
Abstract
Sterol regulatory element-binding protein-2 (SREBP-2) is a transcriptional factor, which mainly regulates cholesterogenesis-associated genes. The metabolism of cholesterol was found to be abnormal in non-small cell lung cancer (NSCLC). However, the clinical relevance of SREBP-2 in NSCLC has yet to be elucidated. Herein, the prognostic significance of SREBP-2 in NSCLC patients, and the functional roles of SREBP-2 in NSCLC proliferation, migration and invasion was examined. Then, therapeutic potential of targeting SREBP-2 in NSCLC was evaluated using mouse xenograft tumor models. We found that SREBP-2 was significantly increased in NSCLC, as compared with pericarcinous lung tissues. High expression of SREBP-2 was associated with poor clinical characteristics, and predicted a shorter overall survival (OS) in NSCLC patients. Cox multivariate regression revealed that high SREBP-2 expression served as an independent predictor for poor OS. Biofunctional analysis showed that gene silencing of SREBP-2 abrogated the proliferation, migration and invasion of NSCLC cells, while enforced SREBP-2 promoted NSCLC cell proliferation, migration and invasion. Mechanistically, SREBP-2 was found to promote cell proliferation, migration and invasion via the mevalonate-Akt pathway in NSCLC cells. Further in vivo treatment experiments showed that SREBP inhibitor decreased NSCLC tumor growth in mouse xenograft models in vivo. In combination, this study dissected the clinical significance and oncogenic role of SREBP-2 in NSCLC progression, providing evidence that have both prognostic and therapeutic implications.
Similar content being viewed by others


Introduction
Non-small cell lung cancer (NSCLC), which constitutes nearly 85% of lung cancers, is recognized as a leading cause of cancer mortality across the world1,2. Despite recent improvements in diagnostic and therapeutic interventions, the long-term prognosis of NSCLC remains dismal and the relative survival at 5 years is below 20%2. Dysregulation of metabolism, such as glucose, lipid, and amino acid metabolism, has been defined as one of the hallmarks in carcinogenesis, including NSCLC3. Therefore, it is imperative to gain a deep understanding of genes underlying the dysregulated metabolism in NSCLC.
Sterol regulatory element-binding protein-2 (SREBP-2) is a member of basic helix-loop-helix leucine zipper transcriptional factor family that preferentially regulates cholesterol metabolism and homeostasis4. When the intracellular cholesterol level is reduced, SREBP-2 can be transferred to the Golgi complex and cleaved by two proteases. Then, the N-terminal of SREBP-2 enters the nucleus and activates the transcription of target genes associated with cholesterol metabolism, such as hydroxymethylglutaryl (HMG)-CoA reductase (HMGCR) and low-density lipoprotein receptor (LDLR)5,6. Recent studies reported a critical role for SREBP-2 in tumorigenesis7,8,9,10,11. SREBP-2 was found to be upregulated in prostate cancer and bone-metastatic tumor tissues, and be associated with poor clinical outcomes7. SREBP-2 enhances prostate cancer cell proliferation, migration, invasion and stem cell-like features via inducing c-Myc transcription and activation. However, the expression alongside clinical relevance of SREBP-2 in NSCLC tissues have never been evaluated. Moreover, the functional role of SREBP-2 in NSCLC tumorigenesis and progression remains to be dissected.
In this study, the expression profile of SREBP-2 in a total of 459 clinical NSCLC samples were examined by immunohistochemistry (IHC). The prognostic value of SREBP-2 was analyzed. Moreover, the biological functions of SREBP-2 in NSCLC progression were determined. The mechanisms underlying the tumor-promoting functions of SREBP-2 in NSCLC was characterized. At last, the therapeutic implications of targeting SREBP-2 in NSCLC were explored both in vitro and in mouse xenograft tumor models. We propose SREBP-2 as a promising prognostic indicator, as well as a future therapeutic target in NSCLC treatment.
Materials and methods
Patients and samples
From 2015 to 2023, 459 NSCLC samples (275 cases for the training cohort, and 184 cases for the validation cohort), of which 439 had matched pericarcinous lung tissues, with complete clinicopathological information and survival data were collected from the Department of Pathology in Shandong Provincial Hospital. The pathological stages were evaluated according to the 8th edition of the lung cancer staging system12. Patients who were treated with neoadjuvant chemo- or radio-therapy, or had other cancers were excluded.
IHC staining and analysis
For IHC staining, NSCLC and matched normal lung tissue cores were punched from paraffin blocks by using 2-mm punch needles, inserted into the recipient blocks, cut by using a microtome, and mounted onto slides to make tissue microarray (TMA). All sections on TMA slides were incubated with rabbit anti-human SREBP-2 antibody (cat. no. ab30682; Abcam, UK, 1:1000 dilution). Meanwhile, isotype control was performed using concentration-matched nonspecific IgG (cat. no. ab65834; Abcam). Then, the slides were incubated with horseradish peroxidase (HRP)-labeled anti-rabbit secondary antibody (cat. no. 31460; ThermoFisher Scientific, 1:1000 dilution). All sections on TMA slices were photographed using the Aperio ScanScope XT Slide Scanner and viewed on Aperio ImageScope software (Aperio Technologies, Vista, CA).
The IHC staining data were independently analyzed by two pathologists, and a final consensus was reached. SREBP-2 nuclear staining in tumor cells was scored for further analysis. The staining intensity was judged as negative (score 0), weak (score 1), moderate (score 2) and intense (score 3) according to the dyeing degree. The percentage of positive cells was defined as 0% (score 0), 1–25% (score 1), 26–50% (score 2), 51–75% (score 3), and 76–100% (score 4). The final IHC score was reached by multiplying the staining intensity and percentage scores, which was classified as low (scores 0–4) or high (scores 6–12).
