
Abstract
The gut microbiome early in life plays a crucial role in development of the host and affects health throughout life. The definition of a healthy microbiome early in life has not been established, and the underlying mechanism of how a young host selects appropriate microbes for colonization remains unclear. Understanding the mechanism may provide insights into novel preventive and therapeutic strategies by correcting dysbiosis early in life. We employed germ-free mice early in life (4 weeks of age) and later in life (10 weeks of age) for fecal microbiota transfer (FMT) from specific pathogen-free mice. We performed age-unmatched FMT between recipients early in life and donors early or later in life, in addition to common age-matched FMT. Age-matched FMT resulted in significantly different bacterial compositions between recipients early vs. later in life. When the gut microbiome from donors early or later in life was transferred to recipients early in life, bacterial compositions of recipients from donors later in life were similar to those of recipients from donors early in life. This finding suggests that the host early in life has mechanisms to select microbes appropriate for age from the exposed microbiome. We hypothesized that the age-specific intestinal environment promotes age-appropriate intestinal microbiome colonization and examined gene expression in the intestinal mucosa of germ-free mice. We observed that gene expression profiles were different between early vs. later in life. Correlation analysis demonstrated that genera Lachnospiraceae NK4A136 group and Roseburia were positively correlated to genes expressed predominantly early in life, but negatively with genes expressed predominantly later in life. We confirmed that the relative abundance of these genera was significantly higher in specific pathogen-free mice early in life compared with mice later in life. The characteristic gene expression of the intestinal mucosa early in life might play roles in selecting specific bacteria in the intestinal microbiome early in life.
Similar content being viewed by others

Introduction
The host and gut microbiome establish a symbiotic relationship through close interactions, contributing to mutual healthy development1. The compositions of the gut microbiome are affected by various factors, such as genetic background2, sex3, lifestyle including diet4, hygiene5, and geography6. Human studies have suggested that even monozygotic twins with identical genetic backgrounds obtain different gut microbiomes when they live in different environments7. The gut microbiome affects various host organs and physiological functions8,9. Bidirectionally, the gut microbiome is regulated by the host10,11,12. The gut microbiome can be affected by the functions of various organs outside the intestinal tract via metabolites13,14,15,16,17. In the context of the interactions between the gut microbiome and humans, associations between dysbiosis (i.e., perturbations of the microbiome) and various diseases have been reported.
Recent human epidemiological studies have demonstrated that the use of antibiotics during pregnancy, peripartum period, and early childhood is associated with an increased risk of various diseases, such as inflammatory bowel disease18, asthma19, and food allergy20. Because antibiotics can cause an imbalance in the gut microbiome, these findings suggest that dysbiosis early in life leads to disease later in life. Several animal studies have provided mechanistic insights into this notion. We reported a murine model in which intestinal dysbiosis caused by maternal perinatal antibiotic exposure increases the risk of colitis in genetically susceptible offspring21. Microbial and functional dysbiosis and the skewed immune system early in life persist later in life in this model22. Moreover, microbial restitution early in life, but not later in life, reduces the risk of colitis in offspring23. These findings support the notion that it is critical to obtain a healthy microbiome at the appropriate time for the host’s health.
Murine models have shown that bacterial compositions early in life contribute to the direction of subsequent development in the gut microbiome24, and the peri-weaning period is a critical window for immune development25. Al Nabhani et al. reported that exposure to the appropriate bacteria during the weaning period (2–4 weeks of age) is crucial for the development of the host immune system25. We previously reported that an intervention restituting a keystone microbial strain in the gut microbiome at 3 weeks of age reduces the risk of spontaneous colitis in a mouse colitis model23. Furthermore, Cahenzli et al. reported that high serum IgE levels due to Th2-dominant immunity in GF mice return to the same levels as those in SPF mice when GF mice are exposed to the SPF environment by 5 weeks of age26. Additionally, the gut microbiome changes with age27, and some commensal, beneficial bacteria in the intestinal tract early in life reduce or disappear later in life28. These studies provide clues that the appropriate timing to obtain and establish eubiosis throughout life may be close to the weaning age. However, the underlying mechanisms of how commensal colonization of important microbes early in life leading to the host’s healthy development is achieved remain unclear. We hypothesized that the intestinal environment provided by the host early in life plays a role in promoting the colonization of keystone microbes that contribute to the healthy development of the host and the gut microbiome and conducted the present study to test this hypothesis.
Methods
Animals
Specific pathogen-free (SPF) C57BL/6 wildtype mice were purchased from CLEA Japan (Tokyo, Japan) (C57BL/6JJcl). The mice were maintained under SPF conditions. Germ-free (GF) C57BL/6 wildtype mice were purchased from Sankyo Labo Service Corporation (Tokyo, Japan) (Tsl:C57BL/6NCr[GF]) and reared in a vinyl isolator. Temperature and humidity were controlled at 23 ± 2 °C and 55 ± 10%, respectively. The light cycle was 12:12 light/dark (8:00 on/20:00 off). Five mice were housed in one plastic cage with wood-shaving bedding. The bedding was changed once a week. SPF and GF mice were fed gamma-sterilized or autoclaved CLEA Rodent Diet CE-2 (CLEA Japan), respectively. All mice were supplied with drinking water ad libitum. The regular health condition check was conducted three times a week. These rearing conditions were consistent with those described in our previous report29. GF mice transferred to Kyorin University for tissue collection were quickly dissected after removal from the sterile transport container. All mice were euthanized at the end of the study by carbon dioxide inhalation and cervical dislocation. Experiments were conducted after review and approval by the Institutional Animal Care and Use Committee for Kitasato University and the Experimental Animal Facility of Kyorin University School of Medicine Collaborative Research Facility Division (Kitasato University approval number: 20-010; Kyorin University approval number: 254). All animal experiments were conducted according to the Japanese guidelines for experimental animal welfare and study protocols. The authors have complied with the ARRIVE guidelines for reporting.
Optical microscopy
Histological analysis was performed as described previously29. Collected tissue samples of the cecum were fixed in a 4% formalin solution and then embedded in paraffin. Tissue sections were stained with hematoxylin and eosin and observed under an optical microscope.
Transmission electron microscopy
Ultrastructural observations were performed using the previously reported protocol29. Collected tissue samples of the cecum were washed with phosphate-buffered saline (PBS) and pre-fixed in 2.5% glutaraldehyde (Nissin EM, Tokyo, Japan) in PBS. Post-fixation was performed with 1% osmium tetroxide (Nissin EM) in 0.1 M phosphate buffer (pH 7.2). The samples were embedded in EPON resin (Nissin EM) and prepared as ultrathin sections (80 nm). The ultrathin sections were stained with a 2% uranium acetate solution, followed by a 1% lead citrate solution. The specimens were then observed under a transmission electron microscope (JEM-1011, JEOL, Tokyo, Japan). Ultrastructural findings, including the microvillus length, were assessed in the captured images.
