
Abstract
Jute endophyte Staphylococcus hominis strain MBL_AB63 was reported to produce a novel antimicrobial peptide, ‘homicorcin’. This exhibits potential activity against a broad spectrum of Gram-positive bacteria. Eight genes were predicted to be involved in the sequential maturation of this peptide antibiotic, which includes structural (homA), dehydratase (homB), cyclase (homC), peptidase (homP), immunity (homI), oxidoreductase (homO), ATP-binding cassette transporter (homT1), and permease (homT2), respectively. Among the modification enzymes, HomB, HomC, and HomP exhibit sequence similarities with class I lantibiotic dehydratase, cyclase, and leader peptidase, respectively. The current study investigated the sequential modifications and secretion of homicorcin by constructing robust computational protein models and analyzing their interaction patterns using protein-protein docking techniques. To enhance comprehension of the protein arrangement, their subcellular localization was also extrapolated. The findings demonstrate a network of proteins that works in a synchronized manner, where HomC functions as an intermediary between HomB and the transporter (HomT). Following its dehydration by HomB and cyclization by HomC, the pro-homicorcin is taken out of the cell by the transporter and processed by HomP, resulting in the production of matured, processed homicorcin. This biosynthesis model for homicorcin will lay the groundwork for the sustainable and efficient production of this peptide antibiotic.
Similar content being viewed by others


Introduction
The emergence of bacterial resistance to existing antibiotics poses an escalating risk to public health. The increasing incidence of drug resistance in numerous pathogenic fungi and bacteria to a diverse array of existing antibiotics has underscored the critical and pressing need for the identification and development of novel therapeutic interventions. Since the inception of lantibiotics over forty years ago, they have been widely employed worldwide without significant emergence of bacterial resistance1. The biotechnological synthesis of lantibiotics, which are peptide-antimicrobials synthesized by ribosomes, is of great interest for enhancing their therapeutic properties by redesigning their natural scaffolds2.
Lantibiotics are ribosomally synthesized peptide antibiotics mostly produced by Gram-positive bacteria as a means of defense against other bacteria3. Prior to achieving full functionality as a lantipeptide, they undergo several post-translational modifications, producing several unusual amino acids, including Lan, MeLan, Dha and Dhb residues4. Several key steps are involved in the production of lantibiotic: the synthesis of the prelantibiotic, glutamylation followed by dehydration, the cyclization process, proteolytic cleavage of the leader peptide, and secretion outside5. Genetic factors responsible for these events are located in certain gene clusters. The products of these genes are the precursor peptide (LanA), post-translational modification enzymes (LanB, LanC/LanM), ABC-superfamily transport proteins involved in peptide translocation (LanT), proteases that remove the leader peptide (LanP), and dedicated self-protection proteins (LanI)6,7. Lantibiotics are classified into several classes based on modification enzymes (LanB, LanC/LanM)8. Modification of class I lantibiotics are mediated by LanB and LanC, while class II lantibiotics undergo modification by LanM9. In the case of class I lantibiotics, nisin and subtilin, a lantibiotic synthetase complex consisting of LanB, LanC, and LanT has been observed to come into contact with LanA across the cell membrane10.
Lantipeptides are in high demand as antimicrobial agents in food preservation and pharmaceutical industries because of their little toxicity in mammalian systems, superior effectiveness compared to antibiotics, absence of lantibiotic resistance in bacteria, and strong activity against drug-resistant strains3.
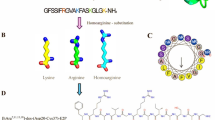
Aftab Uddin et al. reported a novel lantibiotic named ‘homicorcin’ derived from the jute endophyte Staphylococcus hominis strain MBL_AB6311. The homicorcin demonstrated significant antibacterial efficacy against a minimum of eight indicator strains, including methicillin-resistant Staphylococcus aureus (MRSA)11. The genome sequence study of S. hominis MBL_AB63 revealed the presence of a class I lantibiotic gene cluster encoding homicorcin. Other well-known class I lantibiotics such as epicidin 280, pep5, and nisin A have also been shown to contain a similar type of cluster. Homicorcin cluster consists of eight genes comprising homO (reductase gene), homI (immunity gene), homA (structural gene), homP (protease gene), homB (dehydratase gene), homC (cyclase gene), homT1 (ATP-binding cassette domain), and homT2 (permease). The transporter genes (homT1T2) are oriented in the opposite direction. (Fig. 1A).
Due to its recent discovery, the mechanism of modifications for the lantibiotic homicorcin remains unknown. A thorough understanding of the lantibiotic’s post-translational modification is essential for its effective production and application. The purpose of this study was to explore the gradual events of modifications, crucial amino acids implicated in these processes, and propose a computational model of the maturation process of homicorcin. To do this, computational models of the biosynthesis protein structures were created and verified. Protein complexes were modeled using protein-protein docking techniques, and subcellular localization of the proteins and ATP dependence of the reactions were investigated. Simultaneously, the essential residues involved in protein interactions have been revealed. Finally, a model of homicorcin biosynthesis and maturation was created based on the results obtained. This model will facilitate a more profound comprehension of its biosynthesis process and initiate the creation of a separate interface to specifically focus on the engineering of this peptide lantibiotic to build an improved candidate that can effectively counteract the rising issue of antibiotic resistance.
