
Abstract
The oriental fruit fly Bactrocera dorsalis (Hendel) is considered as a quarantine pest in many countries and regions. Challenges remain in distinguishing this species with morphological similarities, especially in relevant development stages. In recent years, CRISPR/Cas12b genetic diagnostics has seen rapid advancements and offers an efficient tool for the identification of pathogens, viruses, and other genetic targets. Here we developed a new and rapid visual detection assay of B. dorsalis using recombinase polymerase amplification (RPA) and the CRISPR/Cas12b system. The system can detect different developmental stages of B. dorsalis within 30–35 min at 43 ℃ and the results are easily observed by the naked eye based on the color change in the tube during the reaction. The specificity and high sensitivity of this method was demonstrated, allowing for detection from 3.2 pg µL− 1 of DNA. With crude DNA, this diagnostic system successfully identified B. dorsalis by holding the reaction tubes in the hand. Our study demonstrates that RPA-CRISPR/Cas12b visualization system is effective to detect B. dorsalis rapidly and accurately. This approach can be applied for monitoring and identification of other pests in border and relevant locations, preventing biological invasions and ensuring pest control.
Similar content being viewed by others


Introduction
The oriental fruit fly Bactrocera dorsalis (Hendel), originally from tropical and subtropical regions of Asia, is considered as a quarantine pest in many countries and regions due to its potential to cause significant harm in fresh fruits1. At present, B. dorsalis is gradually spreading and has been found in 124 regions across 75 countries in Asia, Africa, and North America2. However, challenges remain in distinguishing this species with morphological similarities, especially in relevant development stages. As a result, further research and development is required to obtain highly effective and precise molecular identification techniques of B. dorsalis.
Bactrocera is a type of fruit fly belonging to the family Tephritidae, subfamily Dacinae, and tribe Dacini. This genus is considered a quarantine pest in China because of its varied feeding habits, high tolerance, strong fertility, and quick adaptation to new environments. These traits enable the fly to settle and cause significant damage to the local agricultural economy3,4. Bactrocera dorsalis, known as the oriental fruit fly, has spread to several countries including India, the Philippines, Thailand, the United States, and Australia. The population of B. dorsalis is increasing in South China, and its range is expanding toward the north5. Therefore, rapid and effective detection techniques are needed for use at quarantine ports to prevent and control the spread of B. dorsalis.
When fruits are imported into China, they are inspected on site. Although fly samples are often found during inspections, they can be difficult to identify because the larval and pupal stages are difficult to distinguish visually. To accurately identify flies in these stages, they must be raised in a laboratory until they become adults. Even then, it can be challenging to differentiate between sympatric species with similar adult forms. Molecular identification techniques have evolved rapidly, resulting in improved detection accuracy and efficiency6. Various DNA-based detection and identification methods are available, including traditional polymerase chain reaction (PCR) assay7, reverse transcription-PCR (RT-PCR)8, loop-mediated isothermal amplification (LAMP) assay9,10,11, and recombinase polymerase amplification (RPA). PCR is highly sensitive but difficult to operate and requires large equipment, making it unsuitable for on-site detection12. While the LAMP method is easier to operate than PCR, the primer design can be more complex13, and the reaction temperature is higher14. Therefore, further research and development is required to obtain highly effective and precise molecular identification techniques.
The CRISPR/Cas system has gained attention as a diagnostic tool due to its gene-targeting capabilities. Combining this system with isothermal amplification offers three advantages: increased number of target sequences, improved assay sensitivity, and reduced assay run time and complexity15,16. The low by-product production in CRISPR systems during identification also results in improved specificity17. Moreover, the simple reading device makes this technique user friendly and appropriate for point-of-care testing (POCT)18.
RPA detection has gained popularity as an isothermal amplification technique. This method offers advantages including simple operation, fast amplification, and a mild reaction temperature (37–42 °C)19, making it highly suitable for on-site detection. Based on advances in sample treatment, amplification, and result detection systems, RPA has become increasingly prevalent in molecular diagnosis for insects20, bacteria21, fungi22, parasites23, and viruses24 including SARS-CoV-225. The CRISPR/Cas system is made up of two core components: the enzyme Cas and guide RNA (gRNA). This system can cut target DNA or RNA based on the sequence of the gRNA, making it attractive for genetic engineering. Some Cas proteins such as Cas12 and Cas13 exhibit trans-cleavage activity in addition to targeted binding and cleavage. The CRISPR/Cas system performs numerous functions, including cutting the surrounding single-strand DNA or RNA on Cas-gRNA complexes26. The specific binding of CRISPR/Cas system allows it to play a role in phytosanitary, animal quarantine and sanitary quarantine. Cas12b is a dual RNA-guided nuclease that recognizes simple PAM sequences (5’-TTN-3’) and contains a single RuvC structural domain27,28. Cas12b requires two types of RNA molecules, crRNA and tracrRNA, which provides Cas12b with a wider range of genomic targets while minimizing the risk of off-target effects29. Due to its precision and low risk of error, Cas12b is considered a safe option for therapeutic and clinical applications. Cas12b is commonly used for genome editing in plants30 and mammals31. Cas12b has also been combined with LAMP assay to construct detection systems (e.g., HOLMESv232, TB-QUICK33, and STOPCovid34 and detect human infectious diseases35,36. In addition, Cas12b was combined with reverse transcription-recombinase aided amplification (RT-RAA) to construct the CDetection.v2 detection system for SARS-CoV-2 detection37. However, Cas12b has not yet been used for insect detection.