Fecal microbiota transplantation
A fecal solution was prepared by dissolving a fecal specimen collected from a donor SPF mouse in 1 mL PBS per 100 mg. Fecal microbiota transfer (FMT) was performed in GF recipient mice by gavaging using an oral administration needle. GF mice at 4 weeks of age received 100 µl and those at 10 weeks of age received 200 µl. This protocol was consistent with our previous reports23,29. The health condition of the recipients was checked on the next day of FMT. Recipient mice after FMT were housed in the SPF environment until the end of the experiment.
DNA extraction and 16S rRNA gene amplicon sequencing analysis
Fecal samples were collected and frozen at − 80 °C. DNA was extracted from the fecal samples as described previously30. After amplification of the 16S rRNA gene V4 region, amplicon sequencing analysis was performed by Miseq (Illumina, San Diego, CA, USA). The sequences were processed using the Quantitative Insights into Microbiome Ecology (QIIME) 2 pipeline31. Sequences were denoised and filtered using DADA232. The sampling depth was set to 5,000. Amplicon sequence variants (ASVs) were assigned to taxonomy using Silva database 13_8 (https://docs.qiime2.org/2021.2/data-resources/). Diversity of the bacterial community was assessed by QIIME 2. Alpha-diversity was evaluated using the Shannon diversity index. Beta-diversity was assessed with UniFrac distances and visualized using principal coordinate analysis (PCoA).
Metagenome shotgun sequencing
Metagenome shotgun sequencing was performed as previously reported33. The purity of DNA samples extracted from feces was evaluated by a Nanodrop (Thermo Fisher Scientific, USA). The A260/A280 ratio was confirmed to be > 1.8. The DNA concentration in a sample was determined using a Qubit dsDNA HS Assay Kit (Thermo Fisher Scientific). Libraries were prepared from 100 ng DNA using an Illumina DNA Prep, (M) Tagmentation (Illumina) in accordance with the manufacturer’s protocol. The DNA concentration of each library was measured using the Qubit dsDNA HS Assay Kit. Library quality and DNA fragment size were measured using a High Sensitivity D5000 Reagent Kit (Agilent). A diluted library was loaded into a flow cell containing NextSeq 1000/2000 P2 reagents (300 cycles) (Illumina). Sequencing was performed using a NextSeq2000 (Illumina). Analysis was performed at 150 bp paired ends. The resulting data were merged using BBtools BBmerge. The merged data were then analyzed with Trimmomatic to remove low-quality bases and adapter sequences. The analysis pipeline HUMAnN3.0 (https://huttenhower.sph.harvard.edu/humann) was used to analyze pathways in the obtained data.
Flow cytometry
T cell subpopulations were analyzed by flow cytometry using previously described procedures23. Briefly, spleens and mesenteric lymph nodes were harvested, homogenized, and resuspended in PBS containing 2% fetal bovine serum. For all samples, Fc receptor blocking was performed with Purified Rat Anti-Mouse CD16/CD32 (BD Biosciences, San Jose, CA, USA). Cells were stained using a LIVE/DEAD Fixable Aqua Dead Cell Stain Kit (Thermo Fisher Scientific) to assess viability. Anti-mouse TCRβ (BioLegend, CA, USA) and anti-mouse CD4 (BioLegend) antibodies were used for cell surface staining. A FOXP3/Transcription Factor Staining Buffer Set (Thermo Fisher Scientific) was used to fix and permeabilize cells. Anti-mouse/rat Foxp3 (Thermo Fisher Scientific), anti-mouse/human T-bet (Thermo Fisher Scientific), anti-mouse GATA3 (Biosciences), and anti-Mouse RORγt (Thermo Fisher Scientific) antibodies were used for intranuclear staining. Mouse IgG1 Kappa Isotype Control (Thermo Fisher Scientific), Rat IgG1 Isotype Control (Thermo Fisher Scientific), and Rat IgG2a Kappa Isotype Control (Thermo Fisher Scientific) were used as isotype controls. Samples were analyzed with a CytoFLEX (Beckman Coulter, Brea, CA, USA) and FlowJo version 10.8 (FLOWJO, Ashland, OR, USA).
mRNA microarray analysis
Total RNA extraction from the ileal and colonic mucosal scrapings collected as previously described34,35, using TRIzol Reagent (Thermo Fisher Scientific), following the manufacturer’s protocol. Microarray analysis was performed using a Clariom™ S Assay, mouse (Thermo Fisher Scientific) in accordance with the manufacturer’s protocol. The obtained expressed genes were analyzed using Transcriptome Analysis Console (Thermo Fisher Scientific). Metascape was used as the analysis pipeline for clustering and functional analysis of expressed genes (https://metascape.org/gp/index.html#/main/step1)36.
Correlation analysis
The R package mixOmics (http://mixomics.org/)37 was used for integrated analyses to investigate the correlation between expressed genes in the intestinal mucosa and intestinal bacterial ASVs.
Statistical analysis
The Mann–Whitney U-test was performed using GraphPad Prism version 8.4.3 to compare the Shannon diversity index, cecum length, microvillus length of the cecum, T cell subpopulations, and relative abundances of genera between two groups. The criterion of statistical significance was set at a p < 0.05 or q < 0.05.
Results
Age and sex-matched gut microbiome transfer reproduces the bacterial compositions in germ-free recipients
Age and sex-matched FMT was performed to examine reproducibility of the intestinal microbiome in SPF donors in GF recipients early and later in life. The experimental design is shown in Fig. 1A (5 female and 5 male animals per group). GF mice at 4 and 10 weeks of age (exGF A and exGF B groups, respectively) received the age and sex-matched gut microbiome from donor SPF mice (SPF A and SPF B groups, respectively). Age and sex-matched GF mice were also prepared (GF A and GF B groups, respectively). No significant difference was found in the Shannon diversity index between SPF A and SPF B groups before the gut microbiome transfer (week 0) in both sexes (Fig. 1B and Fig. S1A). Female mice in SPF A and SPF B groups showed significant differences in bacterial compositions in unweighted and weighted UniFrac distances between these groups (unweighted: p = 0.010, weighted: p = 0. 011) (Fig. 1C). Male animals showed a similar tendency (Fig. S1B). These results appeared compatible with previous findings that the intestinal bacterial composition changes with age27. The Shannon diversity index of the exGF A group was significantly higher than that of the exGF B group at week 2 (female: p = 0.0159, male: p = 0.0317), but not at week 4 in both sexes (Fig. 1D and Fig. S1C). At week 2, the bacterial compositions of female animals in exGF A and exGF B groups were significantly different in terms of unweighted and weighted UniFrac distances (unweighted: p = 0.007, weighted: p = 0.013) (Fig. 1C). Significant differences were also found in bacterial compositions in male animals of exGF A and exGF B groups (unweighted p = 0.007, weighted p = 0.016) (Fig. S1B). At week 4, bacterial compositions were significantly different between exGF A and exGF B groups in both sexes (female unweighted: p = 0.007, weighted: p = 0.041; male unweighted p = 0.01, weighted p = 0.005) (Fig. 1C and Fig. S1B). Moreover, SPF A and exGF A groups, and SPF B and exGF B groups clustered closely in PCoA plots of unweighted and weighted UniFrac distances. Relative abundances at phylum and genus levels in cohorts A and B are shown in Figs. S2 and S3, respectively. Overall, despite the methodological limitations of FMT38,39, mice in exGF A and exGF B groups maintained the aging difference in bacterial compositions reproducing the gut microbiome of age and sex-matched SPF donors.

