
Abstract
Parkinson’s disease (PD) and insomnia are prevalent neurological disorders, with emerging evidence implicating tryptophan (TRP) metabolism in their pathogenesis. However, the precise mechanisms by which TRP metabolism contributes to these conditions remain insufficiently elucidated. This study explores shared tryptophan metabolism-related genes (TMRGs) and molecular mechanisms underlying PD and insomnia, aiming to provide insights into their shared pathogenesis. We analyzed datasets for PD (GSE100054) and insomnia (GSE208668) obtained from the Gene Expression Omnibus (GEO) database. TMRGs were obtained from the Molecular Signatures Database (MSigDB) and the Genecards database. Tryptophan metabolism-related differentially expressed genes (TM-DEGs) were identified by intersecting TMRGs with shared differentially expressed genes (DEGs) from these datasets. Through Protein–Protein Interaction (PPI) network analysis, Support Vector Machine-Recursive Feature Elimination (SVM-RFE) , and Extreme Gradient Boosting (XGBoost) machine learning, we identified Cytochrome P4501B1 (CYP1B1) and Electron Transfer Flavoprotein Alpha (ETFA) as key hub genes. Subsequently, we employed CIBERSORT and single-sample gene set enrichment analysis (ssGSEA) to further investigate the association between hub genes and peripheral immune activation and inflammatory response. Additionally, gene interaction, Drug-mRNA, Transcription Factor (TF)-mRNA, and competing endogenous RNA (ceRNA) networks centered on these hub genes were constructed to explore regulatory mechanisms and potential drug interactions. Finally, validation through bioinformatics and animal experiments identified CYP1B1 as a promising biomarker associated with both PD and insomnia.
Similar content being viewed by others


Introduction
PD is a prevalent neurodegenerative disorder, with incidence increasing significantly with age. The primary pathological feature is the early degeneration and loss of dopaminergic neurons in the substantia nigra pars compacta (SNpc), accompanied by widespread intracellular aggregation of α-synuclein (aSyn)1. The clinical presentation of PD includes motor symptoms such as bradykinesia, muscle rigidity, resting tremor, and postural instability, along with a spectrum of non-motor symptoms,including sleep disturbances, depression, autonomic symptoms, sensory symptoms and fatigue2,3.Insomnia is notably prevalent among PD patients, with studies indicating that approximately 60% experience this condition4. Insomnia in PD is mainly characterized by difficulties in maintaining sleep rather than initiating it5 . Insomnia often emerges in the early stages of PD, preceding motor symptoms, and progressively worsens as the disease advances6,7. It significantly affects the quality of life in PD patients and is regarded as one of the most distressing non-motor symptoms in advanced PD8. The precise mechanisms underlying the co-occurrence of PD and insomnia remain unclear but may involve neuronal damage affecting brain regions crucial for sleep regulation and circadian rhythms. These regions include cholinergic, serotonergic, and adrenergic neurons in the brainstem, as well as orexinergic neurons in the posterolateral hypothalamus9,10. Additionally, the chronic use of dopaminergic medications may also contribute to this association11,12,13.
Tryptophan is a vital aromatic amino acid involved in three primary metabolic pathways: the microbiota-related indole pathway, the kynurenine pathway, and the serotonin pathway14. Dysregulated tryptophan metabolism is closely linked to various neurological disorders, including neurodegenerative diseases, traumatic brain injury15, ischemic stroke16, intracerebral hemorrhage17 , and sleep disorders18. Significant alterations in kynurenine pathway metabolite levels have been reported in cerebrospinal fluid and plasma of PD patients, correlating with disease severity19,20. Inhibitors targeting key enzymes involved in tryptophan metabolism are being considered as potential therapeutic drugs for PD21. Dietary TRP supplementation has shown promise in alleviating symptoms in insomnia patients22,and enhancing the dietary ratio of TRP to large neutral amino acids (TRP/LNAAs) has significantly improved sleep duration and quality in specific populations, such as athletes and obese adolescents with sleep disorders23. Although clinical evidence linking kynurenine and serotonin pathway metabolites to both PD and insomnia, the precise mechanisms of tryptophan metabolism in each condition, as well as its role in co-morbid insomnia in PD, remain insufficiently understood.
Recent studies on PD and insomnia have increasingly employed microarray and high-throughput sequencing technologies to identify relevant biomarkers, paving the way for exploring shared tryptophan metabolism-related hub genes in PD and insomnia through bioinformatics approaches24,25. Compared to traditional brain tissue or cerebrospinal fluid samples26, peripheral blood samples are gaining traction for identifying disease-related biomarkers27,28. This trend reflects not only the accessibility of peripheral blood but also a growing appreciation of the peripheral immune system’s role in neurodegenerative disease pathogenesis.
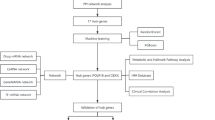
This study represents the first bioinformatics investigation into the shared link between insomnia, PD, and tryptophan metabolism. Through analysis of gene expression data from the GEO database we identified CYP1B1 and ETFA as central hub genes. Subsequently, we explored the associations of these hub genes with peripheral immune inflammation, gene interactions, potential drug interactions, transcription factors, and microRNAs (miRNAs) through various bioinformatics approaches. Finally, through bioinformatics analysis and experimental validation, we propose that CYP1B1 may be a key shared hub gene involved in both PD and insomnia. These findings enhance our understanding of the complex molecular mechanisms connecting PD and insomnia, and provide potential biomarkers for the early diagnosis and prognostic evaluation of PD patients with insomnia symptoms. The bioinformatics analysis workflow is illustrated in Fig. 1.

Workflow of the bioinformatics analysis used in this study. DEGs differentially expressed genes, TM-DEGs tryptophan metabolism related differentially expressed genes, GO gene ontology, KEGG Kyoto encyclopedia of genes and genomes, PPI Protein–Protein Interaction, SVM-RFE support vector machine-recursive feature Elimination, XGBoost extreme gradient boosting, ceRNA competing endogenous RNA, TF transcription factor, ssGSEA single-sample gene set enrichment analysis, qRT-PCR quantitative real-time reverse transcription PCR, ANN artificial neural network.