The aim of this study was to create a rapid and accurate method for identifying B. dorsalis through nucleic acid detection. This was accomplished by designing and testing B. dorsalis-specific RPA primers along with gRNA. A reaction system based on RPA-CRISPR/Cas12b was then established. Reagents were used to obtain a visible color change during the reaction, and the specificity and sensitivity of the system were explored through cross-reactions with sympatric species of B. dorsalis as well as genome gradient dilution. The system was designed to meet the needs of rapid on-site identification while protecting the domestic agricultural economy and facilitating international trade.
Methods
Samples collection
Fruit fly specimens of six fruit fly species were collected from Vietnam and China (Guangdong province, Guangxi Zhuang Autonomous Region, Yunnan province, Zhejiang province, Hainan province), including 7 geographical strains of B. dorsalis, 2 geographical strains of Zeugodacus scutellata and one geographical strain each of Bactrocera correcta, Zeugodacus cucurbitae, Zeugodacus tau and Bactrocera rubiginus (Table S1). All these fly samples were used in specificity test of detection for B. dorsalis based on RPA-CRISPR/Cas12b.
In order to ensure the reliability of RPA/CRISPR visual detection, four insect stages (eggs, larvae, pupae, and adults) were used in effectiveness test of detection for B. dorsalis. The samples in this study were provided by Chinese Academy of Inspection and Quarantine. B. dorsalis was collected from Guangzhou, Guangdong Province and reared for 18 generations in the laboratory (Table S2). The rearing conditions were as follows: a light-dark period of 14 h light and 10 h dark; temperature = 25℃±2℃; and relative humidity = 60-70%.
DNA extraction
Silica spin column-based DNA extraction
The DNA of the experimental fruit flies was extracted using Magen D3129 HiPure Insect DNA Kits (Guangzhou Magen Biotechnology Co., Ltd.), thereby obtaining high-purity DNA. The larval fly samples were lysed and digested with Buffer ITL and protease, releasing DNA into the lysate. After adding Buffer IL and ethanol and transferring the mixture to a filtration column, the DNA was adsorbed onto the membrane of the column; the unadsorbed proteins were removed by filtration. The column was washed with Buffer GW1 to remove proteins and other impurities followed by washing with Buffer GW2 to remove salts. Finally, the DNA was eluted using a low-salt buffer (10 mM Tris, pH 8.5) to obtain the sample genes. The entire process took between 40 min and 1 h for a single sample.
Rapid on-site DNA extraction
Furthermore, DNA rapid extraction kits (Beijing Synsortech Co., Ltd.) and JXMF-06 grinder (Shanghai Jingxin Industrial Development Co., Ltd.) were used for samples’ on-site DNA extraction, thereby obtaining crude DNA. The samples were ground in a lysis solution, rinsed in a washing solution, and transferred to an elution solution to obtain the sample genes. The total time for a single sample was 50–200 s.
RPA primer and gRNA design
The mitochondrial sequence DQ116269 of B. dorsalis was obtained from NCBI (https://www.ncbi.nlm.nih.gov/). Specificity was determined by comparison using the BLAST available from NCBI (https://blast.ncbi.nlm.nih.gov/Blast.cgi). The gRNA sequences were designed using localized CRISPOR tool38, and RPA primers were designed using Oligo_v7.6539(Figure S1). The optimal length of Cas12b-recognized target DNA is 23 bp, with the TTN PAM sequence positioned upstream. The RPA primers were selected based on the DNA sequences recognized by the gRNA. The amplified DNA sequences were approximately 100–200 bp in length and required primers of approximately 30 nt (Fig. S2). They were designed with a GC content of 40–60%, and avoid secondary structures as much as possible. Five gRNAs, two RPA-Fs, and three RPA-Rs were screened based on their scores from highest to lowest.
We selected specific mitochondrial COI gene sequences of 10 B. dorsalis sympatric species (Z. cucurbitae, Trypeta fujianica, Bactrocera proprediaphora, Ceratitis rubivora, Ceratitis rosa, B. correcta, Ceratitis capitata, Bactrocera incisa, Bactrocera ruiliensis, and Z. tau) from NCBI (Table S3) for preliminary determination of RPA primer and sgRNA.
Recombinant plasmids containing the mitochondrial COI gene from 10 sympatric species were obtained from Beijing Tsingke Biotech Co., Ltd. The gRNAs and RPA primers were ordered from GeneScript and Sangon Biotech, respectively. The specificities of the gRNA and RPA primers were evaluated by qPCR fluorescence quantification experiments, and the most effective gRNA and RPA primers were selected.
RPA/CRISPR reaction
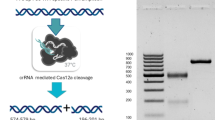
The RPA/CRISPR reaction system uses a two-step process (Fig. 1). First, the extracted sample DNA is added to the RPA system to rapidly amplify the DNA. Second, the CRISPR system is added to the amplified system to observe the reaction, with positive samples becoming green. For negative controls, ddH2O is added to both the RPA and CRISPR reaction systems, resulting in a pink reaction color.

RPA-CRISPR/Cas12b visual detection protocol of B. dorsalis: (1) extraction of DNA from fly samples (four stages); (2) RPA reaction of extracted DNA; (3) CRISPR reaction of ssDNA reporter and RPA products in the Cas12b-gRNA complex; and (4) naked eye observation of color changes in the tubes.