Age and sex-matched gut microbiome transfer to germ-free mice early and later in life. (A) In cohort A, fecal samples were collected from specific pathogen-free (SPF) mice at 4 weeks of age (SPF A group), and fecal microbiota transfer (FMT) was performed in sex-matched germ-free (GF) mice at 4 weeks of age (ex-germ-free [exGF] A group). Age and sex-matched GF mice were used as controls (GF A group). In cohort B, fecal samples were collected from SPF mice at 10 weeks of age (SPF B group), and FMT was performed in sex-matched GF mice at 10 weeks of age (exGF B group). Age and sex-matched GF mice were used as controls (GF B group) (n = 5 per group). Fecal samples were collected at weeks 0, 2, and 4. Mice were sacrificed and tissues were collected at week 4. (B) Shannon diversity index of SPF A and SPF B groups at week 0. (C) Temporal changes in the bacterial compositions of cohorts A and B. Principal coordinate analysis plots based on unweighted and weighted UniFrac distances are shown for cohort A (SPF A and exGF A groups) and cohort B (SPF B and exGF B groups). (D) Shannon diversity index of exGF A and exGF B groups at weeks 2 and 4 after FMT. Data are described as the mean ± SEM (n = 5). *p < 0.05. Male data are shown in Fig. S1.
The cecum is larger in GF mice than in SPF mice40. The cecum length was measured as an index of cecum size. No significant differences were found in the cecum length between exGF A (female: 3.0 ± 0.1 cm, male: 3.1 ± 0.1 cm) vs. SPF A (female: 3.3 ± 0.1 cm, male: 3.1 ± 0.1 cm) groups and exGF B (female: 3.6 ± 0.1 cm, male: 3.6 ± 0.2 cm) vs. SPF B (3.3 ± 0.2 cm, male: 3.6 ± 0.1 cm) groups (Fig. S4A). No apparent histological differences were observed in villi and at the cecum between exGF A vs. SPF A groups and exGF B vs. SPF B groups in both sexes, respectively (Fig. S4B). No significant differences were found in the microvilli length in the cecum between exGF A (female: 0.8 ± 0.07 µm, male: 0.9 ± 0.03 µm) vs. SPF A (female: 0.8 ± 0.04 µm, male: 0.8 ± 0.07 µm) groups and the exGF B (female: 0.7 ± 0.03 µm, male: 0.7 ± 0.03 µm) vs. SPF B (0.7 ± 0.03 µm, male: 0.8 ± 0.07 µm) groups (Fig. S4C). These results suggest that the intestinal tract of GF mice has morphological plasticity regardless of the timing (4 or 10 weeks of age) of colonization by the gut microbiome.
CD4+ T cell subpopulations in the spleen and mesenteric lymph nodes (MLNs) were assessed by flow cytometry. The gating strategy is shown in Fig. S5. The percentage of CD4+T-bet+ T cells among CD4+ T cells in the spleen was significantly lower in the exGF B group (0.25% ± 0.08% in females and 0.13% ± 0.04% in males) than in the SPF B group (1.18% ± 0.33% in females and 0.98% ± 0.43% in males) (p = 0.016 in females and p = 0.032 in males) (Fig. 2A). The percentage of CD4+GATA3+ T cells among CD4+ T cells was significantly higher in the male exGF A group (2.53% ± 0.31%) than in the male SPF A group (1.74% ± 0.16%) (p = 0.040), while it was significantly higher in the exGF B group than in the SPF B group in both sexes (5.20% ± 0.40% vs. 2.65% ± 0.13% [p = 0.008] in females and 4.74% ± 0.13% vs. 3.23% ± 0.48% [p = 0.016] in males) (Fig. 2A). In MLNs, the percentage of CD4+T-bet+ T cells among CD4+ T cells was significantly lower in the male exGF B group (0.13% ± 0.04%) compared with the male SPF B group (1.15% ± 0.21%) (p = 0.008) (Fig. 2B). The percentage of CD4+RORγt+ T cells among CD4+ T cells was significantly higher in the female exGF B group (16.9% ± 1.81%) than in the SPF B group (11.2% ± 0.58%) (p = 0.016) (Fig. 2B). The percentage of CD4+Foxp3+ T cells among CD4+ T cells was significantly higher in the female exGF A group (12.9% ± 0.35%) compared with the female SPF A group (11.1% ± 0.22%) (p = 0.008) (Fig. 2B).

Subpopulations of CD4+ T cells in age-matched ex-germ-free recipients vs. specific pathogen-free donors. Flow cytometric analyses of CD4+ T cells expressing T-bet+, GATA3+, RORγt+, or Foxp3+ were performed in the spleen (A) and mesenteric lymph node (B) in cohorts A and B. Subpopulations of CD4+ T cells were compared between ex-germ-free (exGF) recipients vs. specific pathogen-free (SPF) donors. Data are described as the mean ± SEM (black filled circle: female [n = 5], black filled inverted triangle: male [n = 5]). *p < 0.05, **p < 0.01. The gating strategy for flow cytometric analyses is shown in Fig. S5.
Intestinal bacteria colonized early in life after gut microbiome transfer are determined by the age of germ-free recipients, but not the age of specific pathogen-free donors
Next, the gut microbiome of SPF mice at 4 and 10 weeks of age (SPF C and SPF D groups, respectively) was transferred to GF recipient mice at 4 weeks of age (exGF C group and exGF D group, respectively) (Fig. 3A) (5 female and 5 male animals per group). No significant difference was found in the Shannon diversity index between SPF C and SPF D groups of both sexes before transfer of the gut microbiome (week 0) (Fig. 3B and Fig. S6A). Female mice in SPF C and SPF D groups had significant differences in bacterial compositions in unweighted UniFrac distances between these groups (unweighted p = 0.009, weighted p = 0.092) (Fig. 3C). In male mice, a significant difference was found in both unweighted and weighted UniFrac distances between SPF C and SPF D groups (unweighted p = 0.011, weighted p = 0.011) (Fig. S6B). These results demonstrated that FMT donors at 4 and 10 weeks of age had different intestinal bacterial compositions.

