
Abstract
The prognosis of advanced CRC is poor, and identifying key genes related to CRC is vital for improving CRC prognosis. Our research used univariate Cox analysis and Mendelian randomization (MR) analysis to identify key prognostic genes in CRC. Multiple datasets such as the nomogram model and single-cell sequencing (scRNA-seq) were used to investigate the potential molecular mechanisms of the key genes. The expression levels were confirmed by using quantitative real-time polymerase chain reaction (qRT-PCR). The biological functions and effect on prognosis of the identified prognostic genes were also explored. MMRN1 and SLC6A19 were identified as key prognostic genes for CRC. Subsequently, the nomogram model demonstrated that MMRN1 and SLC6A19 can strongly predict survival. Further examination with multiple datasets elucidated the potential molecular mechanisms of the key prognostic genes, revealing a close association with immune cell infiltration. MMRN1 is enriched in classic CRC signaling pathways, whereas SLC6A19 is enriched in metabolism-related pathways. They are closely linked to immune cell infiltration levels and significantly influence the immune microenvironment in CRC. These key prognostic genes are significantly correlated with chemotherapeutic drug sensitivity and present promising opportunities for CRC therapy. The expression of both key genes was also observed in the scRNA-seq data of CRC. Finally, qRT-PCR validation revealed that MMRN1 is markedly downregulated and SLC6A19 is significantly upregulated in CRC. Lower expression of MMRN1 and higher expression of SLC6A19 significantly promoted the proliferation and metastasis of colorectal cancer cells. Our study identified MMRN1 and SLC6A19 as potential key prognostic genes for CRC, as they can reliably predict the prognosis of CRC. Furthermore, the potential molecular mechanisms of MMRN1 and SLC6A19 were revealed, suggesting new drug targets and therapeutic directions for managing prognosis.
Similar content being viewed by others
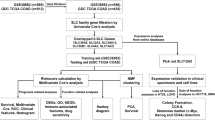


Introduction
Colorectal cancer (CRC) is the third most common cancer globally and the second leading cause of cancer-related mortality1. By 2035, it is estimated that there will be 2.5 million new cases of CRC; thus, CRC poses a major public health challenge2. Surgical treatment continues to be the primary modality for treating CRC, yet more than 30% of stage II or III CRC patients and 60–70% of those who undergo oligometastatic resection experience recurrence3. As many as 50% of patients with localized CRC eventually develop metastases4, and the five-year survival rate is approximately 14%5. Overall, the prognosis remains poor, and effective prognostic management of these patients continues to be a significant challenge4. Therefore, it is imperative to identify reliable prognostic genes to improve the clinical prognosis of CRC.
Recently, Mendelian randomization (MR) has attracted increased amounts of attention. MR employs single nucleotide polymorphisms (SNPs) as instrumental variables (IVs) in nonexperimental data to study the causal relationships between modifiable exposures and outcomes6,7. Thus, it provides accurate forecasts of randomized trial results and is often referred to as "Nature’s randomized trial"8. MR is essential for elucidating cancer risks and predicting cancer progression from an etiological perspective9. Genetic colocalization analysis of exposures and outcomes often provides additional support for the findings from MR analyses9. Expression quantitative trait locus (eQTL) studies are employed to experimentally annotate the human genome10. By integrating data from genome-wide association studies (GWASs) with eQTL study findings, genes associated with expression levels and complex traits can be identified11. These approaches provide powerful tools for studying the causal relationships between gene expression and disease prognosis, offering significant ethical and practical benefits12. MR provides a method to infer causal relationships by using genetic variants as IVs, thus overcoming confounding and reverse causality issues inherent in observational studies6,7. By using eQTLs as representation for gene expression, we attempt to identify prognostic genes and provide insights into their functional mechanisms in CRC.
Thus, our study hypothesized a causal relationship between specific gene expression and CRC prognosis. By employing two-sample MR analysis, we aim to identify these genes and elucidate their role in colorectal cancer prognosis. First, we employed univariate Cox analysis and MR to identify key genes. Next, a nomogram model was constructed to assess the prognostic predictive power of the key genes. Third, the potential molecular mechanisms of key genes were explored through analyses such as immune infiltration, gene set enrichment analysis (GSEA)/gene set variation analysis (GSVA) pathway enrichment, drug sensitivity, and transcriptional regulation. Fourth, single-cell sequencing (scRNA-seq) was used to determine whether key genes exhibit cell type-specific enrichment in CRC, providing substantial evidence for exploring the molecular mechanisms involved. Finally, real-time quantitative polymerase chain reaction (qRT-PCR) was utilized to validate the expression levels of key genes. The biological functions of key genes were explored by cck-8, plate cloning and transwell. Our research revealed the potential molecular mechanisms of two key genes, MMRN1 and SLC6A19, in the prognosis of CRC, providing new targets and therapeutic approaches for the prognostic management of CRC.
