
Abstract
Alport syndrome is a hereditary kidney disease with significant variations in onset and prognosis. While 80–85% of cases are due to pathogenic variants in the COL4A5 gene, there is a notable lack of mouse models with Col4a5 mutations for basic research. Our research presents an 8-year-old child with Alport syndrome, exhibiting facial edema and abnormal urine. Next-generation sequencing revealed a c.1517-1G > T mutation in the intron sequence of the COL4A5 gene. Minigene experiments confirmed that this intronic mutation affects mRNA splicing. Using the CRISPR/Cas9 system, we developed a Col4a5-c.1517-1G > T mutant mouse model. Col4α5-deficient mice exhibited growth retardation and reduced lifespan. Renal function analysis indicated progressive deterioration, with high levels of BUN and creatinine. Histological and ultrastructural analyses revealed abnormalities such as mesangial sclerosis, interstitial fibrosis and severe irregularity in membrane thickness. Additionally, significant immune cell infiltration was observed in the renal interstitium. This mouse model provides a valuable tool for studying the role of immune cells in the pathogenesis and treatment of XLAS. It is also the first reported X-linked Alport syndrome mouse model caused by a splicing mutation.
Similar content being viewed by others


Introduction
Alport syndrome (AS) is a hereditary nephropathy caused by mutations in the genes COL4A3, COL4A4, and COL4A5, which encode the α3, α4, and α5 chains of type IV collagen1,2. Structural anomalies in type IV collagen can lead to dysfunction of the glomerular basement membrane, resulting in proteinuria and hematuria. Additionally, some patients may exhibit extrarenal symptoms, such as sensorineural hearing loss and ocular abnormalities3. The prognosis for Alport syndrome is generally poor, and currently, there is no cure. Early intervention with angiotensin-converting enzyme inhibitors (ACEIs) and angiotensin receptor blockers (ARBs) may slow the progression to renal failure4,5.
Within the general population, mutations in the COL4A5 gene account for 80-85% of Alport syndrome cases, while a smaller proportion is due to mutations in the COL4A3 and COL4A4 genes6,7. Located on the X chromosome, the COL4A5 gene’s position elucidates the higher prevalence and more severe clinical manifestations of X-linked Alport syndrome in males. It is estimated that nearly all male patients with this condition will eventually develop end-stage renal disease. Despite the identification of the genes responsible, the specific molecular mechanisms that determine the disease’s onset and its prognosis have yet to be fully determined8.
Establishing an effective animal model of Alport syndrome is crucial for exploring prognostic differences caused by various types of mutations and for developing new targeted drugs. However, the currently available mouse models mainly involve mutations in the Col4a3 gene, with fewer models specifically related to Col4a5 mutations9. Currently, there are three reported mouse models of Alport syndrome caused by exon mutations in the Col4a5 gene. Rheault et al. and Kentarou Hashikami et al. constructed mouse models with point mutations in the Col4a5 gene exons, resulting in premature stop codons that affect the synthesis of Col4a5 collagen10,11. Wei-qing Wu et al. confirmed that a frameshift mutation caused by an ATGG deletion results in abnormal functionality of Col4a5 collagen12. Different types of gene mutations cause aberrant function of the Col4a5 protein, resulting in Alport syndrome.
Recently, our research group diagnosed a case of Alport syndrome characterized by significant proteinuria. Next Generation Sequencing of both family members and the affected child revealed a COL4A5 gene mutation located in the intronic region, with genotype and phenotype co-segregation. Through minigene experiments, we demonstrated that this intronic mutation can cause splicing variations and cause frameshift mutation. Transcriptional anomalies of the COL4A5 gene result in the generation of shortened COL4A5 protein, which is an important cause of Alport syndrome.
To explore the renal damage characteristics caused by this intronic mutation in Alport syndrome, we established a mouse model using CRISPR/Cas9 gene editing technology, targeting this genetic pattern. The results validated that this mouse model effectively simulates the clinical features of Alport syndrome patients. Furthermore, our studies revealed significant immune cell infiltration in the late-stage kidneys of this mouse model.
This study presents a new mouse model of Alport syndrome caused by a novel Col4a5 gene mutation based on our clinical findings. To our knowledge, this is the first mouse model of Alport syndrome resulting from an intronic sequence variant. This model provides a reliable clinical research tool for further investigating the heterogeneity of Alport syndrome.
Result
Clinical information of the patient of Alport syndrome
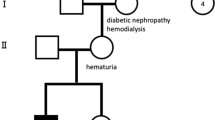
The male patient under discussion is the offspring of unrelated parents. When he was 8 years old, he displayed facial and eyelid swelling and was taken to a local hospital. A urinalysis showed proteinuria (3+), 350 red blood cells/µl, and albumin levels of 15 g/L. Subsequently, he was referred to our hospital. Upon inquiring about the medical history, we discovered that several relatives in the child’s family have a history of kidney disease. Male individuals have more severe symptoms, advancing to end-stage renal failure, whereas female patients have varied degrees of hematuria. Peripheral blood samples were collected from multiple members of the patient’s family, and Next Generation Sequencing analysis revealed the presence of the COL4A5-c.1517-1G > T mutation in the probands’ family, highlighted in black in Fig. 1A.

Clinical manifestations of the patient. (A) Pedigree chart of the proband and Sanger sequencing results of the proband, father, mother, and sister. The black color in the pedigree chart represents the patient with COL4A5-c.1517-1G > T mutation. Half-hatched symbols represent patients or carriers of the mutation. DNA sequencing was performed using Sanger sequencing analysis. The red boxes highlight the gene mutation sites. (B-D) Histological changes in the patient’s kidney tissue. (B) PAS staining (×400); (C) PASM staining (×400); (D) MASSON staining (×400). (E-G) TEM results of the patient. In (E, F), the red arrows indicate relatively thickened basement membrane areas, while the blue arrows indicate relatively thin sections of the basement membrane. In (G), the black arrow indicates the thickening of the dense layer of the basement membrane with fusion in the foot processes.