Effect of age-unmatched gut microbiome transfer on bacterial compositions in young germ-free mice. (A) In cohort C, fecal samples were collected from specific pathogen-free (SPF) mice at 4 weeks of age (SPF C group), and fecal microbiota transfer (FMT) was performed in age and sex-matched germ-free (GF) mice at 4 weeks of age (ex-germ-free [exGF] C group). In cohort D, fecal samples were collected from SPF mice at 10 weeks of age (SPF D group), and FMT was performed in age-unmatched and sex-matched GF mice at 4 weeks of age (exGF D group) (n = 5 per group). Fecal samples were collected at week 0, 2, and 4. Mice were sacrificed and tissues were collected at week 4. (B) Shannon diversity index of SPF C and SPF D groups at week 0. (C) Temporal changes in the bacterial compositions of cohorts C and D. Principal coordinate analysis plots based on unweighted and weighted UniFrac distances are shown for cohort C (SPF C and exGF C groups) and cohort D (SPF D and exGF D groups). (D) Shannon diversity index of exGF C and exGF D groups at weeks 2 and 4 after FMT. Data are described as the mean ± SEM (n = 5). Male data are shown in Fig. S6.
The Shannon diversity index between exGF C and exGF D groups showed no significant difference at weeks 2 and 4 for both sexes (female: Fig. 3D, male: Fig. S6C). Although the bacterial composition of female animals in exGF C and exGF D groups was significantly different in unweighted and weighted UniFrac distances (unweighted p = 0.010, weighted p = 0.010), no significant difference was found in weighted UniFrac distances at week 4 (unweighted p = 0.008, weighted p = 0.553) (Fig. 3C). In male animals, no significant differences were found between exGF C and exGF D groups in weighted UniFrac distances at both weeks 2 and 4 (week 2: unweighted p = 0.01, weighted p = 0.734, week 4: unweighted p = 0.007, weighted p = 0.091) (Fig. S6B). Relative abundances at phylum and genus levels in cohorts C and D are shown in Figs. S7 and S8A, respectively. Unweighted UniFrac distances describe the presence or absence of ASVs in samples, while weighted UniFrac distances take into account the proportions of those ASVs. Overall, the results showed that, regardless of the donor’s age (4 or 10 weeks of age), the colonized bacterial compositions in GF mice at 4 weeks of age (i.e., exGF C and exGF D) became similar at 4 weeks after FMT. Metagenomic shotgun sequencing demonstrated no significant difference in microbial metabolic pathways between exGF C and exGF D groups at week 2 (Fig. S8B). This finding suggests that the microbial functions in GF recipients early after FMT were not affected by the age of SPF donors, and these functions were suitable to colonize the host early in life.
Whereas the exGF A group (4-week-old GF recipients of FMT from 4-week-old SPF donors) and exGF B group (10-week-old GF recipients of FMT from 10-week-old SPF donors) had different bacterial compositions at 4 weeks after FMT, the exGF C group (4-week-old GF recipients of FMT from 4-week-old SPF donors) and exGF D group (4-week-old GF recipients of FMT from 10-week-old SPF donors) had similar bacterial compositions at 4 weeks after FMT. This observation suggests that the age of a recipient (4 weeks) is a determinant of the gut bacterial compositions.
No significant difference was found in the cecum length between exGF C (female: 4.7 ± 0.1 cm, male: 4.9 ± 0.2 cm) and exGF D (female: 4.5 ± 0.1 cm, male: 5.2 ± 0.1 cm) groups of both sexes (Fig. S9A). No apparent histological differences were observed in villi at the cecum between exGF C and exGF D groups of both sexes, respectively (Fig. S9B). No significant differences were found in the length of microvilli in the cecum between exGF C (female: 0.8 ± 0.06 µm, male: 0.8 ± 0.02 µm) and exGF D (female: 0.7 ± 0.03 µm, male: 0.7 ± 0.02 µm) groups of both sexes (Fig. S9C). Overall, 4-week-old GF recipient mice exhibited the same morphological plasticity of the intestinal tract regardless of the age of SPF donors. CD4+ T cell subpopulations in the spleen and MLNs were assessed by flow cytometry. In the spleen, the percentage of CD4+GATA3+ T cells among CD4+ T cells was significantly higher in the male exGF C group (3.17% ± 0.53%) than in the male exGF D group (0.75% ± 0.15%) (p = 0.008) (Fig. 4A). In MLNs, the percentage of CD4+Foxp3+ T cells among CD4+ T cells was significantly higher in the female exGF C group (13.2% ± 0.22%) than in the female exGF D group (11.6% ± 0.35%) (p = 0.008), while it was lower in the male exGF C group (11.6% ± 0.40%) than in the male exGF D group (13.0% ± 0.29%) (p = 0.016) (Fig. 4B).

Subpopulations of CD4+ T cells in young ex-germ-free recipients of age-unmatched fecal microbiota transfer. Flow cytometric analyses of CD4+ T cells expressing T-bet+, GATA3+, RORγt+, or Foxp3+ were performed in the spleen (A) and mesenteric lymph nodes (B) of ex-germ-free (exGF) C (fecal microbiota transfer [FMT] from young donors) and exGF D (FMT from adult donors) groups. Data are described as the mean ± SEM (black filled circle: female [n = 5], black filled inverted triangle: male [n = 5]). *p < 0.05, **p < 0.01. The gating strategy for flow cytometric analyses is shown in Fig. S5.
Gene expression profiles in the intestinal mucosa vary depending on the host age
Regarding the underlying mechanism to select microbes appropriate for early life, we hypothesized that the young host’s intestinal tract provides an environment favorable for desirable bacterial colonization and bacteria with functions advantageous for survival in the young intestinal environment colonize early in life. To investigate host factors involved in the intestinal environment, we performed mRNA microarray analysis of the ileal and colonic mucosa in GF mice at 4 weeks of age [(early life (EL) group] and 10 weeks of age [later life (LL) group]. Expressions of all genes analyzed in this study are publicly available (Supporting file 1). Gene expressions of intestinal mucosal markers, such as mucin, e-cadherin, lysozyme, and villin, were included in the list. Principal component analysis plots showed that EL and LL groups clustered separately (ileum: Fig. 5A, colon: Fig. S10A); that is, EL and LL groups had different gene expression patterns in the ileum and colon. Among the 22,206 genes analyzed, in the ileal mucosa, 120 genes satisfied the criteria: the expression level was different more than twofold with p < 0.05 between EL and LL groups. Volcano plots of genes in the ileum and colon are shown in Fig. 5B and Fig. S10B. The 120 genes included 66 genes predominant in the EL group (Table S1) and 54 genes predominant in the LL group (Table S2). In the colon mucosa, 74 genes out of 22,206 genes satisfied our criteria, and 36 genes (Table S3) and 38 genes (Table S4) were predominantly expressed in EL and LL groups, respectively. Next, enrichment analysis of the ileal and colon mucosa was performed. Enriched functions in the ileal mucosa of EL and LL groups are shown in Fig. 5C,D, respectively. Transport of small molecules was the top function in the EL ileum, whereas the top function in the LL ileum was the defense response to gram-positive bacteria. The lists of genes included in the Gene Ontology (GO) terms of the ileal mucosa in EL and LL groups are shown in Tables S5 and S6, respectively. Enriched functions in the colon mucosa of EL and LL groups are shown in Fig. S10C,D, respectively. Mineral absorption was the top function in the EL colon, whereas the top function in the LL colon was regulation of lipid storage. The lists of genes included in the GO terms of the colon mucosa in EL and LL groups are shown in Tables S7 and S8, respectively. These findings suggest that gene expression profiles in the intestinal mucosa change with age; that is, the environment for microbes in the intestinal tract varies between different ages of the host.

