
Abstract
Antimicrobial peptides (AMPs) are considered one of the most promising new antimicrobial agents to combat antibiotic resistance and have garnered significant attention over the past few decades. However, their development has been hindered by high manufacturing costs, toxicity, and poor enzyme tolerance. In this study, we employed bioinformatics tools to simulate the trypsin cleavage of Esculentin-2P (E2P), a frog-derived AMP with 37 amino acids and performed functional screening to identify its effective active fragments. Building on the best derivative, des-(Asp20-Cys37)-E2P, which demonstrated considerable antimicrobial activity, minimal haemolysis, and low cytotoxicity, we introduced the naturally occurring antimicrobial amino acid homo-arginine for further modification. The results showed that the derivate, [hArg7,11,15,19]-des-(Asp20-Cys37)-E2P, not only optimised the antimicrobial activity of des-(Asp20-Cys37)-E2P but also exhibited lower toxicity with a selectivity index of 40.6, improved tolerance to variable environments such as salts, heat, and trypsin, and a reduced likelihood of resistance development. Mechanism studies revealed that the peptide exerts its bactericidal effects by disrupting the bacterial membrane. Additionally, it demonstrated strong therapeutic efficacy in treating Galleria mellonella models infected with drug-resistant Escherichia coli. These results suggest that [hArg7,11,15,19]-des-(Asp20-Cys37)-E2P held great promise as a novel antimicrobial candidate against resistant pathogens. The cleavage-mimic truncation strategy, combined with the incorporation of homo-arginine, offers valuable insights for developing potent, shorter AMPs with enhanced therapeutic potential. This approach may help address some key challenges in peptide-drug development, paving the way for more effective antimicrobial therapies.
Similar content being viewed by others

Introduction
Antibiotic resistance is an escalating global health crisis, driven by the overuse and misuse of antibiotics, leading to the rise of multidrug-resistant bacteria that render many conventional treatments ineffective1. The World Health Organization (WHO) has identified antibiotic resistance as one of the most significant threats to global health, food security, and development2. This alarming trend highlights the urgent need for alternative therapeutic strategies.
One promising alternative to traditional antibiotics is antimicrobial peptides (AMPs), small naturally occurring molecules with broad-spectrum antimicrobial activity3. AMPs offer several advantages over conventional antibiotics, including rapid bactericidal action, a broad range of pathogen targets, and a lower likelihood of resistance development due to their unique mechanism of action4. These properties make AMPs potential game-changers in the fight against antibiotic-resistant bacteria. However, several challenges hinder the clinical application of AMPs, particularly their instability and susceptibility to protease degradation5,6. This sensitivity to proteolysis limits their therapeutic potential, often restricting their use to localised applications and necessitating modifications to enhance their stability7,8. Additionally, the high cost of synthesis and low selectivity have significantly impeded the broader development of AMPs. As a result, research has frequently focused on finding efficient strategies to balance peptide length with bioactivity to optimise therapeutic potential.
Various strategies have been explored to increase the proteolytic resistance of AMPs, including the incorporation of D-amino acids, N-acetylation, C-amidation, cyclisation, PEGylation, and lipidation9,10,11,12,13,14. However, a recurring challenge in these approaches is that improving protease stability often compromises antibacterial efficacy or increases toxicity. For example, lipidation may enhance cytotoxicity and negatively impact the pharmacokinetics of peptides15, while the addition of D-amino acids may reduce the hydrophobicity of AMPs, diminishing their antibacterial effectiveness16,17,18.
Homoarginine (hArg), a nonproteinogenic amino acid, has emerged as a promising modification for enhancing AMP stability and efficacy19. It has been shown to improve protease resistance20, boost antimicrobial activity21, and reduce cytotoxicity towards mammalian cells22. In this study, Esculentin-2P, a potent AMP with 37 amino acids from the skin secretions of Lithobates pipiens, was used as a template to explore an efficient modification strategy for developing a shorter, enhanced therapeutic AMP. The peptide was synthesized using solid-phase peptide synthesis (SPPS) and purified through reverse-phase high-performance liquid chromatography (RP-HPLC). Bioinformatics tools and biological activity assays were applied to design and evaluate a series of truncated analogues by simulating trypsin cleavage sites, followed by the incorporation of hArg. These analogues were systematically assessed for their antimicrobial activity, cytotoxicity, resistance development, and potential mechanisms of action, with the aim of identifying peptides with strong antimicrobial activity, high selectivity, improved stability, and low toxicity. Furthermore, the relationship between peptide structure and biological function was explored to provide valuable insights for future AMP design and modifications.
Methods and materials
Synthesis and characterisation of peptides
Peptides used in this study were synthesised using a Tribute solid-phase peptide synthesiser (Gyros Protein Technologies, USA). Fmoc-Cys(Trt)-Wang resin (Novabiochem, China) was employed for synthesising Esculentin-2P, while MBHA resin (Novabiochem, China) was used for the synthesis of other peptides, which were amidated at the C-terminus. Fmoc-hArg(Pbf)-OH (Sigma-Aldrich, UK) was utilised in the synthesis of peptide 7. Dimethyl sulfoxide (DMSO) was used to facilitate disulfide bond formation in Esculentin-2P. Peptides were purified via reverse-phase high-performance liquid chromatography (HPLC) (1260 Infinity II HPLC, Agilent, UK) using a C18 column (21.2 × 250 mm, 5 µm, Agilent, UK) with a linear gradient from 100% buffer A (99.5/0.05 (v/v), ddH₂O/TFA) to 100% buffer B (19.95/80.00/0.05 (v/v/v), ddH₂O/acetonitrile/TFA) over 60 min at a flow rate of 4 mL/min. The purified peptides were characterized by matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectrometry (4800 MALDI-TOF/TOF, Applied Biosystems, USA).
Circular dichroism (CD) spectroscopy
A JASCO-815 CD spectrometer (Jasco, UK) was used to analyse the secondary structure of Esculentin-2P and its derivatives23,24. Peptide samples were prepared at a concentration of 50 µM in 10 mM NH4AC and 50% TFE/NH4AC (v/v) solutions. Measurements were conducted in quartz cuvettes with a path length of 1 mm, scanning across a range of 190 to 260 nm. The scanning speed was set at 100 nm/min, with a bandwidth of 1 nm and a data pitch of 0.5 nm. Online server K2D3 (https://cbdm-01.zdv.uni-mainz.de/~andrade/k2d3/) was used to calculate the potential α-helical content of peptides in TFE solutions.
Antimicrobial assay
The minimal inhibitory concentration (MIC) and minimal bactericidal concentration (MBC) assays were performed to evaluate the antibacterial activity of Esculentin-2P and its derivatives25,26. Ten bacterial strains were tested, including three Gram-positive strains: S. aureus ATCC 6538, MRSA NCTC 12,493, E. faecalis NCTC 12,697, and seven Gram-negative strains: E. coli ATCC 8739, E. coli NCTC 13,846, E. coli ATCC 2340, E. coli ATCC 2469, K. pneumoniae ATCC 43,816, A. baumannii ATCC BAA 747, and P. aeruginosa ATCC 27,853. Peptides were prepared in two-fold dilutions ranging from 128 µM to 1 µM and incubated with 5 × 105 CFU/mL bacterial suspensions at 37 °C overnight to determine the MIC values. Mixtures of peptide and bacterial suspensions showing no visible growth were spotted on the MHA agar plate and incubated overnight to determine MBC values. Each experiment was repeated three times, with three replicates for each sample in every experiment.
Time-killing kinetics assay
The time-kill kinetics assay was employed to evaluate the bactericidal efficacy of the peptides against E. coli ATCC 8739. Bacterial cultures at a concentration of 5 × 105 CFU/mL were incubated with peptides at four-fold MIC, two-fold MIC, and MIC concentrations. Viable bacterial cells were assessed by collecting samples at different time intervals (0, 5, 10, 20, 30, 60, 90, 120, and 180 min), followed by overnight incubation at 37 °C for colony quantification27,28. The results were derived from three independent experiments, each performed in triplicate.