Materials and methods
Data set acquisition
We retrieved expression profiles from the GEO database29 and conducted comprehensive analyses on the GSE100054 dataset (10 PD peripheral blood samples and 9 controls) and the GSE208668 dataset (17 insomnia peripheral blood samples and 25 controls). The GSE165082 dataset (12 PD peripheral blood samples and 14 controls) and the GSE49036 dataset (8 substantia nigra samples from PD patients and 8 controls) were used to validate hub genes. Circular RNA expression profiles were obtained from the GSE198273 dataset (3 PD peripheral blood samples and 3 controls). Detailed information about these datasets is available in Table 1. Additionally, 180 TMRGs were gathered from MSigDB30 and GeneCards31, as summarized in Supplementary Table 1.
Identification of TM-DEGs in PD and insomnia
RNA-seq data were initially subjected to standard gene expression normalization and log2 transformation. DEGs were identified using the ‘limma’ package (v3.54.2)32, with selection criteria of p-value < 0.05 and absolute log2 fold change (|log2FC|) > 0.5. Volcano plots and heatmaps were generated to visualize the expression patterns of DEGs33. Shared upregulated and downregulated DEGs from the GSE100054 and GSE208668 datasets were visualized with the ‘ggvenn’ package (v0.1.10). Common DEGs were intersected with TMRGs to define TM-DEGs for further analysis.
Functional enrichment analysis
Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analyses were conducted on TM-DEGs using the ‘clusterProfiler’ package (v4.7.1003)34, with a p-value < 0.05 considered significant.
PPI network construction and analysis
The STRING database35 was used to construct the PPI network, which was subsequently visualized in Cytoscape software (v3.9.1)36. CytoHubba37, a Cytoscape plugin offering 12 topological analysis methods, retained the top 75% of genes for each algorithm. Network modules were screened with the MCODE plugin38 using the default settings : degree cutoff = 2, node score cutoff = 0.2, K-core = 2, and max depth = 100. Four marker genes were identified by intersecting genes filtered through all 13 algorithms.
Machine learning identification of hub genes
SVM-RFE39 and XGBoost40 models were developed using the ‘caret’ package (v6.0–93) and the ‘xgboost’ package (v1.7.5.1) to further screen for hub genes in the PD dataset.
Immune infiltration and signaling pathway analysis
In the PD and insomnia datasets, CIBERSORT41 was used to analyze immune cell differences between control and disease samples and assess correlations between hub genes and immune cells. The ‘GSVA’ package (v1.46.0)42 was used to perform ssGSEA on the HALLMARK_INFLAMMATORY_RESPONSE pathway from MSigDB, followed by an assessment of correlations between pathway scores, pathway genes, and hub genes43.
Gene interaction network of hub genes
We used GENEMANIA software44 to construct a gene interaction network exploring interactions between hub genes and their associated genes, as well as their biological functions.
Construction of Drug-mRNA, TF-mRNA, and ceRNA networks
The Drug-Gene Interaction Database (DGIdb )45 was used to predict small molecule drugs associated with hub genes, and these interactions were visualized as a drug-mRNA interaction network. To understand the regulatory mechanisms, transcription factors linked to hub genes were identified from the JASPAR database46 using the NetworkAnalyst platform47 and visualized accordingly. Differentially expressed circRNAs (DEcircRNAs) were identified from the GSE198273 dataset (p-value < 0.05 and (|LogFC|) > 0.5). The Starbase database48 was used to predict circRNA-miRNA interactions, and the miRNet platform49 was used to predict miRNAs related to hub genes. A ceRNA network involving mRNAs, miRNAs, and circRNAs was then constructed using Cytoscape.
Artificial neural network (ANN)
The ANN model was constructed using the ‘neuralnet’ package (v1.44.2)50, comprising an input layer, a hidden layer, and an output layer. The model calculates the weighted sum of input gene scores to determine the output. Predictor importance was assessed, and the receiver operating characteristic (ROC) curve and area under the curve (AUC) were calculated using the ‘pROC’ package (v1.18.0)51 to evaluate the diagnostic performance of the hub genes.
Animals
All experimental animals were male and age-matched, with all procedures approved by the Institutional Animal Care and Use Committee of the First Affiliated Hospital of Zhengzhou University (approval number: ZZU-LAC20240531[06]). The 3R principles (Reduction, Refinement, and Replacement) were strictly followed to ensure statistical robustness while minimizing animal use and suffering. Six 15-month-old A53T α-synuclein transgenic mice and six age-matched wild-type littermates (from Jackson Laboratory) were used to validate hub gene expression in a PD model. Ten-week-old C57BL/6 J mice were obtained from Beijing Vital River Laboratory Animal Technology Co., Ltd. (Beijing, China). Six of these mice were subjected to a chronic sleep deprivation apparatus (KW-BD, Nanjing Calvin Biotechnology Co., Ltd.) operating at 10 revolutions per minute, with a 5-h operation followed by a 1-h rest cycle, keeping them awake for 20 h per day for one week to model clinical insomnia. Control mice were housed in identical apparatuses to maintain normal sleep. All mice were kept under standard laboratory conditions: 25 °C, 60 ± 5% humidity, with a 12-h light/dark cycle and ad libitum access to food and water.
Mouse euthanasia and tissue collection
Mice were anesthetized with 3% isoflurane, and blood samples were collected via retro-orbital venous plexus puncture. Peripheral blood mononuclear cells (PBMCs) were then isolated using density gradient centrifugation. Following blood collection, mice were perfused with 30 mL of cold 1 × Dulbecco’s phosphate-buffered saline (DPBS) via cardiac perfusion, and the substantia nigra was rapidly dissected for subsequent experiments.
Quantitative reverse transcription polymerase chain reaction (qRT-PCR)
Total RNA was extracted from the substantia nigra and PBMCs using TRIzol reagent (Invitrogen, USA). Reverse transcription was conducted with the PrimeScript™ RT reagent Kit (Takara). Primers (listed in Table 2) were synthesized by Sangon Biotech (Shanghai, China). qRT-PCR was performed on a QuantStudio 5 system (Applied Biosystems) using iQ™ SYBR Green Supermix (Bio-Rad). Target gene expression was normalized to glyceraldehyde 3-phosphate dehydrogenase (GAPDH) and analyzed using the 2−ΔΔCTmethod.
Western blotting analysis
PBMCs and substantia nigra tissues were homogenized in RIPA buffer (Beyotime, China). Proteins were separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) , transferred to polyvinylidene fluoride (PVDF) membranes, and incubated overnight at 4 °C with the following primary antibodies: anti-mouse CYP1B1 (DF6399, 1:1000, affinity), anti-mouse ETFA(ab153722, 1:1000, Abcam), anti-mouse GAPDH (ab9485, 1:5000, Abcam), and anti-mouse β-actin (ab8227, 1:5000, Abcam). After washing, secondary antibody incubation was performed at room temperature with goat anti-rabbit IgG (ab6721, 1:5000). Protein bands were visualized using an ECL chemiluminescence kit (Merck Millipore), and band intensities were quantified using ImageJ software.
