
Abstract
Cancer survivors experience many short- and long-term side effects caused by chemotherapy, including low bone mineral density and deterioration of bone microarchitecture. Administration of chemotherapy drugs to disease free mice causes rapid bone loss. However, whether the bone effects persist throughout life and the mechanisms responsible remain unclear. One plausible cause of chemotherapy-induced bone loss is cellular senescence. Here, female mice were administered doxorubicin, cyclophosphamide and docetaxel, a chemotherapy regimen commonly used in breast cancer patients, in combination with two types of drugs that kill senescent cells (senolytics), namely dasatinib + quercetin or piperlongumine. Mice receiving chemotherapy experienced a rapid decrease in trabecular bone mass, which was detectable two weeks after initiation of treatment and was associated with increased expression of senescence markers. None of the senolytics prevented the effects of chemotherapy on bone mass. In separate experiments, we examined the skeletal effects of chemotherapy six and twelve months after the cessation of treatment. The deleterious effects of chemotherapy on bone mass remained up to 12 months after cessation of treatment, while no markers of senescence could be detected in bone. Together, these results suggest that the deleterious effects of this chemotherapy regimen on bone health are not due to the accumulation of senescent cells.
Similar content being viewed by others


Introduction
Advances in cancer treatment, such as chemotherapy, have contributed to a steady rise in cancer survivors over the past 50 years1. As of January 2022, an estimated 18.1 million people are cancer survivors in the United States alone and this number is expected to continue growing1,2. While improving disease-free survival, chemotherapy has been linked to accelerated biological aging and frailty3,4,5,6,7. A decrease in bone mineral density, deterioration of bone microarchitecture, and an increased risk for fractures are common consequences of chemotherapy8,9,10,11,12,13.
After development and growth have ceased, bone is continually remodeled via the work of osteoclasts, giant multinucleated cells specialized in resorbing bone, and osteoblasts which produce bone matrix and replace the bone removed by the osteoclasts. Bone loss occurs when the amount of bone resorbed is larger than the amount of new bone formed14. The loss of bone with chemotherapy has been attributed to both an increase in osteoclasts and a decrease in osteoblasts15,16,17,18,19,20. Chemotherapy administration to breast cancer patients, the most common cancer among women, often causes bone loss21. Cancer incidence increases with aging when women experience a large amount of bone loss due to aging itself combined with loss of estrogens at menopause8,14,21,22,23,24. However, breast cancer also impacts many young women21,24. Because bone regenerates throughout life, it is unclear whether the consequences of chemotherapy during youth remain with aging1,10,13,25,26. Administration of diverse chemotherapy drugs such as doxorubicin, paclitaxel cyclophosphamide, etoposide, cisplatin, or carboplatin to disease-free young mice decreases bone mass15,16,17,18,19,20. Nonetheless, the mechanisms responsible for chemotherapy-induced bone loss remain poorly understood. Chemotherapy drugs increase inflammation, DNA damage, and apoptosis27,28. Some chemotherapy regimens also cause cellular senescence—a process in which cells stop dividing and become resistant to apoptosis29,30. The cell cycle arrest in senescent cells is due to the upregulation of cyclin-dependent kinase (CDK) inhibitors such as Cdkn2a (p16) and Cdkn1a (p21). Senescent cells also secrete cytokines, chemokines, and proteases, known as the senescence-associated secretory phenotype (SASP)31. Chemotherapy increases the expression of SASP factors in various tissues, including bone, of young mice17,29,32,33,34,35,36,37.
Administration of senolytics—drugs that kill senescent cells—along with genetic approaches in mice have been used to evaluate the contribution of cellular senescence to tissue degeneration due to aging and other causes38,39. Several types of senolytics have been described, including the natural product piperlongumine (PL) and the combination of dasatinib (D), a tyrosine kinase inhibitor and quercetin (Q), a naturally occurring flavonoid with diverse biological activities38,39,40. As in other tissues, the number of senescent cells increases in the aged skeleton including bone, joints, and intervertebral discs41,42. Administration of D + Q to aging mice eliminates senescent bone cells, increases bone mass, and attenuates the degeneration of intervertebral discs41,42. D + Q also attenuates the deleterious consequences of chemotherapy in brain, kidney, ovary, and heart35,37,43,44. In addition, genetic ablation of p16-expressing cells has implicated senescence in chemotherapy-induced bone loss17.
A combination of doxorubicin (Adriamycin®), cyclophosphamide (Cytoxan®), and docetaxel (Taxotere®), known as TAC45,46, is commonly administered as a breast cancer treatment. In this study, we examined whether administration of the senolytics D + Q or PL attenuates the effects of TAC on bone mass. We also determined whether the skeletal consequences of chemotherapy remain long after cessation of treatment.
Materials and methods
Ethical approval and statements
Animal studies reported in this manuscript complied with the Animal Research: Reporting of In Vivo Experiments (ARRIVE) guidelines 2.0 (https://arriveguidelines.org/arrive-guidelines) to ensure methodological rigor and reproducibility of methods and findings. All animal procedures (protocol #4178) were reviewed and approved by the Institutional Animal Care and Use Committee of the University of Arkansas for Medical Sciences in compliance with the Public Health Service Policy on Humane Care and Use of Laboratory Animals, the National Institutes of Health Guide for the Care and Use of Laboratory Animals (2011) as well as the American Veterinary Medical Association Animal Euthanasia Guidelines (2020). All the experiments and methods were performed in accordance with the relevant guidelines and regulations.
Drug preparation
Doxorubicin (2 mg/kg; Hikma Pharmaceuticals, London, UK), cyclophosphamide (50 mg/kg; Amneal Pharmaceuticals, Bridgewater, NJ, USA), and docetaxel (8 mg/kg; AuroMedics Pharma LLC, USA) solutions were prepared with sterile physiological saline (0.9% sodium chloride). On the day of injection drugs were diluted and mixed. Dasatinib (5 mg/kg; LC Laboratories, Woburn, MA, USA) and quercetin (50 mg/kg; Sigma-Aldrich, St. Louis, MO, USA) were prepared in 30% polyethylene glycol (PEG 400) on the day of administration. Piperlongumine (Sigma-Aldrich, St. Louis, MO, USA) was prepared in dimethyl sulfoxide (DMSO; Fisher Scientific™) on the day of administration. Doses of chemotherapeutic agents were adapted from Flanigan et al.47 using the body surface area normalization method. D + Q41 and PL48 doses were based on previous studies.