Upon admission, the patient underwent a renal biopsy and pathological examination. HE staining, PAS staining, and PASM staining revealed mild proliferation of mesangial cells and matrix, along with glomerular atrophy and sclerosis in the patient’s kidneys (Fig. 1B-D). The electron microscopy examination of the renal biopsy revealed diffuse thickening and thinning of the glomerular basement membrane. The thickness of the membrane varied at different locations, with thickening noted in the dense layer of the basement membrane and fusion in the foot processes. (Fig. 1E-G). Immunofluorescence labelling revealed normal α1 expression as a positive control (Supplementary Fig. 1A). The stability of COL4A3 and COL4A5 collagens was compromised by the exon 23 deletion of COL4A5 gene, therefore α3 expression was lost in the glomerular basement membrane and tubular basement membrane (Supplementary Fig. 1B); and α5 expression was lost in glomerular basement membrane, Bowman’s capsule, tubular basement membrane (Supplementary Fig. 1C). Based on the patient’s medical history and laboratory findings, a diagnosis of Alport syndrome was made.
Minigene experiment confirms the impact of COL4A5-c.1517-1G > T on COL4A5 splicing
As early as 2008, literature reported patients with Alport syndrome carrying the point mutation COL4A5-c.1517-1G > T, who belong to the same family as the patient mentioned in this article13. The American College of Medical Genetics and Genomics (ACMG) has developed standards for variant interpretation whereby the COL4A5-c.1517-1G > T mutation site is found at the 3’ end of intron 22, thereby reflecting a conventional splice site − 1 mutation. Classed as the one with highly strong indication of pathogenicity (PVS1), this Variations at splice sites can cause exon loss, shortenings, or even transform intronic sequences into exonic areas. Although the development of several software and computational methods has improved the accuracy of estimating the consequences of such changes, experimental validation is still required to verify the impact generated by this kind of mutation14.
Constructing minigene vectors with mutation sites allows for the observation of mutations’ effects on splicing events, including exon skipping and intron retention. In vitro minigene studies can elucidate the impact of splicing mutations on mRNA splicing mechanisms15,16. To validate the form and mechanism of this splice mutation, we designed a minigene experiment. We constructed wild-type plasmids and COL4A5-c.1517-1G > T mutant plasmids, and transfected them into 293T cells (Fig. 2A-B, supplementary Fig. 2A). By analyzing the band sizes of PCR amplification products and Sanger sequencing results, we found that the mRNA sequence transcribed from the WT plasmid matched the expected pattern, containing complete mRNA products transcribed from exon 22 and exon 23. However, the mutant plasmid introduced into 293T cells exhibited abnormal expression fragments. The COL4A5-c.1517-1G > T mutation causes exon 23 to be skipped, therefore lowering the possible amplification length of the exon22-exon24 of COL4A5 gene from 474 bp to 403 bp. Furthermore, a frameshift mutation results from the number of deleted bases not being a multiple of three, leading to a premature stop codon to show up in exon 24 and hence early translation termination (Fig. 2A). The sequencing of the abnormal mRNA product is shown in Fig. 2B. Alignment of the RT-PCR products from the minigene studies with the CDS sequence of the COL4A5 gene further reveals that the Mutant group, post-splicing, is deficient in a region of exon 23 (71 bp) relative to the WT group (Supplementary Fig. 2B). These results indicate that the COL4A5-c.1517-1G > T mutation leads to abnormal mRNA splicing.

Minigene validation experiment for the splicing pattern of the Col4a5-c.1517-1G > T mutation. (A) Minigene Experimental Workflow: Before the experiment, the contiguous intronic sequences of exons 22 to 24 of the Col4a5 gene, featuring the c.1517-1G mutation, and the WT control were individually cloned into the vector. The Col4a5-c.1517-1G > T mutation leads to skipping of exon 23, resulting in an amplified fragment length of 403 bp. (B) Sequencing results of the wild-type and Col4a5-c.1517-1G > T type transcripts.
CRISPR/Cas9 technology was used to generate Col4a5-c.1517-1G > T mutant mice
The COL4A5 gene is highly conserved across humans and mice, indicating its relevance17. The patient’s mutation site is located at the junction of intron 22 and exon 23, and it shares strong similarities with the corresponding sequence in mice, indicating a classic splicing mutation site (Supplementary Fig. 3A-B). As a result, we used the c.1517-1G > T mutation in this position as the model mouse’s mutation site.
The genetically modified mice used in this study were generated by Jiangsu Jicui Yaokang Biotechnology Co., Ltd. to construct the Col4a5 gene mutation mouse model. The CRISPR/Cas9 system was employed to generate all F0 mice (Fig. 3A). The designed sgRNA, along with a constructed donor vector, Cas9, and sgRNA, was microinjected into the fertilized eggs of C57BL/6JGpt mice for homologous recombination. Heterozygous female mice were crossed with wild-type male mice, and subsequent generations produced the desired mice. Mouse genotypes were identified through PCR and Sanger sequencing (Fig. 3B-C). To verify the presence of a splicing mutation in the produced KI mice, we isolated RNA from the kidney tissues and reverse transcribed it into cDNA. Subsequently, we mapped the cDNA sequences with the Col4a5 mouse coding sequence. The findings demonstrated that the mRNA from the KI mice was devoid of the exon 23 region (Supplementary Fig. 4A-C). Based on the collected data, the research team successfully constructed Col4a5-c.1517-1G > T gene mutant mice, which will be referred to as KI mice in the following text.

Targeting strategy, genotyping, and clinical phenotypes of Col4a5-c.1517-1G > T mutant mice. (A) The target locus was genetically modified through homologous recombination in embryonic stem cells using the CRISPR/Cas9 system. (B) Electrophoresis results of the PCR products in the target region of F1 mice. Number 126, 127, 128 refer to KI mice, while number 129 and C57L/B6J are chose as control. (C) Sequencing results of the target region in F1 mice. The red mark indicates the Col4a5-c.1517-1G > T mutation site.