Statistical analysis
Statistical analyses were conducted in R software (v4.2.2) and GraphPad Prism 8. Student’s t-test was applied for normally distributed data with homogeneous variances, and the Wilcoxon test for heterogeneous variances. Statistical significance was defined as p < 0.05, with results indicated as *p < 0.05, **p < 0.01, ***p < 0.001.
Ethics statement
All animal procedures were approved by the Institutional Animal Care and Use Committee of the First Affiliated Hospital of Zhengzhou University (approval number: ZZU-LAC20240531[06]) and complied with the Animals Research: Reporting of In Vivo Experiments (ARRIVE) guidelines 2.0. All methods adhered to relevant guidelines and regulations.
Results
Identification and functional enrichment analysis of TM-DEGs
We initially identified DEGs between PD and control samples from the GEO database. In the GSE100054 dataset, 1741 DEGs were identified, including 1015 up-regulated genes and 726 down-regulated genes (Fig. 2A,B). Similarly, in the GSE208668 dataset, 6,069 DEGs were identified between insomnia and control samples, with 3,438 upregulated and 2,631 downregulated genes (Fig. 2C, 2D). By intersecting the up-regulated and down-regulated genes from both datasets, we identified 286 shared upregulated genes and 58 shared downregulated genes (Fig. 2E). These 344 DEGs were further intersected with 180 TMRGs, resulting in eight identified TM-DEGs (Fig. 2F, Supplementary Table 2). GO and KEGG enrichment analyses indicated that, in addition to tryptophan metabolism, TM-DEGs are involved in various metabolic processes, including cellular biogenic amine metabolism, xenobiotic metabolism, and estrogen metabolism51. These results suggest that multiple metabolic pathways exhibit common alterations in the peripheral blood of patients with PD and insomnia (Supplementary Figure S1).

Identification of the TM-DEGs. (A) Circular heatmap showing the top 10 upregulated and downregulated genes in the GSE100054 dataset. (B) Circular heatmap showing the top 10 upregulated and downregulated genes in the GSE208668 dataset. (C) Volcano plot showing the DEGs in the GSE100054 dataset. (D) Volcano plot showing the DEGs in the GSE208668 dataset. (E) Intersection of upregulated and downregulated DEGs between PD and Insomnia, resulting in 344 intersecting DEGs. (F) Intersection of DEGs and TMRGs resulting in eight TM-DEGs.
Identification of hub genes using PPI network analysis and machine learning
We used STRING to construct a PPI network for the eight TM-DEGs (Fig. 3A). Subsequently, we analyzed the network using 13 algorithms, including CytoHubba and MCODE plugins. This analysis identified four marker genes (CYP1B1, ETFA, KYNU, SULT1A1) at their intersection (Fig. 3B,C, Table 3). Further, we used the eight TM-DEGs to construct SVM-RFE and XGBoost models, identifying five marker genes (BLVRB, CYP1B1, SULT1A1, WARS1, ETFA) for SVM-RFE and four marker genes (BLVRB, CYP1B1, KYNU, ETFA) for XGBoost. We generated cross-validation curves for SVM-RFE (Fig. 3D) and a gene importance ranking plot for XGBoost (Fig. 3E). After intersecting results, we finally identified 2 hub genes (CYP1B1, ETFA) shared across PPI network analysis, SVM-RFE, and XGBoost algorithms (Fig. 3F).

Identification of hub genes using PPI network analysis and machine learning models. (A) PPI network model constructed from TM-DEGs. (B) Key cluster with 6 genes chosen as hub genes by MCODE. (C) Hub genes identified by intersecting the top 6 genes from 13 algorithms. (D) Cross-Validation accuracy curve for SVM-RFE model. (E) Top genes ranked by feature importance scores in the XGBoost model. (F) Intersection of genes identified by XGBoost, SVM-RFE, and PPI network analysis, resulting in two hub genes.
Analysis of immune infiltration and inflammatory response signaling pathways
Using the CIBERSORT algorithm, we assessed the infiltration of 22 immune cell types in the peripheral blood of PD and insomnia patients (Supplementary Figure S2). After filtering out cell types with zero abundance, we retained 15 immune cell types for analysis. M2 macrophages and monocytes were notably more abundant in PD samples, while naive B cells were more prevalent in controls. In insomnia samples, CD8 + T cells and resting memory CD4 + T cells showed increased abundance, whereas naive and activated CD4 + T cells were lower (Fig. 4A). Spearman correlation analysis revealed a strong association between CYP1B1 expression and monocyte levels, with ETFA showing the strongest positive correlation with monocytes in PD and with NK cells in insomnia (Fig. 4B). The ssGSEA analysis revealed a significant positive correlation between the hub gene and the HALLMARK_INFLAMMATORY_RESPONSE pathway in both diseases, and further correlation analysis showed that CYP1B1 and ETFA were closely associated with multiple genes in this inflammatory pathway (Fig. 4C,D). These findings suggest that CYP1B1 and ETFA influence inflammation and immunity in PD and insomnia through potentially distinct mechanisms, highlighting their value as biomarkers and therapeutic targets.

Immune infiltration analysis and inflammation correlation analysis (A) The bar plot illustrates the differences in immune cell infiltration abundance in GSE1000504 and GSE208668 datasets, with statistical significance indicated by P < 0.05. (B) Correlation analysis between hub genes and infiltrating immune cells. (C) Correlation analysis between hub genes and scores of the HALLMARK_INFLAMMATORY_RESPONSE pathway. (D) Correlation analysis between hub genes and genes within the HALLMARK_INFLAMMATORY_RESPONSE pathway.
Gene interaction network and related functions of the hub genes
To further understand the biological functions of the hub genes, we constructed a gene interaction network using GeneMANIA with automated weighting. The network consists of two central hub genes and 20 peripheral predicted genes, including ETFB, ETFDH, and EPHX2 (Fig. 5A). Functional and downstream pathway analyses indicated that these genes participate in lipid metabolism and oxidation processes. GO enrichment analysis of the hub genes and predicted genes showed significant enrichments in various functions, notably including the fatty acid metabolic process, cellular lipid catabolic process, mitochondrial protein complex, respiratory chain complex, electron transfer activity, and flavin adenine dinucleotide binding. KEGG analysis further indicated associations with multiple metabolic pathways and neurological disorders (Fig. 5B,C, Supplementary Table 3).