Conditional sensitivity assays
The sensitivity of the peptides to physiological salts, serum, and trypsin was evaluated. For salt and serum sensitivity, peptides were incubated with 5 × 105 CFU/mL of E. coli ATCC 8739 in the presence of 0.5 mM KCl, 2.5 mM CaCl2, 150 mM NaCl, 1 mM MgCl2, 8 mM ZnCl2, 4 μM FeCl3, 6 μM NH4Cl, 10% FBS, and 20% FBS28,29. To assess peptide sensitivity toward trypsin, peptides were incubated with trypsin solutions at final concentrations of 0.5 mg/mL, 1 mg/mL, and 2 mg/mL at 37 °C for 1 h. The mixtures were then heated at 80 °C for 10 min to inactivate trypsin. Following this treatment, the MIC/MBC assay was performed to assess any alterations in the antimicrobial activity of the peptides.
Biocompatibility assays
The haemolytic activity of Esculentin-2P and its derivatives was assessed using fresh defibrinated equine blood (E&O Laboratories Ltd, UK)30. Briefly, defibrinated equine erythrocytes were washed with sterile PBS and diluted to a 4% (v/v) suspension. Peptides were prepared in two-fold dilutions ranging from 128 µM to 1 µM and incubated with the erythrocyte suspension at 37 °C for 2 h. PBS and 1% (v/v) Triton X-100 were used as the blank and positive controls, respectively. After incubation, the samples were centrifuged, and the supernatant was transferred to a 96-well plate for absorbance measurement at 570 nm using a Synergy HT plate reader (BioTek, USA). The percentage of haemolysis was calculated using the following formula:
As: sample group; An: negative group; Ap: positive group.
The cytotoxicity of Esculentin-2P and its derivatives toward human healthy cells was evaluated using the human embryonic kidney cell line HEK-293 (Calta Medsystems, UK)25. In brief, HEK-293 cells (2.5 × 104 cells per well) were cultured in MEM medium (Gibco™, USA) supplemented with 1% (v/v) penicillin–streptomycin (PS, Gibco™, USA) and 10% (v/v) fetal bovine serum (FBS, Gibco™, USA). The cells were incubated with peptides in ten-fold dilutions ranging from 10–4 to 10–7 M at 37 °C overnight. Following incubation, MTT reagent (5 mg/mL) was added to each well, and the cells were incubated for an additional 4 h at 37 °C. Afterwards, the medium was replaced with 100 μL of DMSO, the plate was shaken for 15 min, and absorbance was measured at 570 nm.
Resistance development assay
E. coli ATCC 8739 was used as a model for the resistance development study. For twenty consecutive days (twenty passages), bacteria were passaged every 24 h in MHB containing sub-MIC concentrations of the peptide or antibiotics31. The MIC value was determined daily to monitor any changes over the course of the experiment.
LPS-binding assay
The binding affinity of the peptides to lipopolysaccharide (LPS) was evaluated using the BODIPY-TR cadaverine (BC) displacement assay23,25. LPS (25 μg/mL, Sigma-Aldrich, UK) and BC dye (2.5 μg/mL, ThermoFisher, USA) were incubated at room temperature for 4 h. Peptides, including positive control Melittin, were prepared in Tris–HCl buffer (pH 7.4) at concentrations ranging from 0.5 μM to 32 μM. The LPS-BC mixture was then added to a black 96-well plate and incubated at 37 °C for 1 h. Fluorescence measurements were taken using a Synergy HT plate reader (BioTek, USA) with an excitation wavelength of λ = 590 nm and an emission wavelength of λ = 645 nm. Each test was conducted in triplicate, with experiments performed independently.
Outer membrane permeability assay
The NPN (N-phenyl-1-naphthylamine) fluorescent dye assay was used to evaluate the impact of the peptides on the permeability of the Gram-negative bacterial outer membrane30,32. Bacteria in the logarithmic growth phase were centrifuged at 2000 × g for 10 min and washed twice with 5 mM glucose and 5 mM HEPES buffer (pH 7.2). The bacteria were then resuspended in HEPES buffer to a density corresponding to an OD600 of 0.5. A black 96-well plate was prepared with 100 μL of bacterial culture and 50 μL of peptide solutions (at 1 × MIC, 2 × MIC, and 4 × MIC), and incubated for 2 h at 37 °C. Melittin was used as the positive control. After incubation, 50 μL of NPN was added, and fluorescence was measured using a Synergy HT plate reader with excitation and emission wavelengths set at 360 nm and 460 nm, respectively.
Cytoplasmic membrane permeability assay
The cytoplasmic membrane permeability assay was employed to analyse the effect of peptides on membrane integrity33,34. In summary, bacterial strains in the logarithmic growth phase were harvested by centrifugation at 1000 × g for 10 min. The bacterial pellet was then resuspended in 5% TSB (dissolved in 0.85% NaCl) at a density of 1 × 108 CFU/mL after two washes. The bacterial suspensions were treated with peptides at doses of 1 × MIC, 2 × MIC, and 4 × MIC for 2 h at 37 °C. Subsequently, SYTOX™ Green Nucleic Acid Stain (ThermoFisher, USA) was added and incubated for 10 min at 37 °C in a shaking incubator. Fluorescence variations were measured using a Synergy HT plate reader with excitation and emission wavelengths set at 485 nm and 528 nm, respectively.
Cytoplasmic membrane depolarization assay
Changes in cytoplasmic membrane potential were measured using the membrane potential-sensitive fluorescent dye Disc₃(5) (ThermoFisher, USA)25,30. E. coli ATCC 8739 was washed twice with HEPES buffer and then resuspended in HEPES buffer (5 mM, pH 7.4) containing glucose (20 mM) and KCl (100 mM) to an OD600 of 0.05. 10 μL of the bacterial suspension was incubated with 200 µL of Disc₃(5) (20 µM) for 1 h in the dark. In a black 96-well plate, 100 µL of the bacterial mixture was first added, and fluorescence was measured at 1 min intervals over a 5 min period using a plate reader with excitation at 485 nm and emission at 645 nm. Then, 90 µL of the bacterial suspension was added to the plate, along with 10 µL of peptide solutions at various concentrations and the positive control Melittin. Fluorescence was recorded at 1 min intervals over a 15 min period.
Fluorescence imaging
Fluorescent dyes SYTO9 (ThermoFisher, USA) and Propidium iodide (PI, Sigma, UK) were used to stain bacteria treated with peptides as described previously24,25. Briefly, bacteria in the logarithmic growth phase were washed three times with PBS and then incubated with peptides at concentrations ranging from 1 × MIC to 4 × MIC at 37 °C for 2 h. After incubation, the bacteria were collected by centrifugation and stained with SYTO9 for 15 min. Following a wash to remove excess dye, the cells were then stained with PI for an additional 15 min. After a final wash to remove the unbound dye, the bacteria were examined using a fluorescent microscope (DMi8, Leica, Wetzlar, Germany) with a 100 × oil-immersion objective.
Antimicrobial activity in blood
The antimicrobial activity of peptides in a blood environment was assessed following a modified version of Li et al.’s protocol35. Briefly, horse whole blood (E&O Laboratories Ltd, UK) was inoculated with 10⁸ CFU/mL of bacteria, then divided into separate tubes (50 µL per tube) and treated with peptide or antibiotic solutions. The samples were incubated at 37 °C for 6 h. Every hour, the samples were diluted tenfold in PBS, and 10 µL of the diluted mixture was plated on MHB agar. After overnight incubation at 37 °C, the colonies were counted.