Materials and methods
Study design and ethics permit
This study strictly adhered to the standards for Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) checklist13,14. For the MR analysis, the following assumptions were made: Relevance: The SNPs used as IVs are strongly associated with gene expression levels. Independence: The IVs are not associated with any confounders of the exposure-outcome relationship. Exclusion restriction: The IVs affect the outcome solely through the exposure (gene expression) and not through any alternative pathways12. The study was based on publicly available data. In addition, the included study has been approved by The Ethics Committee of Affiliated Hospital of Ningbo University Medical School (No.KY20220101). All human studies were carried out in accordance with the principles of the Declaration of Helsinki.
Patient and tissue sample collection
Thirty pairs of matched human colorectal cancer and adjacent nontumor tissue samples were obtained from the First Affiliated Hospital of Ningbo University. All patients provided informed consent prior to surgery, and samples were obtained during the surgical procedure. The Ethics Committee of the First Affiliated Hospital of Ningbo University granted approval for this project (No.KY20220101). All human studies were carried out in accordance with the principles of the Declaration of Helsinki.
Data acquisition
Using the TCGA database (https://portal.gdc.cancer.gov/), we acquired original mRNA expression data for colorectal cancer samples; there were a total of 701 samples. Of these samples, 51 were from the normal group, and 650 were from the tumor group. The GEO database (https://www.ncbi.nlm.nih.gov/geo/info/datasets.html) was utilized to download the single-cell dataset GSE221575 for subsequent single-cell analysis. Exposure data were derived from the eQTLGen consortium (https://www.eqtlgen.org) through eQTL data, which includes data from 31,684 blood samples. The outcome data in this study were obtained from the latest genome-wide association studies, and the participants were predominantly of European descent. All outcome summary data were obtained from the MR Base database (finngen_R10_C3_COLORECTAL_EXALLC). Data from the GWAS Catalog were mapped onto the Genome Assembly and dbSNP Build, featuring 6,847 positive and 314,193 negative cases of colorectal cancer.
Differential expression analysis
Using differential gene analysis, the Limma package in R language (version 4.2.2, https://www.r-project.org/) was utilized to find genes that substantially differed in expression between the groups. The Limma software was used to examine the variations in the molecular pathways of CRC. Screening criteria included genes that showed a change in expression between the control and CRC samples, with P values < 0.05 and |logFC|> 1.
Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) functional annotations
The “ClusterProfiler” function in R language (version 4.2.2) was employed to performed GO and KEGG pathway enrichment analyses on prognostic genes to further comprehend their functional correlations. GO terms and KEGG15,16,17 pathways with both p-values and q-values below 0.05 were deemed significant.
MR analysis
The MR Base database (http://app.mrbase.org/) hosts 11 billion SNP-trait associations derived from GWAS, with updates conducted routinely. Outcome IDs were obtained via the MR Base database extraction of related causal relationships in eQTLs from GWAS summary data (https://gwas.mrcieu.ac.uk/). The SNPs correlated with each gene at a genome-wide significance threshold (P < 1e−5) were selected as potential IVs, and the linkage disequilibrium (LD) among SNPs was calculated. Those SNPs with R2 < 0.001 (clumping window size = 10,000 kb) and p2 < 5e−5 were used for analysis. The protocol involves using four statistical methods—inverse variance weighted (IVW, integrating meta-analysis with Wald estimates for each SNP), MR egger (predicated on the instrument strength being independent of direct effects (InSIDE), weighted median (enables accurate causal estimates even with up to 50% invalid IVs), and weighted mode (a weighted model estimate offering greater causal effect detection capabilities, reduced bias, and lower Type I error rate than MR‒Egger regression)—to evaluate the reliability of causal relationships, providing a comprehensive estimate of the influence of all cis and certain trans gene expression in whole blood on CRC. Finally, the retention method was used to validate and analyze the identified causal relationships.