The RPA system utilizes a DNA/RNA constant-temperature rapid amplification kit (Beijing Synsortech Co., Ltd.). The RPA system was operated with the following components: 12 µL amplification buffer; 1 µL Mg (CH3COO)2; 2 µL upstream and downstream primers (10 µM); and 1.6 µL nucleic acid template. The reaction was performed using a slan-96P qPCR instrument (Shanghai Hongshi Medical Technology Co., Ltd.) at 43℃ for 20 min. The CRISPR reaction system used an AaCas12b protein kit (Beijing Synsortech Co., Ltd.). The volume of each CRISPR system was 10 µL, and the specific components were as follows: 1 µL AaCas12b (50 ng µL− 1); 0.5 µL sgRNA (250 ng µL− 1); 0.33 µL FAM-BHQ1 (100 µM); 1 µL 10x AaCas12b Buffer; and 7.17 µL ddH2O. The reaction was performed in a slan-96P qPCR instrument (Shanghai Hongshi Medical Technology Co., Ltd.) at 43 °C for 30 min.
Optimization of the RPA/CRISPR visual detection for Oriental fruit fly
Based on the original RPA/CRISPR reaction system, an optimization was performed to visualize the detection in the test tube. The components of the RPA reaction system remained unchanged, while the probe concentration in the CRISPR system increased. In this experiment, the high-purity DNA extracted from the larva of B. dorsalis was used to conduct three replicate experiments (Table S4). The final system consisted of 1 µL AaCas12b (50 ng µL− 1), 0.5 µL sgRNA (250 ng µL− 1), 1.65 µL FAM-BHQ1 (100 µM), 1 µL 10x AaCas12b buffer, and 5.85 µL ddH2O.
To decrease the overall reaction time, we optimized the RPA and CRISPR reactions and conducted control experiments. The RPA reaction was optimized for 10, 15, and 20 min, while the CRISPR reaction was optimized for 15, 20, 25, and 30 min.
Specificity and sensitivity of visual detection test for Oriental fruit fly
To evaluate the specificity of the RPA-CRISPR/Cas12b system, we selected five sympatric species of flies (Table S4) for cross-reactivity (Z. cucurbitae, B. correcta, Z. tau, Z. scutellata, and B. rubiginus), the four insect stages from three species (Z. cucurbitae, Z. tau, and B. correcta) and ten types of recombinant plasmids with COI gene inserts from sympatric species (Z. cucurbitae, T. fujianica, B. proprediaphora, C. rubivora, C. rosa, B. correcta, C. capitata, B. incisa, B. ruiliensis, and Z. tau). For each species of flies, three specimens were selected to conduct the replicate experiments.
To evaluate the sensitivity of the RPA-CRISPR/Cas12b system, we diluted a 10 ng µL− 1 B. dorsalis genome with ddH2O in a 1:5 ratio gradient to determine the lowest detectable concentration of B. dorsalis (Table S4). In this experiment, the high-purity DNA extracted from the larva of B. dorsalis was used to conduct six replicate experiments.
Effectiveness test of visual detection for fruit fly in different stages
In this experiment, we validated the ability of the system to detect different geographical strains and different developmental stages of flies using crude DNA (Table S4). The development process of flies includes four insect stages: egg, larva, pupa, and adult. Three samples were selected for each geographical strain and each insect stage to conduct replicate experiments.
Results
Preliminary determination of RPA primers and gRNA
The gRNA sequences were designed using the localized CRISPOR tool, while the RPA primers were designed using Oligo 7 software. Through qPCR fluorescence quantification experiments, five gRNAs, two RPA-Fs and three RPA-Rs (Table S5) that were screened by software. They were used to further screen for the most efficient gRNA and primers, and then specificity tests were carried out on them. As shown in Fig. 2, gRNA3 was the most effective gRNA (Fig. 2a), and the F1R1 combination was the most effective primers (Fig. 2b). The screened gRNA and RPA primers successfully detected B. dorsalis, which produced fluorescent signals, whereas the other sympatric species did not. Moreover, the specificity of the gRNA and RPA primers was confirmed (Fig. 2c). The final sequences are shown in Table 1.

Validation process for gRNA and RPA primers. (a) Fluorescence quantification results for gRNA screening. (b) Fluorescence quantification results for RPA primer screening. (c) Specificity results for gRNA and RPA primers.
Optimization of the RPA-CRISPR/Cas12b visual detection system
The concentration of the probe in the CRISPR system was optimized to enable observation of the reaction results with the naked eye (Fig. 3a). The CRISPR system volume was kept constant at 10 µL, while the volume of the probe at a concentration of 100 µM was gradually increased to 0.66 µL, 0.99 µL, 1.32 µL, and finally 1.65 µL. The green color of the system deepened as the probe concentration increased. The most significant green color was observed at a probe concentration of 1.65 µL. The negative control appeared pink in color. Fluorescence quantification results showed that the fluorescence value of the system at a reporter volume of 1.65 µL was 2.9 times greater than that of 1.32 µL, exhibiting a statistically significant difference (p < 0.001) (Fig. 3b). Based on the results, subsequent experiments were carried out using a probe concentration of 1.65 µL.

Optimization and validation of ssDNA reporter volume for visual detection: (a) A, B, and C are three groups of replicate experiments. 1, 0.66 µL; 2, 0.99 µL; 3, 1.32 µL; 4, 1.65 µL; and 5, negative control. (b) Fluorescence quantification results for different reporter volumes.