Results and discussion
Comparative analysis of gene clusters involved in class I lantibiotic biosynthesis
Class I lantibiotic gene clusters were compared to determine the extent of their homologous relationships with homicorcin as depicted in Fig. 1A. In order to determine the level of similarity at the gene level, individual genes from the homicorcin gene cluster were compared with the corresponding genes from other lantibiotic clusters (Table 1). The findings demonstrate that the homicorcin biosynthesis genes have the greatest degree of homology with the respective genes of epicidin 280. The percentage identity of homicorcin biosynthesis genes with that of epicidin 280 is greater than 80% with a reliable expect value. This is followed by pep5 and epilancin 15X, respectively. However, nisin A is the most diverged cluster in this instance. Not only with four immunity and two regulatory genes in the cluster, the nisin biosynthesis genes homologous to other lantibiotics also differ significantly. This divergence is reflected with ~ 25% identity of NisB, NisC, and NisP with respective genes of the homicorcin gene cluster.

Comparison of homicorcin biosynthesis genes with other class I lantibiotics. (A) The gene cluster that is responsible for the production of homicorcin was predicted using the whole genome sequence of S. hominis MBL_AB63, and a comparison was made between the gene clusters of other lantibiotics. Homicorcin gene cluster comprises biosynthesis genes – reductase (homO), immunity (homI), homicorcin peptide (homA), protease (homP), dehydratase (homB), cyclase (homC), and two transportation related genes- homT1 (ATP-binding cassette domain) and homT2 (permease). Other clusters of genes involved in the biosynthesis of class I lantibiotics have comparable gene types. (B) Leader peptides and core peptides of respective lantibiotics are represented separately. Red-colored residues of core peptides are dehydrated during post-translational modifications. The so-called FNLDV box in leader peptides is marked (inset). (C) and (D) Chemical structures of homicorcin and nisin A, respectively. Blue circles indicate modified residues. Dha- dehydroalanine; Dhb- dehydrobutyrine; Abu- aminobutyric acid; Abu-S-Ala- Methyllanthionine; Dha-S-Ala- Lanthionine.
Leader peptide mediates the recognition of modification enzymes by homicorcin prepeptide
Homicorcin and nisin biosynthesis gene cluster contains the homA and nisA structural genes, which encode ribosomally synthesized precursor peptides known as “prepeptides”. These prepeptides, in contrast to mature peptides, have an N-terminal extension, also known as a leader peptide, connected to the C-terminal propeptide. The modified propeptide domain, which corresponds to the mature peptide, is only active upon the leader’s proteolytic cleavage. HomA prepeptide has an equal number of positive and negative charged residues, resulting in a net charge neutrality whereas NisA has a net positive charge. The GRAVY index determines both peptides as hydrophilic. There are three Ser residues and four Thr residues in the core peptide of homicorcin. Within this group, it is anticipated that HomB dehydrates all of the Ser residues as well as three of the Thr residues as the first modification11. HomA has three cysteine residues, which are responsible for the synthesis of lanthionine and methyllanthionine crosslinks through the nucleophilic addition of cysteinyl thiols to dehydroalanine (Dha) and dehydrobutyrine (Dhb), respectively12.
HomA and NisA leaders have a significant percentage of charged amino acids. The FNLDV box in nisin is crucial for the synthesis of accurately modified nisin13. Since the similar FDLEI motif in Pep5’s leader peptide has been identified as an essential component for maintaining an adequate rate of its manufacture, it is postulated that the same motif is also essential for homicorcin biosynthesis14 (Fig. 1B).
3D structure analysis of homicorcin and nisin biosynthesis proteins
Due to the unavailability of the 3D structures of the homicorcin biosynthesis peptide and enzymes (HomA, HomB, HomC, HomP, HomT1, and HomT2) in RCSB-PDB, the sequences were utilized to construct the corresponding 3D structures. Although homicorcin shows no significant sequence similarity with nisin, the current study used nisin as a control since it is a well-studied lantibiotic available in the literature to validate the present study. The available structures of nisin biosynthesis peptides and enzymes were retrieved from the RCSB-PDB.
The 3D structures of homicorcin and nisin prepeptides have predominantly loops and turns (Fig. 2A, B) that provide an overall flexibility in the structures15. The coexistence of polar amino acids such as serine, threonine, asparagine, glutamine, lysine, aspartate, and glutamate in HomA promotes the formation of hydrogen bonds between main-chain atoms, resulting in the formation of numerous turns16. The compact C-terminal structure of NisA, which is not present in HomA, is attributed to the presence of hydrophobic residues such as alanine, isoleucine, methionine, and valine17. HomB and NisB are both categorized as dehydratase enzymes responsible for catalyzing the dehydration process of Ser/Thr residues found in homicorcin and nisin prepeptide, respectively. The analysis of HomB and NisB’s structure unveiled a dimeric bifurcated cleft-like configuration. These enzymes are rendered inactive when existing as monomers but become activated in their dimeric state12. Both HomB and NisB contain a glutamylation domain and a glutamate elimination domain, as shown in Fig. 2C, D. The residues from Pro40 - Leu650 of HomB consist of the N-terminal glutamylation domain, while the region from Trp713 - Leu953 includes the carboxy terminal elimination domain. The glutamylation domain of NisB consists of approximately 700 N-terminal residues, while the elimination domain is formed by the remaining C-terminal residues.

Predicted 3D structures of (A) HomA, (B) NisA, (C) HomB dimer, (D) NisB dimer, (E) HomC, (F) NisC, (G) HomT dimer, (H) NisT dimer, (I) HomP, (J) NisP.
