frequency. The BITOLA system developed by Hristovski mainly selects intermediate concepts based on MeSH vocabulary and its semantic types, while Johannes Stegmann and others mainly select intermediate concepts based on centripetality and density. It can be seen that in the Literature-based Knowledge Discovery, researchers have different methods of selecting intermediate concepts, and the purpose is to find a fulcrum to increase the accuracy of knowledge discovery. Although the intermediate concept mentioned above increases the diversity of entries for knowledge discovery, it also improves the accuracy of prediction. However, compared with a large number of concept groups, the prediction target is still large, and it is still not easy to quickly find intermediate concepts with higher accuracy.
In recent years, with the development of artificial intelligence, especially the rise of ChatGPT, AI large models are increasingly widely used in the field of bioinformatics. BioGPT is a domain-specific Transformer language model that is pre-trained on large-scale biomedical literature. Literature abstracts from PubMed database are used as training data to perform relationship prediction (triplet), question and answer (QA), document classification and text generation tasks13. Insiliconsmart Pharma R&D has integrated advanced technology on its AI drug discovery platform PandaOmics. ChatPandaGPT " AI large language model question-and-answer function, which enables researchers to efficiently conduct natural language-based question-and-answer while browsing and analyzing large data sets, promotes easier discovery of potential targets and biomarkers14. MedGPT uses clinical electronic case records as training data to predict a range of medical events, such as the diagnosis of new diseases and complications of existing or future diseases15. GeneGPT takes the description, function and other documents in NCBI as training data, integrates the information in the biomedical database, and provides genomics-related issues such as gene naming, association, function analysis, sequence matching and so on16. It can be seen that the application of AI-driven models in text mining in the biomedical field and the pre-training of a large amount of extracted text data can link diseases, genes and biological processes, which is characterized by multi-source data and technological progress, and its purpose is to quickly identify the biological mechanism of disease occurrence and progression, and discover potential drug targets and biomarkers. This is the same as the goal of the current research on Literature-based Knowledge Discovery. Although multiple sources of data and technological advances have enabled large models to have efficient data processing capabilities and prediction capabilities. However, large models necessarily rely on big data, which brings problems such as data dependence, poor interpretation, and may raise some ethical and social issues. In contrast, biomedical text mining research based on traditional and authoritative literature databases of published scientific research results can ensure the security of data sources, data quality, data management, data update, data access and review, and data ethics.
Function and role of bioinformatics
Bioinformatics is a subject that studies the collection, processing, storage, dissemination, analysis and interpretation of biological information. It reveals the biological laws of a large number of complex biological data through the comprehensive use of biology, computer science and information technology17.Bioinformatics analysis is a method to explore biological related problems through the analysis of biological sequence, protein structure and literature data18. With the development of science and technology, traditional biological data (such as species basic data, physiological and biochemical data, trait genetics, environmental data, etc.) and various omics data (such as genome, transcriptome, proteome, metabolome, epigenome, phenotypic group, etc.) are accumulating, providing a data basis for knowledge discovery from the perspective of Bioinformatics. At the same time, massive data and complex background have led to the rapid development and application of machine learning, statistical data analysis and system description methods in Bioinformatics19,20, which can help researchers better understand gene expression profiles, realize gene function prediction, molecular structure relationship prediction21, and discover the hidden knowledge from massive biological data. Bioinformatics is often used in the biomedical field to study the hidden information of diseases or drugs in organisms.
In this study, based on omics data, Bioinformatics analysis was used to calculate differentially expressed genes, enrichment of Gene ontology(GO)22 functions, enrichment of Kyoto Encyclopedia of Genes and Genomes(KEGG)23,24,25 pathways, etc. by using statistical methods in R language, in order to find the potential knowledge or association hidden in biological genes.
Exploration and practice of using Bioinformatics as an intermediate concept to carry out literature-based Knowledge Discovery
In the process of Literature-based Knowledge Discovery, the core is to determine the intermediate concept, and an accurate intermediate concept is the key to improve the efficiency of knowledge discovery.
Although the text source field in the latest version of Arrowsmith system has been extended to the fields of document title, subject words and abstract, and the text processing time has been shortened, its natural language processing function is relatively limited, and the number of intermediate concept results provided is large, so it is unable to accurately and quickly identify the required biomedical concepts. On this basis, BITOLA system can accurately extract biomedical concepts by introducing MeSH vocabulary and natural language processing technology to support semantic prediction for the discovery of specific relationship types of “disease-gene”. However, due to its wide variety and large number, it is still unable to accurately identify effective biomedical concepts. How to effectively reduce the noise of intermediate concept set has been the goal of researchers for many years. To solve this problem, other systems have also adopted measures. For example, BITOLA uses association rules instead of co-occurrence word frequency to express the relevance of concepts, DAD26system uses concept frequency to sort intermediate concepts in the open discovery process, LitLinker27 uses UMLS semantic network to filter, and uses association rule mining algorithm to determine association concepts, but the fact is that despite this, It is still unable to effectively solve the problem of too many interfering words. Therefore, the efficiency of knowledge discovery cannot be truly solved only through these original unprocessed traditional intermediate concepts.

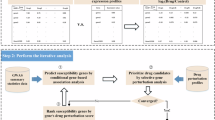
Technical roadmap of the BIOINF-ABC+ Literature-based Discovery Model.
On the basis of traditional methods, if entity information that is crucial, informative, and more directional for a certain disease or drug is used as an intermediate concept, it will undoubtedly be a highly filtered primary traditional intermediate concept, which will greatly improve the credibility and accuracy of knowledge discovery results. These more accurate and reliable entity information can be obtained through Bioinformatics analysis, that is, through the processing, analysis and mining of biomolecular data, the specific Bioinformatics entities in deep level can be extracted. Compared with the traditional intermediate concept, a specific Bioinformatics entity covers more information and has higher directivity. If it is used as an intermediate concept to carry out Literature-based Knowledge Discovery, it will greatly improve the scientificity and efficiency of research and development, such as detecting disease-related genes (i.e., differentially expressed genes) according to the results of gene function analysis. Therefore, based on the BITOLA system, this study combined with Bioinformatics methods to determine the intermediate concept, put forward the knowledge discovery concept of “Swanson framework + Bioinformatics”, and carried out the exploration and practice of knowledge discovery in unrelated literature, in order to improve the prediction efficiency (the technical roadmap is shown in Fig. 1).