Animal studies
Female C57BL/6 and BALB/cJ mice were obtained from the Jackson Laboratory (Bar Harbor, ME, USA). Mice were housed in specific-pathogen-free conditions in groups of up to five per cage and were given one week to acclimatize to the facility before any experimental procedures. Animal facility was maintained at an ambient temperature of 23–25 °C with 30–70% humidity and a 12-h light–dark cycle. The mice had access to water and standard chow, containing 14% protein and 4% fat ad libitum. Four independent experiments were performed.
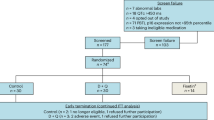
In experiment 1 (Fig. 1), mice were administered a combination of doxorubicin and cyclophosphamide (AC) with or without the senolytic D + Q. Specifically, 16-week-old C57BL/6 mice were randomly distributed into four treatment groups based on spine bone mineral density (BMD) measurements: saline + vehicle, AC + vehicle, saline + DQ, and AC + DQ. BMD was determined in live mice using dual-energy X-ray absorptiometry with a PIXImus densitometer (GE Healthcare Lunar, software v2.0) as previously described49. AC or saline was administered via intraperitoneal (IP) injection once a week for three cycles. D + Q or vehicle solution was administered via oral gavage 24 h after each chemotherapy cycle. Mice were sacrificed one day after the last injection. In experiment 2 (Fig. 3), mice were administered AC, as above, for four cycles, followed by docetaxel once a week for four cycles via IP injection. D + Q or vehicle solution was administered after each AC and T chemotherapy cycle. Weekly administration of D + Q was maintained for an additional five weeks after cessation of chemotherapy. Mice were sacrificed one week after the last D + Q administration. Experiment 3 (Fig. 5) followed a similar experimental protocol to experiment 2, with D + Q being replaced by PL. In experiment 4 (Fig. 7), 19-week-old BALB/cJ mice were administered the TAC regimen, as described in experiment 2, and sacrificed 6 or 12 months after cessation of treatment. All mice were euthanized by exposure to CO2, followed by cervical dislocation. No mice were excluded from the analysis. However, one femur damaged during tissue collection was omitted from Fig. 3b.

D + Q failed to prevent early bone loss caused by AC. (a) Schematic of experimental design created with BioRender.com. (b) Percent change calculated from measurements taken the day before the beginning of treatment and before sacrifice. (c) μCT analysis of trabecular bone volume over tissue volume (BV/TV) and (d) trabecular microarchitecture in fifth lumbar (L5) vertebrae after sacrifice (n = 9/group). (e) Quantitative RT-PCR analysis of mRNA from the sixth lumbar (L6) vertebrae. Bars represent mean ± SD; p-values by 2-way ANOVA with Tukey’s multiple comparisons test. The most relevant comparisons between individual groups are displayed.
Micro-computed tomography (μCT) analysis
Following euthanasia, lumbar vertebrae (L5) and right femora were dissected, cleaned of adherent soft tissues, and fixed overnight in 10% Millonig’s solution (Leica Microsystems, Germany). Bones were then dehydrated in a graded ethanol series (70–100%) and preserved in 100% ethanol until analysis. Micro-CT analysis was performed using a µCT 40 system (Scanco Medical AG, Switzerland) to determine the microarchitecture of the bones, as previously described50,51. 3D voxel images (1024 × 1024 pixels) were acquired using the following scan settings: 12 µm isotropic voxel size, 55 kVp X-ray tube potential, 145 µA X-ray intensity, and 220 ms integration time. Images were processed using Gaussian filtration (sigma = 0.8, support = 1) and a 200 mg/cm3 threshold to segment bone tissue. The thickness of the femoral cortex was measured at the mid-diaphysis (18 slices) and the distal metaphysis (50 slices). Femoral trabecular bone was measured at the distal femur, starting 8–10 slices away from the growth plate and continuing towards the diaphysis (151 slices) to avoid the primary spongiosa. The entire vertebrae were scanned with a transverse orientation and trabecular bone analyses were performed on contours of cross-sectional images, excluding cortical bone and the primary spongiosa. Trabecular bone volume per total bone volume fraction (BV/TV), trabecular thickness, trabecular number, and trabecular separation were calculated using 3D standard microstructural analysis. All nomenclature, symbols, and units follow the American Society for Bone and Mineral Research recommendations52.
Reverse transcription-quantitative polymerase chain reaction (RT-qPCR)
Following euthanasia, the tibiae, right femora, L6 vertebrae, liver, and kidney were dissected. Cortical bone pieces were prepared from the tibiae and femora by removing the periosteum with a scalpel, cutting both bone ends, and flushing the marrow with a 23-gauge needle using sterile PBS. The cortical bone pieces, L6 vertebrae, liver, and kidney were then snap-frozen in liquid nitrogen and stored at − 80 °C for subsequent RNA isolation. Frozen specimens were pulverized with a multi-well tissue pulverizer (BioSpec Products, Inc. USA) followed by total RNA extraction with TRIzol reagent (Thermo Fisher Scientific Inc, USA) according to the manufacturer’s instructions. Complementary DNAs were generated from 1 μg of RNA using the High-Capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific Inc, USA). Real-time qPCRs were performed using TaqMan™ Gene Expression Master Mix and gene specific TaqMan™ (Thermo Fisher Scientific Inc, USA) probe and primer sets in a QuantStudio 3 PCR System (Thermo Fisher Scientific Inc, USA). The TaqMan™ probe and primer sets used were the following: Acp5 (Mm00475698_m1); Alpl (Mm00475831_m1); Bglap (Mm03413826_mH); Ccl2 (Mm00441242_m1); Ctsk (Mm00484039_m1); Cxcl12 (Mm00445553_m1); Cdkn1a (Mm00432448_m1); Cdkn2a (Mm00494449_m1); Cd38 (Mm01220906_m1); Il-1α (Mm99999060_m1); Il-1b (Mm00434228_m1); Il-6 (Mm00446190_m1); Hmgb1 (Mm00849805_gH); Mmp-13 (Mm00439491_m1); Mrsp2 (Mm00475528_m1); and Tnfsf11 (Mm00441908_m1). Transcripts were calculated relative to the housekeeping gene ribosomal protein S2 (Mrps2) and shown as fold change using the 2−∆∆Ct method53.