Antimicrobial activity in vivo
The in vivo antibacterial activity of peptide 7 against three drug-resistant E. coli strains (E. coli NCTC 13,846, E. coli ATCC 2340, and E. coli ATCC 2469) was evaluated using Wax Moth Larvae (Galleria mellonella) (Livefood UK Ltd., UK)29. Ten larvae weighing 250 ± 20 mg were included in each treatment group. The larvae were infected with 10 µL of a bacterial suspension (1 × 10⁷ CFU/mL), and one hour post-infection, they were treated with 10 µL of peptide 7 at MIC, 2 × MIC, and 4 × MIC concentrations (corresponding to 80, 160, and 320 mg/kg). Colistin at 10 × MIC (50 mg/kg) was used as a positive control, while PBS was used as a negative control. The larvae were incubated at 24 °C and observed every 24 h for five days to monitor survival.
Statistical analysis
All data were analysed using GraphPad Prism 9 software (GraphPad, San Diego, CA, USA). Error bars in all graphs represented the standard error of the mean (SEM) from nine replicates across three independent experiments. P values were determined through one-way analysis of variance (ANOVA) to compare the mean values of the specified data. Significant differences were indicated by asterisks (*p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001).
RESULTS
Modification and characterisation of E2P and its derivates
The truncation strategy for the E2P sequence was informed by simulations of trypsin cleavage sites, with the goal of reducing peptide length while identifying the shortest fragment with optimal therapeutic potential. Excluding the possible protective function of the rana box, five cleavage sites were identified within the E2P peptide (Fig. 1a). Based on these cleavage points, five potential derivatives were designed, with amino acid lengths ranging from 7 to 23 residues (Table 1). Following an initial screening for antimicrobial activity and toxicity, des-(Asp20-Cys37)-E2P (peptide 2) emerged as the optimal candidate, exhibiting a balanced profile of sequence length, high activity, minimal haemolysis, and low cytotoxicity. As a result, peptide 2 was selected as the template for subsequent modifications aimed at enhancing its antibacterial efficacy and biocompatibility. One modification involved introducing tryptophan at positions 2, 5, 6, 9, 12, and 17 of the peptide 2 sequence. The bulky side chain of tryptophan may strengthen hydrophobic interactions with bacterial membranes, thereby augmenting the antibacterial properties of the analogue [Trp2,5,6,9,12,17]-des-(Asp20-Cys37)-E2P (peptide 6). In the second modification plan, the positively charged residues in peptide 2 were replaced with the naturally occurring antimicrobial amino acid hArg, to optimise antibacterial activity while also improving peptide tolerance to enzymatic degradation (Fig. 1B–D). MALDI-TOF MS analysis revealed that the measured molecular weights of all peptides were close to their theoretical values, confirming successful synthesis and purification. Additionally, HPLC analysis indicated that the purity of all peptides exceeded 90% (Figure S1).

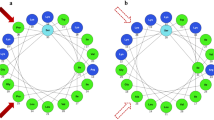
(A) Truncation on Esculentin-2P based on the simulations of trypsin cleavage sites (left). Predicted 3-D model of E2P by PEPFOLD4 server (right). (B) Molecular three-dimensional models of Lysine, Arginine, and Homoarginine. (C) Helical wheel projection of the peptide [hArg7,11,15,19]-des-(Asp20-Cys37)-E2P. (D) The sequence and structural scheme of the peptide [hArg7,11,15,19]-des-(Asp20-Cys37)-E2P.
Secondary structure of E2P and its derivatives
E2P adopted a random structure in an aqueous environment but displayed a strongly α-helical conformation in TFE solution, as indicated by distinct positive peaks around 193 nm and negative peaks at 208 nm and 222 nm (Fig. 2). Although truncation modifications reduced the amino acid length of E2P derivatives, they still exhibited a similar structural pattern, predominantly adopting a random structure in NH4AC solution and an α-helical conformation in TFE. According to calculations from the K2D3 online server, the α-helical content of E2P and its derivatives in TFE consistently exceeded 92%. Compared to the template peptide 2, the structural conformations of peptides 6 and 7 remained largely unchanged, maintaining a high degree of α-helical structure (Table 2).

CD spectra of E2P and its analogues. The measurement was conducted under the environments of 10 mM NH4AC (Shown in blue) and 50% TFE in 10 mM NH4AC (Shown in red).
Antimicrobial activities
As shown in Table 3, E2P demonstrated potent antimicrobial activity against all tested bacterial strains, with a MIC of 1 or 2 µM. The geometric mean (GM) MIC of E2P against all bacteria was 1.2 µM, much lower than that of the other tested peptides. As the peptide length was truncated, the antimicrobial activities of peptides 1, 2, and 3 gradually decreased, with complete loss of activity observed in peptides 4 and 5. During this process, the antimicrobial activity against Gram-positive bacteria was more substantially affected compared to Gram-negative bacteria. Based on peptide 2, the introduction of tryptophan in peptide 6 did not lead to a remarkable improvement in antibacterial activity, with the value of GM MIC of all tested bacteria only slightly decreased from 22.6 µM to 19.7 µM. In contrast, incorporating hArg substantially enhanced the antibacterial efficacy of peptide 7, reducing the GM MIC of all tested bacteria to 5.6 µM, which is markedly stronger than that of the other derivatives.
Time-killing kinetics
E2P and its derivatives exhibited bactericidal activity against E. coli ATCC 8739 at the tested concentrations, with killing rates positively correlated to concentration levels (Fig. 3). For E2P, complete bacterial eradication required 30 min at the MIC, 20 min at 2 × MIC, and 10 min at 4 × MIC. In comparison, peptide 1 showed a slower killing rate, needing 90 min to achieve bactericidal efficacy at the MIC and 30 min at 2 × MIC and 4 × MIC. Further truncation in the peptide 2 resulted in a requirement of 120 min for bacterial elimination at the MIC, although its bactericidal efficacy at higher concentrations was comparable to that of Esculentin-2P. In addition, the peptide 3, the derivative with the shortest sequence, demonstrated the fastest bactericidal activity, achieving complete eradication within just 20 min at the MIC. Peptide 6 was four times faster in killing bacteria compared to peptide 2 at MIC concentrations. Conversely, peptide 7 had a bactericidal rate three times slower than peptide 2 at 2 × MIC but achieved complete bacterial eradication in just 5 min at 4 × MIC.

The kinetic time-kill curves of E2P and its derivatives against E. coli ATCC 8739.
Salts and serum sensitivity
E2P exhibited robust antimicrobial activity at concentrations of 1–2 µM under various conditions, including the presence of salts and FBS, with the exception of calcium ions (Table 4). Similarly, calcium ions greatly compromised the antibacterial properties of the truncated derivatives, resulting in activity levels that exceeded the tested concentrations. These derivatives were also affected by sodium ions, magnesium ions, and 20% FBS, leading to at least a two-fold decrease in activity. Peptide 6 showed no sensitivity to sodium ions, zinc ions, ammonium ions, or 10% FBS, demonstrating a two-fold increase in antimicrobial activity. Peptide 7 remained unaffected by potassium ions, iron ions, or ammonium ions, maintaining its antimicrobial activity at the MIC concentration. Especially, compared to its template peptide (peptide 2), peptide 7 exhibited less sensitivity to conditions such as calcium ions, sodium ions, and 20% serum, which had the most negative impact on the antibacterial activity of peptide 2. While peptide 7 exhibited a four-fold increase in MIC under sodium and serum conditions and a 32-fold increase in the presence of calcium, these changes were still less drastic than those observed for peptide 2.
Cytotoxicity and selectivity
The in vitro cytotoxicity of the synthetic peptides was assessed by evaluating haemolysis of horse erythrocytes and cytotoxicity toward healthy human HEK-293 cells. The therapeutic potential of the peptides was evaluated using the selectivity index (SI). As shown in Table 5, while E2P displayed the strongest antibacterial activity, it also exhibited the highest cytotoxicity, with an MHC value of 7 µM and an IC50 of 1.9 µM, both apparently lower than those of other derivatives. This resulted in a low SI value of 0.3 against all tested bacteria. With truncation, all shorter derivatives (peptides 2, 3, 4, and 5) demonstrated much lower cytotoxicity toward both erythrocytes and HEK-293 cells. Although reduced amino acid length lowered toxicity, it also led to decreased antibacterial activity and, consequently, lower SI values. Among these derivatives, peptide 2 had the highest SI value of 6.6. In peptide 6, the substitution of tryptophan slightly improved antibacterial activity, reducing the GMMIC from 22.6 µM to 19.7 µM (Table 3). However, the MHC and IC50 values increased significantly, leading to a reduced SI value of 1.1. In contrast, the incorporation of hArg in peptide 7 preserved the low cytotoxicity of peptide 2 while greatly enhancing both antibacterial activity and selectivity. Peptide 7 showed the highest SI values, with 14.1 for Gram-positive bacteria and 62.9 for Gram-negative bacteria, resulting in an overall SI value of 40.4.