Gene interaction network and functional enrichment analyses of hub genes. (A) Gene interaction network of hub genes constructed using GeneMANIA. (B) GO enrichment analysis of genes from the interaction network (C) KEGG enrichment analysis of genes from the interaction network.
Drug-mRNA, TF-mRNA and ceRNA network construction
Based on the DGidb database, we predicted small molecule drugs corresponding to two hub genes (CYP1B1, ETFA). We identified 28 small molecule drugs targeting CYP1B1, including 'BETA-NAPHTHOFLAVONE’, ‘ISOPROPYL ALCOHOL’, and ‘FORMESTANE’. However, no corresponding small molecule drugs targeting ETFA were predicted. We generated a Drug-mRNA network using Cytoscape software (Fig. 6A, Table 4). Furthermore, based on the JASPAR database, we constructed a TF-mRNA regulatory network. Our analysis revealed 9 transcription factors regulating ETFA expression and 8 regulating CYP1B1 expression (Fig. 6B). We identified 30 DEcircRNAs (20 up-regulated and 10 down-regulated) in GSE198273, visualized in the Volcano Plots (Fig. 6C). Predictions from the Starbase database helped identify 218 miRNAs targeting these DEcircRNAs. Additionally, a total of 121 miRNAs corresponding to CYP1B1 and ETFA were obtained from the miRNet website. By overlapping these, we identified 18 intersecting miRNAs (Fig. 6D). Finally, we integrated the two hub genes (CYP1B1, ETFA), intersecting miRNAs, and DEcircRNAs to construct a ceRNA network. Our analysis revealed that seven circular RNAs competed with CYP1B1 for binding to 18 miRNAs, including hsa-miR-122-5p, hsa-miR-124-3p, hsa-miR-148a-3p. Similarly, ETFA might be regulated by has_circ_0000943 circRNA through competitive binding with has_mir_18a-5p miRNA (Fig. 6E).

Drug-mRNA, TF-mRNA, and ceRNA network construction. (A) Drug-mRNA interaction networks. (B) TF-mRNA interaction networks. (C) Volcano plot showing the DEcircRNAs in the GSE165082 dataset. (D) Intersection of predicted DecircRNA miRNAs and hub genes predicted miRNAs, resulting in 18 common miRNAs. (E) The ceRNA network of mRNA-miRNA-circRNA.
Bioinformatics validation of hub genes
We validated the hub genes using internal datasets (GSE1000504 and GSE208668) and an external dataset (GSE165082), showing significant upregulation in disease cohorts. To determine whether the expression of hub genes in the substantia nigra was consistent with peripheral blood findings in PD patients, we assessed CYP1B1 and ETFA expression using the substantia nigra dataset (GSE49036). CYP1B1 was significantly upregulated in PD patients, whereas ETFA expression remained unchanged (Fig. 7A). ROC curve analysis further confirmed the diagnostic potential of each hub gene, with all AUC values exceeding 0.7 (Fig. 7B). These findings indicate that the differential expression of CYP1B1 is more reliable compared to ETFA, suggesting its stronger candidacy as a biomarker. Additionally, correlation analysis revealed a positive association between CYP1B1 and ETFA in peripheral blood datasets, suggesting potential synergistic roles in regulating tryptophan metabolism (Fig. 7C). Notably, no such correlation was observed in substantia nigra tissue, implying differential functional roles of CYP1B1 and ETFA between peripheral blood and brain regions.To further validate the significance of CYP1B1, we constructed multiple artificial neural network (ANN) models incorporating both CYP1B1 and ETFA across different datasets (Supplementary Figure S2A). These ANN models demonstrated robust predictive performance (Supplementary Figure S2B). CYP1B1 consistently emerged as the most significant classifier across these datasets (Supplementary Figure S2C), further supporting its role as a potential shared biomarker for both PD and insomnia.

Bioinformatics validation of hub genes. (A) Validation of hub gene expression in the GSE100054, GSE208668, GSE165082, and GSE49036 datasets. (B) ROC curves for the hub genes in the GSE100054, GSE208668, GSE165082, and GSE49036 datasets (C) Correlation analysis between CYP1B1 and ETFA across the GSE100054, GSE208668, GSE165082, and GSE49036 datasets.
Experimental validation of hub genes
To further validate our conclusions, we employed qRT-PCR and Western blotting to assess transcriptional changes in CYP1B1 and ETFA in PD and insomnia mouse models. In both models, qRT-PCR analysis showed significantly elevated mRNA levels of CYP1B1 and ETFA in peripheral blood compared to controls (Fig. 8A). Western blotting similarly confirmed increased protein expression levels of these two hub genes in both disease models (Fig. 8B), aligning with bioinformatics findings.We also used qRT-PCR to assess CYP1B1 and ETFA mRNA expression levels in the substantia nigra of PD and insomnia model mice. In PD mice, CYP1B1 was significantly upregulated, while ETFA remained unchanged. In insomnia model mice, CYP1B1 levels increased, and ETFA levels decreased compared to controls (Fig. 8C). Subsequent Western blotting confirmed these results, consistent with the qRT-PCR findings (Fig. 8D).

Validation of hub genes in mouse models. (A) qRT-PCR validation of differential mRNA expression of hub genes in mouse peripheral blood samples. (B) Western blot validation of differential protein expression of hub genes in mouse peripheral blood samples. (C) qRT-PCR validation of mRNA expression of hub genes in mouse substantia nigra samples. (D) Western blot validation of differential protein expression of hub genes in mouse peripheral blood samples.
Discussion
Patients with PD often experience non-motor symptoms, such as sleep disturbances, autonomic dysfunction, constipation, hyposmia, and cognitive impairment, which typically precede motor symptoms52. Insomnia is the most common sleep disorder among PD patients, typically emerging early and worsening over time. Notably, a bidirectional causal relationship may exist between PD and insomnia. A retrospective study identified sleep disturbances as an independent risk factor for PD53. Furthermore, a previous cohort study confirmed that long-term insomnia increase the risk of developing PD54. Tryptophan, an essential amino acid crucial for protein synthesis, undergoes metabolic conversion to yield immune and neuroactive derivatives such as melatonin, kynurenine, and serotonin. These metabolites are involved in both PD and insomnia pathogenesis55. However, the mechanisms linking tryptophan metabolism with both PD and insomnia remain unclear.
