
Abstract
Periplocin, a tumor-inhibitory compound derived from Cortex periploca, was investigated for its mechanisms of action and therapeutic potential against human papillary thyroid carcinoma cell proliferation. BCPAP/TPC-1 cells were treated with periplocin ± necrostatin-1 (a necroptosis inhibitor) or Z-Val-Ala-Asp(OMe)-fluoromethylketone (an apoptosis inhibitor). Cell proliferation was assessed using the cell counting kit 8/real-time cell analysis assay. Necrotic morphology was quantified using Hoechst 33,342/propidium iodide (PI) staining (PI⁺ rate), and apoptosis/necroptosis pathways were analyzed using Annexin V-FITC/PI flow cytometry. RNA-seq was conducted to compare the transcriptomes of periplocin-treated and untreated TPC-1 cells. The key proteins (phosphorylated mixed lineage kinase domain-like protein [p-MLKL], phosphorylated receptor-interacting protein kinase-3 [p-RIP3], IL6, IL1A, and death receptor 4 [DR4]) were validated by western blotting. In vivo, TPC-1 xenografts from BALB/c nude mice were evaluated for tumor growth, necrosis (PI staining), and expression of protein markers (proliferating cell nuclear antigen, p-MLKL, and p-RIP3) via immunohistochemistry. Periplocin suppressed the growth of BCPAP and TPC-1 thyroid carcinoma cells via concentration-dependent necroptosis. Necrostatin-1 co-treatment reduced PI⁺ cells and necrotic morphology (high Hoechst/PI staining; P < 0.05), confirming necroptosis dependence. Mechanistically, periplocin activated RIP3/MLKL signaling and damage-associated molecular patterns, whereas DR4 knockdown (si-DR4) attenuated p-MLKL expression (P < 0.01), indicating DR4-mediated pathway activation. In vivo, periplocin reduced TPC-1 xenograft volume (P < 0.01), weight, and proliferation (decreased proliferating cell nuclear antigen⁺ cells; P < 0.05), while elevating p-RIP3/p-MLKL (P < 0.01) and PI⁺ necrosis (P < 0.01). Periplocin selectively induces DR4-dependent necroptosis via RIP3/MLKL activation, providing the first evidence of necroptosis induction in papillary thyroid carcinoma. These findings suggest that periplocin is a promising therapeutic candidate, particularly for tumor necrosis factor-related apoptosis-inducing ligand-resistant papillary thyroid carcinoma that evades apoptosis.
Similar content being viewed by others

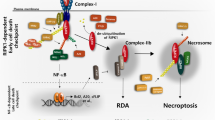
Introduction
Papillary thyroid carcinoma (PTC) accounts for 80% of thyroid cancer cases. Even though surgery, radioactive iodine therapy, and TSH suppression have greatly improved outcomes, about 5–15% of patients still have lower survival rates because their cancer comes back or spreads to other parts of the body1,2. Current treatment options for advanced or radioactive iodine-refractory PTC (such as multi-kinase inhibitors like sorafenib and lenvatinib) face severe challenges of acquired resistance and toxicity3. Tumour necrosis factor-related apoptosis-inducing ligand (TRAIL) has attracted much attention for its property of selectively inducing apoptosis in tumour cells by activating the death receptors DR4/DR54. However, about 60% of PTC cells have intrinsic resistance to TRAIL-mediated apoptosis, which is closely related to the inhibition of death receptor expression and the abnormal activation of the PI3K/AKT pathway5,6. How to overcome this resistance has become a key scientific issue.
The RIPK1/RIPK3/MLKL pathway regulates necroptosis7, a form of lytic cell death, and it offers unique advantages: Overcoming apoptosis resistance: It does not rely on caspases and can get around resistance issues like the silencing of death receptors or the overproduction of BCL-2. Activating anti-tumour immunity: The rupture of the cell membrane releases DAMPs molecules. However, no effective targeted strategy for activating this pathway has been found in PTC so far. Periplocin, a natural cardiac glycoside derived from the bark of Periploca sepium, has broad-spectrum anti-tumour activity: Pathway regulation: It targets pathways such as AKT/NF-κB and AMPK/mTOR, inhibits the proliferation of colorectal cancer and liver cancer, and induces G2/M phase arrest8,9,10,11,12,13,14. Reversing drug resistance: In combination with gemcitabine/oxaliplatin, it overcomes drug resistance by inhibiting Nrf2 and blocking M2 macrophage polarisation8,13. Immunomodulation: It downregulates oncogenes such as survivin/c-myc and inhibits the infiltration of immunosuppressive cells such as MDSCs9,12. Key gap: Although it has been confirmed in gastric cancer that periplocin can upregulate DR4/DR5 and enhance TRAIL sensitivity15, there is no report on its role in inducing necroptosis through the DR4/RIPK3/MLKL pathway to overcome PTC resistance.
This study reveals for the first time that periplocin overcomes TRAIL resistance in PTC by specifically activating DR4/RIPK3/MLKL-mediated necroptosis. We have clarified the mechanism of DR4-dependent necroptosis activation by combining RNA-seq and functional verification. This discovery provides a new strategy for targeting death receptors to reverse PTC resistance and also opens a new direction for natural compounds to induce programmed cell death.