Ileal mucosal gene expression in germ-free mice early vs. later in life. (A) Principal component analysis plots of ileal mucosal gene expression profiles in germ-free (GF) mice at 4 weeks of age [early life (EL) group] and 10 weeks of age [later life (LL) group]. Red and blue dots represent EL and LL groups, respectively (n = 10 per group, black filled circle: female, black filled diamond: male). (B) Volcano plots of gene expression in the ileal mucosa of EL vs. LL groups. Red and blue dots indicate a gene that was expressed predominantly in EL or LL group, respectively (expression ratio > twofold and p < 0.05 between the two groups). A gray dot represents a gene that did not satisfy the criterion. (C) Characteristics annotated by enrichment analysis of genes predominantly expressed in the ileal mucosa in the EL group. (D) Characteristics annotated by enrichment analysis of genes predominantly expressed in the ileal mucosa in the LL group. Colon data are shown in Fig. S10.
Intestinal gene expression characteristic to mice early in life correlates to bacteria that predominantly colonize the host early in life
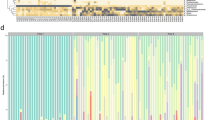
The bacteria that colonized GF recipients as early as 2 weeks after FMT were favorably adapted to grow in the intestinal mucosal environment provided by a GF host. ASVs that can colonize GF recipients at 4 weeks of age after FMT from SPF donors at 10 weeks of age and have positive correlations to characteristic gene expressions early in life can be considered candidates with the favorable potential to colonize early in life. We observed 1387 ASVs at 2 weeks after FMT in GF recipients that were 4 or 10 weeks of age when FMT was performed from SPF donors at 10 weeks of age. These 1387 ASVs should include bacteria that preferred to colonize the intestines at 4 weeks of age, 10 weeks of age, or both. We performed correlation analysis between the genes that were expressed in the ileal mucosa predominantly in the EL group (66 genes) or LL group (54 genes) and the 1387 ASVs (Fig. 6A). Among the 31 ASVs with strong positive correlations to genes predominantly expressed in the EL group, 12 ASVs were annotated to the Lachnospiraceae NK4A136 group, eight ASVs were Muribaculaceae (genus unclassifiable), seven ASVs were Roseburia, and four ASVs were Lachnospiraceae (genus unclassifiable). Correlation analysis was also performed between genes that were expressed in the colonic mucosa predominantly in the EL group (36 genes) or LL group (38 genes) and the 1387 ASVs (Fig. S11). Among the 36 ASVs with strong positive correlations to genes predominantly expressed in the EL group, 12 ASVs were annotated to the Lachnospiraceae NK4A136 group, nine ASVs were Muribaculaceae (genus unclassifiable), seven ASVs were Roseburia, four ASVs were Lachnospiraceae (genus unclassifiable), two ASVs were the Eubacterium coprostanoligenes group, one ASV was RF39, and one ASV was Woesearchaeales. These results suggested that the common Lachnospiraceae NK4A136 group, Muribaculaceae, and Roseburia adapted to the lower intestinal environment of GF mice at 4 weeks of age; that is, they may have the potential to colonize mice early in life as commensal bacteria. Next, we focused on the genera Lachnospiraceae NK4A136 group and Roseburia because the genera Muribaculaceae was unclassified. Among the genes predominantly expressed in the ileal mucosa of the EL group, the top five genes with the highest correlation coefficients to the genus Lachnospiraceae NK4A136 group were Acaa1a, Me1, Paqr7, St3gal6, and Fabp1 and those with the highest correlation coefficients to genus Roseburia were St3gal6, Aqp3, Rab8b, Podn, and Slc2a2 (Table 1). Among the genes predominantly expressed in the colonic mucosa of the EL group, the top five genes with the highest correlation coefficients among those positively correlated to the genus Lachnospiraceae NK4A136 group were Slc30a10, Ggt1, Col1a1, Slc11a2, and Pdk4, and the top five genes with the highest correlation coefficients among those positively correlated to Roseburia were Mt1, Olfr1100, Cma2, Fam69c, and Olfr1316 (Table 1). Moreover, among the genes predominantly expressed in the ileal mucosa of the LL group, the top five genes with the most negative correlation to the genus Lachnospiraceae NK4A136 group were Tnfsfm13, Tnfsf13, Tnfsf12, Rnf180, Tfcp2l1, Defa- rs1, and Pfkfb3 and those with the most negative correlation to genus Roseburia were Oit1, Rnf180, Lyz1, Mmp7, and Nos2 (Table 2). Among the genes predominantly expressed in the colonic mucosa of the LL group, the top five genes with the most negative correlation to the genus Lachnospiraceae NK4A136 group were Grem1, Cdo1, Cnn3, Apob, and Aldh1a1 and those with the most negative correlation to the genus Roseburia were Cnn3, Cdo1, Jchain, Car4, and Pdia6 (Table 2).

Correlations between ileal gene expression and bacterial genera that colonized mice early in life. (A) Correlation analysis of gene expression in the ileal mucosa and bacterial amplicon sequencing variants (ASVs). In the heat map, each row represents a bacterial ASV derived from adult donors (1387 ASVs) and each column represents a gene that was expressed predominantly in the EL group (66 genes, red line) or LL group (54 genes, blue line). (B) Relative abundance of the genus Lachnospiraceae NK4A136 group in specific pathogen-free (SPF) mice at 4 vs. 10 weeks of age. (C) Relative abundance of the genus Roseburia in SPF mice at 4 vs. 10 weeks of age. (D) Total relative abundance of genera Lachnospiraceae NK4A136 group and Roseburia in SPF mice at 4 vs. 10 weeks of age. Data are described as the mean ± SEM (n = 10, black filled circle: female, black filled diamond: male). *p < 0.05, **p < 0.01. Colon data are shown in Fig. S11.
Our findings suggest that the genera Lachnospiraceae NK4A136 group and Roseburia favorably colonize early in life. To examine this hypothesis, we assessed the relative abundances of these genera in SPF mice at 4 weeks of age vs. 10 weeks of age. Among 49 genera identified in SPF mice at 4 weeks of age, Lachnospiraceae NK4A136 group and Roseburia were the top two genera with the highest relative abundances among the gut bacteria. The relative abundance of Lachnospiraceae NK4A136 group was 21.0% ± 2.0% at 4 weeks of age and 16.2% ± 2.1% at 10 weeks of age (Fig. 6B). The relative abundance of Roseburia was 14.8% ± 2.0% at 4 weeks of age and 8.5% ± 1.6% in SPF mice at 10 weeks of age (p = 0.0147) (Fig. 6C). Lachnospiraceae NK4A136 group and Roseburia dominated at 35.7% ± 3.2% of the gut bacteria at 4 weeks of age and 24.8% ± 3.6% at 10 weeks of age (p = 0.0021) (Fig. 6D). These observations in the SPF environment supported that Lachnospiraceae NK4A136 group and Roseburia have a potential to colonize and flourish in the intestines early in life.