Colocalization analysis
If an SNP is situated within the regions of two or more genes, it may hold eQTL information for at least two distinct genes. This means that the impact of the disease (in this case, CRC would be influenced by a combination of various genes. Colocalization analysis is then employed to ascertain the genetic variation potentially sharing a causal relationship with CRC and eQTLs. The coloc method, together with eQTL data and colorectal cancer GWAS, was used for colocalization analysis, employing a 100-kilobase region surrounding the index SNP for posterior probability calculations. The coloc results utilize posterior probabilities (PP) to quantify support for each hypothesis. H3 indicates that gene expression and CRC are associated but with different causal variations. H4 indicates that both gene expression and CRC are associated and share the same causal variations. A threshold of SNP.PP. H4 > 0.6 was used for colocalization. In many genomics studies, a threshold greater than 0.5 is considered possible for colocalization. This value is more common in colocalization analysis and has shown good predictive power in several studies, such as Meritxell18. In practical applications, full causality usually requires a higher threshold, but the 0.6 chosen in this study can already indicate the presence of colocalization. This value can effectively filter out the possibility of low probability, but also can retain the SNPs with strong evidence, so as to screen out the variation with potential biological significance. The limitation is that more complex genetic mechanisms may be overlooked, and there is a risk of false positives and false negatives. Therefore, it is still necessary to combine other analytical methods and more refined experimental design to ensure the reliability of the results of colocalization analysis.
Immune cell infiltration analysis
Based on deconvolution, the CIBERSORT algorithm was used to assess the different immune cells morphologies present in the microenvironment. This included T cells, B cells, plasma cells, and subsets of myeloid cells, among other human immune cell phenotypes. Through the CIBERSORT algorithm, patient data was analyzed to estimate the relative proportions of 22 immune infiltrating cell types, subsequently, a Spearman correlation analysis was conducted for gene expression levels and immune cell contents.
GSEA
GSEA was employed to contrast the distinct signaling pathways between the high and low expression groups, supplying a more thorough comprehension of the fundamental mechanisms of important genes. Using the Molecular Signatures Database (MsigDB), the annotated gene sets were downloaded to conduct differential expression analysis of pathways among subtypes, and the highly enriched gene sets (adjusted P value < 0.05) were arranged based on consistency scores.
GSVA
GSVA was used to assess the enrichment status of key genes by downloading gene sets from MSigDB. This study applied the GSVA method to access each gene set comprehensively, investigating potential variations in biological functions among various samples.
Drug sensitivity analysis
Genomics of Drug Sensitivity in Cancer (GDSC, https://www.cancerrxgene.org/) is the biggest database for pharmacogenomics, gathering data on drug sensitivity and molecular biomarkers of response in tumor cells. Data from the GDSC database were used in this research, and the R package “pRRophetic” was used to forecast the sensitivity of tumor samples to chemotherapy. IC50 estimates for specific chemotherapy drugs were derived via regression, with tenfold cross-validation in the GDSC training set for assessing regression and predictive accuracy.
Nomogram model construction
Constructed from regression analysis data, the nomograms in this study visually present variables such as age, sex, stage, TNM classification, MMRN1, and SLC6A19 on a common scale, illustrating the interrelationships of these variables within the predictive model. Totaling individual scores yields an overall score for predictive outcomes.
Transcriptional regulation analysis of key genes
The “RcisTarget” R package was utilized to forecast transcription factors, with all RcisTarget computations grounded on motifs, which are typical sequences or structures. For each motif, a normalized enrichment score (NES) was computed. Extra annotations are inferred beyond the source data, considering motif similarities and gene sequence information. Estimating the overexpression of each motif within a gene set begins with calculating the area under the curve (AUC) for each motif–gene set pairing, a process based on recovery curve calculations that sort motifs according to gene sets. NES calculations for each motif are derived from the AUC distribution of total motifs within a gene set.
scRNA-seq analysis
scRNA-seq analysis offers novel evidence for addressing challenges in biology and medicine. Genes with low expression were first omitted and expression profiles were arranged using the Seurat program. The data were normalized and standardized. T-distributed stochastic neighbor embedding (t-SNE) analysis elucidates the spatial connections among clusters, followed by annotations of clusters via the celldex package.