Materials and methods
Reagents and antibodies
Periplocin (CAS 13137-64-9) was purchased from Shanghai Yuanye Biotechnology Co., Ltd. (Shanghai, China) and authenticated via high-performance liquid chromatography using a C18 reverse-phase column (4.6 × 250 mm, 5 μm) with an acetonitrile/water (65:35 v/v) mobile phase at a 1.0 mL/min flow rate (UV detection at 220 nm), demonstrating ≥ 98% purity. Stock solutions were prepared at 10 mM in dimethyl sulfoxide (DMSO) in accordance with the compound’s solubility profiles and experimental requirements. Aliquots were stored at − 80 °C for long-term stability and diluted to working concentrations with phosphate-buffered saline (PBS) immediately before use, maintaining final DMSO concentrations below 0.1% to avoid solvent effects.
Primary antibodies against IL6 (clone D3K3N, #12153), IL1A (clone C48C3, #11195), GAPDH (clone 14C10, #2118), phospho-MLKL (Ser345; clone D6M5X, #62287), phospho-RIP3 (Ser227; clone D6W2T, #91702), β-actin (clone 13E5, #4970), DR4 (clone D9S1R, #42533), and proliferating cell nuclear antigen (PCNA) (clone D3H8P, #13110) were obtained from Cell Signaling Technology, Inc. (Danvers, MA, USA). TriZol™ reagent was sourced from Thermo Fisher Scientific, Inc. (Waltham, MA, USA), while BCA protein assay kits and SuperSignal™ West ECL substrates were procured from Pierce Biotechnology (Rockford, IL, USA).
Cell lines and cell culture
Human thyroid carcinoma cell lines BCPAP and TPC-1 were obtained from the American Type Culture Collection (Manassas, VA, USA). Before use, all cell lines were examined for Mycoplasma contamination. Cells were cultured in DMEM supplemented with 10% fetal bovine serum, 100 U/mL penicillin, and 100 U/mL streptomycin. Cultures were maintained at 37 °C in a humidified incubator containing 5% CO₂.
Cytotoxicity assay
The cytotoxicity of periplocin on thyroid cancer cells was assessed using real-time cell analysis (RTCA) and a Cell Counting Kit-8 (CCK-8) assay (Dojindo Molecular Technologies, Inc., Kumamoto, Japan). The xCELLigence RTCA system, outfitted with an RTCA MP station, was positioned inside a CO₂ incubator, whereas the control unit was operated externally using RTCA software (ACEA Biosciences, San Diego, CA, USA). For real-time monitoring, TPC-1 cells were seeded into 16-well E-plates (Roche Diagnostics GmbH, Basel, Switzerland) at a density of 5.0 × 10³ cells per well. Impedance readings were recorded every 15 min. After a 20 h incubation, the cells were treated with periplocin at concentrations of 0, 125, and 250 nM, either alone or in combination with necrostatin-1 (Nec-1) and Z-Val-Ala-Asp(OMe)-fluoromethylketone (ZVAD-FMK). The 150 µL culture system was maintained for up to 72 h, and cell proliferation was analyzed using RTCA-DP software (Roche Diagnostics GmbH). For the CCK-8 assay, BCPAP and TPC-1 cells were seeded in 96-well plates at 2,000 cells per well and treated with periplocin, Nec-1, ZVAD-FMK, or si-DR4-1 for 48 h. Cell viability was evaluated using CCK-8 (Dojindo Molecular Technologies, Inc., Kumamoto, Japan) according to the manufacturer’s instructions.
Annexin V-FITC/PI staining for apoptosis and necroptosis assessment
For cell counting, BCPAP and TPC-1 cells in the logarithmic growth phase were trypsinized, centrifuged, resuspended, and plated in six-well culture plates. The experimental groups received Nec-1 and/or ZVAD-FMK treatments, and control groups were included. The drugs were administered after cell adhesion. The supernatant was discarded, and the cells were washed twice with 2 mL PBS. Subsequently, 1 mL PBS containing 10 µL Hoechst 33,342 solution (Sigma-Aldrich) was added, followed by a 10 min incubation at room temperature in the dark. After incubation, cells were washed with PBS before adding 500 µL binding buffer containing 10 µL PI solution, then incubated for another 10 min in the dark.
For rapid processing of xenograft tumors:
-
Tumors were immersed in a pre-cooled preservation solution and transported on ice to the slicing platform (entire process < 10 min).
-
A total of 200 μm-thick live tissue slices were cut and transferred to a 24-well plate with oxygenated medium (95% O₂/5% CO₂), allowing them to recover metabolically for 1 h at 37 °C.
-
A total of 1 mL Hoechst working solution was added, incubated for 20 min at 37 °C in the dark.
-
Samples were gently washed three times with pre-warmed PBS (2 min each) to remove unbound dye.
-
PI working solution (1 mL) was added and incubated for 5 min at room temperature in the dark (avoiding over-incubation to prevent false positives).
-
Samples were quickly washed twice with PBS and immediately imaged (delay ≤ 10 min).
The cells were imaged using a confocal microscope. Necrotic cells showed Hoechst dark-blue staining, PI positivity, and nuclear disintegration. The necrosis rate was calculated as (PI⁺ cell count/total cell count) × 100. ImageJ software was used for threshold segmentation.