Col4a5-c.1517-1G > T mutant mice develop hematuria, proteinuria, and impaired renal function
First, we evaluated the growth and development of the mice. The results showed that the growth and development of male KI mice were delayed compared to wild-type male mice. The average weight growth of KI mice started to slow down by week 14 compared to wild-type male mice (Fig. 4A).

The biochemical parameters of Col4a5-c.1517-1G > T mice at different ages. (A) Growth curve of mouse body weight. (B) Changes in the urine protein to creatinine ratio in 24-hour urine. (C) Changes in creatinine (Cre) in 24-hour urine. (D) Changes in blood urea nitrogen. Serum biochemistry tests were performed on 5 male Col4a5-c.1517-1G > T and 5 control male mice. P < 0.05 *, P < 0.01 **, unpaired t-test was used for inter-group comparison.
To assess the renal function of KI mice, urine samples were collected for the measurement of hematuria and the urine protein/creatinine ratio. The dipstick test revealed early onset hematuria in the KI group at 7 weeks of age, with a prevalence that increased progressively with age (data not shown). The urine protein/creatinine ratio of the KI group was significantly elevated at 14 weeks of age and continued to increase at 21 weeks (Fig. 4B), indicating impaired renal function. However, the urine protein/creatinine ratio of wild-type male mice remained at a relatively low level (Fig. 4B). Compared to wild-type male mice, the blood urea nitrogen (BUN) and creatinine (Cre) levels in the serum of KI mice were significantly elevated at 28 weeks of age (Fig. 4C and D).
Pathological changes in Col4a5-c.1517-1G > T mutant mice
After euthanizing the mice, we removed the kidneys and stripped off the capsules. Gross observation showed that the kidneys of KI mice exhibited sclerosis and atrophy with increasing age (Data not shown). Our initial focus was to assess the expression of the Col4a5 gene at the RNA level. The frameshift mutation induced by the Col4a5-c.1517-1G > T alteration results in premature termination codons. In our investigation, through qPCR analysis, we identified a 30% reduction in Col4a5 gene expression levels in KI mice compared to WT mice. This observation confirmed a decrease in Col4a5 gene expression at the RNA level (Supplementary Fig. 5A).
We then looked at the pathogenic alterations in the kidneys of KI mice at several ages using H&E, PAS, and Masson staining among other techniques. Kidney pathological staining showed that at 7 weeks of age, the overall morphological structure of the renal tubular interstitium was normal. With increasing age, the renal sclerosis gradually worsened. Renal sclerosis worsened as mice got older. The interstitium was highly inflamed, with glomerular atrophy and sclerosis. Mesangial growth varied from mild to moderate. It was accompanied by interstitial fibrosis, collagen deposition, and interstitium fibrosis. Tubular cells swelled and atrophy. Additionally, persistent interstitial alterations were identified.
By 28 weeks of age, a large number of glomeruli showed abnormal morphology, including proliferation of parietal epithelial cells forming crescentsand global sclerosis. Renal fibrosis was also increased. In the tubular interstitium, partial atrophy and disappearance of the brush border of the renal tubules were observed (Fig. 5A-C).

Pathological changes in the kidney tissue of Col4a5-c.1517-1G > T mutant mice. (A-C) Representative images of H&E(A), PAS(B), and Masson(C) staining of kidney tissue in wild-type and KI mice at 7 weeks, 14 weeks, 21 weeks, and 28 weeks of age (H&E staining magnification ×100, other staining magnification ×200). The yellow arrows indicate infiltration of mononuclear cells (A). The black box highlights abnormal thickening of the glomerular basement membrane, glomerular atrophy, and sclerosis (B). The green arrows represent deposition of collagen fibers, with focal fibrosis present in both the glomeruli and renal tubules (C). (D) Quantification of podocytes in wild-type and KI groups from 7 to 28 weeks, showing a progressive decrease in podocyte number in the KI group compared to the control group. The average podocyte density per glomerulus is the number of WT1 positive cells/number of glomeruli. P < 0.01 **, unpaired t-test was used for inter-group comparison.
Previous studies have suggested that a decrease in the number of podocytes is an important pathological change in the kidneys of Alport syndrome patients18. Our data showed that the number of podocytes in KI mice was significantly lower than that in wild-type mice at 21 weeks of age, and the difference further increased by 28 weeks of age (Fig. 5D).
Ultrastructural changes in the kidneys of Col4a5-c.1517-1G > T mutant mice
We further used transmission electron microscopy (TEM) and scanning electron microscopy (SEM) to evaluate the pathological changes in the kidneys of KI group mice and wild-type littermates (Fig. 6A-B).

Pathological images of the kidneys from Alport syndrome model mice and wild-type mice at 7, 14, 21, and 28 weeks of age, using (A) transmission electron microscopy and (B) scanning electron microscopy. The red arrows indicate that the ultrastructure of the mice in the KI group exhibited displayed uneven thickness of the glomerular basement membrane.
At 28 weeks of age, the ultrastructure of the KI group mice displayed uneven thickness of the glomerular basement membrane and partial foot process effacement. In contrast, the glomerular basement membrane of wild-type control mice at 28 weeks of age was of uniform thickness, and the interdigitating foot processes were closely attached to the outer side of the basement membrane. By 28 weeks of age in the KI group mice, the ultrastructure showed splitting and lamellation of the glomerular basement membrane, severe irregularity in membrane thickness, extensive foot process effacement, and disappearance. SEM observation revealed the disappearance of the tight interdigitation between the podocyte foot processes and the shrinkage of podocyte cell bodies in the KI group mice.
Infiltration of immune cells in the renal interstitium of Col4a5-c.1517-1G > T mutant mice
Increasing evidence suggests that the disruption of the immune microenvironment is another important factor in the deterioration of renal function in Alport syndrome, and the infiltration of immune cells is also an important indicator of the prognosis of Alport syndrome patients19,20. However, existing reports on Alport syndrome mouse models with Col4a5 gene variations have not mentioned the phenomenon of extensive immune cell infiltration in the renal interstitium of these mice.