In this study, we identified CYP1B1 and ETFA as key hub genes linking PD and insomnia by intersecting DEGs from PD and insomnia datasets with TMRGs, followed by PPI network analysis and screening using SVM-RFE and XGBoost models.
CYP1B1, a member of the cytochrome P450 family, plays a role in phase I metabolism of both endogenous and exogenous substrates, including melatonin56. Melatonin has been shown to effectively manage sleep disturbances in PD57,58 alleviate neuroinflammation, enhance neuroprotection, and slow neurodegeneration59,60,61. Melatonin is synthesized in the gastrointestinal tract and brain through the tryptophan-serotonin pathway and degraded by CYP1B1 in the liver and brain to form 6-hydroxymelatonin and N-acetylserotonin62. Elevated CYP1B1 in PD and insomnia may disrupt melatonin metabolism, contributing to sleep disturbances and neurofunctional impairment. Furthermore, gut microbiota dysbiosis, observed in both PD and insomnia63,64,65, may affect tryptophan metabolic pathways and produce tryptophan-derived metabolites that modulate the aryl hydrocarbon receptor (AhR) in the brain. This regulation of CYP enzymes, including CYP1B1, could represent a shared mechanism in PD and insomnia pathogenesis66,67,68. Despite the evidence connecting CYP1B1 to tryptophan metabolism in PD and insomnia, direct evidence remains limited. Genome-wide association studies have linked CYP1B1 to tau protein accumulation in Alzheimer’s disease (AD)69. In CYP1B1 knockout mice, compromised blood–brain barrier integrity results in increased striatal oxidative stress, disrupted dopamine homeostasis, accelerated manganese-induced α-synuclein accumulation, and subsequent dopaminergic neuronal damag69. Studies indicate a synergistic relationship between CYP1B1 and the Wnt/β-catenin signaling pathway in oxidative stress responses70,71,72. Given the established role of the Wnt signaling pathway in PD pathology73, it is plausible that CYP1B1, in conjunction with Wnt signaling, may significantly impact PD pathology. Additionally, CYP1B1 deletion activates Nrf2, mitigating oxidative stress, and Nrf2 activation has been shown to inhibit PD progression74,75. Currently, only one study identifies CYP1B1 as a therapeutic target for ellagic acid in sleep-deprived mice76, underscoring the need for further research on CYP1B1 in PD and insomnia. Research on ETFA is limited, primarily linking it to cellular energy metabolism77. In this study, the potential of ETFA as a biomarker remains unconfirmed and requires further validation.
Recent evidence from both patient studies and animal models suggests a link between PD, insomnia, and peripheral immune inflammation78. immune infiltration analysis indicates significant monocyte activation in the peripheral blood of PD patients. Similarly, previous studies have reported an association between monocytes and insomnia in affected individuals79. Our findings further reveal that CYP1B1 shows the strongest positive correlation with monocytes in both diseases. Furthermore, inflammatory response analysis highlights a close association between CYP1B1 and peripheral inflammatory pathways and genes, such as NOD2 and FPR1, both strongly linked to monocytes80,81. This suggests that elevated CYP1B1 expression in monocytes may play a crucial role in the progression of both insomnia and PD.
We used GeneMANIA to construct a gene interaction network for CYP1B1 and ETFA. Enrichment analysis of the resulting 22 genes highlighted their roles in metabolism, synthesis, redox activities, and mitochondrial function, aligning with CYP1B1 and ETFA functions. We also built drug-mRNA, TF-mRNA, and ceRNA networks to further explore disease mechanisms and identify therapeutic targets. A meta-analysis revealed significant differential expression of hsa-miR-29c-3p in the peripheral blood of PD patients, suggesting its potential as a biomarker82. In vitro studies have shown the therapeutic potential of the CYP1B1 inhibitor beta-naphthoflavone (βNF) for PD treatment.βNF exhibited neuroprotective effects by inhibiting the AKT/Nrf-2/HO-1-NF-κB inflammatory pathway in microglia83 and mitigated MPP + -induced cytotoxicity in SH-SY5Y cells84,85.These findings suggest that CYP1B1 mediates the neuroprotective effects of βNF, warranting further validation. Other drugs, including naringenin86, chrysin87, and acacetin88, have shown therapeutic effects for PD. Naringenin, identified through network pharmacology, may also improve sleep89, indicating potential dual benefits for PD and insomnia by targeting CYP1B1.
To validate the hub genes identified by machine learning, we used an independent PD peripheral blood dataset (GSE165082) that showed good similarity to the original dataset (GSE100054) in sample size, age, sex distribution, and disease state90,91. Analysis of GSE165082 confirmed significant upregulation of the hub genes, with AUC values exceeding 0.7, indicating strong predictive capability. Given the increasing focus on identifying reliable peripheral blood biomarkers in PD patients as alternatives to brain tissue or cerebrospinal fluid27,92,93, we further assessed the reliability of these peripheral blood biomarkers by analyzing substantia nigra samples from PD patients (GSE49036). The results showed consistent expression of CYP1B1 between brain tissue and peripheral blood, whereas ETFA did not show significant differences. Additionally, we constructed multiple ANNs across different datasets, which confirmed the importance of CYP1B1 in diagnosing PD and insomnia. To further validate our findings, we examined the expression of hub genes in peripheral blood and substantia nigra using PD and insomnia animal models. In peripheral blood, hub gene expression was consistently elevated in both models. In the substantia nigra of PD mice, CYP1B1 was significantly upregulated, while ETFA showed no significant changes. In the insomnia model, CYP1B1 was elevated, whereas ETFA expression decreased. These findings reveal a strong concordance between animal experiments and bioinformatics analysis, with consistent upregulation of CYP1B1 in both peripheral and central tissues. This consistency reinforces CYP1B1 as a promising biomarker and potential therapeutic target for PD and insomnia.