To reduce the overall reaction time, we optimized the RPA and CRISPR reactions by conducting three sets of replicate experiments (Fig. 4). By comparing the results of the RPA reaction at 10, 15, and 20 min while keeping the CRISPR reaction unchanged at 30 min (Fig. 4a), we found that the reaction system became green at 10 min, and the color did not deepen with further increasing RPA reaction time. However, the negative control remained pink. Therefore, an RPA reaction time of 10 min was used for subsequent RPA experiments.
While maintaining the RPA reaction time of 10 min, we conducted three replicate experiments with different CRISPR reaction times of 15, 20, 25, and 30 min (Fig. 4b). We observed a slight color change in the tubes at 15 min, although it was not very noticeable. At 20 min, the green color of the system was pronounced. The negative control remained pink. The color change became increasingly apparent as time progressed. To ensure a short reaction time along with a distinct color change, we selected the CRISPR reaction time of 20 min for subsequent experiments.

Optimization of the RPA and CRISPR reaction times for visual detection. (a) Optimization of the RPA reaction time at a CRISPR reaction time of 30 min: 1–3, 10 min; 5–7, 15 min; 9–11, 20 min; and 4, 8, and 12, negative controls. (b) Optimization of the CRISPR reaction time at an RPA reaction time of 10 min: 1–3, 15 min; 5–7, 20 min; 9–11, 25 min; 13–15, 30 min; and 4, 8, 12, and 16, negative controls.
Sensitivity test of the RPA-CRISPR/Cas12b detection system
To test the sensitivity of the system, we diluted 10 ng µL− 1 of the genome of B. dorsalis flies with ddH2O in a 1:5 gradient with concentrations ranging from 10 ng µL− 1 to 0.64 pg µL− 1 (Fig. 5a). The colors of the systems at concentrations of 10 ng µL− 1, 2 ng µL− 1, 0.4 ng µL− 1, 0.08 ng µL− 1, 0.016 ng µL− 1, and 3.2 pg µL− 1 were green. In six replicates of the system with a concentration of 0.64 pg µL− 1, only D7 was green, the other five were light pink. The results indicate that the lowest detectable concentration in the system was 3.2 pg µL− 1. Fluorescence quantification results showed that The fluorescence value of 3.2 pg µL− 1 was found to be significantly different from that of the NTC (p < 0.001), while the fluorescence value of 0.64 pg µL− 1 was not found to be significantly different from that of the NTC, which was consistent with the results of those in vitro visualizations (Fig. 5b).

Sensitivity evaluation of the RPA-CRISPR/Cas12b visual detection system: (a) A, B, C, D, E and F are six groups of replicate experiments. 1, 10 ng µL− 1; 2, 2 ng µL− 1; 3, 0.4 ng µL− 1; 4, 0.08 ng µL− 1; 5, 0.016 ng µL− 1; 6, 3.2 pg µL− 1; 7, 0.64 pg µL− 1; and 8, negative controls. (b) Fluorescence quantification results for different gene concentration.
Specificity test of visual detection for Oriental fruit fly
The RPA-CRISPR system was used to detect adult B. dorsalis samples collected from various geographic regions. As shown in Fig. 6a, all B. dorsalis systems appeared green in color, while the negative control was pink. Fluorescence quantification results were consistent with those of in vitro visualizations (Fig. 6b). The results demonstrate that geographical differences did not affect the accuracy of B. dorsalis detection.

Cas12b detection of B. dorsalis in samples obtained from various geographic regions: (a) A, B, and C are three groups of replicate experiments.1, Guangzhou, Guangdong; 2, Nanning, Guangxi; 3, Dongxing, Guangxi; 4, Jinghong, Yunnan; 5, Hangzhou, Zhejiang; 6, Haikou, Hainan; 7, Vietnam; and 8, negative controls. (b) Fluorescence quantification results for different geographic regions.
To evaluate the specificity of this system, we detected fly samples of five sympatric species of B. dorsalis and recombinant plasmids with COI gene inserts of 10 sympatric species using the developed RPA/CRISPR system (Fig. 7). B. correcta, Z. cucurbitae, and Z. tau were tested in four insect stages: egg, larva, pupa, and adult (Fig. 7a). Z. cucurbitae, T. fujianica, B. proprediaphora, C. rubivora, C. rosa, B. correcta, C. capitata, B. incisa, B. ruiliensis, and Z. tau were tested as recombinant plasmids (Fig. 7b). B. correcta, Z. cucurbitae, Z. tau, Z. scutellata and B. rubiginus were tested as adults (Fig. 7c). The fluorescence quantification results of Fig. 7b showed that B. dorsalis was significantly different from NTC, which is consistent with the results of in vitro visualization (Fig. 7d). All reaction systems for the sympatric species and negative control were pink in color, indicating no cross-reactivity and confirming the system’s specificity.

Specificity evaluation of RPA-CRISPR/Cas12b-based visual detection. (a) present samples: A, B, and C are three groups of replicate experiments. 1–4, B. correcta; 5–8, Z. cucurbitae; 9–12, Z. tau; 13, negative control and 14, B. dorsalis. 1, 5 and 9, eggs; 2, 6, 10 and 14, larvae; 3, 7 and 11, pupae; 4, 8 and 12, adults. (b) Recombinant plasmids: A, B, and C are three groups of replicate experiments. 1, Z. cucurbitae; 2, T. fujianica; 3, B. proprediaphora; 4, C. rubivora; 5, C. rosa; 6, B. correcta; 7, C. capitata; 8, B. incisa; 9, B. ruiliensis; 10, Z. tau; 11, negative control and 12, B. dorsalis. (c) Present samples: A, B, and C are three groups of replicate experiments. 1, Z. cucurbitae; 2–3, Z. tau; 4, B. correcta; 5–6, Z. scutellata; 7, B. rubiginus and 8, negative control. (d) Fluorescence quantification results for Fig. 7b.