Proposing “Swanson framework + Bioinformatics” knowledge discovery (referred to as “BIOINF-ABC+”)
Knowledge discovery based on “Swanson framework + Bioinformatics”, that is, Literature-based Knowledge Discovery based on the intermediate concept of Bioinformatics, refers to the use of important deep-seated information about organisms (such as differentially expressed genes) obtained from Bioinformatics analysis as the intermediate concept of ABC model to explore the potential “disease-drug” relationship, referred to as BIOINF-ABC+. This study selected the disease Autism Spectrum Disorder (hereinafter referred to as ASD or autism) to explore the practice of knowledge discovery in the unrelated literature, in order to evaluate the feasibility of the concept and the accuracy of the prediction results.
BITOLA system has two modes: closed discovery system and open discovery system. The closed discovery pattern allows the input of two established concepts and generates A possible association of the relationship between the two entities, that is, using A and C as a starting point to find a common intermediate concept B. The open discovery mode only allows the input of A given concept A, that is, the process of searching for the middle word B through A, and then searching for C. Compared with the two modes, the closed discovery mode requires clear A and C, while the open discovery mode only requires clear A. For example, in the drug knowledge discovery of A specific disease, it is only necessary to set the specific disease as A, and then the disease (A) seeks disease-related genes (B), and then the gene (B) discovers related drugs (C). It’s an open process of discovery. The BIOINF-ABC+ model proposed in this study adopts this open discovery process to obtain the “disease-gene-drug” link.
“BIOINF-ABC+” result sorting algorithm
The algorithm follows the knowledge discovery algorithm of BITOLA system, that is, based on the association rules representing the known relationships between concepts and considering the background knowledge, a new relationship between concepts is proposed. In order to check the results as easily as possible, the related concepts are sorted. Related concepts Y can be sorted by association rule support (co-occurrence frequency), confidence or semantic type.
The related concepts Z can be sorted by the following calculation formula:
The ranking is calculated based on support, but it can also be calculated based on confidence. In this equation, Zk is the concept of calculating its rank, SXYi and SYiZk are the support of association rule X → Yi and Yi → Zk, and m is the number of intermediate concepts Y.
Calculation of differentially expressed genes
Bioinformatics analysis results include differentially expressed genes, enrichment of GO functions, enrichment of KEGG pathways, etc. Among them, differentially expressed genes (DEGs) refer to genes with significant differences in RNA expression due to environment, time and other factors. Differentially expressed genes are the basis of Bioinformatics analysis and drug research. Researchers can analyze the potential information of diseases and drugs, such as targets and biomarkers through differentially expressed genes. Therefore, differentially expressed genes are a key and necessary element in the research of “disease-drug” potential association. Taking them as intermediate concepts is an important basis for improving the scientificity and accuracy of knowledge discovery research. Therefore, this paper takes one of the results of bioinformatics analysis of differentially expressed genes as an example and takes it as an intermediate concept to explore the practical effect of the new method BIOINF-ABC+ for Literature-based Knowledge Discovery.
The calculation method of differentially expressed genes in this study is FC (fold change) algorithm. The principle of the algorithm is to calculate the multiple of the average expression level of genes in the two types of samples. If the value reaches the preset threshold (generally set to 2, which is greater than 1 or less than − 1 in the logarithmic expression ratio based on 2), the gene is judged to be differentially expressed. The calculation formula is as follows:
FC represents the calculation method of differentially expressed genes; \(\:\overline{\text{X}}\left(\text{i}\right)\) is the average expression value of gene i in X samples; \(\:\overline{\text{Y}}\left(\text{i}\right)\:\)is the average expression value of gene i in Y samples.
The general principle for screening differentially expressed genes based on threshold values is to use two indicators: | Fold Change | ≥ 2, that is, | log2FC | ≥ 1 and FDR < 0.05 or P < 0.0528. The larger the absolute value of Fold Change, the larger the multiple of differences. The smaller the FDR or P value, the more significant the difference. FDR or P-value less than 0.05 indicates a statistically significant difference. The above criteria are relatively accepted in the research. Among them, FDR (False Discovery Rate) refers to the false discovery rate, also known as Q-value. Simply put, FDR is the corrected P value, which is tested and corrected for false positive rates through error control methods. For the case where there are few differentially expressed genes identified through screening, the FDR < 0.05 or P < 0.05 condition remains unchanged, and the multiple of differences can be adjusted to fluctuate appropriately between 1.2 and 2 times, that is, the value of | log2FC | can be between 0.232 and 129. Adjust the threshold appropriately based on the required number of differentially expressed genes.
Determination and analysis steps of literature collection
For the construction of the initial concept set, this study uses the BITOLA system strategy, which extracts the concepts in the title, abstract and MeSH fields of PubMed related literature as the initial concepts.
For intermediate concept sets, the large number of concept sets will cause great interference to the discovery of truly meaningful target concepts. BIOINF-ABC+ knowledge discovery model in order to improve the quality of target concepts, the intermediate concept set is filtered by Bioinformatics methods. After determining the target disease or drug, this method needs to select the appropriate data in the gene expression database to achieve Bioinformatics analysis and obtain a specific intermediate concept set. Choose one of the differential genes, pathways or proteins in the intermediate concept set as the intermediate concept (Y) of this study. At the same time, on the basis of the target concept set, the results are still screened by combining Bioinformatics methods (such as protein interaction network and pathway analysis). Of course, different Bioinformatics analysis methods (such as differentially expressed genes, pathways, proteins or immune infiltrating cells) may be used for different intermediate concepts or target concepts, which greatly improves the efficiency of target concept hit.
Practice of “BIOINF-ABC+” literature-based Knowledge Discovery: taking the discovery of potential relationship of “ASD-drugs” as an example
Differentially expressed genes calculation of ASD
The Gene Expression Omnibus(GEO)30 (https://www.ncbi.nlm.nih.gov/geo/)database was selected as the data source to obtain the experimental genes of ASD. The selected subjects are all human datasets, and the experimental data is derived from brain tissues extracted from the cerebellum, frontal cortex, and temporal cortex of ASD patients and control individuals after death, with a total of 30 sample data. The R language Limma program package was used to calculate the differentially expressed genes31, and the intersection genes with opposite regulatory effects in the differentially expressed genes were removed. The screening conditions were: |log2 (Fold Change) |>0.5, P < 0.05. 105 genes with significant differential expression of ASD were obtained, including 57 up-regulated and 48 down-regulated genes; The clusterprofiler package was used to analyze the KEGG pathway enrichment of significantly differentially expressed genes32, and 60 pathways enriched by up-regulated genes and 79 pathways enriched by down-regulated genes were obtained.