Thermal and trypsin sensitivity
Since hArg has been reported to slow peptide degradation, the sensitivity of peptide 7 to trypsin was evaluated by monitoring changes in MIC and MBC values. During the experiment, peptide-trypsin mixtures were heated to 80℃ to inactivate trypsin, allowing assessment of both enzymatic and thermal sensitivity. As shown in Table 6, heat exposure alone did not alter the MIC or MBC values of peptides 2, 7, or Melittin, indicating that changes observed in the presence of trypsin can reflect their sensitivity to enzymatic degradation. The results showed that 0.25 mg/mL of trypsin did not affect the antimicrobial activity of peptide 7 but remarkably impacted peptide 2 and Melittin. Peptide 2 completely lost its antibacterial function within the tested concentration range, while Melittin’s MIC and MBC values increased 32-fold. At 0.5 mg/mL of trypsin, peptide 7’s MIC and MBC values both increased two-fold, whereas Melittin’s antibacterial activity was completely lost. At 1 mg/mL of trypsin, peptide 7’s MIC and MBC values increased to 16 µM, but it still retained antibacterial potency, demonstrating greatly improved tolerance to trypsin compared to the control peptides.
Drug resistance development
A drug resistance development experiment was conducted to assess the resistance-inducing potential of peptide 7 and several antibiotics (ceftazidime, rifampicin, norfloxacin, and gentamicin), using E. coli ATCC 8739 as the model. The bacteria were continuously exposed to sub-MIC levels of peptide 7 or antibiotics for twenty days (twenty passages). As shown in Fig. 4, E. coli ATCC 8739 remained highly susceptible to peptide 7 after twenty consecutive passages, indicating a low potential for resistance development. In contrast, the MIC values of the control antibiotics increased over time. Cefotaxime and gentamicin showed a 32-fold increase in MIC by the end of the study, while Norfloxacin’s MIC increased 512-fold over twenty passages. The MIC of rifampicin rose 512-fold within five passages and then exceeded the tested concentration range. These results suggest that the control antibiotics led to rapid resistance development, whereas peptide 7 effectively limited resistance development in E. coli ATCC 8739.

Resistance development of E. coli ATCC 8739 to the sub-MIC of Peptide 7, Cefotaxime, Rifampicin, Norfloxacin, and Gentamicin for twenty passages. The MIC/MBC values (μM) were determined as follows: cefotaxime 1/1, rifampicin 8/8, norfloxacin 0.25/0.25, and gentamicin 2/4.
Antibacterial mechanism
LPS-Binding affinity
To investigate the antibacterial mechanism of peptide 7, its LPS-binding affinity was first evaluated. As shown in Fig. 5A, peptide 7 exhibited a concentration-dependent binding affinity with LPS. However, compared to melittin, the binding capacity of peptide 7 was relatively weaker. For melittin, the binding affinity peaked at a concentration of 16 μM. At 32 μM, the BC fluorescent intensity induced by melittin and peptide 7 was similar, indicating comparable binding capacities at this concentration.

(A) LPS-binding affinity of melittin and peptide 7. (B) The outer membrane permeabilisation of E. coli ATCC 8739. (C) The cytoplasmic membrane permeabilisation of E. coli ATCC 8739 induced by Peptide 7. (D) Fluorescent microscope observation of E. coli ATCC 8739 with or without the treatment of Peptide 7. The scale bar is 10 µm.
Outer membrane &cytoplasmic membrane permeability
The effect of Peptide 7 on the outer membrane permeability of E. coli was assessed using the NPN assay. As shown in Fig. 5B, Peptide 7 exhibited significant membrane permeabilisation activity on the outer membrane of E. coli ATCC 8739 compared to the control group, with fluorescence intensity peaking at 2 × MIC concentration. Similarly, for the cytoplasmic membrane, Peptide 7 demonstrated concentration-dependent permeabilisation, significantly increasing membrane permeability (Fig. 5C). At 2 × MIC, fluorescence intensity again reached its maximum. To further validate the effect of Peptide 7 on E. coli ATCC 8739, SYTO9/PI-stained bacteria were observed using fluorescent microscopy. As shown in Fig. 5D, the presence of Peptide 7 induced a noticeable increase in PI fluorescence intensity compared to the control group, indicating enhanced membrane permeabilisation.
Cytoplasmic membrane electrical potential
Disruption of the cytoplasmic membrane leads to dissipation of the membrane’s electric potential. To assess the peptides’ ability to depolarise the membrane, the fluorescent dye Disc3(5) was employed. As shown in Fig. 6, treatment with Peptide 7 resulted in a rapid, concentration-dependent increase in fluorescence intensity. Although the control peptide Melittin induced a stronger fluorescence response, reaching its maximum at 2 µM after 3 min of incubation, Peptide 7 acted more quickly, peaking in fluorescence intensity at 8 µM after just 1 min of incubation.

Cytoplasmic membrane depolarisation of E. coli ATCC 8739 induced by Peptide 7 (left) and melittin (Right).
Antimicrobial activity in blood
The complex composition of blood may interfere with the antibacterial activity of the peptide. To assess this, the antibacterial activity of peptide 7 in blood was tested, with colistin serving as the comparison group. As shown in Fig. 7, treatment with peptide 7 effectively inhibited the growth of all tested E. coli strains in blood, demonstrating potency comparable to colistin, except against E. coli ATCC 2469 at MIC concentration. After 6 h of incubation, a bacterial count of 4 × 105 CFU/mL was observed, remarkably lower than that of the control group. However, at 2 × MIC and 4 × MIC concentrations, complete eradication of bacteria was observed during the incubation period.

Antibacterial activity of Colistin and peptide 7 in horse blood against (A) E. coli ATCC 2340, (B) E. coli NCTC 13,846, and (C) E. coli ATCC 2469. Due to the influence of blood colour, some samples at 10⁻1 dilution concentrations did not show bacterial colonies at 0 h.
In Vivo antimicrobial activity
To evaluate the in vivo antibacterial activity of peptide 7, its toxicity was first assessed by treating larval worms with the highest test concentration of the peptide. As shown in Fig. 8, peptide 7 did not affect the survival of the larvae during a consecutive five-day observation period. In the in vivo antibacterial tests, peptide 7 demonstrated comparable therapeutic effects at 4 × MIC against E. coli ATCC 2340 and at 2 × MIC or 4 × MIC against E. coli NCTC 13,846. Treatment with peptide 7 greatly reduced the mortality rate of infected larvae, increasing the survival rate from 60 to 90% for E. coli ATCC 2340 and from 30 to 55% for E. coli NCTC 13,846. For E. coli ATCC 2469, where the infection led to a mortality rate of 80%, peptide 7, although not as effective as colistin, improved the survival rate to 30–45%.

In vivo toxicity and antibacterial potentials of peptide 7 in infected larvae models. For toxicity, larvae worms were treated with 4 × MIC of peptide 7. For in vivo antibacterial tests, larvae infected with E. coli strains ATCC 2340, NCTC 13,846, and ATCC 2469 were treated with varying doses of peptide 7. Colistin (10 × MIC) was set as control group. The untreated group refers to larvae that received no peptide or antibiotics treatment and were treated solely with the vehicle, PBS.