RNA-seq and pathway enrichment analysis
Total RNA was extracted from TPC-1 cells at 70–80% confluency after a 24 h treatment with either 0 or 125 nM periplocin using TRIzol reagent (Invitrogen, Carlsbad, CA, USA). The RNA samples were preserved on dry ice and transported to UW Genetics (Shenzhen, China) for high-throughput RNA sequencing using a BGISEQ-500 platform (BGI, Shenzhen, China). Differential gene expression analysis was performed, followed by functional annotation via Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway and Gene Ontology (GO) enrichment analyses. Data were processed using Dr. Tom’s online analysis platform (BGI).
Protein expression analysis via Western blotting
BCPAP and TPC-1 cells were treated with varying concentrations of periplocin, Nec-1, ZVAD-FMK, or si-DR4-1 individually for 24 h to reach 70–80% confluency. Cell lysates were centrifuged at 14,000×g for 20 min at 4 °C, and the supernatant was collected. Protein concentrations were determined using a BCA assay kit according to the manufacturer’s instructions. Equal amounts of protein (20 µg per lane) were separated by 12% SDS-PAGE at 100 V for 90 min and transferred onto 0.22 μm PVDF membranes. Membranes were blocked with 5% non-fat milk for 1.5 h at room temperature, then incubated with primary antibodies against IL6, IL1A, phosphorylated MLKL (p-MLKL), phosphorylated RIP3 (p-RIP3), β-actin, and GAPDH (1:1000 dilution). After washing, membranes were incubated with horseradish peroxidase-conjugated secondary antibodies. Protein bands were visualized using the BeyoECL Plus substrate (Beyotime Biotechnology) and imaged using a ChemiDoc MP system (Bio-Rad Laboratories, Hercules, CA, USA). Densitometric analysis was performed using ImageJ (National Institutes of Health, Bethesda, MD, USA).
In vivo tumorigenicity assay in nude mice
All animal experiments were conducted in accordance with the ARRIVE guidelines (Animal Research: Reporting of In Vivo Experiments) and approved by the Animal Ethics Committee of Jiangxi Zhonghong Boyuan Biotechnology Co., Ltd. (Approval No. LL-202404040001). All procedures adhered to relevant ethical regulations.
The experiment involved 4–6-week-old BALB/c nude mice purchased from Beijing Vital River Laboratory Animal Technology Co., Ltd. (Beijing, China). Subcutaneous tumor models were established by injecting 2 × 10⁶ TPC-1 cells into the right dorsal flank. Ten male mice were randomly assigned to two groups: vehicle control (5% DMSO, n = 5) and periplocin-treated (30 mg/kg periplocin in 5% DMSO, n = 5). Treatments were administered via intraperitoneal injection every 3 d for a total of three doses, and tumor dimensions (length and width) were measured every 3 d. After the final treatment, mice were euthanized using CO₂ asphyxiation. Tumors were excised for histological evaluation, including hematoxylin and eosin staining and immunohistochemical analysis.
Immunohistochemical analysis
The expression of PCNA, p-MLKL, and p-RIP3 in xenograft tumor tissues was evaluated. Tissue specimens were fixed in formalin, embedded in paraffin, and sectioned into 4 μm slices. Sections were deparaffinized with xylene and rehydrated through a graded ethanol series. Antigen retrieval was performed by immersing sections in citrate buffer and applying microwave heating at high temperature for 5 min, repeated 2–3 times. To inhibit endogenous peroxidase activity, sections were treated with 3% H₂O₂ for 10 min and then incubated with 5% normal goat serum to reduce nonspecific binding. Sections were then incubated overnight at 4 °C with a primary antibody against PCNA (1:200 dilution). Visualization was performed using a reaction enhancer and DAB chromogenic solution.
siRNA- mediated gene knockdown
Detailed information on siRNA sequences is shown in Table 1 below.
Cells were seeded in 24-well plates at a density of 1 × 10⁴ cells per well and allowed to adhere for 12 h before transfection using Lipofectamine® 3000 (Thermo Fisher Scientific, Waltham, MA, USA) in accordance with the manufacturer’s instructions. Transfection was carried out using 2 µL/well of Lipofectamine® 3000 with 50 nM siRNA, followed by a 48 h incubation. Cells were harvested 48 h post-transfection for subsequent analyses, including western blotting to validate DR4 knockdown efficiency.
Statistical evaluation
Statistical analyses were performed using GraphPad Prism 9 software. Using the RTCA software package 1.2 (N = 3), the Cell Index, a parameter for real-time cytotoxicity assessment, was calculated. Results are presented as mean ± standard deviation relative to control groups. Comparisons between groups were analyzed using Student’s t-test or one-way analysis of variance, with statistical significance set at P < 0.05. All experiments were conducted in triplicate to ensure reproducibility.