Following dehydration, the lantibiotic undergoes cyclization facilitated by lantibiotic cyclases. HomC and NisC both have an α,α barrel structure consisting of several alpha helices arranged in a toroid shape when seen vertically (Fig. 2E, F). Both of them possess an overall negative charge, and their tertiary structure is stabilized by a considerable amount of Leu and Ile. Another similarity between these two LanC is the prevalence of helices over strands. The entire structure operates as a unified domain known as class I lantibiotic biosynthesis cyclase. The mutational research of nisin biosynthesis discovered that the leader peptide also plays a role in facilitating the detection of dehydrated pre-nisin by NisC18. Following cyclization, the prepeptide is subsequently transported outside the cell using an ATP-binding cassette (ABC) transporter. ABC transporters consist of two distinct types of proteins: the nucleotide (ATP) binding domain (NBD), which possesses ATPase activity, and the transmembrane permease domain (TMD), forming a tetrameric transporter complex (Fig. 2G)19,20,21. The highly conserved cytoplasmic HomT1 (NBD) and six transmembrane helices of HomT2 (TMD) are the signature of the ‘cassette-like’ structure of ABC transporters22. In silico ATP-binding motif analysis by ATPbind revealed the potential ATP-binding residues of HomT1 (Ile13, Lys16, Ile18, Asn38- Thr44, Glu77, and His180). The NBD undergoes dimerization upon ATP binding, followed by hydrolysis of ATP. The released energy is utilized to trigger a conformational alteration in the TMD, enabling the export of peptides20. The peptide outside the cell is subsequently cleaved by lantibiotic proteases LanP (leader peptidases). HomP consists of 9 alpha helices and 14 beta strands. The number of beta strands in NisP is significantly higher, specifically 31 (Fig. 2I, J). Serine proteases have an augmented quantity of beta strands involved in the identification of substrates23. The synthesis of mature homicorcin and nisin depends significantly on these leader peptidases.
HomA and HomB interact through the leader peptide, causing dehydration of the Ser/Thr residues in the core peptide, resulting in the formation of Dha and Dhb, respectively
Upon examination of the HomA-HomB complex’s binding energies, a strong interaction was revealed with an estimated value of -73.07 kcal/mol. The NisA- NisB complex retrieved from PDB also showed a binding energy of -64.12 kcal/mol. Interaction analysis between HomA-HomB and NisA-NisB reveals that several residues from both LanA and LanB are involved in the interactions (Fig. 3). The interacting residues of HomA mainly reside in leader peptide including Met1, Asn3, Asn4, Lys5, Asn6, Leu7, Phe8, Asp9, Leu10, Glu11, Asn19, Asn21, Glu22, Leu23, Glu24, Gln26, and Lys36 (Fig. 3B). Phe8, Asp9, Leu10, and Glu11 are positioned in the FDLEI motif (Fig. 1B). The residues of HomB, maintaining the highest degree of interactions with HomA are Asn54, Asn56, Arg128, Leu171, Ile172, Asp302, Ser304, Tyr345, Asn556, Tyr559, Ala642, Asp643, Lys665, and Asn666 (Fig. 3B). These interactions bring the HomA core peptide to the proximity of the HomB active site for glutamylation, followed by dehydration of all Ser/Thr residues except Thr3011. In NisA, the leader peptide residues Asp5, Phe6, Leu8, Asp9, Leu10, Ser12, Ser14, Lys15, Lys16, Asp17, and Ser18 interact with NisB residues of Arg154, Leu166, Glu168, Val169, Asn170, Ile171, Lys172, Val176, Val198, Tyr202, Leu209, Ser210, Tyr213, Leu217, and Glu502 (Fig. 3D). The crystallographic study of NiA-NisB complex also identified these residues as playing a key role during the interaction12. This interaction also brings the core peptide into proximity of the NisB active site for glutamylation. The FNLDV box, which is part of the LanA leader peptide, plays a crucial role in its interactions with the modifying enzymes HomB and NisB. Analysis of the mutation in the nisin leader has revealed that certain conserved amino acids inside the FNLDV box play a critical role in nisin modification, secretion, or both24. HomB acts on Ser/Thr of the core peptide (HomA), adding glutamate to its -OH group, hence called glutamylation, which is performed by the N-terminal glutamylation domain of HomB. Subsequently, the glutamate is eliminated, ultimately dehydrating the Ser/Thr forming Dha and Dhb, respectively12.

HomA-HomB (A, B) and NisA-NisB (C, D) complex formation is mediated by extensive interface interactions. The leader peptide of prehomicorcin (A-blue) plays a vital function in the recognition of HomB (A-green). The interaction relies on residues inside the FNLDV box (B-* marks). NisA leader peptide (C-pink) plays a similar role in recognition of NisB (C-cyan). The thickness of interaction lines is proportionally adjusted based on the strength of the interaction.
Some conserved residues of NisC play a key role in cyclization
Following dehydration, the LanA undergoes cyclization by the LanC in the subsequent step. The calculated binding energy of the dehydrated HomA and HomC complex is -75.09 kcal/mol, while the binding energy of the dehydrated NisA and NisC complex is anticipated to be -54.39 kcal/mol. In dehydrated HomA, eight residues of the leader peptide (Lys13, Val17, Asn19, Asn21, Leu23, Glu24, Ala25, and Gln26) aid in recognizing HomC (see Supplementary Fig. S2). The docked residues once again highlight the important role of the leader peptide of HomA in recognizing HomC. The B ring of HomA is formed by dehydrating Thr47 to Dhb, which is then docked with the HomC active site by interacting Tyr15 of HomC (Fig. 4A).
It has been documented that a zinc ion is essential to the cyclization reaction by NisC. The zinc ligand is situated in the NisC active site and is stabilized by interactions with Cys284, Cys330, and His33125. The zinc ion coordinates with the Cys residue in pre-NisA, which leads to thiol activation and reduction in pKa of deprotonation for the cyclization step25. The cyclization reaction is performed by two key residues, His212 and Arg280 of NisC18. These residues participate in the acid-base chemistry of the reaction, which involves removing a proton from the Cys residue and adding a proton to the enolate intermediate formed by conjugate addition to the β carbon of Dha/Dhb18. Figure 4C shows that the zinc binding residues of NisC are conserved in HomC (Fig. 4D). Therefore, it is highly probable that HomC also includes a zinc ligand in its active site that triggers the cyclization reaction of dehydrated pre-HomA. His212 and Arg280 of NisC are also found conserved in HomC as His204 and Arg261, respectively (Fig. 4D). They might play a similar role in the catalysis of the cyclization reaction of pre-HomA. The COFACTOR metal binding residues prediction tool (https://seq2fun.dcmb.med.umich.edu/COFACTOR2/) has confirmed the zinc-binding of these conserved residues in HomC (Fig. 4E).