Discussion
In this study, we employed a composite modification approach to quickly balance the production costs and therapeutic efficacy of AMPs while enhancing the therapeutic potential of the modified fragments. We used E2P, a 37-amino-acid AMP derived from the skin secretions of the frog Lithobates pipiens, as our modification template. Testing revealed that E2P exhibited strong broad-spectrum antimicrobial activity, likely attributable to its six net positive charges, which enhance its binding affinity to the negatively charged components of bacterial membranes36. Like other peptides in the Esculentin-2 family, E2P adopted an alpha-helical conformation in hydrophobic solutions. This helical structure facilitated interactions with the lipid bilayer of bacterial membranes, contributing to its antimicrobial efficacy37. Additionally, E2P demonstrated resilience to salt and serum sensitivity against E. coli ATCC 8739, maintaining robust antimicrobial activity across various physiological conditions. In subsequent haemolytic activity assays, E2P induced only about 5.6% haemolysis at its MIC. However, when the concentration exceeded 16 µM, haemolytic activity increased significantly (around 16.2%), likely due to peptide accumulation on the cell membrane, reaching a threshold that rapidly disrupted membrane integrity38. In cytotoxicity studies, the IC50 of E2P for the HEK-293 cells was 1.897 µM, indicating high cytotoxicity, which poses challenges for therapeutic use. Although eukaryotic and bacterial membranes differ in composition, both contain phospholipids and other components, leading to potential toxicity due to the peptide’s non-specific membrane interactions39,40.
Natural antimicrobial peptides are susceptible to degradation by in vivo proteases (e.g. trypsin, pepsin, etc.) recognising specific cleavage sites, leading to a loss of activity, especially E2P, which is a long peptide with 37 amino acids and seven cationic residues (lysine and arginine) in the sequence, which is susceptible to preferential cleavage by trypsin41,42. Random N–C truncation has the potential to disrupt the formation of the hydrophobic core or delete important structural elements43,44, which may be further degraded upon entry into the body. Therefore, truncation strategies that mimic enzymatic cleavage reduce the risk of enzymatic degradation by analysing the site of action of the protease and proactively removing or replacing amino acid sequences susceptible to cleavage36,42.
Following a trypsin-cleavage simulation process, five truncated analogues were developed. Functional screening revealed that truncation of the C-terminal sequence (LGVNLVACKISKQC) containing the Rana box, a conserved disulfide-constrained cyclic domain critical for amphibian AMP structure, reduced antimicrobial activity, with pronounced effects against Gram-positive bacteria. This loss of potency correlates with decreased hydrophobicity. Among the analogues, peptides 1 and 2 exhibited minimal differences in antimicrobial activity, suggesting that the amino acids at positions 20 to 23 of Esculentin-2P may have little impact on activity. However, peptide 3 showed reduced activity against Gram-negative bacteria, potentially due to a decrease in cationic residues, as basic charges are essential for effectively targeting Gram-negative bacteria, while increased peptide hydrophobicity is crucial for Gram-positive bacteria12. This difference likely arises from the distinct membrane compositions of Gram-positive and Gram-negative bacteria. The cell wall of Gram-positive bacteria is rich in teichoic and uronic acids, making them more sensitive to peptides with higher hydrophobicity45. Peptide 4 and peptide 5 directly lost their activity against Gram-negative bacteria, probably due to the potential ability of the motifs in the main chain part (GLGKFASKGVAK) to modulate activity against Gram-negative bacteria. In addition, possibly due to the net charge of the truncated peptide drops from + 6 to + 2, which is below the minimum threshold required for Gram-negative bacterial targeting (usually ≥ + 4)46
Under physiological saline and serum conditions, E2P and its truncated analogues showed sensitivity to calcium ions, while the truncated peptides were generally sensitive to sodium ions and 20% FBS. This sensitivity may have resulted from the displacement of divalent Ca2⁺ and Mg2⁺ ions after peptide binding to the outer membrane LPS of Gram-negative bacteria, initiating a self-promoting uptake process that reduced antimicrobial activity47,48,49. Sodium ions, present in the highest concentrations, likely induced peptide aggregation, reducing solubility and activity50. Furthermore, high serum concentrations strongly inhibited AMP bactericidal activity, likely due to AMP binding to abundant serum albumin in plasma, which reduces the effective concentration of AMPs at the bacterial surface51. In terms of haemolytic activity and cytotoxicity, all truncated analogues exhibited minimal haemolytic activity. This reduction can be attributed to the removal of the C-terminal domain containing the Rana box. The cyclic structure and charge distribution of the Rana box likely increased the rigidity of the peptide and enhanced its interaction with cell membranes, facilitating insertion into bacterial membranes and pore formation. This capability also extended to eukaryotic cell membranes, resulting in leakage of cellular contents and cell death52,53.
These results demonstrate that the truncation strategy effectively screens the functions of large protein scaffolds and may also be suitable for modifying short peptides to balance synthetic costs and bio-functional properties, addressing one of the major challenges in developing peptide-based drugs. It is evident that during the truncation process, the therapeutic efficacy of the derivatives peaked, aiding in the identification of the most active fragment within the parent peptide. Subsequent modification strategies, such as introducing unnatural amino acids, lipidation, and cyclisation, could further enhance the stability, specificity, and overall drug efficacy of the screened fragments. Our studies validate the feasibility of this modification strategy and may provide valuable insights for future AMP development.
The assays described above led to the selection of peptide 2 for further modification, owing to its optimal sequence length, superior antimicrobial activity, and reduced cytotoxicity. Subsequent rational design efforts focused on enhancing its antibacterial activity, stability, and minimising cytotoxicity. Initially, all hydrophobic amino acids on the hydrophobic surface of peptide 2 were replaced with tryptophan. It has been reported that the addition of tryptophan typically enhances the affinity of peptides for membranes54,55. Due to the presence of negatively charged π-electron clouds above and below its aromatic ring, tryptophan can interact with positively charged amino acid side chains and the phosphocholine head groups of lipid bilayers through cation-π interactions56,57. By binding to tryptophan, cationic residues such as arginine are less affected by the high hydrophobicity of the lipid bilayer, allowing for easier penetration57,58. Additionally, the large and bulky indole side chain of tryptophan disrupts hydrophobic interactions between lipid acyl chains, facilitating further insertion of the peptide into the lipid bilayer59. This may explain the increased antimicrobial activity of peptide 6 against Gram-negative bacteria, even in the presence of physiological ion concentrations and serum. However, results indicated that both haemolytic activity and cytotoxicity exceeded those of E2P. This could be attributed to tryptophan’s preference for the interfacial region of lipid bilayers, affecting not only bacterial membranes but also eukaryotic membranes60. Moreover, the introduction of tryptophan increased the hydrophobicity and hydrophobic moment of the peptide. Peptides with higher hydrophobicity tend to penetrate more deeply into the hydrophobic core of erythrocyte membranes, forming pores or channels that lead to stronger haemolytic activity61,62,63.
The second modification involved replacing the positively charged amino acids in the peptide 2 sequence with the hArg to enhance stability and efficacy. Antimicrobial assays demonstrated that peptide 7 exhibited at least a twofold increase in antimicrobial activity with respect to the template peptide 2. This enhancement is likely due to the extended side chain of hArg, which facilitates more effective interactions with microbial membranes, thereby increasing the peptide’s ability to disrupt pathogen membrane integrity. This improved interaction potentially broadens the range of pathogens that the AMP can target21. Furthermore, the structurally modified hArg enabled more selective interactions with microbial membranes, likely reducing the AMP’s cytotoxicity toward human cells22. This selectivity could account for the negligible haemolytic activity and very low cytotoxicity toward HEK-293 observed in peptide 7. The enhanced salt and serum tolerance of peptide 7 compared to peptide 2 is attributed to the guanidinium group in hArg, which facilitates strong electrostatic interactions with negatively charged components, including microbial membranes and serum proteins, thus maintaining the structural integrity of the peptide under harsh conditions64. Notably, peptide 7 retained antimicrobial activity against E. coli ATCC 8739 at MIC concentrations after incubation with 0.25 mg/ml trypsin and continued to show good activity even with 1 mg/ml trypsin incubation. In contrast, peptide 2 and Melittin lost their antimicrobial activity against E. coli ATCC 8739 immediately following trypsin treatment. The increased tolerance of peptide 7 may be due to the spatial site resistance provided by the extra methylene group in hArg, which reduces the accessibility of the peptide bond to proteolytic enzymes21. The in vitro experiments further confirmed the potent antibacterial efficacy of peptide 7 against three drug-resistant E. coli strains in blood environments, underscoring its exceptional tolerance to diverse physiological conditions. In the in vivo antibacterial tests, treatment with peptide 7 enhanced the survival rate of E. coli-infected larvae compared to the untreated group. Moreover, a 20-day resistance development assay revealed that peptide 7 exhibited a lower tendency for resistance development of E. coli ATCC 8739 compared to conventional antibiotics. Collectively, these findings highlight the great potential of peptide 7 as a promising candidate for novel antimicrobial agents.