Take the concept of Bioinformatics (differentially expressed gene) as an intermediate concept to carry out knowledge discovery
Log in to the BITOLA website https://ibmi.mf.uni-lj.si/bitola and click Open discovery system to enter the search interface of open discovery system. Enter “Autistic Disorder” as the starting concept in “Concept”, which belongs to the semantic type “Mental or Behavioral Dysfunction”. Then set the semantic group to “Any” and the semantic type to “Gene or Gene Product”. The search results show that there are 340 genes related to Autistic Disorder. Taking the intersection with the previously calculated list of DEGs for autism, the differentially expressed genes we obtained are IFI6, LPL, and BRWD2, which are the selected genes Y. Then, search for relevant Z (semantic type “Organic Chemical or Pharmaceutical Substance”) according to Y, and the result is a list of Z drugs that have potential associations with X disease. The calculation results identified 594 drugs, and this study selected the top 50 drugs with the highest semantic frequency for subsequent research (see Table 1).
The method of Bioinformatics was used to find the “disease-drug” correlation in the results. Select the top 50 drugs for analysis and screening: exclude the results of drugs belonging to class I and drugs without experimental data in the GEO database, and finally get 16 drugs (see Table 2). Bioinformatics analysis of these drugs was carried out and compared with the Bioinformatics analysis results of autism.
Using GEO database as the data source, the experimental genes of 16 drugs were obtained. The R language limma program package was used to calculate the differentially expressed genes31, and excel was used to remove the intersection genes with opposite regulatory effects from the calculated differentially expressed genes, and the R language program was used to average the expression of differentially expressed genes with the same regulatory effect; The clusterprofiler package was used to enrich the KEGG pathway of differentially expressed genes32.
By comparing the DEGs with opposite expression of ASD and their enriched KEGG pathway, we found that the drugs with closer association with ASD, and the specific results are shown in Table 2.
For the above 16 drugs, after comprehensive consideration of the number of experiments in the data set, the complexity of data processing, the number of differentially expressed genes and KEGG pathways, it was found that (1) Although Triiodothyronine, Recombinant Interferon γ,Glucagon, Growth hormone and Bucladesine are dominant in differentially expressed genes, they are not considered due to the small number of experiments; (2) Because the number of differentially expressed genes overlapped with ASD is too small, Dexamethasone, Glycerol, Interferonα,Oleic acid, Methionine, Fenofibrate, Arginine, Rosiglitazone, Retinoic acid and other drugs will not be considered; (3) Both Fish oil and Forskolin have absolute advantages in terms of the number of experiments, differentially expressed genes, pathways and so on. Therefore, based on the Literature-based Knowledge Discovery results of BIOINF-ABC+, this study believes that Fish oil and Forskolin have high potential “drug-disease” association credibility for ASD.
Bioinformatics reverse verification for results of “BIOINF-ABC+” literature-based Knowledge Discovery
On the basis of the above research, this study analyzed the two drugs and ASD respectively by Bioinformatics method, verified the above analysis results from the Bioinformatics level, and made a deeper comparison and analysis of the two drugs.
Construct the protein-protein interaction network of significantly different genes and calculate the key genes
Previously, the opposite part of the Fish oil and Forskolin differentially expressed genes has been removed and screened under the condition of |log2(Fold Change)|>0.5. 1129 significant differentially expressed genes in Fish oil were obtained, including 529 up-regulated genes and 600 down-regulated genes; There were 1164 significant differentially expressed genes for Forskolin, including 715 up-regulated genes and 449 down-regulated genes. Upload significant differentially expressed genes to STRING v11.0 (https://string-db.org/)33, an online analysis website, to conduct Protein-Protein Interaction (PPI) network analysis, and take the confidence > 0.4 as the threshold for screening. Key genes are highly correlated genes in PPI network. In this study, the key genes are the top 10 genes with the highest frequency appear in the PPI network relationship. The CytoHubba plug-in34 of the Cytoscape software35 will rank proteins according to their properties in the network, and provide 12 topological analysis methods, such as Degree, Edge Percolated Component (EPC), Maximum Neighborhood Component (MNC), and score and rank proteins according to the corresponding algorithms. In this study, the CytoHubba plug-in of Cytoscape software was used to analyze the results of PPI network. The top 10 proteins of 12 algorithms were output, and the top 10 proteins of occurrence frequency were counted as core genes (see Fig. 2).

Top 10 hub genes of Fish oil and Forskolin. Frequency refers to the occurrence frequency of the top 10 proteins ranked by the 12 algorithms of CytoHubba plug-in.
Enrichment analysis
When screening KEGG enriched pathways, P < 0.05 is the basic threshold for screening significantly enriched pathways. P adjust and Q value are correction values for P value, which are more stringent than P value. In further strict screening, P < 0.01, or P adjust < 0.05, or Q < 0.05 are used as screening conditions.
Use the R language clusterprofiler package to enrich the KEGG pathway of the significant differentially expressed genes between Fish oil and Forskolin32. The results showed that Fish oil had 285 pathways enriched by up-regulated genes and 294 pathways enriched by down-regulated genes. Screening was performed with a threshold of P < 0.05, with 34 pathways enriched by up-regulated genes and 22 pathways enriched by down-regulated genes (see Fig. 3 for some pathways). Additionally, Forskolin had 300 pathways enriched by up-regulated genes and 236 pathways enriched by down-regulated genes, screened with a threshold of P < 0.05. There are 45 pathways enriched by up-regulated genes and 10 pathways enriched by down-regulated genes (some pathways are shown in Fig. 3).

Results of KEGG pathway enrichment analysis of Fish oil and Forskolin23,24,25.Count refers to the number of genes associated with a certain pathway.Orange indicates the pathways that up-regulated differentially expressed genes involved, and blue indicates the pathways that down-regulated differentially expressed genes involved.