Statistical analysis and reproducibility
Sample numbers were based on previous studies and were not statistically predetermined. Data visualization and statistical analyses were performed using GraphPad Prism 10.0 software (GraphPad Software, Inc. USA). In every graph, each dot represents one mouse. The normality of data distribution was assessed by D’Agostino-Pearson, Anderson–Darling, Shapiro–Wilk, and Kolmogorov–Smirnov tests. Variance homogeneity was tested using F-test (P > 0.05) for two groups and Spearman’s test for more than two. P values between two normally distributed groups were determined by unpaired two-tailed Student’s t-test. Welch’s correction was applied for unequal variances. An unpaired two-tailed Mann–Whitney test was used for non-normal data. Comparisons between multiple groups were done by 2-way analysis of variance (ANOVA) with Tukey’s post-hoc test or Kruskal–Wallis with Dunn’s correction for normal and non-normal distribution, respectively. A p-value threshold to determine significance was not used as recommended by the guidelines (2022) of the American Statistical Association54. We followed the ARRIVE guidelines 2.0 (https://www.nc3rs.org.uk/arrive-guidelines) for transparent, reproducible, and complete reporting. Raw data is available in Supplementary Data S1.
Results
Chemotherapy causes rapid loss of bone mass associated with markers of cellular senescence
Previous studies have shown that genetic ablation of p16-expressing cells attenuates the loss of bone mass following doxorubicin administration to mice17 implicating cellular senescence as mediator of the bone loss with chemotherapy. To examine whether senolytics could impact the early effects of chemotherapy on bone mass, we administered AC once a week for a total of three administrations in combination with vehicle or D + Q (Fig. 1a). Mice were sacrificed two weeks after initiating the treatments. No significant body weight changes were detected with any of the treatments (Fig. 1b). Micro-CT measurements in vertebrae revealed that AC treatment decreased trabecular bone volume by approximately 20%, using individual group comparisons (Fig. 1c) or 2-way ANOVA (Supplementary Table S1). Trabecular bone parameters such as thickness, number, and separation were not affected by AC treatment (Fig. 1d). However, 2-way ANOVA analysis (Supplementary Table S1) indicated that the overall effect of AC treatment on trabecular bone volume was likely due to a subtle decrease in trabecular number and an increase in trabecular separation. D + Q did not alter the decrease in bone mass caused by AC (Fig. 1c).
The loss of bone mass with chemotherapy has been associated with a decrease in osteoblasts and an increase in osteoclasts15,16,17,18,19. Expression of osteoblast and osteoclast markers mRNA from whole vertebral bone showed that Bglap (osteocalcin) and Alpl (alkaline phosphatase) was reduced by AC, while the osteoclast markers Acp5 (Trap) and Ctsk (cathepsin K) were not impacted (Fig. 1e). D + Q did not alter the decrease in Bglap or Alpl caused by AC. Two-way ANOVA analysis (Supplementary Table S1) indicated that, overall, AC decreased the expression of the osteoblast marker genes, while the osteoclast markers were decreased (Acp5) or unchanged (Ctsk). Previous work shows that loss of bone mass with chemotherapy still occurs in the presence of a bisphosphonate, used to halt bone resorption55, indicating that a decrease in osteoblasts and bone formation are critical mediators of the effects of chemotherapy.
We next examined the mRNA levels of several genes associated with senescence and SASP. These markers were chosen based on compilations of senescence gene sets such as SenMayo56 and previous evidence of their up-regulation in different tissues, including bone, in response to other chemotherapy regimens17,29,32,33,34,35,36. In mRNA from whole lumbar vertebrae, Cdkn1a, Ccl2, and CxCl12 were increased with chemotherapy. D + Q did not attenuate the increase in these markers. The expression of other senescence markers such as Cdkn2a, Il-6, Il-1a, Tnfsf11, and Mmp13 was unchanged, while CD38 was decreased with AC (Fig. 2a). We also examined senescence markers in the cortical portion of long bones. These samples are devoid of the majority of bone marrow and enriched in osteocytes, which are former osteoblasts embedded in the bone matrix. Osteocytes are terminally differentiated, long-lived, and the most abundant cells in bone57. The cortical bones of femur and tibiae also contain other cell types, including osteoblasts, osteoclasts, as well as other mesenchymal, hematopoietic, and endothelial cells58,59. In osteocyte cultures from aged mice, an increase in the expression of Cdkn1a and Cdkn2a was correlated with in situ markers of osteocyte senescence, such as senescence-associated distension of satellites60. AC increased the expression of Cdkn1a, Cdkn2a, Mmp13, and Ccl2. D + Q attenuated the upregulation of Cdkn1a but not the other markers (Fig. 2b). AC had no effect on the other markers of senescence except for a decrease in CD38. D + Q attenuated the inhibition of CD38 by AC.

Senescence markers altered by chemotherapy and D + Q. Quantitative RT-PCR analysis of mRNA from (a) sixth lumbar (L6) vertebrae, and (b) cortical bone from femur and tibia (n = 8–9/group). Bars represent mean ± SD; p-values by 2-way ANOVA with Tukey’s multiple comparisons test. The most relevant comparisons between individual groups are displayed.
D + Q had no effect on the loss of bone mass caused by TAC
We next examined whether D + Q could attenuate the loss of bone mass with chemotherapy at later time points. To this end, mice received the full TAC regimen and the administration of D + Q was continued for five additional weeks after termination of the chemotherapy regimen. Mice were sacrificed 14 weeks after the beginning of the treatment (Fig. 3a). No significant changes were seen in femoral cortical thickness in TAC treated mice (Fig. 3b). TAC decreased trabecular bone volume by approximately 28% in vertebrae (Fig. 3c). Together with the findings described in Fig. 1, this indicates that majority of bone loss with this regimen of chemotherapy occurs during the first 2 weeks of treatment. The loss of trabecular bone volume was due to a decrease in trabecular number associated with an increase in trabecular separation (Fig. 3d). Trabecular thickness was not altered by chemotherapy. D + Q had no effect on cortical or trabecular bone mass in mice treated with vehicle or TAC. Analysis of mRNA from whole vertebral bone revealed that the osteoblast and osteoclast markers were not altered by TAC at the end of the experiment. Furthermore, D + Q had no impact on the markers of bone remodeling (Fig. 3e).

D + Q did not alter the loss of bone mass caused by TAC. (a) Schematic of experimental design created with BioRender.com. Quantitative μCT analysis of (b) cortical thickness at the midshaft (diaphysis) or the distal end (metaphysis) of the femur; and (c) trabecular bone volume over tissue volume (BV/TV) and (d) microarchitecture in fifth lumbar vertebrae. (e) Quantitative RT-PCR analysis of mRNA from the sixth lumbar vertebrae. Bars represent mean ± SD; p-values by 2-way ANOVA with Tukey’s multiple comparisons test. The most relevant comparisons between individual groups are displayed.