The mechanism of action of peptide 7 against E. coli ATCC 87,739 was preliminarily investigated. LPS-binding assay indicated that peptide 7 exhibited strong binding affinity to LPS. This binding capacity may be attributed to the helical content observed in peptide 7, as evidenced by CD results. The α-helical structure provides a framework that enables the peptide to align correctly with the negatively charged phosphate groups on LPS molecules, facilitating strong electrostatic and hydrophobic interactions65. Furthermore, peptide 7 possesses an optimal balance of hydrophobic and hydrophilic regions, allowing interaction with both the hydrophobic lipid A region and the negatively charged core polysaccharide of LPS66,67. Upon LPS binding, peptide 7 penetrated the outer membrane, disrupting its integrity and thereby leading to changes in membrane permeability. Following outer membrane permeabilisation, peptide 7 interacted with the cytoplasmic (inner) membrane. Similarly, results from the SYTOX Green assay indicated that the truncated analogue had a potent permeabilisation effect on the inner membrane. Membrane potential studies aligned with these results and showed that peptide could induce a potent membrane depolarisation effect in a concentration-dependent manner, similar with that of melittin. This disruption was critical for bacterial survival, as it affected the membrane’s ability to maintain essential gradients and carry out processes such as ATP synthesis. Ultimately, the loss of membrane potential led to bacterial cell death68,69,70.
To better understand the effect of the enzymatic and amino acid modification strategies employed in this study, we compared them with other common antimicrobial peptide design strategies (see Table 7). The modifications, antimicrobial activity, stability and key findings of the different strategies are summarised in the table.
In conclusion, we provide a feasible and efficient approach for developing shorter and potent AMPs by combining trypsin-mimic truncations with hArg incorporation. Peptide 7, which features a reduced amino acid length and enhanced therapeutic potential, demonstrated effective inhibition and killing of a broad spectrum of bacteria in vitro, particularly Gram-negative strains. It also exhibited resilience to various challenging environmental conditions that typically affect AMP activity. These improvements should be due to the incorporation of hArg. Mechanistic studies confirmed that Peptide 7 acts by targeting bacterial membranes, and both ex vivo and in vivo experiments validated its antibacterial efficacy. The design and optimisation of Peptide 7 offer valuable insights into developing shorter AMPs with improved therapeutic potential and may serve as a foundation for future advancements in AMP modification.
Data availability
The authors confirm that the data supporting the findings of this study are available within the article and the datasets used during the current study are available from the corresponding author on reasonable request.
References
Muteeb, G., Rehman, M. T., Shahwan, M. & Aatif, M. Origin of antibiotics and antibiotic resistance, and their impacts on drug development: A narrative review. Pharmaceuticals 16, 1615. https://doi.org/10.3390/ph16111615 (2023).
Aggarwal, R. et al. Antibiotic resistance: A global crisis, problems and solutions. Crit. Rev. Microbiol. https://doi.org/10.1080/1040841X.2024.2313024 (2024).
Wang, J. et al. Antimicrobial peptides: Promising alternatives in the post feeding antibiotic era. Med. Res. Rev. 39, 831. https://doi.org/10.1002/med.21542 (2019).
Nicolas, P. Multifunctional host defense peptides: Intracellular-targeting antimicrobial peptides. FEBS J. 276, 6483. https://doi.org/10.1111/j.1742-4658.2009.07359.x (2009).
Greco, I. et al. Correlation between hemolytic activity, cytotoxicity and systemic in vivo toxicity of synthetic antimicrobial peptides. Sci. Rep. 10, 13206. https://doi.org/10.1038/s41598-020-69995-9 (2020).
Svendsen, J. S. M., Grant, T. M., Rennison, D., Brimble, M. A. & Svenson, J. Very short and stable lactoferricin-derived antimicrobial peptides: Design principles and potential uses. Acc. Chem. Res. 52, 749. https://doi.org/10.1021/acs.accounts.8b00624 (2019).
Moncla, B. J., Pryke, K., Rohan, L. C. & Graebing, P. W. Degradation of naturally occurring and engineered antimicrobial peptides by proteases. Adv. Biosci. Biotechnol. https://doi.org/10.4236/abb.2011.26059 (2011).
Boöttger, R., Hoffmann, R. & Knappe, D. Differential stability of therapeutic peptides with different proteolytic cleavage sites in blood, plasma and serum. PLoS ONE https://doi.org/10.1371/journal.pone.0178943 (2017).
Menacho-Melgar, R., Decker, J. S., Hennigan, J. N. & Lynch, M. D. A review of lipidation in the development of advanced protein and peptide therapeutics. J. Controll. Release https://doi.org/10.1016/j.jconrel.2018.12.032 (2019).
Zhu, Y. et al. Rational avoidance of protease cleavage sites and symmetrical end-tagging significantly enhances the stability and therapeutic potential of antimicrobial peptides. J. Med. Chem. 63, 9421. https://doi.org/10.1021/acs.jmedchem.0c00583 (2020).
Lu, J. et al. D- and unnatural amino acid substituted antimicrobial peptides with improved proteolytic resistance and their proteolytic degradation characteristics. Front. Microbiol. 11, 563030. https://doi.org/10.3389/fmicb.2020.563030 (2020).
He, S. et al. Boosting stability and therapeutic potential of proteolysis-resistant antimicrobial peptides by end-tagging β-naphthylalanine. Acta Biomater. 164, 175. https://doi.org/10.1016/j.actbio.2023.04.030 (2023).
Yu, W. et al. PEGylation of the antimicrobial peptide PG-1: A link between propensity for nanostructuring and capacity of the antitrypsin hydrolytic ability. J. Med. Chem. 64, 10469. https://doi.org/10.1021/acs.jmedchem.1c00879 (2021).
Wang, C. et al. Binding loop of sunflower trypsin inhibitor 1 serves as a design motif for proteolysis-resistant antimicrobial peptides. Acta Biomater. 124, 254. https://doi.org/10.1016/j.actbio.2021.01.036 (2021).
Bellavita, R., Braccia, S., Galdiero, S. & Falanga, A. Glycosylation and lipidation strategies: Approaches for improving antimicrobial peptide efficacy. Pharmaceuticals 16, 439. https://doi.org/10.3390/ph16030439 (2023).
Li, Y. et al. Antimicrobial activity, membrane interaction and stability of the D-amino acid substituted analogs of antimicrobial peptide W3R6. J. Photochem. Photobiol. B 200, 111645. https://doi.org/10.1016/j.jphotobiol.2019.111645 (2019).
Qiu, S. et al. Antimicrobial activity and stability of protonectin with D-amino acid substitutions. J. Pept. Sci. 23, 392. https://doi.org/10.1002/psc.2989 (2017).
Kapil, S. & Sharma, V. D-amino acids in antimicrobial peptides: A potential approach to treat and combat antimicrobial resistance. Can. J. Microbiol. 67, 119. https://doi.org/10.1139/cjm-2020-0142 (2021).