Undeniably, our study faced several limitations. Firstly, as this research primarily relied on bioinformatics analysis of publicly available transcriptomic data, the results are influenced by the quality and quantity of the sequencing data. Variability during sample collection, processing, and sequencing, as well as inter-individual differences among PD patients, may introduce biases affecting the analysis outcomes. Secondly, this study is an initial exploratory investigation using small datasets to identify potential biomarkers. Due to the limited sample size, we employed multiple datasets for validation, combined with in vivo experiments, to enhance the credibility of our findings and provide a foundation for future in-depth research. Thirdly, due to the limited availability of public datasets, we were unable to extensively validate the hub genes in insomnia patients. Lastly, our experimental validation was conducted solely using animal models, which cannot fully replicate the pathological features observed in humans. For instance, the A53T transgenic mouse model used in this study does not exhibit significant dopaminergic neuron loss, and the locations of pathological changes are not entirely consistent with those in humans94,95. Moreover, the sleep deprivation model simulates only the insomnia induced by external stress, failing to capture the complex psychological and social factors experienced by patients. Additionally, the progression of PD and insomnia in humans is a prolonged, multifactorial process, whereas the disease progression in animal models tends to be rapid and simplified. Therefore, in future studies, we aim to maximize the use of clinical samples and expand the validation cohort size to enhance statistical power and reliability. We also plan to employ a diverse range of bioinformatics and experimental approaches, such as single-cell sequencing, cell-based assays, and gene editing experiments, to further investigate the shared mechanisms of the hub genes in both diseases.
Conclusion
In summary, CYP1B1 is identified as a pivotal factor in both PD and insomnia, highlighting its potential as a valuable target for diagnosis and treatment. Our findings have been experimentally validated in animal models. Furthermore, we emphasize the necessity for further research to fully understand its role and therapeutic opportunities.
Data availability
All datasets used in this study are publicly available. The RNA-seq data were obtained from the following GEO datasets: GSE100054, GSE208668, GSE165082, GSE198273, and GSE49036. These datasets are accessible at the NCBI GEO (https://www.ncbi.nlm.nih.gov/geo/).
References
Kalia, L. V. & Lang, A. E. Parkinson’s disease. Lancet (London, England) 386(9996), 896–912 (2015).
Schapira, A. H. V., Chaudhuri, K. R. & Jenner, P. Non-motor features of Parkinson disease. Nat. Rev. Neurosci. 18(7), 435–450 (2017).
Jankovic, J. Parkinson’s disease: Clinical features and diagnosis. J. Neurol. Neurosurg. Psychiatry 79(4), 368–376 (2008).
Gjerstad, M. D. et al. Insomnia in Parkinson’s disease: Frequency and progression over time. J. Neurol. Neurosurg. Psychiatry 78(5), 476–479 (2007).
Gómez-Esteban, J. C. et al. Sleep complaints and their relation with drug treatment in patients suffering from Parkinson’s disease. Mov. Disorders: Off. J. Mov. Disorder Soc. 21(7), 983–988 (2006).
Xu, Z. et al. Progression of sleep disturbances in Parkinson’s disease: A 5-year longitudinal study. J. Neurol. 268(1), 312–320 (2021).
Bohnen, N. I. & Hu, M. T. M. Sleep disturbance as potential risk and progression factor for Parkinson’s disease. J. Parkinson’s Dis. 9(3), 603–614 (2019).
Politis, M. et al. Parkinson’s disease symptoms: The patient’s perspective. Mov. Disorders: Off. J. Mov. Disorder Soc. 25(11), 1646–1651 (2010).
Leng, Y. et al. Association of circadian abnormalities in older adults with an increased risk of developing Parkinson disease. JAMA Neurol. 77(10), 1270–1278 (2020).
Thangaleela, S. et al. Neurological insights into sleep disorders in Parkinson’s disease. Brain Sci. 13(8), 1202 (2023).
Tholfsen, L. K. et al. Changes in insomnia subtypes in early Parkinson disease. Neurology 88(4), 352–358 (2017).
Schütz, L., Sixel-Döring, F. & Hermann, W. Management of sleep disturbances in Parkinson’s disease. J. Parkinson’s Dis. 12(7), 2029–2058 (2022).
Zhu, K., van Hilten, J. J. & Marinus, J. The course of insomnia in Parkinson’s disease. Parkinsonism Relat. Disorders 33, 51–57 (2016).
Huang, Y. et al. Tryptophan metabolism in central nervous system diseases: Pathophysiology and potential therapeutic strategies. Aging Dis. 14(3), 858–878 (2023).
Yan, E. B. et al. Activation of the kynurenine pathway and increased production of the excitotoxin quinolinic acid following traumatic brain injury in humans. J. Neuroinflamm. 12, 110 (2015).
Mo, X. et al. Serum indoleamine 2,3-dioxygenase and kynurenine aminotransferase enzyme activity in patients with ischemic stroke. J. Clin. Neurosci.: Off. J. Neurosurg.Soc. Australas. 21(3), 482–486 (2014).
Meier, T. B. et al. Prospective study of the effects of sport-related concussion on serum kynurenine pathway metabolites. Brain Behav. Immun. 87, 715–724 (2020).
Bhat, A. et al. Effects of sleep deprivation on the tryptophan metabolism. Int. J. Tryptophan Res.: IJTR 13, 1178646920970902 (2020).
Chang, K.-H. et al. Alternations of metabolic profile and kynurenine metabolism in the plasma of Parkinson’s disease. Mol. Neurobiol. 55(8), 6319–6328 (2018).
Heilman, P. L. et al. Tryptophan metabolites are associated with symptoms and nigral pathology in Parkinson’s disease. Mov. Disorders : Off. J. Mov. Disorder Soc. 35(11), 2028–2037 (2020).
Venkatesan, D. et al. Kynurenine pathway in Parkinson’s disease—an update. ENeurologicalSci 21, 100270 (2020).
Hudson, C. et al. Protein source tryptophan versus pharmaceutical grade tryptophan as an efficacious treatment for chronic insomnia. Nutr. Neurosci. 8(2), 121–127 (2005).
Saidi, O. et al. Randomized double-blind controlled trial on the effect of proteins with different tryptophan/large neutral amino acid ratios on sleep in adolescents: The PROTMORPHEUS study. Nutrients 12(6), 1885 (2020).
Mithani, S. et al. Whole blood transcriptome analysis using RNA sequencing in individuals with insomnia disorder and good sleepers: A pilot study. Sleep Med. 80, 1–8 (2021).
Guvenek, A. et al. Transcriptional profiling in microglia across physiological and pathological states identifies a transcriptional module associated with neurodegeneration. Commun. Biol. 7(1), 1168 (2024).
Fayyad, M. et al. Parkinson’s disease biomarkers based on α-synuclein. J. Neurochem. 150(5), 626–636 (2019).
Xiong, L.-L. et al. Single-cell RNA sequencing reveals peripheral immunological features in Parkinson’s disease. NPJ Parkinson’s Dis. 10(1), 185 (2024).
Sullivan, P. F., Fan, C. & Perou, C. M. Evaluating the comparability of gene expression in blood and brain. Am. J. Med. Genet. Part B, Neuropsychiatric Genet. Off. Publ. Int. Soc. sychiatric Genet. 141B(3), 261–268 (2006).