To this end, we extracted the kidneys from 28-week-old KI group mice and wild-type mice, and used immunohistochemistry to detect the content of immune cell infiltration. The results showed a large number of CD3 positive T cells and CD68 positive macrophages in the renal tubular interstitium of the mutant mice. In contrast, no significant CD3 and CD68 positive cells infiltration was found in the renal interstitium of the control group mice (Fig. 7A). The result is consistent with the immunohistochemical results of renal biopsy in children in our study (Fig. 7B). These data indicate that immune cell infiltration is an important feature of the splicing mutation mouse model, providing a new mouse model for future investigations into the role and mechanisms of immune cells in the progression of Alport syndrome.

(A) Representative images of CD3 and CD68 immunostaining in the wild-type control group mice and KI group mice. (B) Representative images of CD3 and CD68 immunostaining in the kidney of the patient with COL4A5-c.1517-1G > T mutation.
Discussion
Alport syndrome is a hereditary kidney disease caused by mutations in the COL4A3, COL4A4, or COL4A5 genes, primarily characterized by progressive impairment of renal function. Some patients also experience extrarenal organ damage1,21. However, the factors influencing the prognosis of Alport syndrome and their molecular mechanisms are still not well understood.
Previous studies have shown that the prognosis of X-linked Alport syndrome is closely related to the type of gene mutation. Jais et al. found that patients with large segment deletions and nonsense mutations have a 90% probability of progressing to end-stage renal disease (ESRD) by the age of 30, while those with splice site mutations have a 70% risk, and those with missense mutations have a 50% risk22. Bekheirnia et al. found that Alport syndrome patients with missense mutations have an average ESRD onset age of 37 years, those with splice site mutations have an onset age of 28 years, and those with truncating mutations have an onset age of 25 years22.
In a typical adult glomerulus, the glomerular basement membrane and distal tubule basement membrane exhibit the α3α4α5 triple helical configuration of type IV collagen, whereas the Bowman’s capsule and distal tubule basement membrane display the α5α5α6 triple helical structure23. In male individuals with XLAS, 70–80% demonstrate the absence of expression of the α3, α4, and α5 chains in the glomerular basement membrane and the basement membrane of the distal tubule, with the α5 chain being absent in the Bowman’s capsule. A modest minority of XLAS male and female patients may have normal expression of the α3, α4, and α5 chains in the GBM24,25,26.
In this case, we used immunohistochemistry to analyze the patient’s expression of COL4A1, COL4A3, and COL4A5. The results showed that the expression of Col4a1 protein remained unaltered, but the expression of COL4A3 and COL4A5 showed punctate signals. This means that certain COL4A5 genes in the patient’s body may create partly shortened mRNA as a result of splicing mutations, but they may also produce correctly spliced full-length copies of COL4A5 mRNA. In future research, determining the quantity of appropriately spliced COL4A5 mRNA molecules using qRT-PCR might aid in better understanding the COL4A5 gene’s mutation rate. Furthermore, Alport syndrome model mice generated using this mutant location had a longer lifetime than previous published Alport syndrome model mice27. From this perspective, our mouse model is more consistent with the characteristic gradual start and course of Alport syndrome in humans, making it a potentially more acceptable animal model for preclinical investigations.
The alteration of the triple helix configuration o induces structural modifications in the GBM, culminating in anomalies such as thinning, thickening, and splitting. Figure 1E illustrates that this patient has thickening of the dense layer of the basement membrane, with certain regions displaying splitting and a cobweb-like appearance. When the structure of the basement membrane is aberrant, its barrier function is impaired. This intensifies proteinuria and diminishes the adhesion between podocytes and the glomerular basement membrane18. Damage and separation of podocytes further compromise the filtering function of the glomerulus. Chronic proteinuria exacerbates renal impairment. Protein deposition from proteinuria in the tubulointerstitium can induce inflammation and fibrosis, ultimately resulting in the gradual decline of renal function and ending in end-stage renal disease.
The phenotypes of mice with different mutation types show significant differences, and the construction of Alport syndrome mouse models with various genetic inheritance patterns has provided more platforms for exploring the pathogenesis of the disease. The three reported Col4a5 gene mutation mouse models are those carrying nonsense mutations G5X, R471X, and a frameshift deletion ATGG10,11,12. Based on the pathogenic site discovered in clinical work, we developed a mouse model of X-linked Alport syndrome. In comparison to mice models with G5X, R471X and Del-ATGG mutations within the Col4a5 gene established by other researchers10,11,12, our model has a comparable onset timing as previously reported; nevertheless, we have seen a longer survival period. Mutations within the same gene can cause variation in the symptoms and severity of Alport syndrome between people.
The development of this mouse model is motivated not only by the limited availability of Alport syndrome model mice with Col4a5 mutations, as previously mentioned. Diverse mutations in mice yield distinct prognoses and onset durations. At now, there is an absence of Alport syndrome murine models predicated on splicing mutations. Identifying novel mutation sites from clinical patients and creating mice models with these mutations will enhance our comprehension of the disease’s course. Furthermore, as the impacted child is part of a sizable family with several members afflicted by Alport syndrome, the creation of this mouse model not only facilitates the examination of the disease’s pathogenesis but also offers a significant opportunity to explore the diagnosis and treatment for this child and other relatives experiencing the same condition.
Thin basement membrane nephropathy (TBMN) is generally considered a benign illness in terms of clinical progression, however a few investigations have found that some TBMN patients develop proteinuria and renal dysfunction28,29. The basement membrane seems unevenly thick in this electron microscopy picture, with some sections seeming thinner. This finding may indicate that this patient’s clinical manifestations share some parallels with TBMN, emphasising the importance of genetic testing in clinical practice when symptoms, clinical manifestations, and pathological exams are ambiguous.