Analysis of the mechanism of Fish oil acting on ASD
It can be seen that among the significant differentially expressed genes screened by ASD and Fish oil, there are four identical genes, including two genes with opposite regulatory effects: PTPRR and RASD1.PTPRR is an important protein in the MAPK signaling pathway, and the protein encoded by the PTPRR gene belongs to the protein tyrosine phosphatase family36. It is mainly expressed in the brain, especially in Purkinje cells of the hippocampus and cerebellum37. The PTPRR gene can regulate downstream MAPK directed cell proliferation, differentiation, and dephosphorylation functions38. Overexpression of PTPRR leads to decreased ERK phosphorylation, resulting in neuronal apoptosis, reduced cell proliferation, and possible synaptic plasticity damage, causing mice to exhibit depression vulnerability and stress sensitivity, so the dephosphorylation of ERK caused by PTPRR overexpression may be at least partially the mechanism of neuroplasticity damage and depression38.RASD1 is a member of the Ras family of small G proteins, mainly expressed in the brain39. RASD1 plays a crucial role in neuronal signal transduction, and studies have found that dysfunction of RASD1 may lead to neurological and psychiatric disorders40. Research has shown that RASD1 can enhance the mitotic signaling of neural progenitor cells and regulate cell survival, becoming an important stage specific regulatory factor for adult hippocampal motor induced neurogenesis41. Knockdown of RASD1 can improve neurological behavior, glial cell polarization, oxidative stress, neuroinflammation, ferroptosis, and demyelination42. Overexpression of RASD1 increases levels of reactive oxygen species (ROS), inflammatory cytokines, MDA, free iron, and NCOA4, while reducing levels of UCP2, GPX4, ferritin, and GSH42. RASD1 can induce oligodendrocyte differentiation and myelin damage after subarachnoid hemorrhage (SAH) by inhibiting the cAMP-CREB pathway, and can serve as a new therapeutic target for neurological dysfunction43.
Among the pathways enriched by genes with significant differences in Fish oil (P < 0.05), the up-regulated genes enrichment pathways are mainly involved in cAMP signaling pathway, Ras signaling pathway, Cell adhesion molecules and other related functions or processes; Down-regulated genes enrichment pathways are mainly involved in p53 signaling pathway, Fatty acid metabolism, Cell cycle and other related functions or processes. Among the KEGG pathways enriched by the significant differentially expressed genes screened from ASD and Fish oil, 126 pathways were enriched by genes with opposite regulatory effects, among which the pathway satisfying P < 0.05 in ASD and Fish oil was 0, and the pathway satisfying P < 0.1 was 1: Ovarian steroidogenesis. Ovarian steroid hormones include estrogen, androgen, progesterone, etc., which are crucial for normal uterine function, the establishment and maintenance of pregnancy, and the development of the breast. But their role in the growth, development, and regulation of the central nervous system cannot be ignored. Estradiol is an ovarian steroid hormone that regulates the physiological functions of the central nervous system, including emotions, cognition, sleep, and mental state44. In addition to its rapid effect on neuronal electrical activity, it can also rapidly alter the calcium concentration in astrocytes through membrane associated estrogen receptors and regulate synaptic transmission45. Clinical research evidence suggests that estrogen can regulate neurotransmitters related to mental illness in the brain, such as dopaminergic, serotonergic, and glutamatergic, and also contribute to improvements in cognitive, emotional, and behavioral responses46.This pathway contains two ASD genes: PLA2G4B and FSHB; There are four Fish oil genes: ACOT1, ACOT2, ADCY4 and ACOT4. These genes are mainly involved in Reproductive organ development, Fatty acid metabolism and other processes. The above six genes are shown in the ovarian steroidogenesis pathway map, which shows that these genes are mainly involved in GnRH signaling pathway. The ovarian steroidogenesis pathway is shown in the Fig. 4a.


Two signaling pathways that Fish oil and Forskolin involved23,24,25.The red and green colors in the figure represent the positions of upregulated and downregulated differentially expressed genes, respectively. The darker the color, the stronger the expression.a The ovarian steroidogenesis pathway that Fish oil involved. b The MAPK signaling pathway that Forskolin involved.
Analysis of the mechanism of Forskolin acting on ASD
Among the significant differentially expressed genes screened by ASD and Forskolin, there were 8 identical genes, and 2 genes with opposite regulatory effects: RASD1 and DUSP14. DUSP14 is a MAP kinase phosphatase that plays an important role in regulating various cellular processes, including oxidative stress and inflammation, its expression can significantly reduce the activation of glial cells47. A study has found that overexpression of DUSP14 can inhibit cell apoptosis, inflammation, pyroptosis, and brain tissue damage in elderly rats after isoflurane anesthesia, improve cognitive dysfunction, and may have a neuroprotective effect on postoperative cognitive dysfunction by regulating NLRP3 inflammasome mediated pyroptosis48. There is experimental evidence that DUSP14 deficiency in T cells can lead to enhanced T cell proliferation, increased cytokine production after T cell activation, and DUSP14 negatively regulates TCR signaling and immune response by inhibiting Table 1 activation49.DUSP14 negatively regulates the TNF or IL-1 induced NF-κB activation pathway by dephosphorylation of TAK1 at Thr-187, and the transcription factor NF-κB plays a critical role in a wide range of physiological and pathological processes, including cell proliferation, immune regulation, inflammation, and anti apoptosis50.
Among the pathways enriched by genes with significant differences in Forskolin (P < 0.05), the pathways of up-regulated genes enrichment are mainly involved in PPAR signaling pathway, p53 signaling pathway, IL-17 signaling pathway and other related functions or processes; Down -regulated genes enrichment pathways are mainly involved in Biosynthesis of amino acid, Nucleotide metabolism, Glycine, serine and threonine metabolism and other related functions or processes.