Wang, X., Yang, X., Wang, Q. & Meng, D. Unnatural amino acids: Promising implications for the development of new antimicrobial peptides. Crit. Rev. Microbiol. 49, 231. https://doi.org/10.1080/1040841X.2022.2047008 (2023).
Hamamoto, K., Kida, Y., Zhang, Y., Shimizu, T. & Kuwano, K. Antimicrobial activity and stability to proteolysis of small linear cationic peptides with D-amino acid substitutions. Microbiol. Immunol. 46, 741. https://doi.org/10.1111/j.1348-0421.2002.tb02759.x (2002).
Arias, M., Piga, K. B., Hyndman, M. E. & Vogel, H. J. Improving the activity of trp-rich antimicrobial peptides by Arg/Lys substitutions and changing the length of cationic residues. Biomolecules 8, 19. https://doi.org/10.3390/biom8020019 (2018).
Hosseini, M. et al. Elucidation of the contribution of active site and exosite interactions to affinity and specificity of peptidylic serine protease inhibitors using non-natural arginine analogs. Mol. Pharmacol. 80, 585. https://doi.org/10.1124/mol.111.072280 (2011).
Wang, J. et al. Discovery, development and optimisation of a novel frog antimicrobial peptide with combined mode of action against drug-resistant bacteria. Comput. Struct. Biotechnol. J. 23, 3391–3406. https://doi.org/10.1016/j.csbj.2024.09.006 (2024).
Fei, F. et al. A frog-derived antimicrobial peptide as a potential anti-biofilm agent in combating Staphylococcus aureus skin infection. J. Cell. Mol. Med. 27, 1565. https://doi.org/10.1111/jcmm.17785 (2023).
Wang, Z. et al. Functional characterisation and modification of a novel Kunitzin peptide for use as an anti-trypsin antimicrobial peptide against drug-resistant Escherichia coli. Biochem. Pharmacol. 229, 116508. https://doi.org/10.1016/j.bcp.2024.116508 (2024).
Liu, X. et al. Modification and synergistic studies of a novel frog antimicrobial peptide against pseudomonas aeruginosa biofilms. Antibiotics 13, 574. https://doi.org/10.3390/antibiotics13070574 (2024).
Jiang, Y. et al. Brevinin-1GHD: A novel Hylarana guentheri skin secretion-derived Brevinin-1 type peptide with antimicrobial and anticancer therapeutic potential. Biosci. Rep. https://doi.org/10.1042/BSR20200019 (2020).
Zou, W. et al. Exploring the active core of a novel antimicrobial peptide, palustrin-2LTb, from the Kuatun frog, Hylarana latouchii, using a bioinformatics-directed approach. Comput. Struct. Biotechnol. J. 20, 6192–6205. https://doi.org/10.1016/j.csbj.2022.11.016 (2022).
Zou, W. et al. A promising antibiotic candidate, brevinin-1 analogue 5R, against drug-resistant bacteria, with insights into its membrane-targeting mechanism. Comput. Struct. Biotechnol. J. 21, 5719–5737. https://doi.org/10.1016/j.csbj.2023.11.031 (2023).
Ma, X. et al. A novel antimicrobial peptide, dermaseptin-SS1, with anti-proliferative activity, isolated from the skin secretion of phyllomedusa tarsius. Molecules 28, 6558. https://doi.org/10.3390/molecules28186558 (2023).
Gong, Z. et al. Identification and rational design of a novel antibacterial peptide dermaseptin-ac from the skin secretion of the red-eyed tree frog agalychnis callidryas. Antibiotics 9, 243. https://doi.org/10.3390/antibiotics9050243 (2020).
Chen, G. et al. Brevinin-2GHK from sylvirana guentheri and the design of truncated analogs exhibiting the enhancement of antimicrobial activity. Antibiotics 9, 85. https://doi.org/10.3390/antibiotics9020085 (2020).
Zai, Y. et al. Aggregation and its influence on the bioactivities of a novel antimicrobial peptide, temporin-pf, and its analogues. Int. J. Mol. Sci. 22, 4509. https://doi.org/10.3390/ijms22094509 (2021).
Yao, A. et al. A designed analog of an antimicrobial peptide, crabrolin, exhibits enhanced anti-proliferative and in vivo antimicrobial activity. Int. J. Mol. Sci. 24, 14472. https://doi.org/10.3390/ijms241914472 (2023).
Li, Q. et al. Outer-membrane-acting peptides and lipid II-targeting antibiotics cooperatively kill Gram-negative pathogens. Commun. Biol. 4, 31. https://doi.org/10.1038/s42003-020-01511-1 (2021).
Ma, Y. et al. Generation of truncated derivatives through in silico enzymatic digest of peptide GV30 target MRSA both in vitro and in vivo. Comput. Struct. Biotechnol. J. 19, 4984. https://doi.org/10.1016/j.csbj.2021.08.039 (2021).
Bechinger, B. & Lohner, K. Detergent-like actions of linear amphipathic cationic antimicrobial peptides. Biochim. Biophys. Acta Biomembr. 1758, 1529. https://doi.org/10.1016/j.bbamem.2006.07.001 (2006).
Papo, N. & Shai, Y. Can we predict biological activity of antimicrobial peptides from their interactions with model phospholipid membranes?. Peptides (N.Y.) 24, 1693. https://doi.org/10.1016/j.peptides.2003.09.013 (2003).
Teixeira, V., Feio, M. J. & Bastos, M. Role of lipids in the interaction of antimicrobial peptides with membranes. Prog. Lipid Res. 51, 149. https://doi.org/10.1016/j.plipres.2011.12.005 (2012).
Neundorf, I. Antimicrobial and cell-penetrating peptides: How to understand two distinct functions despite similar physicochemical properties. Adv. Exp. Med. Biol. https://doi.org/10.1007/978-981-13-3588-4_7 (2019).
Hedstrom, L. Serine protease mechanism and specificity. Chem. Rev. 102, 4501–4523. https://doi.org/10.1021/cr000033x (2002).
Svenson, J. et al. Antimicrobial peptides with stability toward tryptic degradation. Biochemistry 47, 3777–3788. https://doi.org/10.1021/bi7019904 (2008).
Chen, N. & Jiang, C. Antimicrobial peptides: Structure, mechanism, and modification. Eur. J. Med. Chem. 255, 115377. https://doi.org/10.1016/j.ejmech.2023.115377 (2023).
Zasloff, M., Martint, B. & Chen, H. -C. Antimicrobial activity of synthetic magainin peptides and several analogues (vertebrate peptide antibiotics) https://www.pnas.org (1988).
Omardien, S., Brul, S. & Zaat, S. A. J. Antimicrobial activity of cationic antimicrobial peptides against gram-positives: Current progress made in understanding the mode of action and the response of bacteria. Front. Cell. Dev. Biol. 4, 111. https://doi.org/10.3389/fcell.2016.00111 (2016).
Hellewell, L. et al. Efficacy of natural antimicrobial peptides versus peptidomimetic analogues: A systematic review. Future Med. Chem. 14, 1899–1921. https://doi.org/10.4155/fmc-2022-0160 (2022).
Epand, R. M. & Vogel, H. J. Diversity of antimicrobial peptides and their mechanisms of action. Biochim. Biophys. Acta Biomembr. 1462, 11–28. https://doi.org/10.1016/S0005-2736(99)00198-4 (1999).
Hancock, R. E. Cationic peptides: Effectors in innate immunity and novel antimicrobials. Lancet Infect. Dis. 1, 156. https://doi.org/10.1016/S1473-3099(01)00092-5 (2001).
Zasloff, M. Antimicrobial peptides of multicellular organisms. Nature 415, 389. https://doi.org/10.1038/415389a (2002).
Zapadka, K. L., Becher, F. J., Gomes dos Santos, A. L. & Jackson, S. E. Factors affecting the physical stability (aggregation) of peptide therapeutics. Interface Focus 7, 20170030. https://doi.org/10.1098/rsfs.2017.0030 (2017).