Representation of the protein-protein docking complex and interacting residues of dehydrated HomA-HomC (A), cyclized HomA-HomT (B), and cyclized HomA-HomP (C). Conserved residues of HomC and NisC catalytic sites (D). His212, Arg280, Cys284, Cys330, and His331 of NisC are situated in its catalytic site, playing crucial roles in pre-NisA cyclization. Cys284, Cys330, and His331 of NisC stabilize a zinc ion that activates the thiol group of pre-NisA Cys residues, followed by its deprotonation. His212 and Arg280 involve removing the proton from the Cys residue and adding a proton to the enolate intermediate formed by conjugate addition to the β carbon of Dha/Dhb, ultimately facilitating the cyclization reaction in NisC. Conservation of these residues in HomC may function similarly in HomA cyclization (E).
Cyclized HomA transported outside the cell by ABC transporter and permease complex
Transporters that utilize ATP hydrolysis to actively transport molecules across the membrane are known as ATP-binding cassette (ABC) transporters, owing to their structural resemblance to “cassette-like” structures26. Lantipeptide producing bacteria use ABC transporters to export their lantipeptides outside the cell, hence referred to as ‘transport ATPases’22,27,28,29. Typical ABC transporter consists of four functional units or domains, namely two nucleotide binding domains (NBD1, NBD2) and two transmembrane domains (TMD1, TMD2). The crystal structures of individual nucleotide binding domains (NBDs) bound to ATP revealed that two NBDs formed a dimer, with the two ATP molecules positioned between them at the dimer interface22. The process of protein-protein docking was utilized to create a model of a tetrameric complex of an ABC transporter comprising HomT1 and HomT2. This complex consists of two copies of each protein (Fig. 2G). HomT1 connects to HomT2 through a hinge region. In contrast to the NBDs, the TMDs typically do not exhibit any notable sequence conservation but do have a comparable structure within a given transporter class. The absence of conservation in the primary structure of the transmembrane domains (TMDs) is probably a result of the varied characteristics of the substances being transported22. The combination of two transmembrane domains (TMD) ultimately results in the formation of an inward-facing cavity, which serves as an open space for the binding of HomA propeptide. The cyclic HomA molecule interacts with the exposed region of HomT2, and this interaction is characterized by a binding energy of -90.63 kcal/mol. An in-depth examination of these interactions demonstrates that the leader peptide interacts with HomT1, while the core peptide binds to HomT2. Leu7, Phe8, Asp9, Leu10, Ile12, Lys13 of pre-HomA mainly interacts with Lys243, Gly282, His285, Tyr286, Thr290 and Tyr291 of HomT1 (Fig. 4B). On the other hand, Lys41- Gln51 and Asp54- Gln56 of the core peptide show different interactions with HomT2. In order to transport the prepeptide, the HomT2 protein undergoes a conformational change, transitioning from an inward-facing state to an outward-facing state. The conformational change is initiated by the binding of ATP to HomT1, which results in increased proximity to both NBDs and ultimately alters the inward-facing conformation of HomT2 to an outward-facing conformation. This change facilitates the release of pre-HomA outside the cell.
HomP cleaves leader peptide through an ATP-dependent reaction
To produce mature homicorcin, the leader peptide of the HomA must be eliminated after transportation. Therefore, the leader peptide once again identifies the final modification enzyme (HomP) to undergo its proteolytic cleavage. Similar instances of consecutive modification events have been documented for class I lantibiotics12,18. The estimated binding energies of the HomA-HomP and NisA-NisP complexes are − 79.87 and − 88.34 kcal/mol, respectively (Fig. 4C, see Supplementary Fig. 2D).
The requirement of ATP for the maturation of nisin has been confirmed by Ortega et al., 2015. Nevertheless, experiments using pure glutamyl-tRNAGlu and subsequent MALDI-TOF/MS analysis demonstrated that the dehydration reaction catalyzed by NisB does not require ATP12. In fact, co-crystallographic structure analysis revealed that there is no ATP binding domain in NisC18. Given that NisB and NisC operate without utilizing ATP, it is highly probable that HomB and HomC similarly operate independently of ATP. In order to confirm this hypothesis, possible ATP binding motifs were examined in HomB, HomC, HomP, and NisP (Fig. 5). It is clear that nisin maturation relies on ATP. Neither NisB nor NisC employ ATP for their functions. Therefore, it may be inferred that NisP utilizes ATP for leader peptide cleavage, indicating the presence of putative ATP binding sites in NisP. Thus, NisP has been used as a control for this objective. Figure 5 demonstrates that HomP possesses ATP binding motifs similar to NisP, but HomB and HomC fall well below the threshold utilized by the tool. Therefore, it can be inferred that the function of HomP depends on the presence of ATP.

Determination of ATP binding site in HomB, HomC, HomP and NisP. ATPbind utilizes the pdb file of protein structure and delivers the potential ATP binding sites with the probability of each residue of respective position. The binding site is considered if the probability value exceeds the threshold of 0.430. NisP has been employed as a control for the identification of the binding site. HomP possesses several ATP binding sites with a probability value significantly higher than the threshold, unlike HomB and HomC. This suggests that HomP has several possible ATP binding sites that play a role in its energy usage during the proteolytic cleavage of the HomA leader peptide.