Among the KEGG pathways enriched by the significant differentially expressed genes screened by ASD and Forskolin, 129 pathways were enriched by genes with opposite regulatory effects, among which one pathway satisfying P < 0.05 in ASD and Forskolin simultaneously: MAPK signaling pathway. The MAPK signaling pathway is a signaling cascade that transmits signals from membrane receptors to the cytoplasm and nucleus through downstream phosphorylation of proteins51. The MAPK signaling pathway is involved in regulating cellular physiological and pathological processes such as cell proliferation, differentiation, apoptosis, stress response, activation of immune cells, inflammatory response, etc. (including neural development)52. The MAPK signaling pathway is the main pathway for cell division and proliferation, and plays a key role in central nervous system development. Deviation from the conventional control of the MAPK signaling pathway is associated with various human diseases, including autism spectrum disorder (ASD), Parkinsonism, Alzheimer’s disease, and various forms of cancer53,54.This pathway contains three ASD genes: GADD45G, PTPRR and CSF1R; There are 11 Forskolin genes: FLT3LG, RRAS2, RPS6KA2, EPHA2, CACNB4, ATF4, CSF1, FLNC, ANGPT2, GADD45A and DDIT3. These genes are mainly involved in cellular processes and inflammatory reactions. The above 14 genes are represented in the MAPK signaling pathway map, which shows that these genes are mainly involved in the classical MAP kinase pathway. The MAPK signaling pathway is shown in the Fig. 4b.
In summary, 10 key genes were selected from the differentially expressed genes as the core of subsequent text verification. The results of pathway enrichment analysis showed that Fish oil was involved in a key pathway of autism, namely Ovarian steroidogenesis pathway. Forskolin is also involved in a key pathway of autism, namely MAPK signaling pathway. Fish oil and Forskolin can achieve the goal of treating ASD by acting on multiple genes and pathways. Therefore, the results of knowledge discovery based on BIOINF-ABC+ have achieved good verification results in the level of Bioinformatics analysis.
Text verification of results of “BIOINF-ABC+” literature-based Knowledge Discovery
In this study, the domain knowledge score method was used to verify the effectiveness of Fish oil and Forskolin targets in Chinese and English databases. The specific operation is as follows: take 10 key genes as key targets, search in the English database PubMed with “autism” and “key targets” as the key words, and search in the Chinese database CNKI and Wanfang database with “autism” and “key targets” as the key words. If experimental verification shows that key targets are associated with ASD in literature, 1 paper will be counted as 1 point. Relevant search results will be recorded and its cumulative score will be calculated, and no score will be accumulated for duplicate literatures. Table 3 shows the retrieval results of the above key targets that are mainly involved in inflammatory response, cell cycle progression and other related processes in ASD patients. The results showed that Fish oil and Forskolin were highly correlated with ASD, especially Forskolin.
Tricholaryngin is a direct AC/cAMP/CREB activator, which is isolated from Angelica dahurica and has various neuroprotective properties. A number of studies have shown that the application of Forskolin in the treatment of ASD is feasible. Alharbi, et al.55 have shown that Forskolin has been proved in their laboratory that it can directly activate adenylate cyclase (AC) and reverse neurodegeneration related to the progression of autism, multiple sclerosis, ALS and Huntington’s disease. Mehan, et al.56 have shown that Forskolin can alleviate neuronal mitochondrial dysfunction and improve neurological symptoms in autism rats. Chi57 have shown that the agonist Forskolin may regulate FMR1 gene mainly through the cAMP signaling pathway through the overlapping sites in the promoter region of FMR1, the pathogenic gene of fragile X syndrome.
In addition, mitochondrial dysfunction is one of the important pathological markers of autism, and mitochondrial dysfunction in autism is associated with decreased ATP levels due to decreased levels of cyclic adenylate monophosphate. The diterpenoid tricholaryngins extracted from tricholaryngins can regulate various physiological functions of cells by increasing cyclic adenylate monophosphate and up-regulating adenylate cyclase. In order to study the neuroprotective effect of tricholaryngine on autism, Mehan et al.56 from ISF School of Pharmacy, India, administered adenylate cyclase activator tricholaryngine intragastally to model rats with autism for 15 days at doses of 10, 20 and 30 mg/kg. It was found that tricholaryngine can dose-dependently improve neuronal mitochondrial dysfunction, one of the important pathological markers of autism, and reduce the levels of pro-inflammatory cytokines, oxidative stress, and lipid biomarkers, further demonstrating the potential of adenylate cyclase activators in the treatment of autism57. Fish oil supplementation may improve hyperactivity, lethargy, and stereotyping in people with autism, but the available clinical data is too limited to draw definitive conclusions. The above scientific research results once again demonstrate the potential therapeutic effects of these two drugs on autism.
It can be seen that from the perspective of experimental evidence recorded in the literature, the results of knowledge discovery based on BIOINF-ABC+ have also been well verified in the literature set, which proves that the scientific hypothesis (i.e. disease X-drug Z relationship) obtained by this method has a good experimental basis, so the possibility, feasibility and reliability of using this method to predict the potential drug disease relationship are high.
Discussion
Result analysis of “BIOINF-ABC+” literature-based Knowledge Discovery
This study used the BIOINF-ABC+ model to carry out the Literature-based Knowledge Discovery, and predicted the association between Forskolin and Fish oil in the field of ASD, namely “Forskolin-ASD” and “Fish oil-ASD”. The two groups of “drug-disease” association results showed good scientificity in the Bioinformatics analysis results, and also showed high accuracy and reliability in the text verification. From the above analysis results, BIOINF-ABC+ model has good practicability, applicability and accuracy in the biomedical field of “drug-disease” association prediction. Therefore, it is feasible and efficient to use the results of Bioinformatics analysis as an intermediate concept for knowledge discovery on the basis of Literature-based Knowledge Discovery. Secondly, the BIOINF-ABC+ model proposed in this study is scientific and can be used for knowledge discovery. At the same time, it also provides a new research idea for the future study of “drug-disease” relationship.
Efficiency analysis of “BIOINF-ABC+” literature-based Knowledge Discovery
Among the top 50 drugs retrieved by BITOLA system, Fish oil ranked 29th and Forskolin ranked 50th. Assuming that the full score of 50 points is given to each drug according to the order of drug occurrence, the drug ranking first is 50 points, and the drug ranking 50 is 1 point, including 22 points for Fish oil and 1 point for Forskolin. At the same time, Bioinformatics analysis of these 50 drugs showed that Fish oil and Forskolin ranked the top 2 in the analysis results, and the results of BIOINF-ABC+ model were also scored, with 50 points for the first drug and 49 points for the second drug, 50 points for Fish oil and 49 points for Forskolin (see Table 1).