Discussion
This study demonstrated that a host early in life has mechanisms and flexibility to select age-specific commensal bacteria, leading to healthy development of the host. Our results suggest that the characteristic gene expression profiles of the intestinal mucosa early in life contribute to determining bacteria that colonize during the critical developmental period.
In general, the FMT experiment using GF mice aims to reproduce the microbiome (particularly the bacterial population) of a donor as accurately as possible in a GF recipient. For this purpose, because intestinal bacterial compositions vary with age27, it is common to use age-matched donors and recipients in the FMT experiment29. However, in this study, we inventively examined the gut microbiome of 4-week-old GF recipients after FMT from SPF donors at 4 and 10 weeks of age and demonstrated that the colonized bacterial compositions in the recipients were not affected by the donor age. Our age-unmatched FMT demonstrated that mice early in life chose the normal microbiome for their age. This finding raised a further issue of which underlying mechanisms select commensal bacteria early in life. The host and gut microbiome closely interact in complex manners, and this interaction starts immediately after birth and continues to change over time41,42. Additionally, the gene expression in the intestinal mucosa influences the intestinal environment where microbes flourish43. We need to consider a simplified model depending on the scope of research. Therefore, in the present study, we compared gene expression profiles in the intestinal mucosa and bacterial colonization after FMT between early and later in life using GF mice at 4 and 10 weeks of age. This methodology had an advantage allowing us to focus on a single factor, age, with vigorous control of confounding factors, and to investigate the causal link between host gene expression and bacterial colonization.
In the present study, we identified 66 genes in the ileal mucosa and 36 genes in the colonic mucosa that were expressed significantly more at 4 weeks of age than at 10 weeks of age. Although the differences in mucosal gene expression profiles between early and later in life were more apparent in the ileum than the colon, we speculate this finding does not necessarily indicate a lesser environmental change with aging in the colon than the ileum because the differences in the ileum can affect the colon environment. For example, cryptdin (human α-defensin), an antimicrobial peptide secreted by Paneth cells in the ileum, is detected in the colonic lumen, suggesting that the peptide affects the colonic microbiome44. The expression of predominant genes at 4 weeks of age was strongly correlated to colonization of the family Muribaculaceae, the genus Lachnospiraceae NK4A136 group, and the genus Roseburia. We confirmed that Lachnospiraceae NK4A136 group and Roseburia were the top two genera with the highest relative abundances in SPF mice at 4 weeks of age. These findings support the notion that intestinal mucosal gene expression patterns early in life serve as a selection mechanism to promote colonization of specific bacteria.
In our enrichment analysis of expressed genes in the ileal mucosa, the transport of small molecules was the top feature in mice at 4 weeks of age, and Slc9a3, encoding solute carrier family 9, member 3 (a sodium/hydrogen exchanger), was included in this feature. Interestingly, Roseburia decreases in Slc9a3 knockout mice45. This finding is compatible with our result that Slc9a3 expression was advantageous for colonization of Roseburia. St3gal6, encoding st3 beta-galactoside alpha-2,3-sialyltransferase 6, was included in the top five genes with the strongest correlation to colonization of the Lachnospiraceae NK4A136 group genera and Roseburia. Sialyltransferases are involved in the production of sialic acids on mucosal surfaces, and sialic acids serve as a nutrient source for bacteria. A sialic acid, N-glycolylneuraminic acid, promotes the growth of Roseburia46. Conversely, it is interesting that the top feature in the ileal mucosa at 10 weeks of age was a defense response to gram-positive bacteria, and Roseburia, a gram-positive bacterium, decreased at 10 weeks of age. The genes included in this feature are involved in the production and activation of immunoglobulin A and antimicrobial peptides such as cryptdin, lysozyme, and regenerating islet-derived protein 3-γ (Reg3-γ)47,48,49. It has been reported that knockout mice of these protective factors develop gut dysbiosis48,50. In particular, Reg3-γ is induced by IL-33, and it Il33 knockout mice exhibit an increase in the genera Lachonospiraceae NK4A136 group, Allistipes, Bacteroides, Ruminococcus, and Ruminiclostridium49. Overall, our findings that Lachnospiraceae NK4A136 group and Roseburia were positively correlated to genes predominantly expressed early in life and negatively correlated to those expressed later in life appeared reasonable from the viewpoint of gene functions. Our results suggest that the intestinal mucosal immune/defense system develops with age even without stimuli from the microbiome; that is, aging itself is sensed by cells responsible for the defense system, and bacteria that adapt to the continuously changing intestinal environment are selected as commensal gut microbes. A healthy development process of the immune system might exist along with aging, whereas immunosenescence later in life can cause diseases.
Our study identified the genera Lachnospiraceae NK4A136 group and Roseburia as bacteria that flourish in the host early in life because the young host provides an environment that positively affects the colonization of these genera. Lachnospiraceae NK4A136 group and Roseburia are short-chain fatty acid-producing bacteria that play beneficial roles in the host51,52. Ma et al. reported that Lachnospiraceae NK4A136 group produces butyrate and contributes to reducing the intestinal inflammatory response and improving permeability in diet-induced obese mice53. Xia et al. demonstrated that an increase in Lachnospiraceae NK4A136 group by polyphenol treatment reduces intestinal lipopolysaccharide-mediated inflammation after ethanol treatment54. Additionally, Roseburia intestinalis produces butylate that alleviates dextran sulfate sodium (DSS)-induced enteritis55 and induces regulatory T cells, leading to suppression of trinitrobenzene sulfonic acid-induced enteritis56. These findings support the notion that the host early in life has a mechanism to establish symbiotic relationships with beneficial short chain fatty acid-producing bacteria, such as Lachnospiraceae NK4A136 group and Roseburia, for healthy development. Although further functional studies are needed to investigate the underlying mechanisms of how mucosal gene expression positively or negatively affects the colonization of symbionts early in life, these mechanisms may be targets for microbiome interventions to correct dysbiosis.
The present study has some limitations. FMT has methodological limitations in reproducing the donor microbiome in recipients38,39. Gut microbiome samples are exposed to oxygen during the procedure and impacts the viability of anaerobic bacteria, particularly making it challenging to reproduce the balance of aerobic vs. anaerobic bacteria in FMT recipients. Additionally, the influence of recipient digestive fluids can be a survival bias for colonized microbes in FMT. Furthermore, we employed GF animals to investigate the aging effect on gene expression in the intestinal mucosa, but the intestinal gene expression may be affected by microbes via their components and metabolites, and the gene expression profiles of GF and SPF animals may be different. However, because gut microbes integrally affect the host over time from birth and it is impossible to distinguish the effect of the microbiome and aging on intestinal mucosal gene expression in a host, we believe that our study design using GF animals has scientific merit to demonstrate that aging itself affects gene expression in the intestinal mucosa, excluding the influence of the microbiome. Meanwhile, the limited sample size of this study can be a limitation in interpreting the data analysis, including statistics. Moreover, because gut microbial compositions are different among host species57,58 and the critical development period with morphological/immunological plasticity remains unclear in humans, further studies using human samples are needed to understand microbiome development in humans early in life. Nonetheless, this translational study provides the proof of concept that intestinal gene expression early in life serves as a mechanism to select symbionts for a specific development period to achieve health throughout life.