RNA extraction and qRT-PCR
The GoScript Reverse Transcription (RT) equipment (Promega, Madison, WI, USA) was used to reverse-transcribe total RNA into cDNA after it had been extracted using the TRIzol reagent (Ambion, Carlsbad, CA). Next, according to the manufacturer’s detailed protocols, qRT-PCR was performed using an Mx3005P real-time fluorescence quantitative PCR system (Stratagene, La Jolla, CA, USA). Relative expression levels were computed using the 2-ΔΔCt method, where ΔCt = Ct (target gene)—Ct (housekeeping gene). The data are expressed as the mean ± standard deviation (SD). The primer sequences for the housekeeping gene GAPDH and the key genes MMRN1 and SLC6A19 are listed below: GAPDH(F): 5-ACCCACTCCTCCACCTTTGAC-3, (R):5-TGTTGCTGTAGCCAAATTCGTT-3. MMRN1(F): 5-GGCATTGGGCTTAACAACAG-3, (R): 5-GGAACTGAAGCAGAAGGCATAG-3. SLC6A19(F): 5-GCAGGACGCAAGGGCAGTAAG-3, (R): 5-TCAGGATGGAGCAGGCAGAAGG-3. The primer sequences of TFAM is listed below: TFAM(F): 5- GTGGTTTTCATCTGTCTTGGCAAG-3, (R):5- TTCCCTCCAACGCTGGGCAATT-319.
Cell culture and transfection
Cell lines were obtained from Shanghai Cell Bank, Chinese Academy of Sciences (Shanghai, China). Cell lines were cultured in a 37 °C humidified incubator with CO2.
High Glucose Dulbecco’s Modified Eagle’s Medium (DMEM) (HyClone, Los Angeles, USA) supplemented with 10% FBS (Gibco, Grand Island, USA) was used to culture RKO and HCT-116 cells. Roswell Park Memorial Institute (RPMI) 1640 medium (HyClone) supplemented with 10% FBS was used to culture SW-480 cells. During transfection, cells seeded uniformly into a six-well plate. Cells were incubated with complete culture medium for about 24 h until cell confluency reached 50–70%, the plasmid was transfected into the cells according to the instructions of lipofectamin 2000. Subsequent operations were performed 24–48 h after transfection. The small interfering RNA (siRNA) was purchased from Shanghai Jima Pharmaceutical Co., Ltd. (Shanghai, China).
Cell counting kit‐8 assay (CCK-8)
One thousand cells per well were seeded into 96‐well plates and incubated at 37 °C. Ten microliters of CCK-8 (GLPBIO, Montclair, CA, USA) reagent was added to each well every 24 h. After incubating for 2 h, measuring the absorbance at 450 nm to determine the cell proliferative ability.
Colony formation assay
For the colony formation assay, 600 cells were seeded into each well of a 6‐well plate and cultured for 2 weeks. Cells were fixed with 4% paraformaldehyde (Solarbio, Beijing, China), stained with 0.1% crystal violet (Solarbio) for 15 min. The plates were photographed and the number of colonies was counted.
Transwell migration assays
2 × 104 cells were seeded into each chamber (Corning, NY, USA). Then, 200 μL of FBS‐free DMEM and 500 μL of DMEM supplemented with 30% FBS were added to the upper and lower chambers, respectively. The migrating cells were then fixed, stained and observed under a microscope after they were cultured for 24 h.
Statistical analysis
The R language (version 4.2.2, https://www.r-project.org/), was employed in our study’s analysis. A p value of less than 0.05 was deemed to show statistical significance in all two-tailed statistical tests. For statistical analysis, the Statistical Package for the Social Sciences (SPSS) version 29 was used, with P < 0.05 being regarded as statistically significant.
Results
Differential expression analysis and single factor analysis
Colorectal cancer-related datasets were downloaded from the TCGA database. There were an aggregate of 701 patients, including those in the normal group (n = 51) and those in the tumor group (n = 650). A combined number of 2008 differentially expressed genes were identified; these genes included 746 upregulated genes and 1262 downregulated genes (Fig. 1A,B). A single-factor Cox regression model was established, and a forest plot was drawn based on clinical data as well as the genes with variable expression. The single-factor Cox analysis revealed that 197 genes were related to prognosis (Supplementary Fig. 1). We extended our study by performing pathway analysis on the identified 197 prognostic genes. GO enrichment analysis revealed that these genes were predominantly involved in pathways including complement activation, the classical pathway, the B-cell receptor signaling, and the immunoglobulin complex (Fig. 1C). KEGG enrichment analysis indicated that the prognostic genes were primarily associated with pathways such as cytokine-cytokine receptor interactions, protein digestion and absorption, and the cell cycle, among others (Fig. 1D).