If the drug prediction accuracy is:
Then, the accuracy of BITOLA system for calculating Fish oil and Forskolin is YBITOLA= \(\:\frac{22+1}{50\times\:2}\) = 23%;
The accuracy of BIOINF-ABC+ model in this study was YBIOINF−ABC+= \(\:\frac{50+49}{50\times\:2}\) = 99%.
It can be seen from the comparison of the accuracy of prediction that the accuracy of BIOINF-ABC+ model is 76% higher than that calculated by BITOLA system alone. Therefore, the BIOINF-ABC+ model proposed in this study has high accuracy.
Exploration of the applicability of “BIOINF-ABC+” literature-based Knowledge Discovery and related ethical issues
The BIOINF-ABC+ model in this study is based on the BITOLA system. It is based on Medline database and retrieved by using the results of Bioinformatics analysis as an intermediate concept. Taking the complex disease autism as an example, the prediction results are highly effective, but the model is also applicable to other diseases in the biomedical field. The reason is that autism is a neurodevelopmental disorder, and its pathogenesis is complex, which has not been completely clarified at present. Although some studies have shown that autism may be related to genetic factors, not all patients with autism have clear genetic mutations or genetic patterns. Although some genetic variations related to autism have been found, they are not the only cause of autism. Therefore, this study takes autism as an example to carry out model practice, which has two meanings: first, the model is the best for genotypic diseases with clearly different genes; Second, for complex diseases with scattered differential genes, the knowledge discovery model proposed in this study can scientifically and accurately narrow the scope of drug selection from the biological level and improve the prediction accuracy as much as possible. Therefore, the model is also applicable to other diseases in the biomedical field.
If the data used in this model only comes from the open database, the data has been desensitized, access controlled, data quality guaranteed, informed consent and ethical review before being uploaded to the database, so there is no ethical risk. However, if the data used also contains the gene sequencing data of some individual patients, it is necessary to do the above ethical risk control, especially the informed consent of the patients before it can be used. There is no individual gene sequencing data in this study. At the same time, there is still a long way to go from the predicted research results to clinical use, such as laboratory verification, preclinical research, clinical trial design, regulatory approval, clinical practice guideline designation, education and training, continuous monitoring and evaluation, patient participation and informed consent, interdisciplinary cooperation and financial support, so as to jointly promote the transformation and application of research results. Before entering the human body, it is necessary to verify the safety and effectiveness of the drug in treating diseases through cell tests and animal experiments. A wrong drug prediction may be verified at any stage before entering the human body and stop using it to reduce the risk.
Conclusion
In conclusion, based on Swanson’s Literature-based Knowledge Discovery and Bioinformatics, this study proposed the BIOINF-ABC+ model. This study found that Fish oil and Forskolin had a certain therapeutic effect on ASD, which verified the scientificity and accuracy of the BIOINF-ABC+ model, and provided new research ideas and research directions for future drug research.
Limitations and prospects
In the “BIOINF-ABC+” model, the calculation results are valued according to the probability value, and the sorting method is relatively simple, which is the limitation of this algorithm and also the future need to improve. On the basis of this study, the future work can be done in two directions. First, on the basis of this model, an AI-driven model can be further constructed for differential gene screening and knowledge discovery, and the results of this research model and AI-driven model can be compared to find a more favorable method for drug-disease relationship mining. The team has used machine learning for differential gene screening and expanded data sources to further improve knowledge discovery efficiency. The second is to carry out drug prediction based on established or non-established paths with the help of AI models. In this regard, the team has built an AI-driven semantic knowledge discovery model based on two paths (namely “disease-gene-drug” and “disease-gene-gene-drug”) based on the path of “disease-target-drug”, and the comparison of the results is still being further sorted out.
There is still a long way to go from prediction to clinical application. The potential disease-drug relationship found by the “BIOINF-ABC+” model in this study, such as the potential therapeutic value of tricholaryngeal for autism, needs to be done before entering clinical application. First, laboratory validation and replication of research results are needed to rule out chance factors and errors. Secondly, preclinical studies, that is, animal experiments or in vitro experiments are conducted to evaluate the safety and efficacy of the treatment. Then there is clinical trial design, which involves appropriate clinical trials based on research results and clinical needs, while following scientific principles and ethical standards. In addition, before entering clinical application, it also needs regulatory approval (approval by the drug regulatory agency), clinical practice guideline designation (once the research results have been proven valid, relevant professional organizations and institutions can specify clinical practice guidelines, To guide physicians in applying these results in clinical practice), education and training (educating and training physicians and treatment professionals on new research findings and treatments through professional conferences, training courses, and continuing education activities), ongoing monitoring and evaluation (after clinical application, There is a need for continuous monitoring and evaluation of the efficacy and safety of treatments), patient engagement and informed consent, interdisciplinary collaboration (promoting the translation and application of research findings), and financial support to jointly promote the translation and application of research findings. Before entering the human body, it is necessary to go through cell tests and animal experiments to verify the safety and effectiveness of the drug to treat the disease, and a wrong drug prediction may be verified at any stage before entering the human body and stop the use to reduce the risk.
Data availability
The data used in this study were all from publicly available databases.The bioinformatics data used in this study were sourced from the GEO database, with datasets consisting ofGSE59927, GSE45577, GSE43723, GSE52684, GSE58062, GSE28482, GSE62673, GSE73195, GSE70922,GSE50945, GSE48368, GSE56166,GSE46914, GSE59927, GSE5258, GSE137033, GSE124935, GSE59927,GSE68144, GSE59927, GSE83891, GSE73385, GSE68266, GSE42438, GSE22631.
References
Wang, G. H. & Jiang, P. Data mining review. J. Tongji Univ. (Natural Sci. Edition) 32, 246–252. https://doi.org/10.3321/j.issn:0253-374X.2004.02.023 (2004).
Swanson, D. R. & Fish oil, Raynaud’s syndrome, and undiscovered public knowledge. Perspect. Biol. Med. 30, 7–18. (1986). https://doi.org/10.1353/pbm.1986.0087
Swanson, D. R. Undiscovered public knowledge. Libr. Q. 56, 103–118 (1986).
Swanson, D. R. Migraine and magnesium: Eleven neglected connections. Perspect. Biol. Med. 31, 526–557. https://doi.org/10.1353/pbm.1988.0009 (1988).
Swanson, D. R. Two medical literatures that are logically but not bibliographically connected. J. Assoc. Inf. Sci. Technol. 38, 228–233 (2010).