The main cells in the kidney in charge of synthesis of COL4A3/A4/A5 collagen proteins are podocytes. One important component causing Alport syndrome to advance is podocyte malfunction30,31,32. But several investigations conducted recently have shown that podocyte malfunction is not the only element causing Alport disease to worsen. Although alterations in the glomerular basement membrane arise at birth, most patients do not show notable renal abnormalities at that point. Rather, renal function declines usually gradually as people get older19,20. Emerging evidence indicates that aberrantly activated immune cells and dysregulated immune factors are critical contributors to the progression of Alport syndrome19. Stephan Segerer’s team conducted immunohistochemical staining on kidney biopsy samples from 17 Alport syndrome patients and 21 control patients, revealing a marked increase in T cells within the renal interstitium of Alport syndrome patients33. Other studies have also highlighted macrophage and foam cell infiltration as significant markers of Alport syndrome progression33. In mouse models, research has demonstrated that Col4a3-deficient mice exhibit macrophage infiltration, with significant variations in the extent and timing of renal inflammatory responses and macrophage infiltration depending on genetic background9,34.
We also noted substantial macrophage invasion in our Col4a5-c.1517-1G > T mutant mouse model together with CD3 + T cells. These results support earlier studies and confirm the coherence between our mouse model and clinical data10,35, therefore offering more proof that immune cells can be very important in the course of Alport syndrome. In eukaryotic genomes, non-coding sequences comprise non-coding regions and introns. Introns are excised during mRNA processing, resulting in mature mRNA devoid of these non-coding sequences. Furthermore, introns do not influence the structure of the translated product and are often not subject to natural selection. Consequently, they are more likely to be inherited by subsequent generations compared to exonic variations. In this study, we developed a mouse model of Alport syndrome harboring the Col4a5-c.1517-1G > T mutation, based on clinical observations. Considering that distinct pathogenic mechanisms may be implicated in different mutation types, the Col4a5-c.1517-1G > T mouse model offers a novel tool and platform for investigating the pathogenesis and therapeutic targets of X-linked Alport syndrome. This model represents the first reported instance of Alport syndrome with X-linked inheritance caused by a splicing mutation.
Ultimately, our research possesses certain limitations. Our study is deficient in pertinent evidence about the critical diagnostic significance of detecting Col4a5 mRNA in leukocytes for Alport syndrome. Secondly, while we noted a downregulation of Col4a5 gene expression in the Mutant group mice, the precise mechanism behind this RNA downregulation is ambiguous. Finally, we noted a substantial infiltration of immune cells in the kidneys during the advanced phases of Alport disease. According to the reviewer’s remarks. Although this trait is not unique to our model, our understanding of the role and mechanism of immune cells in disease development is still limited. The precise context of immune cell engagement in the presence of a splice site mutation has not been adequately examined. Future study should assess the differences in the number and functionality of immune cell infiltration across mice models. The way immune cells function may also play an important role in the various prognoses associated with different mutation types.
Conclusion
We identified a splicing variant, c.1517-1G > T, in the COL4A5 gene of patients with X-linked Alport syndrome (XLAS). Through both in vivo and in vitro experiments, we confirmed that this splicing variant can cause Alport syndrome. This discovery expands the spectrum of COL4A5 gene variations associated with X-linked inheritance and enhances our understanding of the molecular pathogenesis of Alport syndrome. Furthermore, the NM_033380.3(COL4A5): c.1517-1G > T variant, which we investigated, is currently classified as a “likely pathogenic” variant in the ClinVar database (https://www.ncbi.nlm.nih.gov/clinvar/RCV000021300/). Our research provides the first evidence of the pathogenicity of this mutation.
Materials and methods
Clinical information of the patient
The patient, a male, presented at a local hospital at the age of 8 due to coughing and facial swelling. Abnormalities were detected in the urine test. Subsequently, the patient was transferred to the Guangzhou Women and Children Medical Center for hospitalization and treatment. Blood tests showed normal results, except for a total plasma protein value of 50.4 g/L [normal range: 64–83 g/L] and an albumin level of 13.4 g/L [normal range: 20–30 g/L]. Urine analysis revealed 6.88 g of protein [normal range: ≤ 0.4 g]. Microscopic examination of the urine revealed the presence of 350 red blood cells/µl. Following a comprehensive renal tissue biopsy and pathological examination, the patient was strongly suspected to have Alport syndrome. Further investigations, including a detailed family history of kidney disease, ocular and auditory examinations, and genetic testing, led to the final diagnosis of Alport syndrome in the patient. The data from patient samples in this study have been obtained with the informed consent of the patients themselves and their parents, as well as the approval of the Ethics Committee of the Guangzhou Women and Children’s Medical Center. The practice was performed in accordance with the Declaration of Helsinki.
Next generation sequencing of the patient
Whole blood samples were initially extracted, and nucleic acids were isolated utilizing the Blood Genomic DNA Mini kit (Kangweishiji, Catalog Number: CW2087S). Following the determination of nucleic acid content and purity assessment, 400 ng of genomic DNA samples were obtained. The IGT® Enzyme Plus Library Prep Kit v2.0 (Agilent, Catalog Number: C10982) was utilized in conjunction with the AIExome® Human Exome Panel V3 - Inherit (Agilent, Catalog Number: PT1008102) for genomic DNA library construction and whole exome capture library preparation according to the provided instructions. The assembled entire exome library was quantified utilizing the dsDNA HS analysis kit (Life, Catalog Number: Q32854) with the Qubit 3.0 fluorometer. The whole exome library was subjected to fragment analysis with the DNA 1000 kit (Agilent, Catalog Number: 5067 − 1504) and assessed with the Agilent 2100 Bioanalyzer. Sublibrary pooling, TruSeq adapter ligation, DNB preparation, and sequencing on the MGI DNB-SEQ T7 platform were conducted. Operations were executed in accordance with the DNBSEQ-T7RS High-Throughput Sequencing Reagent Set (FCL PE150) V3.0 kit guidelines (MGI, Catalog Number 940-000268-00).