Conclusion
This mouse study demonstrated that intestinal bacterial colonization following FMT is determined by the age of the recipient rather than that of the donor. Furthermore, gene expression patterns of the intestinal mucosa characteristic in early life correlated with bacterial genera that preferentially colonize during this period. Given the potential beneficial effects of these genera, our findings suggest that the characteristic gene expression profile of the intestinal mucosa early in life promotes selective bacterial colonization, thereby contributing to the healthy development of both the host and the gut microbiome.
Future directions of this research include elucidating the roles of microbes that preferentially colonize early in life and identifying key factors involved in establishing a healthy gut microbiome and promoting host health. These investigations may provide valuable insights into novel targets for therapeutic and preventive interventions.
Data availability
The accession number of the microbial dataset and the mouse sample information reported in this study is DRA: DRA017536, DRA017537 (DNA Data Bank of Japan [DDBJ]. https://www.ddbj.nig.ac.jp). The datasets generated during and/or analyzed during the current study are provided as supporting files.
Abbreviations
- ASV:
-
Amplicon sequencing variant
- ExGF:
-
Ex-germ free
- FMT:
-
Fecal microbiota transfer
- GF:
-
Germ free
- GO:
-
Gene Ontology
- Ig:
-
Immunoglobulin
- IL:
-
Interleukin
- PBS:
-
Phosphate buffered saline
- PCoA:
-
Principal coordinate analysis
- SPF:
-
Specific pathogen-free
References
Afzaal, M. et al. Human gut microbiota in health and disease: Unveiling the relationship. Front. Microbiol. 13, 999001 (2022).
Chen, C. et al. Contribution of host genetics to the variation of microbial composition of cecum lumen and feces in pigs. Front. Microbiol. 9, 2626 (2018).
Org, E. et al. Sex differences and hormonal effects on gut microbiota composition in mice. Gut Microbes. 7(4), 313–322 (2016).
Bibbò, S. et al. The role of diet on gut microbiota composition. Eur. Rev. Med. Pharmacol. Sci. 20(22), 4742–4749 (2016).
Vatanen, T. et al. Variation in microbiome LPS immunogenicity contributes to autoimmunity in humans. Cell 165(4), 842–853 (2016).
Suzuki, T. A. & Worobey, M. Geographical variation of human gut microbial composition. Biol. Lett. 10(2), 20131037 (2014).
Vilchez-Vargas, R. et al. Gut microbial similarity in twins is driven by shared environment and aging. EBioMedicine 79, 104011 (2022).
Claus, S. P. et al. Colonization-induced host-gut microbial metabolic interaction. MBio 2(2), e00271-e310 (2011).
Strandwitz, P. Neurotransmitter modulation by the gut microbiota. Brain Res. 1693(Pt B), 128–133 (2018).
Moor, K. et al. High-avidity IgA protects the intestine by enchaining growing bacteria. Nature 544(7651), 498–502 (2017).
Grasset, E. K. et al. Gut T cell-independent IgA responses to commensal bacteria require engagement of the TACI receptor on B cells. Sci. Immunol. 5(49), eaat7117 (2020).
Bunker, J. J. et al. Innate and adaptive humoral responses coat distinct commensal bacteria with immunoglobulin A. Immunity 43(3), 541–553 (2015).
Nishida, A., Miyamoto, J., Shimizu, H. & Kimura, I. Gut microbial short-chain fatty acids-mediated olfactory receptor 78 stimulation promotes anorexigenic gut hormone peptide YY secretion in mice. Biochem. Biophys. Res. Commun. 557, 48–54 (2021).
Wahlström, A., Sayin, S. I., Marschall, H. U. & Bäckhed, F. Intestinal crosstalk between bile acids and microbiota and its impact on host metabolism. Cell Metab. 24(1), 4–50 (2016).
Tahara, Y. et al. Gut microbiota-derived short chain fatty acids induce circadian clock entrainment in mouse peripheral tissue. Sci. Rep. 8(1), 1395 (2018).
Takakura, W. et al. Exhaled methane is associated with a lower heart rate. Cardiology 147(2), 225–229 (2022).
Alexeev, E. E. et al. Microbiota-derived indole metabolites promote human and murine intestinal homeostasis through regulation of interleukin-10 receptor. Am. J. Pathol. 188(5), 1183–1194 (2018).
Kamphorst, K. et al. Early life antibiotics and childhood gastrointestinal disorders: A systematic review. BMJ Paediatr. Open. 5(1), e001028 (2021).
Zou, Z., Liu, W., Huang, C., Sun, C. & Zhang, J. First-Year antibiotics exposure in relation to childhood asthma, allergies, and airway illnesses. Int. J. Environ. Res. Public Health. 17(16), 5700 (2020).
Metzler, S. et al. Association between antibiotic treatment during pregnancy and infancy and the development of allergic diseases. Pediatr. Allergy Immunol. 30(4), 423–433 (2019).
Miyoshi, J. et al. Peripartum antibiotics promote gut dysbiosis, loss of immune tolerance, and inflammatory bowel disease in genetically prone offspring. Cell Rep. 20(2), 491–504 (2017).
Miyoshi, J. et al. Metagenomic alterations in gut microbiota precede and predict onset of colitis in the IL10 gene-deficient murine model. Cell Mol. Gastroenterol. Hepatol. 11(2), 491–502 (2021).
Miyoshi, J. et al. Early-life microbial restitution reduces colitis risk promoted by antibiotic-induced gut dysbiosis in interleukin 10−/− mice. Gastroenterology 161(3), 940-952.e15 (2021).
Martínez, I. et al. Experimental evaluation of the importance of colonization history in early-life gut microbiota assembly. Elife 7, e36521 (2018).
Al Nabhani, Z. et al. A weaning reaction to microbiota is required for resistance to immunopathologies in the adult. Immunity 50(5), 1276-1288.e5 (2019).
Cahenzli, J., Köller, Y., Wyss, M., Geuking, M. B. & McCoy, K. D. Intestinal microbial diversity during early-life colonization shapes long-term IgE levels. Cell Host Microbe. 14(5), 559–570 (2013).
Xu, C., Zhu, H. & Qiu, P. Aging progression of human gut microbiota. BMC Microbiol. 19(1), 236 (2019).
Ragonnaud, E. & Biragyn, A. Gut microbiota as the key controllers of “healthy” aging of elderly people. Immun. Ageing. 18(1), 2 (2021).