Cell culture and cell proliferation evaluation
Human NSCLC cells (H1299 and PC-9) were purchased from the American Type Culture Collection (ATCC) and cultured with RPMI-1640 medium supplemented with 10% FBS. All cells were authenticated by short tandem repeat profiling and confirmed be mycoplasma-free. MK-2206 (cat. no. HY-108232, MedChemExpress), simvastatin (cat. no. S1792, Selleck) and fatostatin (cat. no. S9785, Selleck) were free of endotoxins, and dissolved using 5% dimethyl sulfoxide (DMSO), 40% PEG300, 5% Tween-80 and 50% ddH2O.
For Cell Counting Kit-8 (CCK-8) assay, cells were seeded in 96-well plates in triplicate for 0, 24, 48 and 72 hours. Then, the complete medium was replaced by 100 µl/well medium with 10 µl/well CCK-8 reagent (Dojindo, Tokyo, Japan). After 2 hours, absorbance at 450 nm was determined with a micrometer. For crystal violet staining assay, the cultured cells were photographed after staining with 0.1% crystal violet. Then, the crystal violet dye was dissolved with 30% acetic acid, and the OD value at 570 nm was measured by a microplate reader.
SiRNA transfection
For siRNA transfection, 6 × 104 H1299 cells or 1 × 105 PC-9 cells were seeded in each well of 6-well plates for adherence. After 24 hours, the cells reached about 30% degree of confluence and transfected with 25 nM negative control (NC) or SREBP-2-targeted siRNAs by using Lipofectamine RNAiMAX (cat. no. 13778100; Invitrogen) following the manufacturer’s instructions. The target sequences of double-stranded nucleotides were 5’-GCAAGAGAAAGTGCCCATT-3’ (siRNA-1) and 5’-GCACATCACAGGGAAGCTT-3’ (siRNA-2) for SREBP-2 knockdown, and 5’-TTCTCCGAACGTGTCACGT-3’ as NC. The transfection efficiency was determined via qRT-PCR and Western Blot.
Lentiviral vectors and cell infection
Lentiviral vectors were generated by OBiO Biotechnology (Shanghai, China) by using pSLenti-EF1-EGFP-P2A-Puro-CMV-3Flag-WPRE plasmids, which were incorporated with the full-length human SREBP-2 coding sequence (GenBank accession NM_004599.4). For cell infection, 1 × 104 PC-9 cells were seeded in each well of 48-well plates for adherence. After 24 hours, the cells were infected with mock or SREBP-2-overexpressed lentiviral vectors at an MOI of 200 in the presence of 1 µg/µl polybrene. Twenty-four hours later, the infected PC-9 cells were cultured in fresh medium for 48 hours. Then, the infected PC-9 cells were cultured in medium containing puromycin (2 µg/ml; BasalMedia) for 2 weeks for the selection of stable clones. The infection efficiency was evaluated via qRT-PCR and Western Blot.
Western blot analysis
For Western blot analysis, gels were loaded with 15 µg of protein. SDS-PAGE was conducted to separate proteins by molecular weight, which were then transferred to PVDF membranes (Millipore, Burlington, MA). The membranes with separated proteins were incubated with primary antibodies, including goat anti-human SREBP-2 (cat. no. AF7119, R&D, 2 µg/ml in application), rabbit anti-human phosphorylated (Ser473)-Akt mAb (cat. no. 4060T, Cell Signaling Technology, 1:2000 dilution), rabbit anti-human phosphorylated (Thr202/Tyr204) -p44/p42 MAPK (Erk1/2) mAb (cat. no. 4376T, Cell Signaling Technology, 1:1000 dilution), rabbit anti-human Akt mAb (cat. no. 4691T, Cell Signaling Technology, 1:1000 dilution), rabbit anti-human p44/p42 MAPK mAb (cat. no. 4695 S, Cell Signaling Technology, 1:1000 dilution), and GAPDH rabbit mAb (cat. no. 5174T, Cell Signaling Technology, 1:2000 dilution), at 4 oC overnight, and then incubated with HRP-conjugated secondary antibodies, including anti-goat IgG (cat. no. SA00001-4, Proteintech, 1:5000 dilution) or anti-rabbit IgG HRP-linked antibody (cat. no. 7074 S, Cell Signaling Technology, 1:2000 dilution) at room temperature for 1 hour. An Enhanced Chemiluminescence (ECL) Prime kit (cat. no. AL010-01, ACE) was used to visualize immunoreactive bands according to the manufacturer’s instruction.
Quantitative real-time PCR (qRT-PCR) analysis
Total RNA was isolated from SREBP-2-overexpressed PC-9 cells using TaKaRa MiniBEST Universal RNA Extraction Kit (cat. no. 9767, TaKaRa) and subjected to reverse transcription using PrimeScript™ RT Master Mix (cat. no. RR036A, TaKaRa) according to the manufacturer’s instructions. The primers used are listed in Table S1. Real-time PCR was performed using an ABI Prism 7500 System (Applied Biosystems, Foster City, CA) with SYBR Green PCR Master Mix (cat. no. RR820, TaKaRa). Data were normalized to GAPDH and represented as the average fold of three independent duplicates.