Modification enzymes primarily adhere to or are integrated into the cellular membrane
To elucidate the entire modification process of homicorcin, it is crucial to ascertain the precise locations of the modification enzymes. The subcellular localization prediction results indicated that the dehydratase (HomB and NisB), cyclase (HomC and NisC), ABC transporter (HomT and NisT), and leader peptidase (HomP and NisP) are localized in different interphases of the membrane. HomB and NisB predominantly localize to the cytoplasmic side of the membrane, with just a minor fraction being linked to it. HomC and NisC consist of 3–5 transmembrane segments alternating with extracellular regions (Table 2). While HomT2 is located in the cell membrane, HomT1 resides exclusively in the cytoplasm. The leader peptidase (HomP) in our study exhibits a transmembrane topology, with a tiny section of its C-terminal region remaining entrenched in the cell membrane while the rest of the domain is located outside (see Supplementary Fig. S3A). Khusainov et al. reported that during the purification of NisB, substantial quantities of NisC were also co-purified, indicating that NisB and NisC interact with one another in close proximity31. Such interaction of NisB-NisC is replicated in their complex formation in a protein-protein docking study with a binding energy of -36.33 kcal/mol. The same study conducted with HomB and HomC resulted in a complex with a binding energy of -97.55 kcal/mol. Hence, it is predicted that HomB and HomC interact in a similar way.
This study also examined the interaction between HomB and HomC, as well as HomC and HomT and Hom T with HomP. The glutamate elimination domains of HomB form a dimer and interact with the HomC, as shown in Supplementary Fig. S3A. The binding free energy of this interaction is -97.55 kcal/mol higher than that of NisB-NisC (-58.95 kcal/mol). Subcellular localization of HomC predicts its position embedded in the cell membrane, interacting with the transporter with several transmembrane helices in a lateral fashion (ΔG -68.72 kcal/mol) (see Supplementary Fig. S3B). Therefore, it is hypothesized that HomB and HomC remain in close proximity to the ABC transporter in order to facilitate the effective release of cyclized pro-HomA from the cell. The HomP protein, which is anchored to the membrane by a transmembrane helix, remains in close proximity to the transporter in order to capture the pro-HomA protein and subsequently remove its leader peptide through cleavage.
Molecular dynamics simulations study
The root-mean-square deviation (RMSD) is a statistical measure that determines the average distance between atoms in superimposed proteins or molecular structures. It offers valuable information about structural deviations observed during simulations32. In molecular dynamics simulations, RMSD is used to monitor the stability of proteins, as demonstrated in the present study of the HomA peptide, where RMSD highlighted significant conformational changes due to interactions with modification enzymes. Figure 6A illustrates that the stability of the HomA-HomB complex remained consistent, reaching a fixed RMSD value of approximately 0.75 nm after 35 ns, with a small deviation around 70 ns. The dHomA + HomC complex exhibited sustained stability at a RMSD of 0.3 nm after 50 ns, making it the most stable among the four complexes (Fig. 6B). In contrast, the cHomA + HomT and cHomA + HomP complexes maintained stability at 0.5 nm after 60 ns and 40 ns, respectively (Fig. 6C, D). The cHomA + HomT complex exhibits a higher degree of dynamism compared to the others, characterized by a little rise in the RMSD towards the conclusion of the simulation period (Fig. 6C). An analogous dynamic character is also evident in cHomA + HomP, with minor fluctuations seen toward the end of the graph (Fig. 6D). Nevertheless, the comparative stability of all four complexes is far superior to that of the control in all instances.

Root mean square deviation (RMSD) analysis of the Cα atoms of the protein complexes and corresponding peptide as control. HomA- ribosomally synthesized raw prehomicorcin, dHomA- dehydrated HomA, cHomA- cyclized HomA. RMSD of HomA + HomB (A), dHomA + HomC (B), cHomA + HomT (C), and cHomA + HomP (D). The enzyme-peptide complex shows lower RMSD in all cases compared to that of the control. Atomic trajectories were collected during the manufacturing run extending 100 ns.
The root mean square fluctuation (RMSF) is used to identify the protein regions that are accountable for variations by assessing the time-dependent flexibility of particular residues. Higher scores correspond to greater levels of instability and flexibility in the bonds, while lower scores indicate a stable region in the complexes33. The RMSF data analysis illustrates the stability of the interacting residues in all the complexes represented in Fig. 7. Figure 7A shows distinct differences in the flexibility of HomA when employed alone (green) and when combined (red) with HomB (HomA + HomB). Upon independent analysis, HomA demonstrates notably greater fluctuations in residues 1–50, indicating higher flexibility or disorder in this site. Nevertheless, the combination of HomA with HomB significantly decreases the fluctuations. The HomA + HomB combination exhibits comparable minimal variations over the remaining region (50–900), indicating that the bulk of the structure remains stable. In Fig. 7B, dHomA alone shows significant fluctuations, which are further reduced when HomC binds. Similar interaction stability is observed in cHomA + HomT and cHomA + HomP complexes (Fig. 7C, D) with a slightly increased RMSF of cHomA (residue 30–45) when bound to HomP (Fig. 7D).

Root mean square fluctuation (RMSF) analysis of the protein complexes and corresponding peptide as control. RMSF of HomA + HomB (A), dHomA + HomC (B), cHomA + HomT (C), and cHomA + HomP (D) depict the fluctuations of individual residues in all four complexes compared to respective controls. There has been a marked decrease in peptide residue fluctuations during complex formation compared to when the compounds were alone in all instances.
Proposed model for homicorcin biosynthesis
A model is proposed to enhance the comprehension of the biosynthesis process of homicorcin (Fig. 8). The biosynthesis gene cluster responsible for the production of homicorcin is expressed as a unified operon. The HomA precursor peptide, together with its leader peptide, is designed for extracellular transportation. The prehomicorcin undergoes dehydration at the Ser and Thr residues catalyzed by the enzyme HomB dehydratase. This is followed by three consecutive cyclization reactions facilitated by the enzyme HomC. The cyclic HomA is subsequently transported through a dedicated ABC transporter and subjected to proteolytic cleavage by HomP, employing ATP, resulting in the production of mature homicorcin.