Edgar, R., Domrachev, M. & Lash, A. E. Gene expression omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 30(1), 207–210 (2002).
Liberzon, A. et al. The molecular signatures database (MSigDB) hallmark gene set collection. Cell Syst. 1(6), 417–425 (2015).
Fishilevich, S. et al. GeneHancer: genome-wide integration of enhancers and target genes in GeneCards. Database J. Biol. Databases Curation 2017, bax028 (2017).
Ritchie, M. E. et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43(7), e47 (2015).
Ito, K. & Murphy, D. Application of ggplot2 to pharmacometric graphics. CPT Pharmacomet. Syst. Pharmacol. 2(10), e79 (2013).
Yu, G. et al. clusterProfiler: an R package for comparing biological themes among gene clusters. Omics: J. Integr. Biol. 16(5), 284–287 (2012).
Szklarczyk, D. et al. The STRING database in 2021: Customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 49(D1), D605–D612 (2021).
Shannon, P. et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13(11), 2498–2504 (2003).
Chin, C.-H. et al. cytoHubba: Identifying hub objects and sub-networks from complex interactome. BMC Syst. Biol. 8(Suppl 4), S11 (2014).
Bader, G. D. & Hogue, C. W. V. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinform. 4, 2 (2003).
Xia, J. et al. A model using support vector machines recursive feature elimination (SVM-RFE) algorithm to classify whether COPD patients have been continuously managed according to GOLD guidelines. Int. J. Chronic Obstr. Pulm. Dis. 15, 2779–2786 (2020).
Sharma, A. & Verbeke, W. J. M. I. Improving diagnosis of depression with XGBOOST machine learning model and a large biomarkers dutch dataset (n = 11,081). Front. Big Data 3, 15 (2020).
Chen, B. et al. Profiling tumor infiltrating immune cells with CIBERSORT. Methods Mol. Biol. 1711, 243–259 (2018).
Hänzelmann, S., Castelo, R. & Guinney, J. GSVA: Gene set variation analysis for microarray and RNA-seq data. BMC Bioinform. 14, 7 (2013).
Subramanian, A. et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A. 102(43), 15545–15550 (2005).
Warde-Farley, D. et al. The GeneMANIA prediction server: Biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res. 38, W214–W220 (2010).
Griffith, M. et al. DGIdb: Mining the druggable genome. Nat. Methods 10(12), 1209–1210 (2013).
Rauluseviciute, I. et al. JASPAR 2024: 20th anniversary of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 52(D1), D174–D182 (2024).
Zhou, G. et al. NetworkAnalyst 3.0: A visual analytics platform for comprehensive gene expression profiling and meta-analysis. Nucleic Acids Res. 47(W1), W234–W241 (2019).
Li, J.-H. et al. starBase v2.0: Decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Res. 42, D92–D97 (2014).
Fan, Y. et al. miRNet - dissecting miRNA-target interactions and functional associations through network-based visual analysis. Nucleic Acids Res. 44(W1), W135–W141 (2016).
Guerriero, S. et al. Artificial intelligence (AI) in the detection of rectosigmoid deep endometriosis. Eur. J. Obstet. Gynecol. Reprod. Biol. 261, 29–33 (2021).
Kanehisa, M. & Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28(1), 27–30 (2000).
Al-Kuraishy, H. M. et al. Sleep disorders cause Parkinson’s disease or the reverse is true: Good GABA good night. CNS Neurosci. Ther. 30(3), e14521 (2024).
Hsiao, Y.-H. et al. Sleep disorders and an increased risk of Parkinson’s disease in individuals with non-apnea sleep disorders: A population-based cohort study. J. Sleep Res. 26(5), 623–628 (2017).
Chen, H. et al. A prospective study of night shift work, sleep duration, and risk of Parkinson’s disease. Am. J. Epidemiol. 163(8), 726–730 (2006).
Xue, C. et al. Tryptophan metabolism in health and disease. Cell Metab. 35(8), 1304–1326 (2023).
Dutkiewicz, Z. & Mikstacka, R. Structure-based drug design for cytochrome P450 family 1 inhibitors. Bioinorg. Chem. Appl. 2018, 3924608 (2018).
Sugumaran, R. et al. Melatonin on sleep in Parkinson’s disease: A randomized double blind placebo controlled trial. Sleep Med. 124, 502–509 (2024).
Reiter, R. J. et al. Dysfunctional mitochondria in age-related neurodegeneration: Utility of melatonin as an antioxidant treatment. Ageing Res. Rev. 101, 102480 (2024).
Xu, G. et al. Melatonin ameliorates spinal cord injury by suppressing the activation of inflammasomes in rats. J. Cell. Biochem. 120(4), 5183–5192 (2019).
Wang, H. et al. Melatonin attenuates spinal cord injury in mice by activating the Nrf2/ARE signaling pathway to inhibit the NLRP3 inflammasome. Cells 11(18), 2809 (2022).
Bocheva, G. et al. The vital role of melatonin and its metabolites in the neuroprotection and retardation of brain aging. Int. J. Mol. Sci. 25(10), 5122 (2024).
Haduch, A. et al. The engagement of cytochrome P450 enzymes in tryptophan metabolism. Metabolites 13(5), 629 (2023).
Sun, M.-F. & Shen, Y.-Q. Dysbiosis of gut microbiota and microbial metabolites in Parkinson’s disease. Ageing Res. Rev. 45, 53–61 (2018).
Sampson, T. R. et al. Gut microbiota regulate motor deficits and neuroinflammation in a model of Parkinson’s disease. Cell 167(6), 1469 (2016).
Wang, Z. et al. The microbiota-gut-brain axis in sleep disorders. Sleep Med. Rev. 65, 101691 (2022).
Iwaniak, P., Owe-Larsson, M. & Urbańska, E. M. Microbiota, tryptophan and aryl hydrocarbon receptors as the target triad in Parkinson’s disease-a narrative review. Int. J. Mol. Sci. 25(5), 2915 (2024).
Coretti, L., Buommino, E. & Lembo, F. The aryl hydrocarbon receptor pathway: A linking bridge between the gut microbiome and neurodegenerative diseases. Front. Cell. Neurosci. 18, 1433747 (2024).
Miyamoto, K., Sujino, T. & Kanai, T. The tryptophan metabolic pathway of the microbiome and host cells in health and disease. Int. Immunol. 36, 601–616 (2024).