No markers of cellular senescence are seen in bone after long-term chemotherapy treatment
We next examined whether the markers of cellular senescence caused by chemotherapy remained or were altered after long-term chemotherapy. Surprisingly, we found that senescence markers in mRNA from L6 vertebrae were unchanged in TAC treated mice compared to saline treated controls (Fig. 4a). Similar findings were obtained when we examined mRNA from cortical bone shafts (Fig. 4b). It has been proposed that senescent cells in some tissues can indirectly impact other tissues via systemic effects61. Because in the long-term treatments, we could not find senescent markers in bone, we examined whether these could be present in other tissues such as liver and kidney, which have been shown to exhibit senescence cells with chemotherapy29,32,33,34,35,36,37. Levels of Cdkn1a were increased in both tissues from mice treated with TAC, with a higher magnitude in liver (Fig. 5a,b). None of the other senescence-related genes examined were impacted. However, examination of the impact of TAC in the liver immediately after the termination of treatment revealed a sevenfold higher expression of Cdkn1a, while a twofold increase was seen for Cdkn2a. Cd38, Il-6, and Hmgb1 also showed modest increases (Fig. 5c). These, along with the results obtained in bone tissue, suggest that the presence of senescent cells with TAC is transient.

Markers of cellular senescence in bone 7 weeks post TAC. (a) Quantitative RT-PCR of cellular senescence markers in cortical bone and (b) whole sixth lumbar vertebrae from mice treated with saline + vehicle and TAC + vehicle of the experiment described in Fig. 3 (n = 5–6/group). Bars represent mean ± SD; p-values by unpaired two-tailed Student’s t-test; with Welch’s t-test for Tnfsf11 (a) and Mann–Whitney test for Il-1a (a).

Markers of cellular senescence in soft tissues following TAC. (a) Quantitative RT-PCR of mRNA isolated from kidney of mice treated with saline + vehicle and TAC + vehicle of the experiment described in Fig. 3, and (b) in livers from mice of all 4 groups. (c) mRNA isolated from livers of mice sacrificed 1 day after the last TAC administration (n = 6/group). Bar graphs represent mean ± SD; p-values by unpaired two-tailed Student’s t-test; with Welch’s correction for Cdkn1a (c) and Cdkn2a (a,c); Mann–Whitney test for Il-1α (a); and two-way ANOVA with Tukey’s test for (b).
Piperlongumine had no effect on the loss of bone caused by TAC
To investigate whether senolytics, other than D + Q, could prevent the bone loss caused by TAC, we administered PL to 16-week-old female mice, similar to the experiment described in Fig. 3. Briefly, PL was administered with each chemotherapy cycle and continued for five additional weeks after termination of the chemo regimen. TAC caused loss of body weight and this effect was attenuated by PL (Fig. 6a). Consistent with the studies above, TAC decreased trabecular bone volume; however, no changes in trabecular microarchitecture could be detected, most likely due to the smaller sample size (Fig. 6b). Nonetheless, 2-way ANOVA analysis indicated that the overall effect of AC treatment on trabecular microarchitecture was similar to the one seen in the previous experiments (Supplementary Table S1). PL did not attenuate the damaging effects caused by TAC on trabecular bone.

Piperlongumine did not alter the loss of bone mass caused by TAC. (a) Body weight of mice during treatment. (b) Quantitative μCT analysis of trabecular bone volume over tissue volume (BV/TV) in the fifth lumbar vertebrae, and (c) trabecular microarchitecture. Bars represent mean ± SD; p-values by two-way ANOVA followed by Tukey’s multiple comparisons test. The most relevant comparisons between individual groups are displayed.
Chemotherapy during adulthood does not accelerate skeletal aging
Chemotherapy causes both short- and long-term side effects. While it is well-established that chemotherapy causes rapid effects on bone mass in young mice15,16,17,18,19,20, it remains unknown whether these effects persist throughout life or whether chemotherapy early in life can exacerbate skeletal aging. Here, we examined bone mass up to 1 year after cessation of TAC administration when mice were 20-month-old. At this age, female C57BL/6 exhibit extensive age-associated bone loss62. To avoid possible confounding effects caused by aging, we used female BALB/cJ mice which are resistant to age-associated bone loss at least up to 20 months of age63,64. Nineteen-week-old mice were administered saline or TAC, as described above (Fig. 3a). Half of the mice were sacrificed after 35 weeks and the other half after 62 weeks of the initiation of treatment (Fig. 7a). As seen in the previous experiments with C57BL/6J, TAC administration had no effect on cortical bone mass of BALB/cJ mice (Fig. 7b). Both the individual group comparisons and the 2-way ANOVA, indicate that TAC caused loss of trabecular bone volume both at the distal femoral metaphysis (Fig. 7b) and vertebrae (Fig. 7c) due to a decrease in trabecular number (Fig. 7d) (Supplementary Table S1). The effects on bone mass were milder compared to the ones seen in C57BL/6J mice. Nonetheless, the magnitude of bone loss due to TAC was similar between the mice examined at the two time points.

Low bone mass persists up to twelve months after cessation of TAC. (a) Schematic of experimental design created with BioRender.com. Quantitative μCT analysis of femur (b) cortical thickness at midshaft (left), and trabecular bone volume over tissue volume (BV/TV) at the distal end (right); (c) fifth lumbar vertebrae BV/TV, and (d) trabecular microarchitecture. Data represents mean ± SD; p-values using two-way ANOVA followed by Tukey’s multiple comparisons test for (b, left) and (d). Kruskal–Wallis with Dunn’s correction for (b, right) and (c). The most relevant comparisons between individual groups are displayed.
We also examined markers of cellular senescence in mRNA collected from the L6 vertebrae, cortical bone of the femur and tibiae, kidney, and liver in mice sacrificed 62 weeks after initiation of treatment. TAC treatment did not alter the expression of the majority of senescence-related genes (Fig. 8a–d), with the exception of an increase in Cd38 in L6 vertebrae and Mmp13 in kidney.

Cellular senescence was not detected up 12 months after cessation of TAC. Quantitative RT-PCR of senescence marker genes in (a) sixth lumbar (L6) vertebrae, (b) femur and tibia cortical bone, (c) kidney, and (d) liver from mice described in Fig. 7a (n = 5/group). Bar graphs represent mean ± SD; p-values by unpaired two-tailed Student’s t-test; with Welch’s correction for Il-6 (d) and Mann–Whitney test for Cdkn2a (b,c), Hmgb1 (c), and Mmp13 (d).