To evaluate the detectability of the four stages during the development of B. dorsalis, experimental validation was conducted on eggs, larvae, pupae, and adults (Fig. 8). The DNA concentrations detected were as follows: eggs (1.55 ng µL− 1), larvae (34.4 ng µL− 1), pupae (43.2 ng µL− 1), and adults’ abdomen (29.25 ng µL− 1). Figure 8a shows the system’s ability to detect all the four insect stages of B. dorsalis. The reaction systems for all insect stages appeared green, while the negative control appeared pink. Fluorescence quantification results were consistent with those of in vitro visualization (Fig. 8c). The weight of each egg was measured to be approximately 30 µg, and DNA was extracted from a single egg at a concentration of 3.22 ng µL− 1. As shown in Fig. 8b, the reaction system derived from the single egg was green in color, while the negative control was pink. The fluorescence quantification result showed that an egg system differed significantly from NTC (p < 0.05) (Fig. 8d). The results indicate that the system is sufficiently sensitive to detect a single egg of B. dorsalis.

Cas12b detection of B. dorsalis in samples obtained from different developmental stages. (a) A, B, and C are three groups of replicate experiments. 1, eggs; 2, larva; 3, pupa; 4, adult and 5, negative controls. (b) 1–3, a single egg; and 4–6, negative controls. (c-d) Fluorescence quantification results for different developmental stages and an egg.
Discussion
Bactrocera dorsali is a species that is known to seriously harm fruits and vegetables. Larvae and adults can both result in crop failure and fruit abscission. Through infestation, the polyphagous fruit fly B. dorsalis, which belongs to the Bactrocera genus, poses a serious threat to more than 250 fruit and vegetable species40. Imported fruits at border ports may contain a variety of fruit fly species at varying stages of development (e.g., eggs and larvae), which can be challenging to classify based on appearance. The widespread spread of fruit flies can be caused by mistakes and omissions in quarantine, which will ultimately destabilize agricultural economies by lowering fruit yields and quality. Numerous techniques for the quick identification of flies in the field have been developed recently. For example, Andrews et al. detected five species of Drosophila simultaneously using a multiplex PCR assay7. Koohkanzade et al. utilized TaqMan real-time PCR assay to identify Bactrocera zonata12. For the isothermal program in this study, a slan-96P qPCR instrument was used because it was more user-friendly than the PCR instrument. Sabahi et al. combined PCR with LAMP and designed two primer pairs to identify the Ethiopian fruit fly41. For detection in this study, only one pair of RPA primers was required. Dermauw et al. employed LAMP assay, which requires a minimum concentration of 10 pg to detect the Mediterranean fruit fly42. In the research, B. dorsalis was detected at a concentration as low as 3.2 pg µL− 1. Blaser et al. successfully identified Bemisia tabaci, Thrips palmi, and several Drosophila genera, including Bactrocera and Zeugodacus, using the LAMP method14. The reaction was carried out at 65 °C followed by heating the reaction system to 98 °C and then cooling it to 75 °C. For isothermal system in this study, a reaction temperature of 43 °C was easily maintained.
To date, a variety of molecular identification techniques for B. dorsalis have been developed, including Random Amplification of Polymorphic DNA (RAPD)43, Restriction Fragment Length Polymorphism (RFLP)44,45, DNA barcoding46, and species-specific primer PCR47,48 In our study, the RPA-CRISPR technology was employed. Although RPA-CRISPR technology exhibits slightly higher costs (Table S6), its rapid detection capability renders it particularly suitable for on-site testing. Compared with RAPD, this methodology demonstrates reduced susceptibility to exogenous DNA contamination and enhanced experimental reproducibility. Relative to RFLP and DNA barcoding approaches, it achieves higher accuracy through precise identification of intra-sequence variations within DNA. When contrasted with specific primer PCR techniques, RPA-CRISPR offers simplified thermal regulation of amplification reactions. Furthermore, it presents advantages over LAMP technology through more straightforward primer design requirements and improved detection sensitivity. RPA has been used to bind with Cas12a for the detection of insects. Zeng et al., for instance, using RPA-CRISPR/Cas12a, the Khapra beetle (Trogoderma granarium Everts) was identified in 35 min, exhibiting a systematic discoloration reaction under blue light 40 and a minimum detectable concentration of 0.1 ng µL− 149. In contrast, our method presented in the current study relies on observation with the naked eye, making our system more intuitive. Shashank et al. detected Keiferia lycopersicella, Phthorimaea absoluta, and Scrobipalpa atriplicella by RPA-CRISPR/Cas12a in 1 h50, while Alon et al. detected B. zonata and C. capitata using RPA-CRISPR/Cas12a in 2.25 h20. These long detection times can be attributed to the lengthy DNA extraction and incubation of RPA and Cas12a. DNA extraction in our study was accomplished in less than five minutes, and the RPA/CRISPR system did not require prior incubation, greatly reducing the reaction time. Li et al. detected B. correcta using RPA-CRISPR/Cas12a and multienzyme isothermal rapid amplification with lateral flow dipstick (MIRA-LFD) with a minimum detectable concentration of 0.1 ng µL− 1; the detection time for RPA-CRISPR/Cas12a was 30 min, while MIRA-LFD took 10 min51. Compared to Cas12a, the Cas12b enzyme used in the present study is smaller and has a wider range of temperature tolerance (31–59 °C)31. Cas12b also recognizes shorter PAM sequences and has higher spacer specificity (20 nt) than Cas12a, allowing it to bind to the target DNA under the guidance of a single-stranded gRNA. After cleaving target sequences, Cas12b can significantly increase the targeting range of the genome and cut other single-stranded nucleic acids in the system32. In addition, Cas12b has minimal off-target effects and is considered safer than other enzymes. RPA has been used in conjunction with Cas12b to detect SARS-CoV-225. Our study is the first time RPA-CRISPR/Cas12b has been used to detect insects.