Cell migration and invasion assays
For Transwell assay, cells were starved in 200 µl FBS-free medium and seeded into the upper chambers of 8.0 μm-pore Transwell (cat. no. 3422; Corning Inc., Corning, NY) pre-coated with or without 2.5% Matrigel (cat. no. 3356234; BD, Franklin Lakes, NJ). The lower chambers were filled with 600 µl medium supplemented with 10% FBS, serving as chemoattractant. After being incubated at 37 oC for 24 hours, the cells migrated or invaded through the membrane were fixed with 4% paraformaldehyde for 15 minutes, stained with 0.1% crystal violet (cat. no. C0121; Beyotime, China), imaged by using a microscope (Olympus BX51; Olympus Corporation, Japan) and counted. The results were obtained and averaged based on five randomly selected fields (magnification, ×200). For wound-healing assay, cells were cultured in 6-well plates. When cells were grown to about 90–100% confluence, a scratch was made with a 10 µl pipette tip in each well. After washing with PBS, cells were maintained in medium with 1% FBS. Pictures were taken before and after the 24 hours’ culture with a phase-contrast microscope (ECLIPSE TS100, Nikon, Japan). The cell migration rate in the scratch distance was quantified by ImageJ software.
Animal experiments
The animal experiments took place in Fujian Medical University Laboratory Animal Center. Mock or SREBP-2-overexpressed PC-9 cells or blank PC-9 cells (5 × 106 in 100 µl phosphate buffered saline, PBS, with 50% Matrigel) were inoculated subcutaneously into the right flanks of BALB/c nude mice to establish mouse xenograft tumor model. For in vivo SREBP-2 inhibition study, when the tumors reached a volume of ~ 100mm3, mice were randomly grouped and injected intraperitoneally with increasing concentrations of fatostatin (0, 10, and 30 mg/kg) every day for 29 days. The tumor growth and body weight were monitored every 3 days. Tumor volume was calculated as (length × width2)/2. At the endpoint of study, the mice were euthanized by carbon dioxide asphyxiation, their organs of heart, liver, spleen, lung and kidney from mice in each group were harvested for H&E staining. The animal experiments was approved by the Institutional Animal Care and Use Committee at Fujian Medical University (Fuzhou, China; approval no. IACUC FJMU 2023-Y-0432). All animal procedures complied with the guidelines of the Institutional Animal Care and Use Committee at Fujian Medical University.
Statistical analysis
Quantitative data were shown as the mean ± standard deviation (SD), and analyzed using Student’s t-test for two-group comparisons. Categorical data were compared using chi-square test or Fisher’s exact test. The survival data were analyzed by Kaplan-Meier method using the log-rank test. Univariate and multivariate Cox regression analysis was employed to identify independent predictors. Statistical analysis was performed using SPSS Statistics 26.0 (IBM Corp.) and GraphPad Prism 8.0 (GraphPad Software, Inc.). All tests were two-sided and p < 0.05 was considered as statistically significant.
Results
SREBP-2 is upregulated in NSCLC and predicts poor clinical outcomes
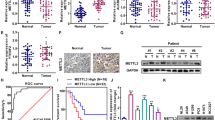
To define the clinical relevance of SREBP-2 in NSCLC, IHC was performed to examine SREBP-2 expression in a training cohort of 275 NSCLC and 263 matched normal lung tissues. Representative IHC images were shown in Fig. 1A. Results indicated that SREBP-2 was significantly increased in NSCLC than in pericarcinous lung tissues (Fig. 1B).

SREBP-2 is upregulated in NSCLC and predicts poor OS. IHC was performed in 275 NSCLC tissues and 263 matched pericarcinous lung tissues. (A) Representative IHC images of SREBP-2 expression in NSCLC tissues are shown (low expression, scores 0–4; high expression, scores 6–12). Isotype control: The primary antibody was replaced by nonspecific rabbit antibody. Scale bar, 50 μm. (B) The IHC score of SREBP-2 was higher in 263 NSCLC tissues than in corresponding pericarcinous lung tissues (p < 0.001, Student’s t-test). (C) Kaplan-Meier analysis shows that “SREBP-2 high” patients (n = 105) had a shorter OS than “SREBP-2 low” patients (n = 170) (p < 0.001, log-rank test).
For further analysis, NSCLC samples were divided into two groups defined as “SREBP-2 low” (scores of 0–4) and “SREBP-2 high” (scores of 6–12). Consistently, the proportion of “SREBP-2 high” specimens was higher in NSCLC samples than in matched pericarcinous lung samples (Table 1). Moreover, a chi-square analysis indicated that high SREBP-2 expression was associated with the elder age (> 60 years at diagnosis), more advanced pTNM stage (stage IIB-V), larger tumor size (> 3.0 cm for longer diameter), T status (T2-T4), and lymph node metastasis in NSCLC patients (Table 2).
The prognostic value of SREBP-2 was also analyzed. Our results revealed that “SREBP-2 high” NSCLC patients had a significantly shorter overall survival (OS) than those expressing lower levels of SREBP-2 (Fig. 1C). Cox regression analysis demonstrated that high SREBP-2 expression, as well as advanced pTNM stage, were independent predictors for shorter OS in patients with NSCLC (Table 3).
To verify the above findings, the expression of SREBP-2 was examined in an independent validation cohort, which includes 184 NSCLC and 176 matched normal lung tissues. As expected, SREBP-2 was upregulated in NSCLC tissues (Fig. S1A and B, and Table S2), and associated with aggressive clinicopathological characteristics, including advanced pTNM stage, larger tumor size, advanced T status and lymph node metastasis (Table S3). Moreover, OS was significantly worse in patients with a high than in patients with a low SREBP-2 expression (Fig. S1C and Table S4), demonstrating the prognostic significance of SREBP-2 in NSCLC.