Discussion
To investigate the contribution of cellular senescence to the deleterious effects of chemotherapy on bone mass, we conducted studies employing the TAC regimen, widely used to treat breast cancer38, combined with administration of senolytics such as D + Q or PL38,39,40. Both D + Q and PL have been extensively used to eliminate senescent cells in multiple tissues35,37,43,44,48,65. However, neither D + Q nor PL were effective in antagonizing the effects of chemotherapy on bone. Our findings also revealed that the mechanisms that cause early loss of bone with chemotherapy do not persist long-term.
We show here that senescence markers in both vertebra and long bones were transiently up-regulated early during chemotherapy treatment. Anti-cancer therapies, such as chemotherapy, can cause senescence in both cancer and healthy cells66,67,68,69. These drugs cause DNA damage which, in turn, promotes ATM-CHK2 and ATR-CHK1 kinase-mediated activation of the interconnected p53-RB pathways to cause senescence70,71. For example, the topoisomerase inhibitor doxorubicin, one of the drugs included in the present studies and widely utilized for the treatment of a variety of cancers, dysregulates re-ligation of DNA strands after supercoil unwinding72. This leads to massive DNA damage and increased expression of p53 and its downstream target Cdkn1a (p21), subsequently inducing senescence71. Administration of doxorubicin alone to healthy mice increases markers of senescence in multiple tissues such as skin, lung, liver, heart, and bone. To date, no effects of docetaxel or cyclophosphamide in inducing senescence in non-cancer cells have been described in mice.
We found that D + Q attenuated some markers of senescence in bone. However, the two distinct types of senolytics used in our studies could not protect bone from the deleterious effects of TAC. In contrast, administration of D + Q is equally effective in clearing senescent cells and improving bone mass with aging and radiation-induced bone loss41,73. Furthermore, D + Q has been used extensively in pre-clinical models to show that intermittent treatment attenuates age-related pathologies such as neurodegeneration, hepatic steatosis, insulin resistance, pulmonary fibrosis, and skeletal muscle dysfunction, among others38. While much less is known about effects of PL, administration of this senolytic enhances cognitive function in aging mice65, in a murine model of Alzheimer’s disease47, and in mouse models of cognitive impairment induced by chemotherapy. A plausible explanation for the lack of effect of senolytics is that the increase in senescent cells by TAC is modest and does not constitute the primary cause of bone loss.
In all the experiments performed, we could not detect changes in bone mass in the femoral cortex. Similar to our findings, the vast majority of studies showing effects of chemotherapy in rodents report decreases in trabecular but not cortical bone mass15,16,17,18,19,55,74. The reasons for the different responses of the two bone compartments to chemotherapy remain unclear but are unlikely to be related to cellular senescence. Indeed, genetic ablation of p16 expressing cells or administration of D + Q to old mice increases both cortical and trabecular bone, suggesting that the presence of senescent cells in bone is deleterious to both bone compartments41. A possible explanation might rely on the rapid but transient effects of chemotherapy in suppressing bone formation, causing an early loss of trabecular bone microarchitecture. We show here that most of the bone loss occurs within two weeks of treatment. Likewise, two weeks of administration of cisplatin to mice is sufficient to decrease trabecular bone mass55. This is in line with the well-established findings that chemotherapy causes rapid and severe disruption of bone marrow populations, followed by acute myelopoiesis in both human and mice. These effects are associated with a transient increase in pro-inflammatory cytokine and chemokines which occurs during the first two weeks after drug administration and returns to baseline afterwards20,75. Importantly, the effects of TAC do not seem to accelerate bone loss in aged mice, supporting the idea that long-lived cells in bone, such as osteocytes and osteoblast precursor cells are not overtly impacted.
A caveat to our studies is that up-regulation of cell cycle inhibitors and SASP components do not conclusively define cellular senescence30,76. Nonetheless, up-regulation of Cdkn2a has been associated with bone cell senescence caused by aging60,77,78, radiation17,73, and chemotherapy17. Furthermore, the elimination of senescent bone cells using genetic or pharmacological approaches is always associated with a decrease in the expression of Cdkn2a41,79,80. It remains possible that TAC causes a type of cellular senescence that is characterized by markers distinct from the ones we used. This is, however, unlikely due to previous findings that DNA damage caused by doxorubicin alone or by focal radiation up-regulates Cdkn2a; and that ablation of p16-expressing cells or treatment with D + Q, respectively, attenuates the skeletal effects caused by these agents17,41,73. Elimination of p16-expressing cells also attenuates cardiac toxicity and the decline in physical activity caused by doxorubicin81.
Our results support the idea that cellular senescence is not a major contributor to the loss of bone mass with TAC administration. It is important to note that the apoptotic response to chemotherapy is dominant in many cancers and that most chemotherapies are unable to induce senescence in a significant fraction of cancer cells in vivo27,82. Thus, a decrease in osteoblast number due to apoptosis is a likely explanation for the rapid effect of chemotherapies. In support of this premise is the evidence that many TUNEL-positive cells are present on the bone surface as early as 2 days after administration of 5-FU83. Moreover, mice in which apoptosis is inhibited by genetic ablation of Bax and Bak in cells of the osteoblast lineage have increased trabecular bone mass50. Interestingly, cortical bone is unaffected in these mice indicating that osteoblast/osteocyte apoptosis has a distinct contribution to trabecular versus cortical bone mass homeostasis. Thus, a pro-apoptotic effect in osteoblastic cells due to DNA damage could potentially explain both the decrease in bone formation and the predominant effect of chemotherapy on trabecular bone. Future research with mice in which osteoblastic cell apoptosis is inhibited should elucidate the contribution of cell death to the skeletal effects of chemotherapy.
Data availability
All data generated or analyzed during this study are included in this published article and its supplementary information files.
Abbreviations
- AC:
-
Doxorubicin (Adriamycin®) and cyclophosphamide (Cytoxan®)
- D + Q:
-
Dasatinib + quercetin
- PL:
-
Piperlongumine
- SASP:
-
Senescence-associated secretory phenotype
- TAC:
-
Docetaxel (Taxotere®), doxorubicin (Adriamycin®) and cyclophosphamide (Cytoxan®)
- μCT:
-
Micro-computed tomography
References
Miller, K. D. et al. Cancer treatment and survivorship statistics, 2022. CA Cancer J. Clin. 72, 409–436. https://doi.org/10.3322/caac.21731 (2022).
Tonorezos, E. et al. Prevalence of cancer survivors in the United States. J. Natl. Cancer Inst. https://doi.org/10.1093/jnci/djae135 (2024).
Smitherman, A. B. et al. Accelerated aging among childhood, adolescent, and young adult cancer survivors is evidenced by increased expression of p16(INK4a) and frailty. Cancer 126, 4975–4983. https://doi.org/10.1002/cncr.33112 (2020).