In summary, we designed RPA primers and gRNAs for B. dorsalis, leading to the development of a detection platform for this species based on RPA-CRISPR/Cas12b. The stability of the system for detecting B. dorsalis in various geographical regions was verified by identifying fly samples from Vietnam, Guangdong Province, Guangxi Zhuang Autonomous Region, Zhejiang Province, Yunnan Province, and Hainan Province. The reported system was highly specific and did not exhibit cross-reactivity when tested on the recombinant plasmids with COI gene inserts of 10 sympatric species and fly samples of five sympatric species. The system showed a high detection sensitivity; a trace concentration of 3.2 pg µL− 1 was sufficient to detect the four insect stages of B. dorsalis. The system was designed to achieve a balance between detection time and sensitivity, taking a total of 30 min to detect the presence of B. dorsalis. A 10-min RPA reaction and a 20-min CRISPR reaction are included in this time. The system is easy to use, and blue light is not necessary to verify the presence of B. dorsalis because the color shift that occurs during the reaction can be observed with the naked eyes.
In order to quickly identify B. dorsalis for pest monitoring and control, we developed a nucleic acid identification system for the rapid visual detection based on RPA-CRISPR/Cas12b. With a portable metal bath device, this system can be used for POCT in quarantine ports, allowing for rapid, on-site and highly specific identification. Although the visual detection results in our system with naked eyes are easy, we will make the judgment of the results more convenient in subsequent study by observing the presence or absence of the detection line using the MIRA-LFD technology51. In addition, research should continue to refine the technology for early and rapid identification of other pests in the field and in border controls. Our findings provide an innovative approach to identifying pests.
Data availability
The datasets generated and analyzed during the current study are available in the National Center for Biotechnology Information (NCBI) repository, NCBI accession number is DQ116269.
References
Gong, Q. T. et al. The occurrence and control of Bactrocera dorsalis. Deciduous Fruits. 54, 49–52. https://doi.org/10.13855/j.cnki.lygs.2022.01.015 (2022).
Gong, Q. T. et al. Overview of the occurrence and prevention of Bactrocera dorsalis in the fruit and vegetable areas of Northern China. Deciduous Fruits. 56, 8–15. https://doi.org/10.13855/j.cnki.lygs.2024.04.002 (2024).
Li, Z. H. et al. Review on prevention and control techniques of Tephritidae invasion. Plant Quarantine. 27, 1–10 (2013).
Li, Z. H., Nopparat, B., Hu, J. T., Zhang, Q. & Fang, Y. Review on invasion origin and invasion mechanism of Tephritidae. P 27lant Quarantine. 12, 1–12 (2013).
Liang, G. H., Chen, J. H., Yang, J. Q., Huang, J. C. & Ji, Q. E. Advances in Research of Bactrocera dorsalis (Hendel) in China. Entomol. J. East China. 90–98 (2003).
Ye, M. H. Research on the development and application of plant quarantine testing technology. Agricultural Dev. Equipments. 1, 86–88. https://doi.org/10.3969/j.issn.1673-9205.2023.01.035 (2023).
Andrews, K. J., Bester, R., Manrakhan, A. & Maree, H. J. A multiplex PCR assay for the identification of fruit flies (Diptera: Tephritidae) of economic importance in South Africa. Sci. Rep. 12, 13089. https://doi.org/10.1038/s41598-022-17382-x (2022).
Wu, Z. et al. Discovery of chemosensory genes in the Oriental fruit fly, Bactrocera dorsalis. PloS One. 10, e0129794. https://doi.org/10.1371/journal.pone.0129794 (2015).
Huang, C. G., Hsu, J. C., Haymer, D. S., Lin, G. C. & Wu, W. J. Rapid identification of the mediterranean fruit fly (Diptera: Tephritidae) by loop-mediated isothermal amplification. J. Econ. Entomol. 102, 1239–1246. https://doi.org/10.1603/029.102.0350 (2009).
Blacket, M. J. et al. A LAMP assay for the detection of Bactrocera tryoni Queensland fruit fly (Diptera: Tephritidae). Sci. Rep. 10, 9554. https://doi.org/10.1038/s41598-020-65715-5 (2020).
Starkie, M. L. et al. Loop-mediated isothermal amplification (LAMP) assays for detection of the new Guinea fruit fly Bactrocera trivialis (Drew) (Diptera: Tephritidae). Sci. Rep. 12, 12602. https://doi.org/10.1038/s41598-022-16901-0 (2022).