Immunohistochemical staining of collagen protein in the patient’s kidney tissue
Immunofluorescence staining of COL4A1, COL4A3, and COL4A5 collagens was performed using collagen type IV antibodies according to the Alport Immunofluorescence Assay Kit manual. This kit was purchased from Jiangsu Lanou Medical Technology Co., Ltd. (Catalog number: LO-5100). The antibodies in question target the NC1 domains of COL4A1, COL4A3, and COL4A5 collagens. The immunohistochemical staining procedure was carried out in accordance with the manufacturer’s instructions.
Construction of the Col4a5 gene mutation mouse model
The genetically modified mice used in this study were generated by Jiangsu Jicui Yaokang Biotechnology Co., Ltd. to construct the Col4a5 gene mutation mouse model. The research group specifically targeted the transcript Col4a5 (ENSMUST00000112931.7, NM_001163155.1) for editing. The gRNA sequences used were gRNA1: ATGTGTGTCTTAGGATCTC and gRNA2: TTAGGATCTCTTGGAATCCC. The designed sgRNA, along with a constructed donor vector, Cas9, and sgRNA, was microinjected into the fertilized eggs of C57BL/6JGpt mice for homologous recombination. The manipulated embryos were then transferred into the oviduct of pseudopregnant ICR females. F0 mice were born 19–21 days after transplantation, and PCR and Sanger sequencing were performed to identify the offspring carrying the Col4a5-c.1517-1G > T gene mutation (F0 mice). Animal protocols were approved by the Guangdong Huawei Testing Co., Ltd.
Breeding and maintenance of mice
The Col4a5-c.1517-1G > T gene mutation mouse model was established using CRISPR/Cas9 technology. The mutation site for this model was referenced from the sequencing results of the Next-Generation Sequencing of the patient at our hospital. The genotype was determined by PCR and DNA sequencing. Genomic DNA was extracted from mouse tail samples. The primers used for genotype identification by PCR were as follows: Col4a5-mut-tF (5’-AGAACTCATGCCCAACCTCACAG-3’) and Col4a5-mut-tR (5’-CTCTCCAGGGATTCCCAAAGATCA-3’). The PCR amplification conditions involved in this study are detailed in Table 1. The mice involved in this study were all male mice. The mice were also housed and maintained at the same facility. All mice had access to water and standard rodent chow ad libitum and were housed in a temperature- and humidity-controlled animal facility. All methods were carried out in accordance with relevant guidelines and regulations. The study was conducted in accordance with the ARRIVE guidelines. (https://arriveguidelines.org). The study was approved by the Ethics Committee of Guangdong Huawei Testing Co., Ltd. (Approval No. (HWT-BG-117). In this research, 40 male mice were used, including 20 wild-type mice and 20 mutant mice. However, one mutant mouse died at the age of 26 weeks.
Detection of Col4a5 gene transcript in KI mice
Kidney tissues were collected separately from 21-week-old WT and KI mice. The mice were anesthetized with pentobarbital sodium via intraperitoneal injection and euthanized by carbon dioxide asphyxiation after reaching full anesthesia. The study was approved by the Ethics Committee of Guangdong Huawei Testing Co., Ltd. (Approval No. (HWT-BG-117)). RNA was isolated from renal tissues with a Trizol extraction kit (Sangon Biotech B511321). The extracted products were reverse transcribed into cDNA sequences, and the target sequences were detected using gel electrophoresis. The PCR products were then recovered using the Evo M-MLV One Step RT-PCR kit (Aidlab AG11606). After purification and cloning, the target DNA pieces were ligated into the pUC18-T vector. The pMD18-T vector was subjected to PCR using universal primers: agggttttcccagtcacg and gagcggataacaatttcacac. Finally, the resultant products were sequenced and mapped to the Col4a5 coding sequence.
Measurement of proteinuria/creatinine, blood creatinine, and blood Urea nitrogen
For the collection of 24-hour urine samples, mice were placed in metabolic cages. Urine protein (Alb) and urine creatinine (U-Cre) levels were measured using an automatic analyzer (Hitachi 7600-020). To measure blood creatinine and blood urea nitrogen, mice were anesthetized using a mixture of medetomidine, midazolam, and butorphanol (at doses of 0.15, 3.0, and 3.75 mg/kg, respectively). After anesthesia, a laparotomy was performed, and blood samples were collected from the heart. The whole blood was centrifuged, and the upper layer of serum was collected. Serum creatinine and blood urea nitrogen levels were measured using an Automatic Analyzer (Chemray 800, Rayto Co., Ltd., China).
mRNA expression analysis of Col4a5
Four KI and five wild-type male mice aged 28 weeks were chosen. Total RNA was extracted from renal tissues. Following RNA extraction, a reverse transcription reaction and real-time quantitative PCR (qPCR) were performed with the One-Step qRT-PCR SYBR Green Kit (Vazyme, China). This kit employs the SYBR Green I intercalating fluorescent dye method, utilizes gene-specific primers, and integrates reverse transcription and PCR reactions in a single tube, thereby eliminating additional tube opening and pipetting steps. The primers used to detect Col4a5 gene expression in this experiment are as follows: Forward - GTCCACCAGGTACAGAAGGTC; reverse - CTCCTTTCAAACCAGGTAAGCC. The expression levels of the Col4a5 gene were then evaluated. β-actin gene was designated as the internal reference in this investigation, utilizing the subsequent primers: Forward Primer: GTGACGTTGACATCCGTAAAGA; Reverse Primer: GCCGGACTCATCGTACTCC. The relative expression of Col4a5 levels were estimated using the 2^(-ΔΔCt) technique.
Optical microscopy and Electron microscopy analysis
After standard procedures, kidney samples were prepared for examination under optical microscopy, transmission electron microscopy (TEM), and scanning electron microscopy (SEM). The tissues were fixed in 4% paraformaldehyde, embedded in paraffin, and sectioned. Following the instructions provided in the protocol, staining was performed on paraffin sections using hematoxylin and eosin (HE), Periodic acid Schiff (PAS), periodic acid-silver methenamine (PASM), and Masson trichrome (MT). For electron microscopy analysis, the tissue samples were fixed in 2.5% glutaraldehyde and observed under an electron microscope at an accelerating voltage of 15 kV and a pressure of 30 Pa.