Gilmore, N. et al. The longitudinal relationship between immune cell profiles and frailty in patients with breast cancer receiving chemotherapy. Breast Cancer Res. 23, 19. https://doi.org/10.1186/s13058-021-01388-w (2021).
Rentscher, K. E. et al. Epigenetic aging in older breast cancer survivors and noncancer controls: Preliminary findings from the Thinking and Living with Cancer Study. Cancer 129, 2741–2753. https://doi.org/10.1002/cncr.34818 (2023).
Gehle, S. C. et al. Accelerated epigenetic aging and myopenia in young adult cancer survivors. Cancer Med. 12, 12149–12160. https://doi.org/10.1002/cam4.5908 (2023).
Ahles, T. A. et al. Relationship between cognitive functioning and frailty in older breast cancer survivors. J. Geriatr. Oncol. 13, 27–32. https://doi.org/10.1016/j.jgo.2021.07.011 (2022).
Rees-Punia, E. et al. Fracture risk among older cancer survivors compared with older adults without a history of cancer. JAMA Oncol. 9, 79–87. https://doi.org/10.1001/jamaoncol.2022.5153 (2023).
Chen, Z. et al. Fracture risk among breast cancer survivors: Results from the Women’s Health Initiative Observational Study. Arch. Intern. Med. 165, 552–558. https://doi.org/10.1001/archinte.165.5.552 (2005).
Hadji, P., Ziller, M., Maskow, C., Albert, U. & Kalder, M. The influence of chemotherapy on bone mineral density, quantitative ultrasonometry and bone turnover in pre-menopausal women with breast cancer. Eur. J. Cancer 45, 3205–3212. https://doi.org/10.1016/j.ejca.2009.09.026 (2009).
Oostra, D. R. et al. Association of osteoprotegerin and bone loss after adjuvant chemotherapy in early-stage breast cancer. Mol. Cell Endocrinol. 402, 51–56. https://doi.org/10.1016/j.mce.2014.12.028 (2015).
Go, J. et al. Risk of osteoporosis and fracture in long-term breast cancer survivors. Korean J. Clin. Oncol. 16, 39–45. https://doi.org/10.14216/kjco.20007 (2020).
Vehmanen, L., Elomaa, I., Blomqvist, C. & Saarto, T. Tamoxifen treatment after adjuvant chemotherapy has opposite effects on bone mineral density in premenopausal patients depending on menstrual status. J. Clin. Oncol. 24, 675–680. https://doi.org/10.1200/JCO.2005.02.3515 (2006).
Manolagas, S. C. Birth and death of bone cells: Basic regulatory mechanisms and implications for the pathogenesis and treatment of osteoporosis. Endocr. Rev. 21, 115–137. https://doi.org/10.1210/edrv.21.2.0395 (2000).
Rana, T., Chakrabarti, A., Freeman, M. & Biswas, S. Doxorubicin-mediated bone loss in breast cancer bone metastases is driven by an interplay between oxidative stress and induction of TGFbeta. PLoS ONE 8, e78043. https://doi.org/10.1371/journal.pone.0078043 (2013).
Park, H. J., Yoon, S. Y., Park, J. N., Suh, J. H. & Choi, H. S. Doxorubicin induces bone loss by increasing autophagy through a mitochondrial ROS/TRPML1/TFEB axis in osteoclasts. Antioxidants (Basel). 11. https://doi.org/10.3390/antiox11081476 (2022).
Yao, Z. et al. Therapy-induced senescence drives bone loss. Cancer Res. 80, 1171–1182. https://doi.org/10.1158/0008-5472.CAN-19-2348 (2020).
Koh, A. J., Sinder, B. P., Entezami, P., Nilsson, L. & McCauley, L. K. The skeletal impact of the chemotherapeutic agent etoposide. Osteoporos Int. 28, 2321–2333. https://doi.org/10.1007/s00198-017-4032-1 (2017).
Zhao, D. et al. Cyclophosphamide causes osteoporosis in C57BL/6 male mice: suppressive effects of cyclophosphamide on osteoblastogenesis and osteoclastogenesis. Oncotarget 8, 98163–98183. https://doi.org/10.18632/oncotarget.21000 (2017).
Quach, J. M. et al. Myelosuppressive therapies significantly increase pro-inflammatory cytokines and directly cause bone loss. J. Bone Miner. Res. 30, 886–897. https://doi.org/10.1002/jbmr.2415 (2015).
Siegel, R. L., Miller, K. D., Wagle, N. S. & Jemal, A. Cancer statistics, 2023. CA Cancer J. Clin. 73, 17–48. https://doi.org/10.3322/caac.21763 (2023).
Parfitt, A. M. et al. Relationships between surface, volume, and thickness of iliac trabecular bone in aging and in osteoporosis. Implications for the microanatomic and cellular mechanisms of bone loss. J. Clin. Investig. 72, 1396–1409. https://doi.org/10.1172/JCI111096 (1983).
Khosla, S. Update on estrogens and the skeleton. J. Clin. Endocrinol. Metab. 95, 3569–3577. https://doi.org/10.1210/jc.2010-0856 (2010).
Johnson, R. H., Anders, C. K., Litton, J. K., Ruddy, K. J. & Bleyer, A. Breast cancer in adolescents and young adults. Pediatr. Blood Cancer 65, e27397. https://doi.org/10.1002/pbc.27397 (2018).
Ramin, C. et al. Evaluation of osteopenia and osteoporosis in younger breast cancer survivors compared with cancer-free women: a prospective cohort study. Breast Cancer Res. 20, 134. https://doi.org/10.1186/s13058-018-1061-4 (2018).
Bruning, P. F. et al. Bone mineral density after adjuvant chemotherapy for premenopausal breast cancer. Br. J. Cancer 61, 308–310. https://doi.org/10.1038/bjc.1990.58 (1990).
Kaufmann, S. H. & Earnshaw, W. C. Induction of apoptosis by cancer chemotherapy. Exp. Cell Res. 256, 42–49. https://doi.org/10.1006/excr.2000.4838 (2000).
Maccormick, R. E. Possible acceleration of aging by adjuvant chemotherapy: a cause of early onset frailty?. Med. Hypotheses 67, 212–215. https://doi.org/10.1016/j.mehy.2006.01.045 (2006).
Demaria, M. et al. Cellular senescence promotes adverse effects of chemotherapy and cancer relapse. Cancer Discov. 7, 165–176. https://doi.org/10.1158/2159-8290.CD-16-0241 (2017).
Gorgoulis, V. et al. Cellular senescence: Defining a path forward. Cell 179, 813–827. https://doi.org/10.1016/j.cell.2019.10.005 (2019).