Koohkanzade, M., Zakiaghl, M., Dhami, M. K., Fekrat, L. & Namaghi, H. S. Rapid identification of Bactrocera zonata (Dip.: Tephritidae) using TaqMan real-time PCR assay. PloS One. 13, e0205136. https://doi.org/10.1371/journal.pone.0205136 (2018).
Gandelman, O., Jackson, R., Kiddle, G. & Tisi, L. Loop-mediated amplification accelerated by stem primers. Int. J. Mol. Sci. 12, 9108–9124. https://doi.org/10.3390/ijms12129108 (2011).
Blaser, S. et al. From laboratory to point of entry: development and implementation of a loop-mediated isothermal amplification (LAMP)-based genetic identification system to prevent introduction of quarantine insect species. Pest Manag Sci. 74, 1504–1512. https://doi.org/10.1002/ps.4866 (2018).
Feng, W. et al. CRISPR technology incorporating amplification strategies: molecular assays for nucleic acids, proteins, and small molecules. Chem. Sci. 12, 4683–4698. https://doi.org/10.1039/d0sc06973f (2021).
Kim, S., Ji, S. & Koh, H. R. CRISPR as a diagnostic tool. Biomolecules 11, 1162. https://doi.org/10.3390/biom11081162 (2021).
Chan, K. G., Ang, G. Y., Yu, C. Y. & Yean, C. Y. Harnessing CRISPR-Cas to combat COVID-19: from diagnostics to therapeutics. Life Basel Switz. 11, 1210. https://doi.org/10.3390/life11111210 (2021).
Sashital, D. G. Pathogen detection in the CRISPR-Cas era. Genome Med. 10, 32. https://doi.org/10.1186/s13073-018-0543-4 (2018).
Li, J., Macdonald, J. & von Stetten, F. Review: a comprehensive summary of a decade development of the recombinase polymerase amplification. Analyst 144, 31–67. https://doi.org/10.1039/c8an01621f (2018).
Alon, D. M., Partosh, T., Burstein, D. & Pines, G. Rapid and sensitive on-site genetic diagnostics of pest fruit flies using CRISPR‐Cas12a. Pest Manag. Sci. 79, 68–75 (2023). https://doi.org/10.1002/ps.7173
Zhang, K. et al. RPA-CRISPR/Cas12a-Based detection of Haemophilus parasuis. Anim. Open. Access. J. MDPI. 13, 3317. https://doi.org/10.3390/ani13213317 (2023).
Guo, Y., Xia, H., Dai, T. & Liu, T. RPA-CRISPR/Cas12a mediated isothermal amplification for visual detection of Phytophthora Sojae. Front. Cell. Infect. Microbiol. 13, 1208837. https://doi.org/10.3389/fcimb.2023.1208837 (2023).
Li, S. et al. Establishment and application of a CRISPR-Cas12a-based RPA-LFS and fluorescence for the detection of Trichomonas vaginalis. Parasit. Vectors. 15, 350. https://doi.org/10.1186/s13071-022-05475-5 (2022).
Miao, J. et al. Rapid detection of Nipah virus using the one-pot RPA-CRISPR/Cas13a assay. Virus Res. 332, 199130. https://doi.org/10.1016/j.virusres.2023.199130 (2023).
Aman, R. et al. iSCAN-V2: A One-Pot RT-RPA-CRISPR/Cas12b assay for point-of-care SARS-CoV-2 detection. Front. Bioeng. Biotechnol. 9 (800104). https://doi.org/10.3389/fbioe.2021.800104 (2021).
van Dongen, J. E. et al. Point-of-care CRISPR/Cas nucleic acid detection: recent advances, challenges and opportunities. Biosens. Bioelectron. 166, 112445. https://doi.org/10.1016/j.bios.2020.112445 (2020).
Shmakov, S. et al. Discovery and functional characterization of diverse class 2 CRISPR-Cas systems. Mol. Cell. 60, 385–397. https://doi.org/10.1016/j.molcel.2015.10.008 (2015).
Shmakov, S. et al. Diversity and evolution of class 2 CRISPR-Cas systems. Nat. Rev. Microbiol. 15, 169–182. https://doi.org/10.1038/nrmicro.2016.184 (2017).
Makarova, K. S. et al. Evolutionary classification of CRISPR-Cas systems: a burst of class 2 and derived variants. Nat. Rev. Microbiol. 18, 67–83. https://doi.org/10.1038/s41579-019-0299-x (2020).
Ming, M. et al. CRISPR-Cas12b enables efficient plant genome engineering. Nat. Plants. 6, 202–208. https://doi.org/10.1038/s41477-020-0614-6 (2020).
Teng, F. et al. Repurposing CRISPR-Cas12b for mammalian genome engineering. Cell. Discov. 4, 63. https://doi.org/10.1038/s41421-018-0069-3 (2018).
Li, L. et al. HOLMESv2: A CRISPR-Cas12b-Assisted platform for nucleic acid detection and DNA methylation quantitation. ACS Synth. Biol. 8, 2228–2237. https://doi.org/10.1021/acssynbio.9b00209 (2019).
Sam, I. K. et al. TB-QUICK: CRISPR-Cas12b-assisted rapid and sensitive detection of Mycobacterium tuberculosis. J. Infect. 83, 54–60. https://doi.org/10.1016/j.jinf.2021.04.032 (2021).