SREBP-2 promotes cell proliferation, migration and invasion in NSCLC
As SREBP-2 predicted adverse clinical outcomes in patients with NSCLC, we hypothesized that SREBP-2 acts as an oncogene in tumorigenesis of NSCLC. Biofunctional experiments were then conducted. As indicated in Fig. 2A–D, SREBP-2 silencing with small interfering RNAs significantly reduced the proliferation rate of H1299 and PC-9 cells. Crystal violet staining also showed that SREBP-2 knockdown significantly decreased the proliferation of H1299 and PC-9 cells (Fig. S2A–D). The effect of SREBP-2 in NSCLC migration and invasion was evaluated by Transwell and wound healing assays. Results showed that SREBP-2 knockdown abrogated the migration and invasion of both H1299 and PC-9 cells (Fig. 2E and F, and Fig. S2E and F).

SREBP-2 knockdown impairs the proliferation, migration and invasion of NSCLC cells. H1299 and PC-9 cells were transfected with NC- or SREBP-2-siRNAs. Expression levels of SREBP-2 in H1299 (A) and PC-9 (B) cells were measured using Western blot analysis. GAPDH served as an internal control. Cell proliferation of H1299 (C) and PC-9 (D) was measured using a CCK-8 assay. Cell migratory and invasive abilities of H1299 (E) and PC-9 (F) were examined using the Transwell method at 48 hour after transfection. Data are presented as the mean ± SD of at least 3 independent experiments. * p < 0.05, ** p < 0.01, *** p < 0.001, compared with the NC group.
The functional role of SREBP-2 was validated in SREBP-2-overexpressed NSCLC cells. Firstly, the mRNA levels of HMGCS1, HMGCR and LDLR, three well-recognized SREBP-2 target genes, were examined in SREBP-2-overexpressed PC-9 stable cells5,6,13. As expected, enforced expression of SREBP-2 in PC-9 led to significantly increased mRNA levels of both SREBP-2 and target genes, including HMGCS1, HMGCR and LDLR (Fig. 3A and B). Importantly, SREBP-2 overexpression significantly promoted the proliferation, migration and invasion of PC-9 cells (Fig. 3C and D, and Fig. S3). At the same time, fatostatin, a specific inhibitor of SREBP activation14dose-dependently abrogated the promotion of proliferation, migration and invasion by SREBP-2 overexpression in PC-9 cells (Fig. 3C and D, and Fig. S3). Together, these data demonstrated an oncogenic role of SREBP-2 in the proliferation, migration and invasion of NSCLC.

SREBP-2 overexpression promotes NSCLC cell proliferation, migration and invasion. PC-9 cells were infected with lentiviral vectors to establish mock or SREBP-2-overexpressed stable cell lines. (A) Expression levels of SREBP-2 in SREBP-2-overexpressed PC-9 stable cells were measured using Western blot analysis. (B) Relative mRNA levels of SREBP-2, HMGCS1, HMGCR and LDLR in SREBP-2-overexpressed PC-9 stable cells were examined by qRT-PCR. GAPDH served as an internal control. (C) SREBP-2-overexpressed PC-9 stable cells were treated with increasing concentrations of fatostatin (0, 10, 20, and 40 µM) for 72 hours. Cell proliferation rate of the treated PC-9 stable cells was tested by crystal violet staining method. (D) Cell migratory and invasive abilities of the treated PC-9 stable cells were examined using the Transwell method. Data are presented as the mean ± SD of at least 3 independent experiments. ** p < 0.01, *** p < 0.001, compared with the mock group. ## p < 0.01, ### p < 0.001, compared with the control group.
SREBP-2 promotes cell proliferation, migration and invasion via mevalonate-Akt pathway in NSCLC
The oncogenic PI3K/Akt and Erk1/2 signals have been implicated in both tumorigenesis and the progression of NSCLC15,16. We sought to determine whether there is a potential correlation between SREBP-2 and these two intracellular signaling pathways in NSCLC. As shown in Fig. 4A and B, SREBP-2 knockdown reduced the phosphorylation of Akt (Ser473) in both H1299 and PC-9 cells, suggested the inactivation of Akt signals. In comparison, the expression levels of phosphorylated Erk1/2 (Thr202/Tyr204) remained unchanged upon SREBP-2 silencing either in H1299 cells or in PC-9 cells (Fig. 4A and B). By contrast, SREBP-2 overexpression increased the phosphorylated Akt (Ser473) without apparently affecting Erk1/2 phosphorylation (Thr202/Tyr204) in PC-9 cells (Fig. 4C). The functional relevance of Akt signaling pathway was then evaluated. Results showed that MK-2206, a selective Akt inhibitor, decreased SREBP-2-promoted proliferation, migration and invasion in PC-9 cells in a dose-dependent manner (Fig. 4D–F, and Fig. S4). Together, these data demonstrated that SREBP-2-promoted NSCLC proliferation, migration and invasion are mediated by Akt signaling pathway.

SREBP-2-promoted NSCLC proliferation, migration and invasion are mediated by Akt signaling pathway. H1299 and PC-9 cells were transfected with NC- or SREBP-2-siRNAs for 48 hours. Expression levels of SREBP-2, p-Akt, t-Akt, p-Erk1/2, t-Erk1/2 protein in H1299 (A) and PC-9 (B) cells were measured using Western blot analysis. (C) Expression levels of SREBP-2, p-Akt, t-Akt, p-Erk1/2, t-Erk1/2 protein in SREBP-2-overexpressed PC-9 stable cells were measured by Western blotting. GAPDH served as an internal control. (D) SREBP-2-overexpressed PC-9 stable cells were treated with increasing concentrations of MK2206 (0, 20, 50, and 100 nM) for 24 hours. Expression levels of SREBP-2, p-Akt, t-Akt protein in the treated PC-9 stable cells were measured by Western blotting. (E) Cell proliferation rate of the treated PC-9 stable cells was tested by crystal violet staining method. (F) Cell migratory and invasive abilities of the treated PC-9 stable cells were examined using the Transwell method. Data are presented as the mean ± SD of at least 3 independent experiments. *** p < 0.001, compared with the mock group. ### p < 0.001, compared with the control group.