Campisi, J. & di d’Adda Fagagna, F. Cellular senescence: when bad things happen to good cells. Nat. Rev. Mol. Cell Biol. 8, 729–740. https://doi.org/10.1038/nrm2233 (2007).
Englund, D. A. et al. Senotherapeutic drug treatment ameliorates chemotherapy-induced cachexia. JCI Insight https://doi.org/10.1172/jci.insight.169512 (2024).
Perkins, D. W. et al. Therapy-induced normal tissue damage promotes breast cancer metastasis. iScience 27, 108503. https://doi.org/10.1016/j.isci.2023.108503 (2024).
Gao, Y. et al. Increased cellular senescence in doxorubicin-induced murine ovarian injury: Effect of senolytics. Geroscience 45, 1775–1790. https://doi.org/10.1007/s11357-023-00728-2 (2023).
Lerida-Viso, A. et al. Pharmacological senolysis reduces doxorubicin-induced cardiotoxicity and improves cardiac function in mice. Pharmacol. Res. 183, 106356. https://doi.org/10.1016/j.phrs.2022.106356 (2022).
Budamagunta, V. et al. Senolytic treatment alleviates doxorubicin-induced chemobrain. Aging Cell 23, e14037. https://doi.org/10.1111/acel.14037 (2024).
Li, S. et al. Tubular cell senescence promotes maladaptive kidney repair and chronic kidney disease after cisplatin nephrotoxicity. JCI Insight. https://doi.org/10.1172/jci.insight.166643 (2023).
Chaib, S., Tchkonia, T. & Kirkland, J. L. Cellular senescence and senolytics: The path to the clinic. Nat. Med. 28, 1556–1568. https://doi.org/10.1038/s41591-022-01923-y (2022).
Zhang, L., Pitcher, L. E., Prahalad, V., Niedernhofer, L. J. & Robbins, P. D. Targeting cellular senescence with senotherapeutics: Senolytics and senomorphics. FEBS J. 290, 1362–1383. https://doi.org/10.1111/febs.16350 (2023).
Wang, Y. et al. Discovery of piperlongumine as a potential novel lead for the development of senolytic agents. Aging (Albany NY). 8, 2915–2926. https://doi.org/10.18632/aging.101100 (2016).
Farr, J. N. et al. Targeting cellular senescence prevents age-related bone loss in mice. Nat. Med. 23, 1072–1079. https://doi.org/10.1038/nm.4385 (2017).
Novais, E. J. et al. Long-term treatment with senolytic drugs Dasatinib and Quercetin ameliorates age-dependent intervertebral disc degeneration in mice. Nat. Commun. 12, 5213. https://doi.org/10.1038/s41467-021-25453-2 (2021).
Ahire, C. et al. Accelerated cerebromicrovascular senescence contributes to cognitive decline in a mouse model of paclitaxel (Taxol)-induced chemobrain. Aging Cell 22, e13832. https://doi.org/10.1111/acel.13832 (2023).
Du, D. et al. Senotherapy protects against cisplatin-induced ovarian injury by removing senescent cells and alleviating DNA damage. Oxid. Med. Cell Longev. 2022, 9144644. https://doi.org/10.1155/2022/9144644 (2022).
Martin, M. Docetaxel, doxorubicin and cyclophosphamide (the TAC regimen): An effective adjuvant treatment for operable breast cancer. Womens Health (Lond.) 2, 527–537. https://doi.org/10.2217/17455057.2.4.527 (2006).
Jones, S. E. et al. Phase III trial comparing doxorubicin plus cyclophosphamide with docetaxel plus cyclophosphamide as adjuvant therapy for operable breast cancer. J. Clin. Oncol. 24, 5381–5387. https://doi.org/10.1200/JCO.2006.06.5391 (2006).
Flanigan, T. J., Anderson, J. E., Elayan, I., Allen, A. R. & Ferguson, S. A. Effects of cyclophosphamide and/or doxorubicin in a murine model of postchemotherapy cognitive impairment. Toxicol. Sci. 162, 462–474. https://doi.org/10.1093/toxsci/kfx267 (2018).
Ntagwabira, F. et al. Piperlongumine as a neuro-protectant in chemotherapy induced cognitive impairment. Int. J. Mol. Sci. https://doi.org/10.3390/ijms23042008 (2022).
Bartell, S. M. et al. FoxO proteins restrain osteoclastogenesis and bone resorption by attenuating H2O2 accumulation. Nat. Commun. 5, 3773. https://doi.org/10.1038/ncomms4773 (2014).
Jilka, R. L. et al. Dysapoptosis of osteoblasts and osteocytes increases cancellous bone formation but exaggerates cortical porosity with age. J. Bone Miner. Res. 29, 103–117. https://doi.org/10.1002/jbmr.2007 (2014).
Warren, A. et al. The NAD salvage pathway in mesenchymal cells is indispensable for skeletal development in mice. Nat. Commun. 14, 3616. https://doi.org/10.1038/s41467-023-39392-7 (2023).
Dempster, D. W. et al. Standardized nomenclature, symbols, and units for bone histomorphometry: a 2012 update of the report of the ASBMR Histomorphometry Nomenclature Committee. J. Bone Miner. Res. 28, 2–17. https://doi.org/10.1002/jbmr.1805 (2013).
Livak, K. J. & Schmittgen, T. D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25, 402–408. https://doi.org/10.1006/meth.2001.1262 (2001).
Amrhein, V., Greenland, S. & McShane, B. Scientists rise up against statistical significance. Nature 567, 305–307. https://doi.org/10.1038/d41586-019-00857-9 (2019).
Essex, A. L. et al. Bisphosphonate treatment ameliorates chemotherapy-induced bone and muscle abnormalities in young mice. Front. Endocrinol. (Lausanne) 10, 809. https://doi.org/10.3389/fendo.2019.00809 (2019).
Saul, D. et al. A new gene set identifies senescent cells and predicts senescence-associated pathways across tissues. Nat. Commun. 13, 4827. https://doi.org/10.1038/s41467-022-32552-1 (2022).
Jilka, R. L. & O’Brien, C. A. The role of osteocytes in age-related bone loss. Curr. Osteoporos. Rep. 14, 16–25. https://doi.org/10.1007/s11914-016-0297-0 (2016).
Nookaew, I. et al. Refining the identity of mesenchymal cell types associated with murine periosteal and endosteal bone. J. Biol. Chem. 300, 107158. https://doi.org/10.1016/j.jbc.2024.107158 (2024).
Zhong, L. et al. Single cell transcriptomics identifies a unique adipose lineage cell population that regulates bone marrow environment. Elife https://doi.org/10.7554/eLife.54695 (2020).