Differential expression and univariate analyses. (A) The differentially expressed genes between the normal group and the tumor group were visualized through volcano plot. (B) Heatmap illustrating the differentially expressed genes between the normal and tumor groups. (C) Clustered bar chart of GO enrichment analysis revealed that the prognosis genes were predominantly involved in pathways including complement activation, the classical pathway, the B-cell receptor signaling, and the immunoglobulin complex. (D) Clustered bar chart of KEGG15,16,17 enrichment analysis revealed that the prognostic genes were primarily associated with pathways such as cytokine-cytokine receptor interactions, protein digestion and absorption, and the cell cycle.
MR analysis
To further pinpoint the key genes affecting CRC, the outcome ID was obtained through the summary statistics of 321,040 colorectal cancer-related samples (314,193 controls and 6,847 cases; finngen_R10_C3_COLORECTAL_EXALLC). Extract_instruments and extract_outcome_data were used in sequence to extract 131 pairs of causal relationships between genes and outcomes. Furthermore, through MR analysis, the causal relationships of 5 pairs of genes corresponding to the positive outcome of eQTLs were obtained (Fig. 2A–E), IVW pval < 0.05). The corresponding genes were C1QB, GNG8, MMRN1, SLC6A19, and SUCLG2. Among these 5 genes, the presence of GNG8 (0.908; 0.826–0.999; P = 0.049), MMRN1 (0.936; 0.886–0.988; P = 0.020) and SLC6A19 (0.606; 0.378–0.970; P = 0.037) may be associated with a lower risk of CRC. The genes C1QB (1.102; 1.010–1.201; P = 0.028) and SUCLG2 (1.099; 1.030–1.172; P = 0.004) are linked to an increased risk of CRC. To further validate the causal relationships of these five genes, we performed a sensitivity analysis using the leave-one-out method. The findings demonstrated that removing any single SNP did not significantly alter the overall error bars, suggesting that the causal relationships we identified are robust (Fig. 3A–E). The MR-Egger intercept test did not show significant horizontal pleiotropy (P > 0.05), and heterogeneity was not observed across SNPs (Cochran’s Q P > 0.05). In addition, colocalization analysis was simultaneously conducted for 5 genes at the eQTL-GWAS level, among which the colocalized SNP.PP.H4 of the MMRN1 and SLC6A19 genes was greater than 0.6 (Fig. 4A,B). Therefore, these two genes were the key genes for our later research.

MR analysis of exposure and outcome factors. (A) Scatter plot demonstrating the causal relationship between C1QB and its corresponding eQTL-positive outcome. (B) Scatter plot showing the causal relationship between GNG8 and its corresponding eQTL-positive outcome. (C) Scatter plot illustrating the causal relationship between MMRN1 and its corresponding eQTL-positive outcome. (D) Scatter plot demonstrating the causal relationship between SLC6A19 and its corresponding eQTL-positive outcome. (E) Scatter plot showing the causal relationship between SUCLG2 and its corresponding eQTL-positive outcome.

Validation of MR analysis through the leave-one-out method. (A) Leave-one-out forest plot for C1QB with its corresponding positive result. (B) Leave-one-out forest plot for GNG8 with its corresponding positive result. (C) Leave-one-out forest plot for MMRN1 with its corresponding positive result. (D) Leave-one-out forest plot for SLC6A19 with its corresponding positive result. (E) Leave-one-out forest plot for SUCLG2 with its corresponding positive result.

eQTL-GWAS colocalization analysis. (A) The colocalization analysis of MMRN1 was conducted at the eQTL-GWAS level, and the results showed the colocalization of MMRN1 with SNP.PP. H4 exceeded 0.6. (B) The colocalization analysis of SLC6A19 was conducted at the eQTL-GWAS level, and the colocalization of SLC6A19 with SNP.PP. H4 exceeded 0.6.