Immunohistochemical staining
The kidneys were extracted and fixed in 4% paraformaldehyde for 4 h, followed by immersion in an 18% sucrose solution for more than 16 h. The fixed kidneys were then frozen and sectioned. The following antibodies were used: WT1 (Rabbit monoclonal, Cambridge, UK, ×300 dilution), CD3 (Servicebio, GB13014-50, ×200 dilution), CD68 (Servicebio, GB113109-100, ×200 dilution), and Type IV collagen α5 (Rabbit polyclonal, Cambridge, UK, ×200 dilution). Immunohistochemical staining was conducted following the instructions provided by the manufacturer. To quantify WT1-positive glomeruli in the kidneys, 40 glomeruli were randomly picked from each group of mice and counted to determine the number of WT1-positive cells. The average was determined. The p-value was calculated using an unpaired T-test.
Minigene experiment
In this study, the pcDNA3.1 vector was used to construct pcDNA3.1-COL4A5-WT and pcDNA3.1-COL4A5 minigenes. The NM 000495.4: Exon23:c.1517-1G > T mutation is located in the 22nd intron. Due to the mutation site located within intron 22, we selected a DNA sequence spanning from exon 22 to exon 24, which includes introns, totaling 2060 base pairs in length. The schematic diagram of the minigene experiment can be found in Supplementary Fig. 2A. The wild-type and mutant target gene fragments were obtained through double enzyme digestion and recombinant reactions, and then inserted into the cloning vector. The recombinant products were transformed into competent cells, cultured, and selected for PCR amplification. Sanger sequencing and RT-PCR were performed to confirm the correct insertion of the COL4A5 gene target fragments into the vector. The correctly recombined wild-type and mutant minigene plasmids were selected. The plasmids were then transfected into 293T cells. RNA from receptor cells was extracted using the Trizol method, and the target segment was amplified using RT-PCR. The primer sequences used were pcDNA3.1-F: CTATAGGGAGACCCAAGCT and pcDNA3.1-R: CAACTAGAAGGCACAGTCG. Gel electrophoresis was performed to determine the size of the target fragment. The original gel images involved in this study are all presented in Supplementary Fig. 6.
Statistical analysis
Data analysis was performed using Prism software (version 8, GraphPad Software) with the one-way analysis of variance (ANOVA) method. All results were presented as mean scatter plots, and statistical significance was indicated by *p < 0.05 and **p < 0.01.
Data availability
The sequencing data have been deposited in the NCBI Sequence Read Archive (SRA) with the accession number SRR32281774. The data can be accessed via the following link: https://www.ncbi.nlm.nih.gov/sra/SRR32281774. Other data generated and analyzed during this study are available from the corresponding author upon reasonable request.
References
Gubler, M. et al. Alport’s syndrome. A report of 58 cases and a review of the literature. Am. J. Med. 70, 493–505. https://doi.org/10.1016/0002-9343(81)90571-4 (1981).
Gibson, J. et al. Prevalence estimates of predicted pathogenic COL4A3-COL4A5 variants in a population sequencing database and their implications for Alport syndrome. J. Am. Soc. Nephrol. 32, 2273–2290. https://doi.org/10.1681/ASN.2020071065 (2021).
Pedrosa, A. L. et al. Alport syndrome: A comprehensive review on genetics, pathophysiology, histology, clinical and therapeutic perspectives. Curr. Med. Chem. 28, 5602–5624. https://doi.org/10.2174/0929867328666210108113500 (2021).
Gross, O. et al. Early angiotensin-converting enzyme Inhibition in Alport syndrome delays renal failure and improves life expectancy. Kidney Int. 81, 494–501. https://doi.org/10.1038/ki.2011.407 (2012).
Kashtan, C. E. & Gross, O. Clinical practice recommendations for the diagnosis and management of Alport syndrome in children, adolescents, and young adults-an update for 2020. Pediatr. Nephrol. 36, 711–719. https://doi.org/10.1007/s00467-020-04819-6 (2021).
Feingold, J. et al. Genetic heterogeneity of Alport syndrome. Kidney Int. 27, 672–677. https://doi.org/10.1038/ki.1985.63 (1985).
Barker, D. F. et al. Identification of mutations in the COL4A5 collagen gene in Alport syndrome. Science 248, 1224–1227. https://doi.org/10.1126/science.2349482 (1990).
Kashtan, C. E. Alport syndrome: achieving early diagnosis and treatment. Am. J. Kidney Dis. 77, 272–279. https://doi.org/10.1053/j.ajkd.2020.03.026 (2021).
Nikolaou, S. & Deltas, C. A. Comparative presentation of mouse models that recapitulate most features of Alport syndrome. Genes (Basel). 13. https://doi.org/10.3390/genes13101893 (2022).
Hashikami, K. et al. Establishment of X-linked Alport syndrome model mice with a Col4a5 R471X mutation. Biochem. Biophys. Rep. 17, 81–86. https://doi.org/10.1016/j.bbrep.2018.12.003 (2019).
Rheault, M. N. et al. Mouse model of X-linked Alport syndrome. J. Am. Soc. Nephrol. 15, 1466–1474. https://doi.org/10.1097/01.asn.0000130562.90255.8f (2004).
Wu, W. Q. et al. A mouse model for X-linked Alport syndrome induced by Del-ATGG in the Col4a5 gene. Front. Med. (Lausanne). 10, 1086756. https://doi.org/10.3389/fmed.2023.1086756 (2023).
Tang, Z. et al. [A novel COL4A5 splicing mutation causing Alport syndrome in a Chinese family]. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 25, 649–652 (2008).
Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet. Med. 17, 405–424. https://doi.org/10.1038/gim.2015.30 (2015).