A synchronized protein interaction network transforms prehomicorcin into a fully developed and functional lantibiotic. (A) The prehomicorcin (pre-HomA) attaches to the glutamylation domains of the HomB dimer and undergoes glutamylation at certain Ser/Thr residues (1). Following that, the glutamate is eliminated, resulting in the dehydration of the peptide (green residues) through the glutamate elimination domains of HomB (2). The HomC dimer forms three thioether ring structures within the pre-HomA molecule (3), which is in close proximity to the transporter, facilitating transportation of the peptide (4). HomP enzymatically cleaves the leader peptide, employing ATP molecules to release active homicorcin (5). (B) Molecular model showing the protein-protein interactions of the biosynthesis machinery drawn by PyMol 2.5.5.
Materials and methods
Prediction of physicochemical parameters and the sub-cellular localization of Homicorcin biosynthesis proteins
The nucleotide sequences of studying homicorcin and nisin A (as control) biosynthesis genes (LanA, LanB, LanC, LanT, and LanP) were collected from NCBI GenBank (https://www.ncbi.nlm.nih.gov/nucleotide/) (Table 1) and subsequently translated into appropriate amino acid sequences using the Expasy translator (https://web.expasy.org/translate/)34. The physicochemical parameters of the proteins were theoretically measured using Expasy’s Protparam server (https://web.expasy.org/protparam/). The predicted properties such as molecular mass, total number of positive and negative residues, aliphatic index, instability index, and grand average of hydropathicity (GRAVY) were determined.
Assessing the subcellular localization of proteins aids in determining their site of activity and function. In this study, PSORTb version 3.0.3 (https://www.psort.org/psortb/index.html) and DeepTMHMM 2.0 (https://dtu.biolib.com/DeepTMHMM) were used to predict the sub-cellular localization of target proteins, identifying potential regions35,36.
Protein 3D structure prediction
The Robetta server (https://robetta.bakerlab.org/) was employed to perform ab initio modeling for the purpose of protein structure prediction. The algorithmic precision and reliability of ‘Robetta’ led to its selection as the favored solution37. The structures were further validated using AlphaFold2 (https://colab.research.google.com/github/sokrypton/ColabFold/blob/main/AlphaFold2.ipynb). AlphaFold2 employs advanced deep learning methodologies that predict the three-dimensional structure of a protein, utilizing its amino acid sequence only38. The structures predicted by Robetta and AlphaFold2 were compared by RCSB pairwise structure alignment server (www.rcsb.org/alignment). No significant difference was found based on their structural alignments, except the HomA (Table 3).
Seeing the poor TM-score of HomA, we evaluated both HomA models by QMEANDisCo and Ramachandran plot. QMEANDisCo global score for HomA (Robetta) is 0.31, while that of HomA (AlphaFold2) is 0.18. Ramachandran plot analysis also showed similar result. The underlying explanation may stem from the operational principle of AlphaFold2, which necessitates approximately 30 homologous sequences to predict the structure. Nevertheless, the technique identified only six structures for predicting HomA. This may lead to low TM-score.
The conformational quality of the protein structures was evaluated using Ramachandran plot analysis by PROCHECK (https://saves.mbi.ucla.edu/) (see Supplementary Fig. S1). The structural models have been submitted to the protein model database ModelArchive, developed by Swiss Institute of Bioinformatics (modelarchive.org), which archives computationally predicted protein structure models (see Supplementary Table S1).
In order to confirm the results of the study, a well-studied class I lantibiotic, nisin, has been chosen as the control. The structures of nisin and its associated modification enzymes have been retrieved from the Protein Data Bank (https://www.rcsb.org/). The use of these reference structures allows for the comparison and evaluation of modeled protein structures against validated templates, hence enhancing the overall reliability of this study.
Protein-protein docking analysis of biosynthesis proteins
The web server ClusPro 2.0 software was utilized to perform protein-docking interactions. ClusPro (https://cluspro.bu.edu/) utilizes a rigid-body methodology to do protein-protein docking. This method entails placing one of the proteins, known as the receptor, at the origin of the coordinate system on an immobile grid. The second protein, referred to as the ligand, is positioned on a movable grid. The energy of their contact is quantified using a correlation function, which might be a composite of multiple correlation functions. The tool is commonly used for protein-protein docking investigations39,40,41. Due to the lack of prior knowledge regarding the dominant forces in the complex, balanced coefficients were employed for model selection from ClusPro output. The model with the largest cluster size (number of members) among peptide combinations with the lowest score was chosen40.The complexes’ binding energies were computed utilizing the end-point free energy. The complexes obtained from the docking process were prioritized according to their binding energy and assessed using the MM/GBSA free energy analysis offered by the HawkDock tool42. Calculations based on continuum electrostatics (GBSA) and molecular mechanics (MM) are combined in the MM/GBSA method, which is used to determine the binding free energy by taking into consideration the molecular interactions (van der Waals, electrostatic, and solvation interactions) that occur between the two proteins. This approach allows to identify the most sustained and biologically important complex structures and organize them based on their strength of binding. Following this, the compound that exhibited the highest binding affinity was selected, and the specific residues that were involved in the interaction were determined.
To determine the interaction pattern closer to reality, HomA was modified to generate dehydrated Ser and Thr residues prior to docking with HomC utilizing the Molecular Operating Environment (MOE) platform (Chemical Computing Group). Subsequently, the dehydrated HomA was cyclized at specific points (as determined by the pioneer group) prior to docking with HomP11. The protein complex models have been submitted to the ModelArchive database (see Supplementary Table S1).