Since SREBP-2 served as a key transcriptional regulator involved in cholesterol metabolism and homeostasis17we questioned whether SREBP-2-activated Akt signals is mediated by SREBP-mevalonate pathway. Simvastatin, a pharmacological inhibitor of the mevalonate pathway in cholesterol biosynthesis18 was applied in further experiments. Results showed that simvastatin treatment dose-dependently inhibited SREBP-2-induced Akt (Ser473) phosphorylation, cell proliferation, migration and invasion in PC-9 stable cells (Fig. 5A–C, and Fig. S5), implying that SREBP-2-induced Akt signals, cell proliferation and invasion are mediated by the mevalonate pathway in NSCLC. Altogether, the above data demonstrated that SREBP-2 promotes cell proliferation, migration and invasion via the SREBP-2-mevalonate-Akt pathway in NSCLC.

SREBP-2 promotes NSCLC proliferation, migration and invasion via the mevalonate-Akt pathway. SREBP-2-overexpressed PC-9 stable cells were treated with increasing concentrations of simvastatin (0, 2, 5, and 10 µM) for 24 hours. (A) Expression levels of p-Akt and t-Akt protein in the treated PC-9 stable cells were measured by Western blotting. GAPDH served as an internal control. (B) Cell proliferation rate of the treated PC-9 stable cells was tested by crystal violet staining method. (C) Cell migratory and invasive abilities of the treated PC-9 stable cells were examined using the Transwell method. Data are presented as the mean ± SD of at least 3 independent experiments. *** p < 0.001, compared with the mock group. ### p < 0.001, compared with the control group.
SREBP inhibitor suppresses NSCLC tumor growth in mouse xenograft models
Firstly, we evaluated the role of SREBP-2 in tumor growth in vivo. A xenograft NSCLC model was established in nude mice using PC-9 stable cells with or without SREBP-2 overexpression. Results showed that enforced expression of SREBP-2 in PC-9 significantly promoted tumor growth in vivo, confirming the tumor-promoting role of SREBP-2 in NSCLC (Fig. 6A). No marked difference in body weight was observed between mice bearing mock or SREBP-2-overexpressing PC-9 tumors (Fig. 6B). Then, the effect of fatostatin on tumor growth was determined in xenografted SREBP-2-overexpressing PC-9 mice. As shown in Fig. 6C, fatostatin significantly reduced the in vivo tumor growth in this xenograft NSCLC model in a dose-dependent manner. However, we observed that 30 mg/kg fatostatin led to an apparent body weight reduction in mice bearing SREBP-2-overexpressing PC-9 tumors (Fig. 6D), implying a side effect of high-dose fatostatin. On the other hand, while the solvent of fatostatin resulted in morphological changes in the liver, spleen and kidney of mice, 30 mg/kg fatostatin had no significant effect on the morphology of the above organs (Fig. S6). Together, these results suggested that SREBP inhibitor decreased NSCLC tumor growth in mouse xenograft models in vivo.

SREBP inhibitor suppresses NSCLC tumor growth in mouse xenograft models. Mock or SREBP-2-overexpressed PC-9 stable cells or blank PC-9 cells were injected subcutaneously into the right flanks of nude mice. The tumor volume (A) and body weight (B) of mice in each group were monitored every three days. n = 8 for blank group, n = 14 for mock and SREBP-2-overexpressing PC-9 tumor groups. As in A and B, mouse xenograft tumor models were established. When the tumors reached a volume of ~ 100mm3mice were randomly grouped and injected intraperitoneally with increasing concentrations of fatostatin (0, 10, and 30 mg/kg) every days for 29 days. The tumor volume (C) and body weight (D) of mice in each group were monitored every three days. n = 5 for mock and SREBP-2-overexpressing PC-9 tumor groups, n = 9 for 10 mg/kg and 30 mg/kg fatostatin-treated SREBP-2-overexpressing PC-9 tumor groups. Data are presented as the mean ± SD. ** p < 0.01, *** p < 0.001, compared with the mock or SREBP-2-overexpressing PC-9 tumors. ns, not significant.
Discussion
SREBP-2 is a metabolic transcriptional factor that preferentially regulates genes involved in cholesterol metabolism and homeostasis4. Recent studies have documented an important role of SREBP-2 in numerous cancers, including prostate, esophagus, ovarian, breast and liver carcinomas7,8,9,10,11. However, the expression alongside clinical relevance of SREBP-2 in NSCLC patients have never been elucidated. Here, we showed that SREBP-2 was upregulated in NSCLC tissues, and correlated with poor clinical characteristics. High SREBP-2 expression predicted a shorter OS and acted as an independent predictor for adverse OS in NSCLC. Moreover, biofunctional experiments demonstrated an oncogenic role of SREBP-2 in NSCLC proliferation, migration and invasion. Mechanistical study suggested that SREBP-2 promoted cell proliferation, migration and invasion via the mevalonate-Akt pathway in NSCLC. In vivo treatment experiments demonstrated that SREBP inhibitor decreased NSCLC tumor growth in mouse xenograft models. This study represents the first to evaluate SREBP-2 expression in a large cohort of NSCLC samples and provides evidence on tumor-promoting functions of SREBP-2 in NSCLC progression.