The MR-Egger intercept test is an important tool for detecting horizontal pleiotropy20. If the P-value of the MR-Egger intercept term is less than 0.05, it indicates that horizontal pleiotropy may exist. The MR-Egger intercept test in our results did not show horizontal pleiotropy (P > 0.05), indicating that the causal relationship between instrumental variables and CRC was not significantly affected by horizontal pleiotropy. In our research, the leave-one-out method was used to examine changes in causal inference results after the removal of a SNP. Our results show that removing any single SNP did not significantly alter the overall results, suggesting that the identified causal relationships are robust. In addition, heterogeneity was tested using Cochran’s Q test (Cochran’s Q < 0.05). Therefore, sensitivity analysis was sufficient to rule out horizontal pleiotropy. Our criteria for selecting SNPs as instrumental variables are SNPs that have a significant association with gene expression and that have a P-value of less than 1e−5 across the genome. This criterion guarantees a strong association of the selected SNP in gene expression, thereby increasing its effectiveness as an instrumental variable. SNPs with P-values less than 1e−5 were selected as instrumental variables in order to ensure a strong enough association between them and gene expression. If the P-value is set too high (such as 1e−3 or 1e−2), it may contain SNPs that are weakly related to gene expression, which can affect the accuracy of causal inference. However, if the P-value is set too low (such as 1e−8 or lower), it may result in the selection of SNPs that are too limited to cover the diversity of gene expression. In general, a P-value range of 1e−5 to 1e−8 balances the accuracy of the selection with the breadth of the instrumental variables.
Nomogram model construction and ROC curve
The nomograms based on the regression analysis of the key gene expression levels are presented. In our samples, the clinical indicators of CRC and the expression levels of MMRN1 and SLC6A19 distributed variably to the scoring process (Fig. 5A). We also performed a survival prediction analysis for overall survival (OS) at three and five years (Fig. 5B). The predicted OS closely matched the observed OS, demonstrating the nomogram model’s robust predictive performance. In addition, we evaluated the predictive performance of key genes using ROC curve for diagnostic accuracy. A higher AUC indicates better predictive performance. The AUCs for MMRN1 and SLC6A19 were 0.974 (0.962–0.986) and 0.953 (0.929–0.977), respectively, suggesting that both genes effectively predict disease occurrence and progression (Fig. 5C,D).

Analysis of drug sensitivity and prognosis prediction in CRC patients. (A) Nomogram for predicting the prognosis of CRC. (B) Calibration curve for survival rate prediction in CRC patients. The nomogram-predicted overall survival (OS) is on the x-axis, with the observed OS on the y-axis. (C) ROC curve of MMRN1 for predicting the onset and progression of CRC. The peak ROC value was 0.974 (95% CI, 0.962–0.986). (D) ROC curve of SLC6A19 for predicting the onset and progression of CRC. The peak ROC value was 0.953 (95% CI, 0.929–0.977). (E) Analysis of MMRN1 sensitivity to common chemotherapy drugs. (F) Analysis of SLC6A19 sensitivity to common chemotherapy drugs.
Drug sensitivity analysis
The efficacy of combining surgery and chemotherapy for early-stage CRC is well established. Our study utilized drug sensitivity data from the GDSC database and the R package “pRRophetic” to predict the chemotherapy sensitivity of individual tumor samples. We aimed to investigate the relationship between key genes and common chemotherapy drugs. The results indicated that MMRN1 is significantly related to sensitivity to AICAR and BI.D1870, MS.275, and JNK.Inhibitor.VIII (Fig. 5E). Similarly, SLC6A19 showed significant correlations with sensitivity to AICAR, BI.D1870, MS.275, JNK.Inhibitor.VIII., CGP.082996, and lapatinib (Fig. 5F).
Immune infiltration analysis
By examining the relationship between key genes and immune infiltration in the CRC dataset and delving into the potential molecular mechanisms by which these key genes influence CRC progression, the proportion of immune cells in each patient and the correlations among different immune cell types were determined (Fig. 6A,B). Additionally, compared to patients in the normal group, the numbers of activated memory CD4 T cells, resting NK cells, M0 macrophages, M1 macrophages, and mast cells in the CRC group were markedly greater (Fig. 6C). The relationships between key genes and immune cells were further investigated. Moreover, our analysis revealed that MMRN1 was strongly positively correlated with M2 macrophages, resting mast cells, and naive B cells and was significantly negatively correlated with activated mast cells, M0 macrophages, and resting NK cells (Fig. 6D). Similarly, SLC6A19 was found to be significantly positively correlated with resting mast cells, plasma cells, naive B cells, etc., and significantly negatively correlated with M0 macrophages, resting NK cells, activated mast cells, etc. (Fig. 6E). The correlations between key genes and various immune factors, including immunosuppressive factors, immunostimulatory factors, chemokines, and receptors, were derived from the TISIDB database. These findings indicate that the key genes MMRN1 and SLC6A19 are closely related to the level of immune cell infiltration and play critical roles in the immune microenvironment (Fig. 7A–E).