Results
Periplocin inhibits the growth of human PTC cells
To assess the cytotoxic efficacy of periplocin in PTC, BCPAP and TPC-1 cells were exposed to a periplocin concentration gradient (0–500 nM for 48 h) determined by preliminary dose–response analyses (0–1000 nM). As illustrated in Fig. 1A, periplocin elicited a concentration-dependent reduction in cell viability, with IC₅₀ values of 189 nM (BCPAP) and 203 nM (TPC-1). Co-administration of Nec-1 (20 µM), a selective RIP1 inhibitor, substantially restored viability to 89.57% (BCPAP) and 60.91% (TPC-1) (##P < 0.01 vs. periplocin alone; Fig. 1B–D). By contrast, the pan-caspase inhibitor ZVAD-FMK (50 µM) showed no cytoprotective activity, whereas co-treatment with Nec-1 significantly increased cell viability, unequivocally confirming necroptosis as the predominant cell death mechanism.

Periplocin inhibits papillary thyroid cancer cell proliferation via necroptosis. (A) BCPAP and TPC-1 cells were treated with 0, 62.5, 125, 250, and 500 nM periplocin for 48 h, and the CCK-8 assay was performed to assess cell viability. Results are shown as mean ± standard deviation (SD) from three replicates. (B) BCPAP and TPC-1 cells were treated with 0 or 125 nM periplocin, 125 nM + Nec-1 (20 µM), or 125 nM + ZVAD-FMK (50 µM) for 24 h, and viability was assessed using the CCK-8 assay. (C) TPC-1 cells were treated with 0, 125, or 250 nM periplocin for 0–72 h, and viability was measured using RTCA. (D) TPC-1 cells were treated with 0 or 125 nM periplocin, 125 nM + Nec-1 (20 µM), or 125 nM + ZVAD-FMK (50 µM) for 0–72 h, and viability was assessed by RTCA. P < 0.05 was considered significant; **P < 0.01 vs. control; ***P < 0.001 vs. control; #P < 0.05 vs. 125 nM + Nec-1; ###P < 0.001 vs. 125 nM + Nec-1. CCK-8, Cell Counting Kit-8; Nec-1: necrostatin-1; RTCA, Real-Time Cell Analysis; ZVAD-FMK: Z-Val-Ala-Asp(OMe)-fluoromethylketone.
The selected concentration range (0–500 nM) was consistent with previous reports in pancreatic cancer models, where periplocin exhibited IC50 values of 331 nM (CFPAC1) and 71.6 nM (PANC1)16. The Nec-1 and ZVAD-FMK concentrations were adapted from protocols optimized in colorectal cancer studies. Subsequent mechanistic investigations prioritized 125 and 250 nM periplocin concentrations, which balanced robust necroptotic induction (Fig. 2A and B) with translational relevance for therapeutic development.

Periplocin induces necrotic-like changes in papillary thyroid carcinoma cells. (A and B) Hoechst 33,342/PI staining was used to detect morphological changes in TPC-1 and BCPAP cells treated with 0 or 125 nM periplocin, 125 nM periplocin + Nec-1 (20 µM), or 125 nM periplocin + ZVAD-FMK (50 µM) for 24 h. P < 0.05 was considered significant; ***P < 0.001 vs. control; ###P < 0.001 vs. 125 nM + Nec-1. Hoechst 33,342, Bisbenzimide H 33,342; Nec-1: necrostatin-1; PI: Propidium Iodide; ZVAD-FMK: Z-Val-Ala-Asp(OMe)-fluoromethylketone.
Periplocin induces necroptosis in PTC cells
To determine whether periplocin induces necroptosis (programmed necrosis) in PTC cells, we performed morphological examinations using the Hoechst 33,342/PI double fluorescent staining kit to observe changes in cell membranes and nuclei after treatment. In the periplocin-treated group, many cells exhibited dark blue and red staining (indicative of necrosis), whereas others showed bright blue and red staining (characteristic of apoptosis). By contrast, the Nec-1 pretreatment group displayed only a few cells with necrotic features (Fig. 2A and B). Pretreatment with the apoptosis inhibitor ZVAD-FMK did not reduce necrotic characteristics. Using the Annexin-FITC/PI double staining kit to examine cell death pathways, the results showed that the percentage of cells in the necrotic region (Q2-2) was significantly reduced in the Nec-1 pretreatment group. However, in the ZVAD-FMK pretreatment group, the percentage in the necrotic region remained high, whereas the early apoptotic region (Q2-4) showed no significant change (Fig. 3A and B). These findings demonstrate that periplocin primarily induces PTC cell death through necroptosis, which is specifically inhibited by Nec-1.

Periplocin-induced thyroid cancer cell death can be inhibited by Nec-1. (A) BCPAP and TPC-1 cells were stained with an Annexin V/PI double staining kit and analyzed by flow cytometry. Total cell death (including early apoptosis, late apoptosis, and necrosis) was quantified after 24 h of treatment. (B) Histograms display early and late apoptosis/necroptosis rates in TPC-1 and BCPAP cells. Quadrant definitions: Q2-3: Annexin V⁻/PI⁻ (viable cells); Q2-4: Annexin V⁺/PI⁻ (early apoptotic cells); Q2-2: Annexin V⁺/PI⁺ (necroptotic cells); Q2-1: Annexin V⁻/PI⁺ (nonspecific staining). Apoptosis rate (%) = Q2-4/(Q2-3 + Q2-4 + Q2-2 + Q2-1) × 100%. Necroptosis rate (%) = Q2-2/(Q2-3 + Q2-4 + Q2-2 + Q2-1) × 100%. P < 0.05 was considered significant; *P < 0.05 vs. control; ***P < 0.001 vs. control; ##P < 0.01 vs. 125 nM + Nec-1; ###P < 0.001 vs. 125 nM + Nec-1; Not significant vs. 125 nM + Nec-1. Nec-1, necrostatin-1; PI, Propidium Iodide.