In the present study, a total of 459 NSCLC tissues and 439 matched pericarcinous lung tissues were screened for SREBP-2 expression to dissect its expression pattern and clinical significance. We found that SREBP-2 was upregulated in NSCLC than in matched normal lung tissues. High SREBP-2 expression correlated with aggressive clinical features, such as elder age, advanced pTNM stage, larger tumor size, advanced T status and lymph node metastasis, and predicted a shorter OS in NSCLC. Notably, it was demonstrated by Cox regression that high SREBP-2 expression acted as an independent predictor for adverse OS in NSCLC patients. Consistently, Li et al. reported that SREBP-2 was increased in advanced pathological grade and bone-metastatic prostate cancer tissues, and correlated with shorter recurrence-free survival and OS7. Another study revealed that the increased expression of SREBP-2 predicted a shorter metastasis-free survival rates in patients with breast cancer10. Although there is no literature on the mechanism of SREBP-2 upregulation in lung cancer, numerous studies have reported the regulatory mechanisms of SREBP2 in other cancers. For instance, SREBP-2 can be transcriptionally upregulated by activated STAT3 (Tyr705) in triple-negative breast cancer19. In squamous cancer, USP28, a deubiquitinating enzyme, can bind to mature SREBP-2, leading to its deubiquitination and stabilisation20. Further studies are warranted to uncover the mechanism by which SREBP-2 is up-regulated in NSCLC.
SREBP-2 was reported to correlate with tumor proliferation and metastasis in numerous types of cancer8,21,22,23. In the present study, we provide agreeable data that SREBP-2 knockdown impaired the proliferation, migration and invasion of NSCLC cells, while enforced SREBP-2 promoted NSCLC proliferation, migration and invasion. Recently, it has been recognized that aberrant lipid and cholesterol anabolism is highly involved in carcinogenesis, including ovarian, prostate, and colorectal cancers24,25,26. Cholesterol biosynthesis and uptake are discovered to be enhanced in NSCLC and infiltrating immunosuppressive cells, consequently promoting cancer progression27,28,29. In this regard, SREBP-2 serves as a key regulator in elevating intracellular cholesterol via upregulating the expression of HMGCR and LDLR. etc., in response to low intracellular cholesterol levels5,6. This study suggests an oncogenic role for SREBP-2 in NSCLC, which is quite reasonable since lipid and cholesterol constitute chief membrane components and lipid raft microdomains in tumor cells30. SREBP-2 activates the transcription of cholesterol biosynthetic genes, sustaining intracellular cholesterol levels and contributing to the metabolic dysregulation in cancer, which is necessary for tumor proliferation, migration, trafficking and signal transduction31. This study verified the upregulation of HMGCS1, HMGCR and LDLR mRNA in SREBP-2-overexpressed PC-9 stable cells. In accordance with this hypothesis, knockdown of ATP citrate lyase, an enzyme responsible for lipid synthesis from glucose, decreased cell proliferation stimulated by cytokines in vitro and impair tumorigenesis in vivo in hematopoietic tumors32. Moreover, cholesterol-lowering drugs were demonstrated anti-cancer and anti-metastatic effects for treating renal caicinoma, malignant melanoma and ovarian cancer33,34,35. Consistently, our in vivo treament experiments found that SREBP inhibitor fatostatin dose-dependently suppressed NSCLC tumor growth in mouse xenograft models.
It has been widely acknowledged that SREBPs serve as downstream molecules of the PI3K/Akt/mTOR pathway in tumors36. Herein, we found that SREBP-2 knockdown reduced, while SREBP-2 overexpression increased, Akt phosphorylation (Ser473) in NSCLC cells, facts that can be interpreted as follows: SREBP-2 knockdown leads to a dysregulated cholesterol metabolism, thereby impairing lipid raft formation in cancer cells. As a result, growth factor signaling, including the oncogenic Akt pathway which is predominantly mediated by lipid rafts, can be attenuated in SREBP2-silenced cancer cells. This hypothesis was validated by mevalonate inhibition experiments that simvastatin dose-dependently inhibited SREBP-2-induced activation of Akt signals in PC-9 stable cells. Consistently, Yamauchi et al. have reported that inhibiting SREBP reduced Akt activation in lipid rafts and inhibited human melanoma cell proliferation and tumor growth37. From this standpoint of view, we speculate that there should be a feed-forward loop between the Akt signals and SREBP-2 in NSCLC cells, which is essential for cancer development. Further studies are warranted to unravel the interplay between the Akt signals and SREBP-2, which might have therapeutic implications in cancer.
There are several limitations in the present study. First, although it was shown that fatostatin dose-dependently reduced NSCLC tumor growth in vivo, transient dermatitis and body weight reduction were observed in high-dose fatostatin-treated mice. Moreover, since no specific SREBP-2 inhibitor exists, fatostatin, an inhibitor to impair the activation of both SREBP-1 and SREBP-2, was used in this study as previously described38,39. Future studies are warranted to investigate the well-tolerated doses of fatostatin, as well as the anti-tumor activities of more specific SREBP-2 inhibitors. Second, SREBP2 activation and cholesterol synthesis was found to be required in triggering IL-1β release in bacteria or LPS-activated macrophages18, suggesting that SREBP-2 may affect the tumor microenvironment. However, only the effects of SREBP-2 on NSCLC cell proliferation, migration and invasion were determined in this study. More efforts are required to comprehensively elucidate the oncogenic role of SREBP-2 in NSCLC progression.