Exploring the biological significance of immune cell infiltration pattern. (A) Stacked bar plots depict the relative proportions of the 22 tumor-infiltrating immune cell subtypes in each sample. (B) Associations between 22 tumor-infiltrating immune cells. (C) The number of active memory CD4 T cells, resting NK cells, M0 macrophages, M1 macrophages, and mast cells was significantly increased in CRC patients compared with the normal group. (D) The lollipop chart showed that MMRN1 was strongly positively correlated with M2 macrophages, static mast cells and naive B cells, and significantly negatively correlated with activated mast cells, M0 macrophages and static NK cells. (E) The lollipop chart showed that SLC6A19 was significantly positively correlated with static mast cells, plasma cells and naive B cells, and significantly negatively correlated with M0 macrophages, static NK cells and activated mast cells.

Correlation analysis of key genes and various types of immune factors. (A) MMRN1 and SLC6A19 are significantly correlated with chemokines. (B) MMRN1 and SLC6A19 are deeply correlated with immunostimulatory factors. (C) MMRN1 and SLC6A19 are correlated with immune inhibitory factors. (D) MMRN1 and SLC6A19 are significantly correlated with MHC molecules. (E) MMRN1 and SLC6A19 are greatly correlated with receptors.
GSEA pathway enrichment analysis
Next, we investigated the specific signaling pathways enriched in the two key genes to explore the potential molecular mechanisms through which these genes influence CRC progression. The GSEA results indicated that MMRN1 is enriched in the oxytocin signaling pathway, relaxin signaling pathway, TGF-β signaling pathway and others (Fig. 8A,B). Similarly, SLC6A19 is associated with pathways including galactose metabolism, retinol metabolism, tyrosine metabolism and other pathways (Fig. 8C,D).

Enrichment analysis of key gene specific signaling pathways. (A) The GSEA results of MMRN1 showed significant enrichment of the oxytocin signaling pathway, relaxin signaling pathway and TGF-β signaling pathway, and showed high activity in CRC group. (B) Oxytocin signaling pathway, relaxin signaling pathway and TGF-β signaling pathway were enriched gene sets, DCN, GREM1 and other genes were more prominent. (C) GSEA results of SLC6A19 showed significant enrichment of galactose metabolism, retinol metabolism and tyrosine metabolism pathways, and showed high activity in CRC group. (D) Gene sets enriched in the pathways of galactose metabolism, retinol metabolism and tyrosine metabolism, with ADH1C and other genes prominent. (E) The GSVA pathway enrichment analysis of MMRN1 showed significant enrichment of IL2/STAT5 signaling pathway, IL6/JAK/STAT3 signaling pathway and Notch signaling pathway. (F) The GSVA pathway enrichment analysis of SLC6A19 showed significant enrichment of bile acid metabolism, xenobiotic metabolism, and allograft rejection pathways.
GSVA pathway enrichment analysis
The GEVA analysis revealed that the pathways enriched in MMRN1 included the IL2/STAT5 signaling pathway, IL6/JAK/STAT3 signaling pathway, and Notch signaling pathway (Fig. 8E); the pathways enriched in SLC6A19 included bile acid metabolism, xenobiotic metabolism, and allograft rejection, among others (Fig. 8F). These findings suggest that key genes may influence disease progression via these pathways.
Transcriptional regulation analysis of key genes
For our analysis, the two key genes were used as the gene set, both regulated by common mechanisms, including multiple transcription factors. Therefore, we conducted an enrichment analysis of these transcription factors using cumulative recovery curves. The motif-TF annotation and selection analysis of important genes identified cisbp_M2575 as having the highest normalized enrichment score (NES: 5.93). All enriched motifs and their corresponding transcription factors of key genes are presented in Supplementary Fig. 2A-B.
Effects of silencing TFAM on MMRN1 and SLC6A19
Transcriptional regulation analysis showed that cisbp_M2575 may be the common mechanism of regulating these two key genes, and the corresponding key gene transcription factor is TFAM. In order to further study the regulatory effects of TFAM on MMRN1 and SLC6A19, we commissioned Shanghai Jima Pharmaceutical Co., Ltd. to design the siRNA of TFAM, a key gene transcription factor corresponding to cisbp_M2575, to silence TFAM. TFAM siRNA was transfected in RKO and HCT-116 cell lines, and it was found that after silencing TFAM (Supplementary Fig. 2C), the expression level of MMRN1 was increased (RKO: P = 0.0275, HCT-116: P = 0.0001) (Supplementary Fig. 2C), and the expression level of SLC6A19 was downregulated (RKO: P = 0.0286, HCT-116: P = 0.0011) (Supplementary Fig. 2C).