Swanson, D. R. A second example of mutually isolated medical literatures related by implicit, unnoticed connections. J. Am. Soc. Inf. Sci. 40, 432–435 (1989).
Gordon, M. D. & Lindsay, R. K. Toward discovery support systems: A replication, re-examination, and extension of swanson’s work on literature-based discovery of a connection between Raynaud’s and Fish oil. J. Assoc. Inf. Sci. Technol. 47, 116–128 (1996).
Lindsay, R. K. & Gordon, M. D. Literature-based discovery by lexical statistics. J. Am. Soc. Inf. Sci. 50, 574–587 (1999).
Weeber, M., Klein, H., Berg, L. T. & Vos, R. Using concepts in literature-based discovery: Simulating Swanson’s raynaud-fish oil and migraine-magnesium discoveries. J. Assoc. Inf. Sci. Technol. 52, 548–557 (2001).
Stegmann, J. & Grohmann, G. Hypothesis generation guided by co-word clustering. Scientometrics 56, 111–135 (2003).
Hristovski, D., Peterlin, B., Mitchell, J. A. & Humphrey, S. M. Using literature-based discovery to identify disease candidate genes. Int. J. Med. Inf. 74, 289–298. https://doi.org/10.1016/j.ijmedinf.2004.04.024 (2005).
Smalheiser, N. R., Swanson, D. R. & Using, A. R. R. O. W. S. M. I. T. H. A computer-assisted approach to formulating and assessing scientific hypotheses. Comput. Methods Programs Biomed. 57, 149–153. https://doi.org/10.1016/s0169-2607(98)00033-9 (1998).
Luo, R. Q. et al. BioGPT: Generative pre-trained transformer for biomedical text generation and mining. Brief. Bioinform. 23, bbac409. https://doi.org/10.1093/bib/bbac409 (2022).
Kamya, P. et al. PandaOmics: An AI-Driven platform for therapeutic target and Biomarker Discovery. J. Chem. Inf. Model. 64, 3961–3969. https://doi.org/10.1021/acs.jcim.3c01619 (2024).
Kraljevic, Z. et al. MedGPT: Medical Concept Prediction from Clinical Narratives (Cornell University, 2021).
Jin, Q., Yang, Y. F., Chen, Q. Y. & Lu, Z. Y. GeneGPT: Augmenting large language models with domain tools for improved access to biomedical information. Bioinformatics. 40, btae075. https://doi.org/10.1093/bioinformatics/btae075 (2024).
Hodgman, T. C. et al. Bioinformatics (Chen,J. L., translate) 1–239 (Science Press, 2010).
Fang, Y. Application of data mining in bioinformatics. Microcomput. Dev. 14, 1–3. https://doi.org/10.3969/j.issn.1673-629X.2004.04.001 (2004).
Du, W. Application of Machine Learning and Data Mining in Bioinformatics (Jilin Univ., 2011).
Wen, Z. L. et al. Comprehensive Genetic Analysis of Tuberculosis and identification of candidate biomarkers. Front. Genet. https://doi.org/10.3389/fgene.2022.832739 (2022).
Zeng, M. et al. Exploring drug usage patterns and pharmacological mechanisms of diabetes treatment based on data mining and bioinformatics. World Sci. Technology-Modernization Traditional Chin. Med. 24, 597–609. https://doi.org/10.11842/wst.20210429008 (2022).
Ashburner, M. et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 25, 25–29. https://doi.org/10.1038/75556 (2000).
Kanehisa, M. & Goto, S. K. E. G. G. Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30. https://doi.org/10.1093/nar/28.1.27 (2000).
Kanehisa, M. Toward understanding the origin and evolution of cellular organisms. Protein Science: Publication Protein Soc. 28, 1947–1951. https://doi.org/10.1002/pro.3715 (2019).
Kanehisa, M., Furumichi, M., Sato, Y., Kawashima, M. & Ishiguro-Watanabe, M. KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res. 51, D587–D592. https://doi.org/10.1093/nar/gkac963 (2023).
Weeber, M. et al. Text-based discovery in biomedicine: The architecture of the DAD-system. Proc. Amia Symp. 7, 903–907 (2000).
Pratt, W., Yetisgen-Yildiz, M. & LitLinker Capturing Connections across the Biomedical Literature. Proceedings of the 2nd International Conference on Knowledge Capture (K-CAP 2003) (2003).
Shu, J. et al. Deep sequencing microRNA profiles associated with wooden breast in commercial broilers. Poult. Sci. 100, 101496. https://doi.org/10.1016/j.psj.2021.101496 (2021).
Zhang, L. et al. Revealing the pathogenic changes of PAH based on multiomics characteristics. J. Transl Med. 17, 231. https://doi.org/10.1186/s12967-019-1981-5 (2019).
Edgar, R., Domrachev, M. & Lash, A. E. Gene expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 30, 207–210. https://doi.org/10.1093/nar/30.1.207 (2002).
Ritchie, M. E. et al. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. https://doi.org/10.1093/nar/gkv007 (2015).
Yu, G., Wang, L. G., Han, Y. & He, Q. Y. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS 16, 284–287. https://doi.org/10.1089/omi.2011.0118 (2012).
Szklarczyk, D. et al. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 47, D607–D613 (2019).
Shannon, P. et al. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504. https://doi.org/10.1101/gr.1239303 (2003).
Chin, C. H. et al. cytoHubba: Identifying hub objects and sub-networks from complex interactome. BMC Syst. Biol. https://doi.org/10.1186/1752-0509-8-S4-S11 (2014).
Thalmeier, A. et al. Gene expression profiling of post-mortem orbitofrontal cortex in violent suicide victims. Int. J. Neuropsychopharmacol. 11, 217–228. https://doi.org/10.1017/S1461145707007894 (2008).
Lein, E. S. et al. Genome-wide atlas of gene expression in the adult mouse brain. Nature 445, 168–176. https://doi.org/10.1038/nature05453 (2007).
Li, X. R. et al. PTPRR regulates ERK dephosphorylation in depression mice model. J. Affect. Disord. 193, 233–241. https://doi.org/10.1016/j.jad.2015.12.049 (2016).
Fang, M. et al. Dexras1: A G protein specifically coupled to neuronal nitric oxide synthase via CAPON. Neuron 28, 183–193. https://doi.org/10.1016/s0896-6273(00)00095-7 (2000).