In conclusion, this study provided novel results that SREBP-2 was increased in NSCLC tissues, and correlated with poor clinical features. High SREBP-2 expression acted as an independent predictor for shorter OS in patients with NSCLC. Functionally, SREBP-2 possessed an oncogenic role in NSCLC cell proliferation, migration and invasion. Mechanistical study showed that SREBP-2 promoted cell proliferation, migration and invasion via the mevalonate-Akt pathway in NSCLC. Importantly, SREBP inhibitor fatostatin effectively suppressed NSCLC tumor growth in mouse xenograft models in vivo. The present study recommends SREBP-2 as a candidate prognostic factor and therapeutic target for patients with NSCLC.
Data availability
The data used and analyzed in the present study are available from the corresponding author on reasonable request.
Abbreviations
- NSCLC:
-
Non-small cell lung cancer
- SREBP-2:
-
Sterol regulatory element-binding protein-2
- HMGCR:
-
Hydroxymethylglutaryl (HMG)-CoA reductase
- LDLR:
-
Low-density lipoprotein receptor
- IHC:
-
Immunohistochemistry
- TMA:
-
Tissue microarray
- HRP:
-
Horseradish peroxidase
- ATCC:
-
American type culture collection
- DMSO:
-
Dimethyl sulfoxide
- CCK-8:
-
Cell counting kit-8
- NC:
-
Negative control
- ECL:
-
Enhanced chemiluminescence
- qRT-PCR:
-
Quantitative real-time PCR
- HMGCS1:
-
HMG-CoA synthase 1
- SD:
-
Standard deviation
- OS:
-
Overall survival
References
Sung, H. et al. Global Cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 3 (71), 209–249 (2021).
Duma, N., Santana-Davila, R. & Molina, J. R. Non-small cell lung cancer: Epidemiology, screening, diagnosis, and treatment. Mayo Clin. Proc. 8 (94), 1623–1640 (2019).
Zhang, C., Quinones, A. & Le, A. Metabolic reservoir cycles in cancer. Semin. Cancer Biol. (2022).
Sato, R. Sterol metabolism and SREBP activation. Arch. Biochem. Biophys. 2 (501), 177–181 (2010).
Eberlé, D. et al. SREBP transcription factors: master regulators of lipid homeostasis. Biochimie 11 (86), 839–848 (2004).
DeBose-Boyd, R. A. & Ye, J. SREBPs in lipid metabolism, insulin signaling, and beyond. Trends Biochem. Sci. 5 (43), 358–368 (2018).
Li, X. et al. SREBP-2 promotes stem cell-like properties and metastasis by transcriptional activation of c-Myc in prostate cancer. Oncotarget 11 (7), 12869–12884 (2016).
Zhong, C. et al. SREBP2 is upregulated in esophageal squamous cell carcinoma and co–operates with c–Myc to regulate HMGCR expression. Mol. Med. Rep. 4 (20), 3003–3010 (2019).
Casella, C. et al. Oxysterols synergize with Statins by inhibiting SREBP-2 in ovarian cancer cells. Gynecol. Oncol. 2 (135), 333–341 (2014).
Jie, Z. et al. SREBP-2 aggravates breast cancer associated osteolysis by promoting osteoclastogenesis and breast cancer metastasis. Biochim. Biophys. Acta Mol. Basis Dis. 1 (1865), 115–125 (2019).
Moon, S. H. et al. p53 represses the mevalonate pathway to mediate tumor suppression. Cell 3 (176), 564–580e519 (2019).
Detterbeck, F. C. et al. The eighth edition lung Cancer stage classification. Chest 1 (151), 193–203 (2017).
Pandyra, A. A. et al. Genome-wide RNAi analysis reveals that simultaneous Inhibition of specific mevalonate pathway genes potentiates tumor cell death. Oncotarget 29 (6), 26909–26921 (2015).
Kamisuki, S. et al. A small molecule that blocks fat synthesis by inhibiting the activation of SREBP. Chem. Biol. 8 (16), 882–892 (2009).
Lee, M. W. et al. Roles of AKT1 and AKT2 in non-small cell lung cancer cell survival, growth, and migration. Cancer Sci. 10 (102), 1822–1828 (2011).
Tan, A. C. Targeting the PI3K/Akt/mTOR pathway in non-small cell lung cancer (NSCLC). Thorac. Cancer. 3 (11), 511–518 (2020).
Miao, J. et al. Hepatic insulin receptor deficiency impairs the SREBP-2 response to feeding and Statins. J. Lipid Res. 4 (55), 659–667 (2014).
Dang, E. V. et al. Oxysterol restraint of cholesterol synthesis prevents AIM2 inflammasome activation. Cell 5 (171), 1057–1071e1011 (2017).
Chen, Y. Y. et al. Copy number amplification of ENSA promotes the progression of triple-negative breast cancer via cholesterol biosynthesis. Nat. Commun. 1 (13), 791 (2022).
Maier, C. R. et al. USP28 controls SREBP2 and the mevalonate pathway to drive tumour growth in squamous cancer. Cell. Death Differ. 7 (30), 1710–1725 (2023).
Yu, Y. et al. LncRNA SNHG16 induces the SREBP2 to promote lipogenesis and enhance the progression of pancreatic cancer. Future Oncol. 33 (15), 3831–3844 (2019).
Li, X. et al. MicroRNA-185 and 342 inhibit tumorigenicity and induce apoptosis through blockade of the SREBP metabolic pathway in prostate cancer cells. PLoS ONE 8 (8), e70987 (2013).