Periplocin induces programmed necrosis in PTC cells via RIP3/MLKL signaling
For RNA-seq analysis, differentially expressed mRNA transcripts were identified between periplocin-treated and control groups. Genes were considered significantly differentially expressed if Q < 0.05 and |logFC| > 0.5. Functional enrichment analyses using GO and KEGG were performed. Results from GO-Biological Process analysis revealed enrichment in terms, such as negative regulation of the extrinsic apoptotic signaling pathway in the absence of a ligand and negative regulation of the epithelial cell apoptotic process. Key KEGG pathways included the TNF signaling pathway, Ras signaling pathway, and cellular senescence (Fig. 4A and B). Figure 4C presents a cluster heatmap of TNF signaling and necroptosis-related genes, highlighting representative genes, such as CXCL1, CXCL2, CCL2, TNFRSF10A (DR4), CASP8, MLKL, and RIP1. Figure 4D-F shows the activation of RIP3/MLKL signalling components (p-RIP3 and p-MLKL) and elevated levels of DAMPs in periplocin-treated cells. Collectively, these results demonstrate that periplocin induces necroptosis in PTC cells via the RIP3/MLKL signalling axis.

Periplocin induces necroptosis of thyroid cancer cells via RIP3/MLKL signaling. (A and B) GO and KEGG analyses of differentially expressed genes between periplocin-treated and control groups based on RNA-seq. Lower p-values indicate stronger enrichment. (C) Heatmap of RIP3/MLKL pathway-related genes. Red, green, and intermediate shades represent high, low, and upregulated expression, respectively. (D and E) Western blotting of RIP3/MLKL pathway proteins and DAMPs (p-MLKL, p-RIP3, IL6, and IL1A) in BCPAP and TPC-1 cells treated with periplocin. (F) ImageJ was used to quantify gray values of RIP3/MLKL pathway proteins and DAMPs, normalized to internal reference proteins; data from three replicates were used to generate histograms. *P < 0.05 vs. control; **P < 0.01 vs. control; ***P < 0.001 vs. control. DAMPs: Damage-Associated Molecular Patterns; GO: Gene Ontology; KEGG: Kyoto Encyclopedia of Genes and Genomes; MLKL, Mixed Lineage Kinase Domain-Like Protein; RIP3: Receptor-Interacting Protein Kinase 3.
Periplocin targets DR4/RIP3/MLKL signaling to induce necroptosis in PTC cells
As shown in Fig. 5A and B, western blot analysis revealed that DR4 protein expression increased in a dose-dependent manner (0–250 nM, 24 h treatment) in both BCPAP and TPC-1 cells following periplocin exposure. To validate DR4’s functional role, we used siRNA-mediated knockdown (si-DR4), with results shown in Fig. 5C and D. CCK-8 tests showed that reducing DR4 with siRNA (si-DR4) lessened the growth-inhibiting effects of periplocin (P < 0.001 compared to periplocin alone; Fig. 6A), proving that DR4 is important. Mechanistically, periplocin treatment alone increased p-MLKL levels by 2.8-fold (P < 0.001), whereas co-treatment with si-DR4 reduced p-MLKL levels by 53.74% (P < 0.001; Fig. 6B). This inverse relation confirmed DR4-dependent activation of the RIP3/MLKL pathway (Fig. 6C).

Periplocin regulates DR4 expression in thyroid cancer cells. (A and B) Western blotting was used to detect DR4 expression in BCPAP and TPC-1 cells treated with periplocin for 24 h. ImageJ analysis of gray values was normalized to internal controls. (C and D) A Western blot (WB) test was done to check for DR4 levels in BCPAP and TPC-1 cells after they were treated with siRNA. DR4 expression in BCPAP and TPC-1 cells transfected with siRNA was quantified using ImageJ (the greyscale values were normalised to β-actin) and presented as a histogram. *P < 0.05 vs. control; **P < 0.01 vs. control; ***P < 0.001 vs. control. DAMPs: Damage-Associated Molecular Patterns; DR4: Death Receptor 4; MLKL, Mixed Lineage Kinase Domain-Like Protein; RIP3: Receptor-Interacting Protein Kinase 3.

Periplocin targets DR4 to induce necroptosis in papillary thyroid cancer cells. (A) CCK-8 assay used to determine cell viability. (B) TPC-1 cells treated with periplocin; p-MLKL detected by western blot, with GAPDH as the internal control. Results expressed as mean ± standard deviation (SD). ***P < 0.001 vs. control; ###P < 0.001 vs. periplocin + si-DR4. (C) Schematic diagram showing the proposed mechanism. CCK-8, Cell Counting Kit-8; DR4, Death Receptor 4; MLKL, Mixed Lineage Kinase Domain-Like Protein.