Joung, J. et al. Detection of SARS-CoV-2 with SHERLOCK one-pot testing. N Engl. J. Med. 383, 1492–1494. https://doi.org/10.1056/NEJMc2026172 (2020).
Wang, R. et al. Engineering of the LAMP-CRISPR/Cas12b platform for Chlamydia psittaci detection. J. Med. Microbiol. 72 https://doi.org/10.1099/jmm.0.001781 (2023).
Wu, F. et al. Rapid visual detection of Vibrio parahaemolyticus by combining LAMP-CRISPR/Cas12b with heat-labile uracil-DNA glycosylase to eliminate carry-over contamination. J. Zhejiang Univ. Sci. B. 24, 749–754. https://doi.org/10.1631/jzus.B2200705 (2023).
Wang, X. et al. CDetection.v2: One-pot assay for the detection of SARS-CoV-2. Front. Microbiol. 14, 1158163. https://doi.org/10.3389/fmicb.2023.1158163 (2023).
Concordet, J. P. & Haeussler, M. CRISPOR: intuitive guide selection for CRISPR/Cas9 genome editing experiments and screens. Nucleic Acids Res. 46, W242–W245. https://doi.org/10.1093/nar/gky354 (2018).
Rychlik, W. OLIGO 7 primer analysis software. Methods Mol. Biol. Clifton NJ. 402, 35–60. https://doi.org/10.1007/978-1-59745-528-2_2 (2007).
Zhan, G. H. et al. Study on quarantine and trapping technique for Bactrocera dorsalis. J. Southwest. Forestry Univ. 30, 1–3. https://doi.org/10.11929/j.issn.2095-1914.2010.S1.001 (2010).
Sabahi, S., Fekrat, L., Zakiaghl, M. & Moravej, G. H. Loop-Mediated isothermal amplification combined with PCR for rapid identification of the Ethiopian fruit fly (Diptera: Tephritidae). Neotrop. Entomol. 47, 96–105. https://doi.org/10.1007/s13744-017-0522-2 (2018).
Dermauw, W. et al. A loop-mediated isothermal amplification (LAMP) assay for rapid identification of Ceratitis capitata and related species. Curr. Res. Insect Sci. 2, 100029. https://doi.org/10.1016/j.cris.2022.100029 (2022).
Zhang, L. & Zhang, Z. Y. Random amplified polymorphic DNA identification of six Bactrocera(Diptera: Tephritidae)species in Yunnan Province of Southwest China. Chinese J. Appl. Ecol. 1165–1168 (2007).
Wu, J. J., Hu, X. N., Zhao, J. P., Liang, F. & Liang, G. Q. Rapid identification among 9 species of quarantine fruit flies (Diptera: Tephritidae) by PCR-RFLP. Plant Quarantine 2–6 (2005).
Wang, W. X., Yu, F., Zhang, Z. & Lin, X. H. Advances in rapid identification methods for the quarantined fruit flies. Plant Protection 36, 39–43, 55 (2010).
Xu, L. et al. Sequencing and analysis of the complete mitochondrial genome of the Oriental fruit fly, Bactrocera dorsalis (Hendel) diptera: Tephritidae. Acta Entomologica Sinica. 755–761. https://doi.org/10.16380/j.kcxb.2007.08.004 (2007).
Wang, Z. H. et al. Identification and differentiation of Bactrocera citri and Bactrocera dorsalis by PCR. Plant Quarantine 155–157 (2008).
Ding, S. M., Wang, S. P., He, K., Li, F. & Jiang, M. X. PCR-based identification of fruit-flies using specific SSR sequences. Chinese J. Appl. Entomol. 55, 759–765 (2018).
Zeng, L. et al. New and rapid visual detection assay for Trogoderma granarium everts based on recombinase polymerase amplification and CRISPR/Cas12a. Pest Manag Sci. 79, 5304–5311. https://doi.org/10.1002/ps.7739 (2023).
Shashank, P. R. et al. CRISPR-based diagnostics detects invasive insect pests. Mol. Ecol. Resour. 24, e13881. https://doi.org/10.1111/1755-0998.13881 (2024).
Li, W. et al. Application of recombinase polymerase amplification with CRISPR/Cas12a and multienzyme isothermal rapid amplification with lateral flow dipstick assay for Bactrocera correcta. Pest Manag Sci. ps.8035 https://doi.org/10.1002/ps.8035 (2024).
Acknowledgements
We thank Prof. Guoping Zhan, Dr. Qingying Zhao, Dr. Yong Zhong, Dr. Xinguo Wang, Dr. Bo Cai, Dr. Rui Meng, Dr Bingyang Liu and Dr Zhiyi Wu for their kind help in sample collection. This work was supported in part by the National Natural Science Foundation of China (no. 62272060 and 62272061), the earmarked fund for Binzhou Medical University.We thank AiMi Academic Services (www.aimieditor.com) for English language editing and review services.
Author information
Authors and Affiliations
Contributions
B. Liu, F.Y. Zhang and L.L. Ren conceived the project, C.Y. Lv, P.Y. Zhu, C.J. Chen and B. Liu designed the experiments and wrote the manuscript, and C.Y. Lv, X.J. Cheng and X.Y. Yang performed the experiments.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Lv, C., Zhang, F., Ren, L. et al. Rapid visual detection assay for Bactrocera dorsalis (Hendel) using recombinase polymerase amplification and CRISPR/Cas12b. Sci Rep 15, 17328 (2025). https://doi.org/10.1038/s41598-025-02441-w
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-02441-w