Mastrangelo, A., Madeira, C., Castorina, P., Giani, M. & Montini, G. Heterozygous COL4A3/COL4A4 mutations: the hidden part of the iceberg? Nephrol. Dial Transpl. 37, 2398–2407. https://doi.org/10.1093/ndt/gfab334 (2022).
Fallerini, C. et al. Alport syndrome: impact of digenic inheritance in patients management. Clin. Genet. 92, 34–44. https://doi.org/10.1111/cge.12919 (2017).
Savige, J. et al. Consensus statement on standards and guidelines for the molecular diagnostics of Alport syndrome: refining the ACMG criteria. Eur. J. Hum. Genet. 29, 1186–1197. https://doi.org/10.1038/s41431-021-00858-1 (2021).
Ding, F. et al. Accelerated podocyte detachment and progressive podocyte loss from glomeruli with age in Alport syndrome. Kidney Int. 92, 1515–1525. https://doi.org/10.1016/j.kint.2017.05.017 (2017).
Gyarmati, G. et al. Intravital imaging reveals glomerular capillary distension and endothelial and immune cell activation early in Alport syndrome. JCI Insight. 7 https://doi.org/10.1172/jci.insight.152676 (2022).
Ryu, M. et al. Bacterial CpG-DNA accelerates Alport glomerulosclerosis by inducing an M1 macrophage phenotype and tumor necrosis factor-alpha-mediated podocyte loss. Kidney Int. 79, 189–198. https://doi.org/10.1038/ki.2010.373 (2011).
Savige, J. et al. Ocular features in Alport syndrome: pathogenesis and clinical significance. Clin. J. Am. Soc. Nephrol. 10, 703–709. https://doi.org/10.2215/CJN.10581014 (2015).
Bekheirnia, M. R. et al. Genotype-phenotype correlation in X-linked Alport syndrome. J. Am. Soc. Nephrol. 21, 876–883. https://doi.org/10.1681/ASN.2009070784 (2010).
Hudson, B. G., Tryggvason, K., Sundaramoorthy, M. & Neilson, E. G. Alport’s syndrome, Goodpasture’s syndrome, and type IV collagen. N Engl. J. Med. 348, 2543–2556. https://doi.org/10.1056/NEJMra022296 (2003).
Jais, J. P. et al. X-linked Alport syndrome: natural history in 195 families and genotype- phenotype correlations in males. J. Am. Soc. Nephrol. 11, 649–657. https://doi.org/10.1681/ASN.V114649 (2000).
Jais, J. P. et al. X-linked Alport syndrome: natural history and genotype-phenotype correlations in girls and women belonging to 195 families: a European community Alport syndrome concerted action study. J. Am. Soc. Nephrol. 14, 2603–2610. https://doi.org/10.1097/01.asn.0000090034.71205.74 (2003).
Heidet, L. & Gubler, M. C. The renal lesions of Alport syndrome. J. Am. Soc. Nephrol. 20, 1210–1215. https://doi.org/10.1681/ASN.2008090984 (2009).
Widjaja, A. A. et al. A neutralizing IL-11 antibody improves renal function and increases lifespan in a mouse model of Alport syndrome. J. Am. Soc. Nephrol. 33, 718–730. https://doi.org/10.1681/ASN.2021040577 (2022).
Daga, S. et al. Slowly progressive autosomal dominant Alport syndrome due to COL4A3 splicing variant. Eur. J. Hum. Genet. https://doi.org/10.1038/s41431-024-01706-8 (2024).
Imafuku, A. et al. Autosomal dominant form of type IV collagen nephropathy exists among patients with hereditary nephritis difficult to diagnose clinicopathologically. Nephrol. (Carlton). 23, 940–947. https://doi.org/10.1111/nep.13115 (2018).
Cosgrove, D., Liu, S. & Collagen IV diseases: A focus on the glomerular basement membrane in Alport syndrome. Matrix Biol. 57–58, 45–54. https://doi.org/10.1016/j.matbio.2016.08.005 (2017).
Frank, C. N. et al. Effect of disease progression on the podocyte cell cycle in Alport syndrome. Kidney Int. 101, 106–118. https://doi.org/10.1016/j.kint.2021.08.026 (2022).
Fukuda, R. et al. Podocyte p53 limits the severity of experimental Alport syndrome. J. Am. Soc. Nephrol. 27, 144–157. https://doi.org/10.1681/ASN.2014111109 (2016).
Jedlicka, J. et al. Interstitial inflammation in Alport syndrome. Hum. Pathol. 41, 582–593. https://doi.org/10.1016/j.humpath.2009.08.024 (2010).
Beirowski, B., Weber, M. & Gross, O. Chronic renal failure and shortened lifespan in COL4A3+/- mice: an animal model for thin basement membrane nephropathy. J. Am. Soc. Nephrol. 17, 1986–1994. https://doi.org/10.1681/ASN.2005101044 (2006).
Yamamura, T. et al. Development of an exon skipping therapy for X-linked Alport syndrome with truncating variants in COL4A5. Nat. Commun. 11, 2777. https://doi.org/10.1038/s41467-020-16605-x (2020).
Funding
This research was supported by grants from Science and Technology Program of Guangzhou (Grant No. 202102010222), Guangzhou Women and Children’s Medical Center Clinical Doctor Initiation Scientific Research Fund (Grant No. 2023BS008).
Author information
Authors and Affiliations
Contributions
Study concept and design were performed by Xia Gao. Zhitao Ye and Di Lu designed and performed most experiments. Drafting of the manuscript was performed by Zhitao Ye. Shumin Zhou, Guanyu Li and Lili Long performed some experiments and revision of manuscript. Ming Liu was responsible for data analysis. Jiayi Zhang evaluated histology/pathology of kidney sections. Xia Gao had full access to all the data in the study and takes responsibility for its integrity and the data analysis.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ye, Z., Lu, D., Zhou, S. et al. A novel mouse model for X-linked Alport syndrome induced by splicing mutation in the Col4a5 gene. Sci Rep 15, 17236 (2025). https://doi.org/10.1038/s41598-025-01663-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-01663-2