Periplocin inhibits PTC xenograft growth in nude mice by inducing necroptosis
A subcutaneous xenograft model was established by injecting BALB/c nude mice with TPC-1 cells. When tumors reached 50 mm³, mice were randomly assigned to receive intraperitoneal injections of either vehicle control (5% DMSO) or periplocin (30 mg/kg) every 3 d for three cycles. The periplocin-treated group showed significantly reduced tumor growth compared with observations in controls, as evidenced by decreased tumor dimensions (Fig. 7A and B), weight (Fig. 7C), and volume (Fig. 7E). Immunohistochemical analysis revealed a reduction in PCNA-positive cells (Fig. 7D) and increased p-MLKL and p-RIP3. Furthermore, PI staining indicated significantly enhanced necrotic cell death in the treated tumors (Fig. 7F). These findings collectively demonstrate that periplocin exerts antitumor effects by activating necroptotic signaling pathways.

Effects of periplocin and its mechanism in BALB/c nude mice. (A and B) Representative images of mice and tumors in control and periplocin-treated groups. (C) Tumor weights. (D) Immunohistochemical detection of PCNA, p-MLKL, and p-RIP3 expression; IOD calculated using ImageJ. Data expressed as mean ± standard deviation (SD). (E) Mean tumor volume over time. (F) PI-positive rate detected using Hoechst 33,342/PI staining. ***P < 0.001. IOD: Integrated Optical Density; MLKL: Mixed Lineage Kinase Domain-Like Protein; PCNA: Proliferating Cell Nuclear Antigen; PI: Propidium Iodide; RIP3: Receptor-Interacting Protein Kinase 3.
Discussion
In the present study, periplocin inhibited the growth of BCPAP and TPC-1 cells in a concentration-dependent manner by inducing necroptosis, leading to necrotic-like changes. With the addition of Nec-1, few cells exhibited high-intensity blue/red staining, and the PI-positive staining rate significantly decreased. Using the Annexin-FITC/PI kit, we observed high cell death rates in the necroptosis quadrants of periplocin-treated cells, which were significantly reduced in the Nec-1 pretreated group. Immunohistochemistry revealed increased expression of p-MLKL and p-RIP3 in tumor tissues from the periplocin-treated group. Additionally, the rate of PI-positive staining increased, suggesting that periplocin inhibits papillary thyroid cancer cell growth through necroptosis17,18,19,20. Periplocin inhibits pancreatic cancer growth via AMPK–mTOR signaling16, reduces protein expression in lymphoma cells21, kills liposarcoma cells22, sensitizes ESCC cells to TRAIL-induced apoptosis23, and induces apoptosis in MM cells24. It induces apoptosis in tumor cells to inhibit growth, which differs mechanistically from the necroptosis observed in this study. Apoptosis is a tightly regulated cell death process characterized by intact membranes, chromatin condensation, DNA fragmentation, and formation of apoptotic bodies. By contrast, necroptosis is a caspase-independent form of regulated necrosis triggered by TNF-α in the absence of caspase-8, marked by major necrotic features. Subsequent studies have elucidated the molecular mechanisms underlying necroptosis, involving key molecules, such as MLKL, which activates TRPM7, leading to ionic imbalances, calcium accumulation, and increased membrane permeability25,26,27. This results in the leakage of DAMPs, which trigger inflammatory responses. In this study, periplocin increased DAMP expression in a concentration-dependent manner, consistent with previous findings28. Other studies have reported that lysicamine alkaloids exert antitumor effects via ROS-mediated necroptosis in thyroid carcinoma cells. The differing mechanisms of periplocin-induced cell death may stem from specific tumor cell characteristics and inter-pathway crosstalk29,30. Further studies are warranted to investigate upstream regulators and signaling nodes. This study relied solely on cell lines and xenograft tumor models, lacking proper evaluation of immune microenvironment interactions, such as those assessable using organoid systems or humanized mouse models. In summary, periplocin induces necroptosis in thyroid carcinoma cells and inhibits tumor growth.
RNA-seq analysis showed that TNF signalling was the most enriched GO and KEGG pathway in TPC-1 cells treated with periplocin compared with observations in controls. The cluster heatmap of the TNF pathway and related genes prominently featured TNFRSF10A. Western blotting revealed that periplocin upregulated DR4 and increased p-MLKL levels, which were suppressed following si-DR4 treatment. These findings indicate that periplocin induces necrosis in thyroid cancer cells and suppresses tumour growth by activating the RIP3/MLKL signalling pathway via DR4. TRAIL initiates apoptosis and necroptosis by binding to DR4 and DR531. AICAR upregulates DR4, DR5, and necroptosis-related proteins, inhibits ATL tumour xenograft growth, and may represent a potential therapeutic agent32. Sirtinol or SIRT1 knockdown increases DR4/DR5 expression and enhances TRAIL-sensitive necroptosis in A549 cells33, indicating that natural compounds can induce necroptosis through DR4-mediated RIP3/MLKL activation. However, MLKL deletion has been shown to enhance p-ERK and p-p38 signalling, delay TRAIL lysosomal degradation, and augment DR4/5 signalling and necroptosis34, findings that contradict the conclusion that natural compounds uniformly activate RIP3/MLKL via DR4. Recent research by Wang et al. demonstrates that the 4EBP1-eIF4E axis forms a self-amplifying loop with RIP3-MLKL during necroptosis35. Mitochondrial voltage-dependent anion channel 1 (VDAC1) oligomerisation was also found to play a critical role in regulating PANoptosis—an integrated cell death modality that combines pyroptosis, apoptosis, and necroptosis36. One explanation is that MLKL also participates in extracellular vesicle generation independently of RIP3. Decreased MLKL slows DR4/5 degradation in response to TRAIL. MLKL’s involvement in endosomal transport doesn’t need its working N-terminal domain, which is important for necroptosis and changes related to extracellular vesicles, indicating that it works in different ways37,38. In conclusion, periplocin induces necroptosis in thyroid carcinoma cells by targeting DR4 to activate the RIP3/MLKL signalling cascade.