Durmaz, C. D. et al. Genetic analysis of RASD1 as a candidate gene for Schizophrenia. Balkan Med. J. 39, 422–428. https://doi.org/10.4274/balkanmedj.galenos.2022.2022-5-90 (2022).
Bouchard-Cannon, P., Lowden, C., Trinh, D. & Cheng, H. M. Dexras1 is a homeostatic regulator of exercise-dependent proliferation and cell survival in the hippocampal neurogenic niche. Sci. Rep. 8, 5294. https://doi.org/10.1038/s41598-018-23673-z(2018).
Fu, W. Q. et al. Rasd1 is involved in white matter injury through neuron-oligodendrocyte communication after subarachnoid hemorrhage. CNS Neurosci. Ther. 30, e14452. https://doi.org/10.1111/cns.14452 (2024).
Xin, Y. J. et al. Dexras1 induces dysdifferentiation of oligodendrocytes and myelin Injury by inhibiting the cAMP-CREB pathway after subarachnoid hemorrhage. Cells. 11, 2976. https://doi.org/10.3390/cells11192976 (2022).
Li, C. Y. et al. Estradiol suppresses neuronal firing activity and c-Fos expression in the lateral habenula. Mol. Med. Rep. 12, 4410–4414. https://doi.org/10.3892/mmr.2015.3942 (2015).
Rao, S. P. & Sikdar, S. K. Acute treatment with 17beta-estradiol attenuates astrocyte-astrocyte and astrocyte-neuron communication. Glia. 55, 1680–1689. https://doi.org/10.1002/glia.20564 (2007).
Hwang, W. J., Lee, T. Y., Kim, N. S. & Kwon, J. S. The role of Estrogen Receptors and their signaling across psychiatric disorders. Int. J. Mol. Sci. 22, 373. https://doi.org/10.3390/ijms22010373 (2020).
Jianrong, S., Yanjun, Z., Chen, Y. & Jianwen, X. DUSP14 rescues cerebral ischemia/reperfusion (IR) injury by reducing inflammation and apoptosis via the activation of Nrf-2. Biochem. Biophys. Res. Commun. 509, 713–721. https://doi.org/10.1016/j.bbrc.2018.12.170 (2019).
Que, Y. Y., Zhu, T., Zhang, F. X. & Peng, J. Neuroprotective effect of DUSP14 overexpression against isoflurane-induced inflammatory response, pyroptosis and cognitive impairment in aged rats through inhibiting the NLRP3 inflammasome. Eur. Rev. Med. Pharmacol. Sci. 24, 7101–7113. https://doi.org/10.26355/eurrev_202006_21704 (2020).
Yang, C. Y. et al. Dual-specificity phosphatase 14 (DUSP14/MKP6) negatively regulates TCR signaling by inhibiting table 1 activation. J. Immunol. 92, 1547–1557. https://doi.org/10.4049/jimmunol.1300989 (2014).
Zheng, H. et al. The dual-specificity phosphatase DUSP14 negatively regulates tumor necrosis factor- and interleukin-1-induced nuclear factor-κB activation by dephosphorylating the protein kinase TAK1. J. Biol. Chem. 288, 819–825. https://doi.org/10.1074/jbc.M112.412643 (2013).
Iroegbu, J. D., Ijomone, O. K., Femi-Akinlosotu, O. M. & Ijomone, O. M. ERK/MAPK signalling in the developing brain: Perturbations and consequences. Neurosci. Biobehav. Rev. 131, 792–805. https://doi.org/10.1016/j.neubiorev.2021.10.009 (2021).
Asl, E. R. et al. Interplay between MAPK/ERK signaling pathway and MicroRNAs: A crucial mechanism regulating cancer cell metabolism and tumor progression. Life Sci. 278, 119499. https://doi.org/10.1016/j.lfs.2021.119499 (2021).
Nussinov, R. et al. Neurodevelopmental disorders, like cancer, are connected to impaired chromatin remodelers, PI3K/mTOR, and PAK1-regulated MAPK. Biophys. Rev. 15, 163–181. https://doi.org/10.1007/s12551-023-01054-9 (2023).
Aluko, O. M., Lawal, S. A., Ijomone, O. M. & Aschner, M. Perturbed MAPK signaling in ASD: Impact of metal neurotoxicity. Curr. Opin. Toxicol. 26, 1–7. https://doi.org/10.1016/j.cotox.2021.03.009 (2021).
Alharbi, M. et al. Effect of natural Adenylcyclase/cAMP/CREB signalling activator forskolin against Intra-striatal 6-OHDA-Lesioned Parkinson’s rats: Preventing mitochondrial, motor and histopathological defects. Molecules 27, 7951. https://doi.org/10.3390/molecules27227951 (2022).
Mehan, S. et al. Adenylate cyclase activator Forskolin alleviates intracerebroventricular propionic acid-induced mitochondrial dysfunction of autistic rats. Neural Regen. Res. 15, 1140–1149. https://doi.org/10.4103/1673-5374.270316 (2020).
Chi, X. F. Mechanism Research: Re-expression of Fragile X Mental Retardation 1 Gene Induced by Adenylate Cyclase Activator (Master’s Thesis) (Guangdong: Southern Medical University, 2012).
Acknowledgements
We would like to express our deep appreciation to the participants who responded to this research.We also would like to express our deep appreciation to all the databases and researchers who provided the research data.
Funding
This study was supported by the National Social Science Foundation of China (Nos.20BTQ064), Special Funding for Shanxi Province Science and Technology Innovation Talent Team (Nos.202304051001017), and Shanxi Key Laboratory of Big Data for Clinical Decision Research.
Author information
Authors and Affiliations
Contributions
L.Y.H. analyzed the data, wrote the paper, and revised the paper. Y.Y.Y. analyzed the data, and wrote the paper. Z.X.Y. analyzed the data. Y.Q. provided strategic design and methods guidance. L.X.C. provided methods guidance. Q.B.Q. analyzed the data. Z.H.X. organized original data. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Lv, Y., Yuan, Y., Zhong, X. et al. Exploration and practice of potential association prediction between diseases and drugs based on Swanson framework and bioinformatics. Sci Rep 14, 29643 (2024). https://doi.org/10.1038/s41598-024-79587-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-79587-6