Prediction of ATP dependency
The necessity of ATP dependency of the modification enzymes was determined by screening ATP binding sites in the proteins. ATPbind server (https://zhanggroup.org/ATPbind/) can determine ATP binding sites efficiently43. ATPbind employs a protein structure pdb file to identify and provide the potential ATP binding sites. It also calculates the likelihood of each residue at its respective location. The binding site is regarded when the probability value exceeds the threshold of 0.430.
Molecular dynamics simulations study
The objective of the molecular dynamics simulations was to evaluate the stability of the presented complexes. The GROMACS version 2024.2 was utilized, and the OPLS-AA/L all-atom force field (2001 aminoacid dihedrals) was applied in this study. Prior to commencing the simulation, the docked structures were immersed in an ionized water box. The establishment of a cubic enclosure was imperative in order to resolve the docked complexes and achieve system neutralization. To achieve this, a sufficient quantity of sodium and chloride ions were introduced. Using a steep descent approach with a cutoff of up to 1,000 kJ/mol and 500,000 iterations or reduction steps, the energy minimization (EM) technique was employed to resolve any steric conflicts. Calculations of long-distance interactions were performed using the particle mesh Ewald (PME) truncation approach. The electromagnetic approach proved to be effective in functioning as a robust simulation framework. A two-phase equilibration process was conducted to achieve equilibrium between the solvent and ions around the complex. In the initial phase, the temperature was stabilized by achieving equilibrium between the number of particles, volume, and temperature (NVT) ensemble at a temperature of 300 K. In the subsequent phase, the pressure of the system was stabilized by achieving equilibrium with the ensemble of particles, pressure, and temperature at a constant number of particles (NPT). The completion time for both phases was 1000 picoseconds. Furthermore, the production run was carried out to collect atomic trajectories over a period of 100 ns with a time step of 2 fractional seconds.
Conclusion
Modeling antibiotic biosynthesis provides guidance for synthetic biology strategies that attempt to create microbes for the production of antibiotics with improved characteristics or novel functionalities. The study’s biosynthesis modeling has practical uses in advancing the development of large-scale antibiotic production methods, which could enhance the efficiency and cost-effectiveness of homicorcin production. Simultaneously, this model will enable researchers to optimize the parameters for homicorcin and other class I lantibiotic synthesis. This will establish a foundation for the sustainable and efficient production of lantibiotics while also contributing to endeavors aimed at addressing antibiotic resistance.
Data availability
All data generated or analysed during this study are included in this published article and its supplementary information files.
References
Lobo-Ruiz, A. & Tulla-Puche, J. Synthetic approaches of naturally and rationally designed peptides and peptidomimetics. Pept. Appl. Biomed. Biotechnol. Bioeng. 23–49. https://doi.org/10.1016/B978-0-08-100736-5.00002-8 (2018).
Lakshmi, S. A., Arunachalam, K., Chunlei, S. & Davoodbasha, M. A. Methods for identification of the modes of action of lantibiotics. Lantibiotics as Altern. Ther. 319–335. https://doi.org/10.1016/B978-0-323-99141-4.00007-2 (2023).
Chakraborty, H. J., Gangopadhyay, A. & Datta, A. Prediction and characterisation of lantibiotic structures with molecular modelling and molecular dynamics simulations. Sci. Rep. 9, 7169 (2019).
Chatterjee, C., Paul, M., Xie, L. & van der Donk, W. A. Biosynthesis and mode of action of lantibiotics. ChemInform 36, (2005).
McAuliffe, O., Ross, R. P. & Hill, C. Lantibiotics: Structure, biosynthesis and mode of action. FEMS Microbiol. Rev. 25, 285–308 (2001).
Siezen, R. J. Comparison of lantibiotic gene clusters and encoded proteins. Antonie Van Leeuwenhoek Int. J. Gen. Mol. Microbiol. 69, 171–184 (1996).
Jack, R., Bierbaum, G., Heidrich, C. & Sahl, H. -G. The genetics of lantibiotic biosynthesis. BioEssays 17, 793–802 (1995).
Arnison, P. G. et al. Ribosomally synthesized and post-translationally modified peptide natural products: Overview and recommendations for a universal nomenclature. Nat. Prod. Rep. 30, 108–160 (2012).
Bierbaum, G., Sahl, H. G. & Lantibiotics mode of action, biosynthesis and bioengineering. Curr. Pharm. Biotechnol. 10, 2–18 (2009).
Kiesau, P. et al. Evidence for a multimeric subtilin synthetase complex. J. Bacteriol. 179, 1475–1481 (1997).
Aftab Uddin, M. et al. A plant endophyte Staphylococcus hominis strain MBL_AB63 produces a novel lantibiotic, homicorcin and a position one variant. Sci. Rep.. 111 (11), 1–12 (2021).
Ortega, M. A. et al. Structure and mechanism of the tRNA-Dependent lantibiotic dehydratase NisB. Nature 517, 509 (2015).
Van Der Meer, J. R. et al. Influence of amino acid substitutions in the nisin leader peptide on biosynthesis and secretion of nisin by Lactococcus lactis. J. Biol. Chem. 269, 3555–3562 (1994).
Neis, S. et al. Effect of leader peptide mutations on biosynthesis of the lantibiotic Pep5. FEMS Microbiol. Lett. 149, 249–255 (1997).
Papaleo, E. et al. The role of protein loops and linkers in conformational dynamics and allostery. Chem. Rev. 116, 6391–6423 (2016).
Dhar, J. & Chakrabarti, P. Defining the loop structures in proteins based on composite β-turn mimics. Protein Eng. Des. Sel. 28, 153–161 (2015).