Wada, H. et al. 5-Aminosalicylic acid alters the gut microbiota and altered microbiota transmitted vertically to offspring have protective effects against colitis. Sci. Rep. 13(1), 12241 (2023).
Wang, Y. et al. 16S rRNA gene-based analysis of fecal microbiota from preterm infants with and without necrotizing enterocolitis. ISME J. 3(8), 944–954 (2009).
Bolyen, E. et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 37(8), 852–857 (2019).
Callahan, B. J. et al. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods. 13(7), 581–583 (2016).
Oguri, N. et al. Akkermansia muciniphila in the small intestine improves liver fibrosis in a murine liver cirrhosis model. NPJ Biofilms Microbiomes. 10(1), 81 (2024).
Martinez-Guryn, K. et al. Small intestine microbiota regulate host digestive and absorptive adaptive responses to dietary lipids. Cell Host Microbe. 23(4), 458-469.e5 (2018).
Frazier, K. et al. High-fat diet disrupts REG3γ and gut microbial rhythms promoting metabolic dysfunction. Cell Host Microbe. 30(6), 809-823.e6 (2022).
Zhou, Y. et al. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 10(1), 1523 (2019).
Rohart, F., Gautier, B., Singh, A. & Lê Cao, K. A. mixOmics: An R package for ’omics feature selection and multiple data integration. PLoS Comput. Biol. 13(11), e1005752 (2017).
O’Callaghan, J. L., Willner, D., Buttini, M., Huygens, F. & Pelzer, E. S. Limitations of 16S rRNA gene sequencing to characterize lactobacillus species in the upper genital tract. Front. Cell Dev. Biol. 9, 641921 (2021).
Caudill, M. T. & Brayton, K. A. The use and limitations of the 16S rRNA sequence for species classification of anaplasma samples. Microorganisms. 10(3), 605 (2022).
Litichevskiy, L. & Thaiss, C. A. Microbiome complexity shapes metabolism. PLoS Biol. 20(9), e3001793 (2022).
Daniel, N., Lécuyer, E. & Chassaing, B. Host/microbiota interactions in health and diseases-Time for mucosal microbiology!. Mucosal. Immunol. 14(5), 1006–1016 (2021).
Yatsunenko, T. et al. Human gut microbiome viewed across age and geography. Nature 486(7402), 222–227 (2012).
Brodziak, F., Meharg, C., Blaut, M. & Loh, G. Differences in mucosal gene expression in the colon of two inbred mouse strains after colonization with commensal gut bacteria. PLoS ONE 8(8), e72317 (2013).
Nakamura, K., Sakuragi, N. & Ayabe, T. A monoclonal antibody-based sandwich enzyme-linked immunosorbent assay for detection of secreted α-defensin. Anal. Biochem. 443(2), 124–131 (2013).
Xue, J., Dominguez Rieg, J. A., Thomas, L., White, J. R. & Rieg, T. Intestine-specific NHE3 deletion in adulthood causes microbial dysbiosis. Front. Cell Infect Microbiol. 12, 896309 (2022).
Zaramela, L. S. et al. Gut bacteria responding to dietary change encode sialidases that exhibit preference for red meat-associated carbohydrates. Nat. Microbiol. 4(12), 2082–2089 (2019).
Johansen, F. E., Braathen, R. & Brandtzaeg, P. Role of J chain in secretory immunoglobulin formation. Scand. J. Immunol. 52(3), 240–248 (2000).
Salzman, N. H. Paneth cell defensins and the regulation of the microbiome: détente at mucosal surfaces. Gut Microbes. 1(6), 401–406 (2010).
Xiao, Y. et al. Interleukin-33 promotes REG3γ expression in intestinal epithelial cells and regulates gut microbiota. Cell Mol. Gastroenterol. Hepatol. 8(1), 21–36 (2019).
Yu, S. et al. Paneth cell-derived lysozyme defines the composition of mucolytic microbiota and the inflammatory tone of the intestine. Immunity 53(2), 398-416.e8 (2020).
Molino, S., Lerma-Aguilera, A., Jiménez-Hernández, N., Rufián Henares, J. Á. & Francino, M. P. Evaluation of the effects of a short supplementation with tannins on the gut microbiota of healthy subjects. Front. Microbiol. 13, 848611 (2022).
Nie, K. et al. Roseburia intestinalis: A beneficial gut organism from the discoveries in genus and species. Front. Cell Infect. Microbiol. 11, 757718 (2021).
Ma, L. et al. Spermidine improves gut barrier integrity and gut microbiota function in diet-induced obese mice. Gut Microbes. 12(1), 1–19 (2020).
Xia, T. et al. Polyphenol-rich vinegar extract regulates intestinal microbiota and immunity and prevents alcohol-induced inflammation in mice. Food Res. Int. 140, 110064 (2021).
Ruan, G. et al. Roseburia intestinalis and its metabolite butyrate inhibit colitis and upregulate TLR5 through the SP3 signaling pathway. Nutrients 14(15), 3041 (2022).
Shen, Z. et al. Roseburia intestinalis stimulates TLR5-dependent intestinal immunity against Crohn’s disease. EBioMedicine 85, 104285 (2022).
Ley, R. E. et al. Evolution of mammals and their gut microbes. Science 320(5883), 1647–1651 (2008).
Ley, R. E., Lozupone, C. A., Hamady, M., Knight, R. & Gordon, J. I. Worlds within worlds: Evolution of the vertebrate gut microbiota. Nat. Rev. Microbiol. 6(10), 776–788 (2008).
Acknowledgements
We thank Mitchell Arico from Edanz (https://jp.edanz.com/ac) for editing a draft of this manuscript. We also thank Sachie Matsubara and Junri Hayakawa (Laboratory for Electron Microscopy, Kyorin University School of Medicine) and Eriko Nozaki (Core Laboratory for Proteomics and Genomics, Kyorin University School of Medicine) for their technical contribution to electron microscopic analysis, metagenomic shotgun sequencing, and mRNA microarray.
Funding
This study was supported by a Grant-in-Aid for Scientific Research (KAKENHI: 19K23977 and 21K07900) and in part by grants from the Japan Sciences Research Grant for Research on Intractable Diseases (Japanese Inflammatory Bowel Disease Research Group) affiliated with the Japan Ministry of Health Labour and Welfare.
Author information
Authors and Affiliations
Contributions
Y.N., J.M., M.M., and T.H. conceived the study, designed experiments, and prepared the manuscript. Y.N., J.M., S.K., H.W., N.O., N.H., O.T., Y.A., and S.T.M.L. performed experiments and analyzed data. S.T.M.L. particularly led the microbiome analysis. M.M., T.K., T.H., and T.H. supervised the manuscript. J.M. and T.H. oversaw the entire project. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Nishinarita, Y., Miyoshi, J., Kuronuma, S. et al. Characteristic gene expression profile of intestinal mucosa early in life promotes bacterial colonization leading to healthy development of the intestinal environment. Sci Rep 15, 18437 (2025). https://doi.org/10.1038/s41598-025-03661-w
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-03661-w