Farr, J. N. et al. Identification of senescent cells in the bone microenvironment. J. Bone Miner. Res. 31, 1920–1929. https://doi.org/10.1002/jbmr.2892 (2016).
Xu, M. et al. Senolytics improve physical function and increase lifespan in old age. Nat. Med. 24, 1246–1256. https://doi.org/10.1038/s41591-018-0092-9 (2018).
Almeida, M. et al. Skeletal involution by age-associated oxidative stress and its acceleration by loss of sex steroids. J. Biol. Chem. 282, 27285–27297. https://doi.org/10.1074/jbc.M702810200 (2007).
Harris, D., Garrett, K., Uppuganti, S., Creecy, A. & Nyman, J. S. The BALB/c mouse as a preclinical model of the age-related deterioration in the lumbar vertebra. Bone 137, 115438. https://doi.org/10.1016/j.bone.2020.115438 (2020).
Willinghamm, M. D. et al. Age-related changes in bone structure and strength in female and male BALB/c mice. Calcif. Tissue Int. 86, 470–483. https://doi.org/10.1007/s00223-010-9359-y (2010).
Go, J. et al. Piperlongumine decreases cognitive impairment and improves hippocampal function in aged mice. Int. J. Mol. Med. 42, 1875–1884. https://doi.org/10.3892/ijmm.2018.3782 (2018).
Schmitt, C. A. et al. A senescence program controlled by p53 and p16INK4a contributes to the outcome of cancer therapy. Cell 109, 335–346. https://doi.org/10.1016/s0092-8674(02)00734-1 (2002).
Chang, B. D. et al. Role of p53 and p21waf1/cip1 in senescence-like terminal proliferation arrest induced in human tumor cells by chemotherapeutic drugs. Oncogene 18, 4808–4818. https://doi.org/10.1038/sj.onc.1203078 (1999).
Wang, X. et al. Evidence of cisplatin-induced senescent-like growth arrest in nasopharyngeal carcinoma cells. Cancer Res. 58, 5019–5022 (1998).
Prasanna, P. G. et al. Therapy-induced senescence: Opportunities to improve anticancer therapy. J. Natl. Cancer Inst. 113, 1285–1298. https://doi.org/10.1093/jnci/djab064 (2021).
Marechal, A. & Zou, L. DNA damage sensing by the ATM and ATR kinases. Cold Spring Harb. Perspect. Biol. https://doi.org/10.1101/cshperspect.a012716 (2013).
Abuetabh, Y. et al. DNA damage response revisited: the p53 family and its regulators provide endless cancer therapy opportunities. Exp. Mol. Med. 54, 1658–1669. https://doi.org/10.1038/s12276-022-00863-4 (2022).
Nitiss, J. L. Targeting DNA topoisomerase II in cancer chemotherapy. Nat. Rev. Cancer 9, 338–350. https://doi.org/10.1038/nrc2607 (2009).
Chandra, A. et al. Targeted reduction of senescent cell burden alleviates focal radiotherapy-related bone loss. J. Bone Miner. Res. 35, 1119–1131. https://doi.org/10.1002/jbmr.3978 (2020).
Hain, B. A., Xu, H., Wilcox, J. R., Mutua, D. & Waning, D. L. Chemotherapy-induced loss of bone and muscle mass in a mouse model of breast cancer bone metastases and cachexia. JCSM Rapid Commun. 2 (2019).
Mitchell, C. A. et al. Stromal niche inflammation mediated by IL-1 signalling is a targetable driver of haematopoietic ageing. Nat. Cell Biol. 25, 30–41. https://doi.org/10.1038/s41556-022-01053-0 (2023).
Cohn, R. L., Gasek, N. S., Kuchel, G. A. & Xu, M. The heterogeneity of cellular senescence: insights at the single-cell level. Trends Cell Biol. 33, 9–17. https://doi.org/10.1016/j.tcb.2022.04.011 (2023).
Kim, H. N. et al. DNA damage and senescence in osteoprogenitors expressing Osx1 may cause their decrease with age. Aging Cell 16, 693–703. https://doi.org/10.1111/acel.12597 (2017).
Piemontese, M. et al. Old age causes de novo intracortical bone remodeling and porosity in mice. JCI Insight https://doi.org/10.1172/jci.insight.93771 (2017).
Kim, H. N. et al. Osteocyte RANKL is required for cortical bone loss with age and is induced by senescence. JCI Insight https://doi.org/10.1172/jci.insight.138815 (2020).
Kim, H. N. et al. Elimination of senescent osteoclast progenitors has no effect on the age-associated loss of bone mass in mice. Aging Cell 18, e12923. https://doi.org/10.1111/acel.12923 (2019).
Zumerle, S. et al. Targeting senescence induced by age or chemotherapy with a polyphenol-rich natural extract improves longevity and healthspan in mice. Nat. Aging https://doi.org/10.1038/s43587-024-00663-7 (2024).
Ewald, J. A., Desotelle, J. A., Wilding, G. & Jarrard, D. F. Therapy-induced senescence in cancer. J. Natl. Cancer Inst. 102, 1536–1546. https://doi.org/10.1093/jnci/djq364 (2010).
Xian, C. J., Howarth, G. S., Cool, J. C. & Foster, B. K. Effects of acute 5-fluorouracil chemotherapy and insulin-like growth factor-I pretreatment on growth plate cartilage and metaphyseal bone in rats. Bone 35, 739–749. https://doi.org/10.1016/j.bone.2004.04.027 (2004).
Acknowledgements
We thank Olivia Reyes-Castro and Kimberly Richardson for tissue collection and Stuart Berryhill for support with μCT analysis. This work was supported by the U.S. National Institutes of Health (R01 AG068449, R01 AR56679, R01 CA258673, and P20 GM125503) and the UAMS Bone and Joint Initiative.
Author information
Authors and Affiliations
Contributions
A.R.A. conceived and designed the experiments; P.S., M.M.A., T.M., L.B.G. and A.W. performed all animal experimentation; M.M.A. and A.R.C. generated data; M.M.A. and M.A. analyzed data; A.R.A. and M.A. provided funding; M.M.A. and M.A. wrote the manuscript; all authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ali, M.M., Simmons, P., Warren, A. et al. The adverse effects of chemotherapy on bone mass are not prevented by senolytics. Sci Rep 15, 17279 (2025). https://doi.org/10.1038/s41598-025-01717-5
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-01717-5
This article is cited by
-
Chemotherapy-induced adipo-lineage cell senescence drives bone loss
Nature Communications (2025)