Mornon, J. P. Hydrophobic compaction, curvature of space and deciphering protein sequences. Europhys. News. 34, 16–19 (2003).
Li, B. et al. Structure and mechanism of the Lantibiotic Cyclase involved in Nisin Biosynthesis published by: American Association for the Advancement of Science Linked references are available on JSTOR for this article: Structure and mechanism of the Lantibiotic Cyc. 311, 1464–1467 (2016).
Alkhatib, Z., Abts, A., Mavaro, A., Schmitt, L. & Smits, S. H. J. Lantibiotics: How do producers become self-protected? J. Biotechnol. 159, 145–154 (2012).
Beis, K. & Rebuffat, S. Multifaceted ABC transporters associated to microcin and bacteriocin export. Res. Microbiol. 170, 399–406 (2019).
Biswas, S. & Biswas, I. SmbFT, a putative ABC transporter complex, confers protection against the lantibiotic smb in Streptococci. J. Bacteriol. 195, 5592–5601 (2013).
Wilkens, S. Structure and mechanism of ABC transporters. F1000Prime Rep. 7, (2015).
Chen, X. et al. Potent, multi-target serine protease inhibition achieved by a simplified β-sheet motif. PLoS One 14, (2019).
de Vos, W. M., Kuipers, O. P., van der Meer, J. R. & Siezen, R. J. Maturation pathway of nisin and other lantibiotics: Post-translationally modified antimicrobial peptides exported by Gram-positive bacteria. Mol. Microbiol. 17, 427–437 (1995).
Repka, L. M., Chekan, J. R., Nair, S. K. & Van Der Donk, W. A. Mechanistic understanding of Lanthipeptide Biosynthetic enzymes. Chem. Rev. 117, 5457–5520 (2017).
Albers, R. W. & Siegel, G. J. ATP-Binding Cassette Proteins. (1999).
Smits, S. H. J., Schmitt, L. & Beis, K. Self-immunity to antibacterial peptides by ABC transporters. FEBS Lett. 594, 3920–3942 (2020).
Nishie, M., Shioya, K., Nagao, J., Jikuya, H. & Sonomoto, K. ATP-dependent leader peptide cleavage by NukT, a bifunctional ABC transporter, during lantibiotic biosynthesis. J. Biosci. Bioeng. 108, 460–464 (2009).
Clemens, R., Zaschke-Kriesche, J., Khosa, S. & Smits, S. H. J. Insight into two ABC transporter families involved in lantibiotic resistance. Front. Mol. Biosci. 4, (2018).
Hu, J. et al. Accurate prediction of protein-ATP binding residues using position-specific frequency matrix. Anal. Biochem. 626, 114241 (2021).
Khusainov, R., Heils, R., Lubelski, J., Moll, G. N. & Kuipers, O. P. Determining sites of interaction between prenisin and its modification enzymes NisB and NisC. Mol. Microbiol. 82, 706–718 (2011).
Rani, N., Boora, N., Rani, R., Kumar, V. & Ahalawat, N. Molecular dynamics simulation of RAC1 protein and its de novo variants related to developmental disorders. J. Biomol. Struct. Dyn. 1–10. https://doi.org/10.1080/07391102.2023.2275188 (2023).
Murmu, S. & Archak, S. In-silico study of protein-protein interactions in wheat blast using docking and molecular dynamics simulation approach. J. Biomol. Struct. Dyn. 42, 5747–5757 (2024).
Gasteiger, E. et al. Protein identification and analysis tools on the ExPASy server. Proteom. Protoc. Handb. 571–607. https://doi.org/10.1385/1-59259-890-0:571 (2005).
Yu, N. Y. et al. PSORTb 3.0: Improved protein subcellular localization prediction with refined localization subcategories and predictive capabilities for all prokaryotes. Bioinformatics 26, 1608–1615 (2010).
Hallgren, J. et al. DeepTMHMM predicts alpha and beta transmembrane proteins using deep neural networks. bioRxiv 2022.04.08.487609 https://doi.org/10.1101/2022.04.08.487609 (2022).
Kim, D. E., Chivian, D. & Baker, D. Protein structure prediction and analysis using the Robetta server. Nucleic Acids Res. 32, (2004).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
Vajda, S. et al. New additions to the ClusPro server motivated by CAPRI. Proteins 85, 435 (2017).
Kozakov, D. et al. The ClusPro web server for protein-protein docking. Nat. Protoc. 12, 255–278 (2017).
Kozakov, D. et al. How good is automated protein docking? Proteins Struct. Funct. Bioinforma. 81, 2159–2166 (2013).
Weng, G. et al. HawkDock: A web server to predict and analyze the protein-protein complex based on computational docking and MM/GBSA. Nucleic Acids Res. 47, W322–W330 (2019).
Hu, J., Li, Y., Zhang, Y., Yu, D. J. & ATPbind Accurate Protein-ATP binding site prediction by combining sequence-profiling and structure-based comparisons. J. Chem. Inf. Model. 58, 501–510 (2018).
Acknowledgements
Al Amin, Molecular Biology Laboratory, Department of Biochemistry and Molecular Biology, Faculty of Biological Sciences, University of Dhaka, Dhaka 1000, Bangladesh. The authors also acknowledge University of Dhaka, Dhaka for supporting this project.
Author information
Authors and Affiliations
Contributions
M.A.H., Md.R.I., O.F., M.R.I.: Conceptualization, designing, investigations, data analysis and writing the manuscript; M.R.I., T.Z., M.A.U., H.K.: Supervision, writing the manuscript and all authors critically revised the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Hossain, M.A., Islam, M.R., Faruk, O. et al. Unveiling the biosynthesis mechanism of novel lantibiotic homicorcin: an in silico analysis. Sci Rep 14, 28893 (2024). https://doi.org/10.1038/s41598-024-80514-y
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-024-80514-y