Wu, J. et al. CYP1B1 affects the integrity of the blood-brain barrier and oxidative stress in the striatum: An investigation of manganese-induced neurotoxicity. CNS Neurosci. Ther. 30(3), e14633 (2024).
Lei, T. et al. ZNF276 promotes the malignant phenotype of breast carcinoma by activating the CYP1B1-mediated Wnt/β-catenin pathway. Cell Death Dis 13(9), 781 (2022).
Chen, X. et al. Activation of the Wnt/β-catenin/CYP1B1 pathway alleviates oxidative stress and protects the blood-brain barrier under cerebral ischemia/reperfusion conditions. Neural Regen. Res. 19(7), 1541–1547 (2024).
Ziegler, N. et al. β-catenin is required for endothelial Cyp1b1 regulation influencing metabolic barrier function. J. Neurosci.: Off. J. Soc. Neurosci. 36(34), 8921–8935 (2016).
Serafino, A. et al. Developing drugs that target the Wnt pathway: Recent approaches in cancer and neurodegenerative diseases. Expert Opin. Drug Discov. 12(2), 169–186 (2017).
Yang, Z. et al. CYP1B1 deiciency ameliorates learning and memory deficits caused by high fat diet in mice. Am. J. Transl. Res. 11(4), 2194–2206 (2019).
Yim, J. et al. Inflachromene ameliorates Parkinson’s disease by targeting Nrf2-binding Keap1. Chem. Sci. 15(10), 3588–3595 (2024).
Wang, W., Liu, T. & Zhang, Y. An integrated targeted metabolomics and network pharmacology approach to exploring the mechanism of ellagic acid against sleep deprivation-induced memory impairment and anxiety. Digit. Health 9, 20552076231169850 (2023).
Schiff, M. et al. Electron transfer flavoprotein deficiency: Functional and molecular aspects. Mol. Genet. Metab. 88(2), 153–158 (2006).
Roodveldt, C. et al. The immune system in Parkinson’s disease: What we know so far. Brain J. Neurol. 147(10), 3306–3324 (2024).
Han, Y. et al. Analysis of the correlation between immune cell characteristics and insomnia: A Mendelian randomization study. J. Neurophysiol. 131(2), 176–186 (2024).
Ogura, Y. et al. Nod2, a Nod1/Apaf-1 family member that is restricted to monocytes and activates NF-kappaB. J. Biol. Chem. 276(7), 4812–4818 (2001).
Li, J. et al. Critical role of FPR1 in splenocyte migration into brain to worsen inflammation and ischemic brain injury in mice. Theranostics 12(7), 3024–3044 (2022).
Schulz, J. et al. Meta-analyses identify differentially expressed micrornas in Parkinson’s disease. Ann. Neurol. 85(6), 835–851 (2019).
Gao, X. et al. Beta-naphthoflavone inhibits LPS-induced inflammation in BV-2 cells via AKT/Nrf-2/HO-1-NF-κB signaling axis. Immunobiology 225(4), 151965 (2020).
Fernandez-Abascal, J. et al. β-Naphthoflavone and ethanol reverse mitochondrial dysfunction in a Parkinsonian model of neurodegeneration. Int. J. Mol. Sci. 21(11), 3955 (2020).
Fernandez-Abascal, J. et al. β-hrome P450 and Protect towards MPP⁺ toxicity in human neuroblastoma SH-SY5Y cells. Int. J. Mol. Sci. 19(11), 3369 (2018).
Giradkar, V., Mhaske, A. & Shukla, R. Naringenin nanocrystals mitigate rotenone neurotoxicity in SH-SY5Y cell line by modulating mitophagy and oxidative stress. AAPS PharmSciTech 25(7), 227 (2024).
Muhammad, F. et al. Anti-α-synuclein toxicity and anti-neurodegenerative role of chrysin in transgenic caenorhabditis elegans models of Parkinson’s disease. ACS Chem. Neurosci. 13(4), 442–453 (2022).
Kim, S. M. et al. Acacetin inhibits neuronal cell death induced by 6-hydroxydopamine in cellular Parkinson’s disease model. Bioorg. Med. Chem. Lett. 27(23), 5207–5212 (2017).
Kong, X. et al. Sleep-improving effect and the potential mechanism of Morus alba L. on mice. Fitoterapia 179, 106205 (2024).
Miki, Y. et al. Alteration of autophagy-related proteins in peripheral blood mononuclear cells of patients with Parkinson’s disease. Neurobiol. Aging 63, 33–43 (2018).
Henderson, A. R. et al. DNA methylation and expression profiles of whole blood in Parkinson’s disease. Front. Genet. 12, 640266 (2021).
Atashrazm, F. et al. Reduced glucocerebrosidase activity in monocytes from patients with Parkinson’s disease. Sci. Rep. 8(1), 15446 (2018).
Papagiannakis, N. et al. Lysosomal alterations in peripheral blood mononuclear cells of Parkinson’s disease patients. Mov. Disorders Off. J. Mov. Disorder Soc. 30(13), 1830–1834 (2015).
Masliah, E. et al. Dopaminergic loss and inclusion body formation in alpha-synuclein mice: implications for neurodegenerative disorders. Science (New York, N.Y.) 287(5456), 1265–1269 (2000).
Dovonou, A. et al. Animal models of Parkinson’s disease: Bridging the gap between disease hallmarks and research questions. Transl. Neurodegener. 12(1), 36 (2023).
Acknowledgements
We sincerely thank the National Natural Science Foundation of China for funding.
Funding
This study was supported by grants from a major project co-constructed by the Ministry of Medical Science and Technology in Henan Province (Grant No. SBGJ202101016) and National Natural Science Foundation of China (Grant No. 82171300).
Author information
Authors and Affiliations
Contributions
L.X.Y and L.Y.S conceived the study and were responsible for the study design. L.X.Y and Y.W.K wrote the origin manuscript. L.X.Y, W.J.H, and C.Y.N performed experiments. G.K, H.W.J, and W.Y.H were responsible for data collection and study supervision. All co-authors have read and approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval and consent to participate
All experimental procedures were approved by the Institutional Animal Care and Use Committee of the First Affiliated Hospital of Zhengzhou University (ZZU-LAC20240531[06]).
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Li, XY., Yu, WK., Wu, JH. et al. Tryptophan metabolism-related gene CYP1B1 serves as a shared biomarker for both Parkinson’s disease and insomnia. Sci Rep 15, 1362 (2025). https://doi.org/10.1038/s41598-024-84362-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-024-84362-8