Chakraborty, P. K. et al. Role of cystathionine beta synthase in lipid metabolism in ovarian cancer. Oncotarget 35 (6), 37367–37384 (2015).
Jacobs, R. J. et al. Cholesterol metabolism and colorectal cancers. Curr. Opin. Pharmacol. 6 (12), 690–695 (2012).
Hager, M. H., Solomon, K. R. & Freeman, M. R. The role of cholesterol in prostate cancer. Curr. Opin. Clin. Nutr. Metab. Care. 4 (9), 379–385 (2006).
Tania, M., Khan, M. A. & Song, Y. Association of lipid metabolism with ovarian cancer. Curr. Oncol. 5 (17), 6–11 (2010).
Dai, Y. B. et al. Ablation of liver X receptors α and β leads to spontaneous peripheral squamous cell lung cancer in mice. Proc. Natl. Acad. Sci. U S A. 27 (113), 7614–7619 (2016).
Mohammadalipour, A. et al. Cholesterol depletion decreases adhesion of non-small cell lung cancer cells to E-selectin. Am. J. Physiol. Cell. Physiol. 2 (325), C471–C482 (2023).
Lagunas-Rangel, F. A. Cholesterol effects on the tumor immune microenvironment: from fundamental concepts to mechanisms and implications. Front. Oncol. 15, 1579054 (2025).
Swinnen, J. V., Brusselmans, K. & Verhoeven, G. Increased lipogenesis in cancer cells: new players, novel targets. Curr. Opin. Clin. Nutr. Metab. Care. 4 (9), 358–365 (2006).
Chang, C. H. et al. Metabolic competition in the tumor microenvironment is a driver of Cancer progression. Cell 6 (162), 1229–1241 (2015).
Bauer, D. E. et al. ATP citrate lyase is an important component of cell growth and transformation. Oncogene 41 (24), 6314–6322 (2005).
Fang, Z. et al. Simvastatin inhibits renal cancer cell growth and metastasis via akt/mtor, ERK and JAK2/STAT3 pathway. PLoS ONE 5 (8), e62823 (2013).
Stine, J. E. et al. The HMG-CoA reductase inhibitor, simvastatin, exhibits anti-metastatic and anti-tumorigenic effects in ovarian cancer. Oncotarget 1 (7), 946–960 (2016).
Chen, Y. C. et al. Targeting cholesterol transport in Circulating melanoma cells to inhibit metastasis. Pigment Cell. Melanoma Res. 6 (30), 541–552 (2017).
Yi, J. et al. Oncogenic activation of PI3K-AKT-mTOR signaling suppresses ferroptosis via SREBP-mediated lipogenesis. Proc. Natl. Acad. Sci. U S A. 49 (117), 31189–31197 (2020).
Yamauchi, Y. et al. Positive feedback loop between PI3K-Akt-mTORC1 signaling and the lipogenic pathway boosts Akt signaling: induction of the lipogenic pathway by a melanoma antigen. Cancer Res. 14 (71), 4989–4997 (2011).
Zhu, L. et al. Fatostatin promotes anti-tumor immunity by reducing SREBP2 mediated cholesterol metabolism in tumor-infiltrating T lymphocytes. Eur J. Pharmacol (971), 176519 (2024).
Codenotti, S. et al. Synthetic Inhibition of SREBP2 and the mevalonate pathway blocks rhabdomyosarcoma tumor growth in vitro and in vivo and promotes chemosensitization. Mol. Metab. 92, 102085 (2025).
Acknowledgements
We would like to acknowledge the help from Dr Jiawen Xu and Dr Zhenhui Su at Shandong Provincial Hospital (Jinan, China) for evaluating the IHC staining.
Funding
This work was supported in part by grants 82104203, 81930073 and 81902325 from the National Natural Science Foundation of China, grants 202019201 and 202225051 from the Jinan Science and Technology Plan Project (China), grant 2023M740684 from the China Postdoctoral Science Foundation, grant ZR2023QH021 from the Shandong Provincial Natural Science Foundation (China), grant 20JC1417200 from the Shanghai Technology Innovation Action Project (China), grant SHSLCZDZK02104 from the Shanghai Municipal Key Clinical Specialty Project (China), and grant XHLHGG202101 from the Cooperation Project of Conquering Major Diseases in Xuhui District (China).
Author information
Authors and Affiliations
Contributions
Wenjie You, Lili Su and Shuping Weng: data curation, software, formal analysis, investigation, resources, visualization and methodology. Wenjie You: writing-original draft. Jing Li and Xingguang Wang: resources, methodology, visualization, formal analysis and validation. Bin Liang and Daowei Li: resources, software and validation. Lijun Li and Haiquan Chen: conceptualization, data curation, supervision, funding acquisition, validation, project administration and writing-review and editing. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics
The study protocol was approved by the Ethics Committee of Shandong Provincial Hospital affiliated to Shandong First Medical University (Jinan, China; approval no. NSFC-2019-05) and complied with the Helsinki declaration and the approved guidelines of our institution. Written informed consent was obtained from all participants. The animal experiments was approved by the Institutional Animal Care and Use Committee at Fujian Medical University (Fuzhou, China; approval no. IACUC FJMU 2023-Y-0432). The authors complied with the ARRIVE guidelines.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
You, W., Su, L., Weng, S. et al. SREBP-2 promotes cancer progression through the mevalonate-Akt pathway in non-small cell lung cancer. Sci Rep 15, 23103 (2025). https://doi.org/10.1038/s41598-025-07437-0
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-07437-0