The incidence of thyroid cancer continues to rise, and its recurrence and disease burden necessitate novel treatment strategies. Botanical natural products, including resveratrol, curcumin, isoflavones, and glucosinolates, have demonstrated antithyroid cancer activity in experimental models. However, most studies remain confined to in vitro and in vivo preclinical assessments, with few proceeding to clinical trials39. Periplocin induces necroptosis in thyroid cancer cells and inhibits tumor growth in TPC-1 xenograft mouse models. Therefore, it may represent a viable therapeutic alternative for human thyroid cancer.
Conclusion
In both in vitro and in vivo studies, periplocin targeted DR4 and activated RIP3/MLKL signaling, inducing necroptosis in PTC cells and inhibiting their growth.
Data availability
No datasets were generated during the current study.
References
Doubleday, A. & Sippel, R. S. Surgical options for thyroid cancer and post-surgical management. Expert Rev. Endocrinol. Metab. 13 (3), 137–148 (2018).
Wang, H. et al. Physiological and pathophysiological roles of ion Transporter-Mediated metabolism in the thyroid gland and in thyroid Cancer. Onco Targets Ther. 13, 12427–12441 (2020).
Fallahi, P. et al. Molecular targets of tyrosine kinase inhibitors in thyroid cancer. Semin Cancer Biol. 79, 180–196 (2022).
Mitsiades, N., Poulaki, V., Tseleni-Balafouta, S., Koutras, D. A. & Stamenkovic, I. Thyroid carcinoma cells are resistant to FAS-mediated apoptosis but sensitive to tumor necrosis factor-related apoptosis-inducing ligand. Cancer Res. 60 (15), 4122–4129 (2000).
Gunda, V. et al. Blocks to thyroid cancer cell apoptosis can be overcome by Inhibition of the MAPK and PI3K/AKT pathways. Cell. Death Dis. 5 (3), e1104 (2014).
Zhu, J. & Li, Z. Overexpression of miR-101 promotes TRAIL-induced mitochondrial apoptosis in papillary thyroid carcinoma by targeting c-met and MCL-1. Oncotarget 8 (65), 108665–108675 (2017).
Yan, W. T. et al. Research trends, hot spots and prospects for necroptosis in the field of neuroscience. Neural Regen Res. 16 (8), 1628–1637. https://doi.org/10.4103/1673-5374.303032 (2021).
Bae, E. S. et al. Periplocin exerts antitumor activity by regulating Nrf2-mediated signaling pathway in gemcitabine-resistant pancreatic cancer cells. Biomed. Pharmacother. 157, 114039 (2023).
Lin, J. P. et al. Periplocin inhibits hepatocellular carcinoma progression and reduces the recruitment of MDSCs through AKT/NF-kappaB pathway. Life Sci. 324, 121715 (2023).
Zhang, H. et al. Periplocin Induces Apoptosis of Pancreatic Cancer Cells through Autophagy via the AMPK/mTOR Pathway. J Oncol. 2022: 8055004. (2022).
Lohberger, B. et al. Periplocin mediates TRAIL-induced apoptosis and cell cycle arrest in human Myxofibrosarcoma cells via the ERK/p38/JNK pathway. Phytomedicine 76, 153262 (2020).
Zhao, L. et al. Periplocin from cortex periplocae inhibits cell growth and down-regulates survivin and c-myc expression in colon cancer in vitro and in vivo via beta-catenin/TCF signaling. Oncol. Rep. 24 (2), 375–383 (2010).
Weng, J. et al. Periplocin improves the sensitivity of oxaliplatin-resistant hepatocellular carcinoma cells by inhibiting M2 macrophage polarization. Biomol Biomed. 25(4), 857–868 (2025).
Liu, X. et al. Study of the PI3K/Akt/mTOR signaling pathway in vitro and molecular Docking analysis of periplocin inhibits cell cycle progression and induces apoptosis in MDA-MB-231. Environ. Toxicol. 39 (1), 444–456 (2024).
Zhao, L. M. et al. Antitumor effect of periplocin in TRAIL-Resistant gastric cancer cells via upregulation of death receptor through activating ERK1/2-EGR1 pathway. Mol. Carcinog. 58 (6), 1033–1045 (2019).
Xie, G., Sun, L., Li, Y., Chen, B. & Wang, C. Periplocin inhibits the growth of pancreatic cancer by inducing apoptosis via AMPK-mTOR signaling. Cancer Med. 10 (1), 325–336 (2021).
Tong, X. et al. Targeting cell death pathways for cancer therapy: recent developments in necroptosis, pyroptosis, ferroptosis, and Cuproptosis research. J. Hematol. Oncol. 15 (1), 174 (2022).