Nguyen, L. T. et al. Serum stabilities of short tryptophan- and arginine-rich antimicrobial peptide analogs. PLoS ONE 5, e12684. https://doi.org/10.1371/journal.pone.0012684 (2010).
Islas-Rodrìguez, A. E. et al. Esculentin 1–21: A linear antimicrobial peptide from frog skin with inhibitory effect on bovine mastitis-causing bacteria. J. Pept. Sci. https://doi.org/10.1002/psc.1148 (2009).
Chen, X. et al. A novel antimicrobial peptide, Ranatuerin-2PLx, showing therapeutic potential in inhibiting proliferation of cancer cells. Biosci. Rep. https://doi.org/10.1042/BSR20180710 (2018).
Staubitz, P. et al. Structure-function relationships in the tryptophan-rich, antimicrobial peptide indolicidin. J. Pept. Sci. 7, 552. https://doi.org/10.1002/psc.351 (2001).
Vogel, H. J. et al. Towards a structure-function analysis of bovine lactoferricin and related tryptophan- and arginine-containing peptides. Biochem. Cell Biol. https://doi.org/10.1139/o01-213 (2002).
Takahashi, D., Shukla, S. K., Prakash, O. & Zhang, G. Structural determinants of host defense peptides for antimicrobial activity and target cell selectivity. Biochimie 92, 1236. https://doi.org/10.1016/j.biochi.2010.02.023 (2010).
Chan, D. I., Prenner, E. J. & Vogel, H. J. Tryptophan- and arginine-rich antimicrobial peptides: Structures and mechanisms of action. Biochim. Biophys. Acta Biomembr. 1758, 1184. https://doi.org/10.1016/j.bbamem.2006.04.006 (2006).
Dougherty, D. A. Cation-π interactions in chemistry and biology: A new view of benzene Phe, Tyr, and Trp. Science 271, 163. https://doi.org/10.1126/science.271.5246.163 (1996).
Yau, W. M., Wimley, W. C., Gawrisch, K. & White, S. H. The preference of tryptophan for membrane interfaces. Biochemistry 37, 14713. https://doi.org/10.1021/bi980809c (1998).
Gopal, R. et al. Effect of repetitive lysine-tryptophan motifs on the eukaryotic membrane. Int. J. Mol. Sci. 14, 2190. https://doi.org/10.3390/ijms14012190 (2013).
Tachi, T., Epand, R. F., Epand, R. M. & Matsuzaki, K. Position-dependent hydrophobicity of the antimicrobial magainin peptide affects the mode of peptide-lipid interactions and selective toxicity. Biochemistry 41, 10723. https://doi.org/10.1021/bi0256983 (2002).
Chen, Y. et al. Role of peptide hydrophobicity in the mechanism of action of α-helical antimicrobial peptides. Antimicrob. Agents Chemother. 51, 1398. https://doi.org/10.1128/AAC.00925-06 (2007).
Chen, Y. et al. Rational design of α-helical antimicrobial peptides with enhanced activities and specificity/therapeutic index. J. Biol. Chem. 280, 12316. https://doi.org/10.1074/jbc.M413406200 (2005).
Wender, P. A., Galliher, W. C., Goun, E. A., Jones, L. R. & Pillow, T. H. The design of guanidinium-rich transporters and their internalization mechanisms. Adv. Drug Deliv. Rev. 60, 452. https://doi.org/10.1016/j.addr.2007.10.016 (2008).
Li, P., Wohland, T., Ho, B. & Jeak, L. D. Perturbation of lipopolysaccharide (LPS) micelles by Sushi 3 (S3) antimicrobial peptide: The importance of an intermolecular disulfide bond in S3 dimer for binding, disruption, and neutralization of LPS. J. Biol. Chem. 279, 50150. https://doi.org/10.1074/jbc.M405606200 (2004).
Hancock, R. E. W. & Chapple, D. S. Peptide antibiotics. Antimicrob. Agents Chemother. 43, 1317. https://doi.org/10.1128/aac.43.6.1317 (1999).
Hancock, R. E. Alterations in outer membrane permeability. Annu. Rev. Microbiol. 38, 237. https://doi.org/10.1146/annurev.mi.38.100184.001321 (1984).
Bakker, E. P. & Mangerich, W. E. Interconversion of components of the bacterial proton motive force by electrogenic potassium transport. J. Bacteriol. 147, 820. https://doi.org/10.1128/jb.147.3.820-826.1981 (1981).
Hurdle, J. G., O’Neill, A. J., Chopra, I. & Lee, R. E. Targeting bacterial membrane function: An underexploited mechanism for treating persistent infections. Nat. Rev. Microbiol. 9, 62. https://doi.org/10.1038/nrmicro2474 (2011).
Farha, M. A., Verschoor, C. P., Bowdish, D. & Brown, E. D. Collapsing the proton motive force to identify synergistic combinations against staphylococcus aureus. Chem. Biol. 20, 1168. https://doi.org/10.1016/j.chembiol.2013.07.006 (2013).
Bucki, R., Leszczyńska, K., Namiot, A. & Sokołowski, W. Cathelicidin LL-37: A multitask antimicrobial peptide. Arch. Immunol. Ther. Exp. (Warsz) 58, 15–25. https://doi.org/10.1007/s00005-009-0057-2 (2010).
Chen, K., Gong, W., Huang, J., Yoshimura, T. & Wang, J. M. The potentials of short fragments of human anti-microbial peptide LL-37 as a novel therapeutic modality for diseases. Front. Biosci. Landmark 26, 1362–1372. https://doi.org/10.52586/5029 (2021).
Zeth, K. & Sancho-Vaello, E. Structural plasticity of ll-37 indicates elaborate functional adaptation mechanisms to bacterial target structures. Int. J. Mol. Sci. 22, 5200. https://doi.org/10.3390/ijms22105200 (2021).
Wang, G., Mishra, B., Epand, R. F. & Epand, R. M. High-quality 3D structures shine light on antibacterial, anti-biofilm and antiviral activities of human cathelicidin LL-37 and its fragments. Biochim. Biophys. Acta Biomembr. 2014, 2160–2172. https://doi.org/10.1016/j.bbamem.2014.01.016 (1838).
Akbari, R. et al. Action mechanism of melittin-derived antimicrobial peptides, MDP1 and MDP2, de novo designed against multidrug resistant bacteria. Amino Acids 50, 1231–1243. https://doi.org/10.1007/s00726-018-2596-5 (2018).
Grieco, P. et al. Alanine scanning analysis and structure-function relationships of the frog-skin antimicrobial peptide temporin-1Ta. J. Pept. Sci. 17, 358–365. https://doi.org/10.1002/psc.1350 (2011).
Conibear, A. C. & Craik, D. J. The chemistry and biology of theta defensins. Angew. Chem. Int. Ed. 53, 10612–10623. https://doi.org/10.1002/anie.201402167 (2014).
Almaaytah, A. et al. The design and functional characterization of the antimicrobial and antibiofilm activities of BMAP27-Melittin, a rationally designed hybrid peptide. Int. J. Pept. Res. Ther. 21, 165–177. https://doi.org/10.1007/s10989-014-9444-6 (2015).
Author information
Authors and Affiliations
Contributions
K. Y.: Investigation, Data curation, writing original draft. J. L.: Investigation, Data curation, writing original draft. R. S.: Investigation, Software, Methodology, Data curation. Y. W.: Methodology, Software. Y. J.: Methodology, Supervision. T. W.: Methodology, Validation, Writing review & editing. C. M.: Methodology. X. C.: Methodology. T. C.: Conceptualisation, Supervision. C. W.: Writing review & editing. M. Z.: Conceptualisation, Supervision, Resources. L. W.: Methodology, Supervision, Writing review & editing.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Yao, K., Liu, J., Sun, R. et al. Enhancing the selectivity and conditional sensitivity of an antimicrobial peptide through cleavage simulations and homoarginine incorporation to combat drug-resistant bacteria. Sci Rep 15, 21798 (2025). https://doi.org/10.1038/s41598-025-06522-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-06522-8