Zhang, T. et al. ADAR1 masks the cancer immunotherapeutic promise of ZBP1-driven necroptosis. Nature 606 (7914), 594–602 (2022).
Meier, P., Legrand, A. J., Adam, D. & Silke, J. Immunogenic cell death in cancer: targeting necroptosis to induce antitumour immunity. Nat. Rev. Cancer. 24 (5), 299–315 (2024).
Baik, J. Y. et al. ZBP1 not RIPK1 mediates tumor necroptosis in breast cancer. Nat. Commun. 12 (1), 2666 (2021).
Zhao, R. et al. Inhibitory effects of periplocin on lymphoma cells: A network Pharmacology approach and experimental validation. Drug Des. Devel Ther. 15, 1333–1344 (2021).
Lohberger, B. et al. Periplocin, the most anti-proliferative constituent of Periploca sepium, specifically kills liposarcoma cells by death receptor mediated apoptosis. Phytomedicine 51, 162–170 (2018).
Han, L. et al. Combination of the natural compound periplocin and TRAIL induce esophageal squamous cell carcinoma apoptosis in vitro and in vivo: implication in anticancer therapy. J. Exp. Clin. Cancer Res. 38(1), 501 (2019).
Aziz, A. et al. Periplocin overcomes bortezomib resistance by suppressing the growth and Down-Regulation of cell adhesion molecules in multiple myeloma. Cancers 15(5), 1526 (2023).
Cai, Z. et al. Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat. Cell. Biol. 16 (1), 55–65 (2014).
Lee, J., Lee, S., Min, S. & Kang, S. W. RIP3-Dependent accumulation of mitochondrial superoxide anions in TNF-alpha-Induced necroptosis. Mol. Cells. 45 (4), 193–201 (2022).
Siegmund D, Wagner J, Wajant H. TNF Receptor Associated Factor 2 (TRAF2) Signaling in Cancer. Cancers. 14(16), 4055 (2022).
Murao, A., Aziz, M., Wang, H., Brenner, M. & Wang, P. Release mechanisms of major damps. Apoptosis 26 (3–4), 152–162 (2021).
Rodrigues, M. T. et al. Lysicamine reduces protein kinase B (AKT) activation and promotes necrosis in anaplastic thyroid Cancer. Pharmaceuticals 16(12), 1687 (2023).
Jones, N. S. et al. A phase I, randomized, ascending-dose study to assess safety, pharmacokinetics, and activity of GDC-8264, a RIP1 inhibitor, in healthy volunteers. Clin. Transl Sci. 16 (10), 1997–2009 (2023).
Maji, A. et al. Significance of TRAIL/Apo-2 ligand and its death receptors in apoptosis and necroptosis signalling: implications for cancer-targeted therapeutics. Biochem. Pharmacol. 221, 116041 (2024).
Aikawa, A., Kozako, T., Kato, N., Ohsugi, T. & Honda, S. I. Anti-tumor activity of 5-aminoimidazole-4-carboxamide riboside with AMPK-independent cell death in human adult T-cell leukemia/lymphoma. Eur. J. Pharmacol. 961, 176180 (2023).
Pal, S., Shankar, B. S. & Sainis, K. B. Cytokines from the tumor microenvironment modulate sirtinol cytotoxicity in A549 lung carcinoma cells. Cytokine 64 (1), 196–207 (2013).
Park, S. Y. et al. Reduction in MLKL-mediated endosomal trafficking enhances the TRAIL-DR4/5 signal to increase cancer cell death. Cell. Death Dis. 11 (9), 744 (2020).
Wang, S. et al. Noncanonical feedback loop between RIP3-MLKL and 4EBP1-eIF4E promotes neuronal necroptosis. MedComm 2025; 6(3): e70107. (2020).
Wan, H. et al. Voltage-dependent anion channel 1 oligomerization regulates PANoptosis in retinal ischemia-reperfusion injury. Neural Regen Res. https://doi.org/10.4103/NRR.NRR-D-24-00674 (2025). [NNR-D-24-00674].
Yang, C. et al. Regulation of RIP3 by the transcription factor Sp1 and the epigenetic regulator UHRF1 modulates cancer cell necroptosis. Cell. Death Dis. 8 (10), e3084 (2017).
Morgan, M. J. & Kim, Y. S. The Serine threonine kinase RIP3: lost and found. BMB Rep. 48 (6), 303–312 (2015).
Sharifi-Rad, J. et al. Plant natural products with anti-thyroid cancer activity. Fitoterapia 146, 104640 (2020).
Acknowledgements
This work was supported by the Wu Jieping Medical Foundation (Grant No. 320.6750.2023-18-20). We sincerely acknowledge their financial contribution to this research.
Author information
Authors and Affiliations
Contributions
All the authors have read and approved the manuscript. Li Ding and Fengmei Wen designed the experiments. Gangyin Xie and Ziwei Li conducted the experiments, wrote the manuscript, and analyzed the results. Liang Zeng participated in the experimental procedures and analyzed the data.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ding, L., Wen, F., Zeng, L. et al. Periplocin induces necroptosis in papillary thyroid carcinoma through DR4 mediated RIPK3 and MLKL signaling. Sci Rep 15, 20383 (2025). https://doi.org/10.1038/s41598-025-08977-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-08977-1