Correlations between key genes and disease regulatory genes
Regulatory genes related to CRC were identified using the GeneCards database. We analyzed the expression differences of these disease-related genes between the groups and found that the expression levels of 17 genes, including RAD51C, ALK, and CCND1, varied significantly between the two groups. There was a substantial difference in gene expression among the groups. Next, correlation analysis was performed for key genes and tumor regulatory genes. Subsequently, we conducted a correlation analysis between key genes and tumor regulatory genes. The results revealed that the expression levels of the key genes were significantly correlated with those of the disease-related genes. Specifically, MMRN1 showed a strong positive correlation with ADRB2 (cor = 0.516), while SLC6A19 exhibited a significant negative correlation with TERF1 (cor = − 0.216) (Supplementary Fig. 2D).
scRNA-seq analysis
We downloaded the single-cell dataset GSE221575 and performed single-cell analysis using the Seurat package. Then, we used the t-distributed stochastic neighbor embedding (tSNE) algorithm to cluster the cells and obtained 15 subtypes (Fig. 9A). Each subtype was annotated through the R package SingleR, categorizing the 15 clusters into 7 cell types: CD8 + T cells, epithelial cells, B cells, monocytes, HSCs, fibroblasts, and endothelial cells (Fig. 9B). The expression of the key genes in 7 types of cells was assessed (Fig. 9C,D). In addition, we examined the coexpression of key genes and tumor progression genes within single cells, identifying the top 5 genes (Supplementary Fig. 3A-J).

Single-cell sequencing. (A) tSNE map of the single-cell profile, color-coded according to subtype. (B) t-SNE plot for seven postannotation cell types, color-coded based on cell type. (C) Expression of MMRN1 and SLC6A19 cell type marker genes. (D) Expression levels of MMRN1 and SLC6A19 across seven cell types.
Downregulation of MMRN1 and upregulation of SLC6A19 in CRC
To confirm the expression of MMRN1 and SLC6A19 in CRC, we analyzed their expression levels in NCM-460 normal colon epithelial cells, the CRC cell lines RKO, HCT-116, and SW-480 and paired CRC tissues with adjacent nontumor tissues. The qRT-PCR results indicated that MMRN1 was significantly downregulated in CRC cells compared with NCM-460 cells and SLC6A19 was significantly upregulated in CRC cells (P < 0.001) (Fig. 10A). Similarly, the qRT-PCR results indicated that among 30 pairs of CRC and adjacent normal tissues, MMRN1 levels were lower in tumor tissues in 23 pairs, while SLC6A19 levels were higher in tumor tissues in 24 pairs. These findings indicate a downregulation of MMRN1 (P = 0.0175) and upregulation of SLC6A19 (P = 0.0115) in the 30 pairs of CRC tissues (Fig. 10B).

Expression and biological function of MMRN1, SLC6A19 in CRC. (A) qRT-PCR analysis of MMRN1 and SLC6A19 level in NCM-460 cells and different CRC cells. (B) qRT-PCR analysis of MMRN1 and SLC6A19 level in CRC tissues (n = 30) and adjacent-normal tissues (n = 30). (C) qRT-PCR analysis of MMRN1 and SLC6A19 level in RKO and HCT-116 cells transfected with si-NC or si-MMRN1 and si-SLC6A19. (D) CCK-8 assays were applied for evaluating cell viability. (E) Colony formation assays were applied for evaluating cell proliferation. (F) Transwell assays were performed to assess cell migration. (*P < 0.05; **P < 0.01; ***P < 0.001).
Relationship between tissue MMRN1 and SLC6A19 levels and clinicopathological factors of CRC patients
As shown in Supplementary Table 1, MMRN1 expression levels in CRC tissue were closely associated with distal metastasis (P = 0.030). Patients in group of lower MMRN1 expression have poorer distant metastasis (Supplementary Table 1). Similarly, SLC6A19 expression levels in CRC tissue were closely associated with distal metastasis (P < 0.001). Patients in group of higher SLC6A19 expression have poorer distant metastasis (Supplementary Table 2).
Effects of MMRN1 and SLC6A19 on cell proliferation and metastasis
To further investigate the potential roles of MMRN1 and SLC6A19 in CRC, we downregulated the expression of MMRN1 and SLC6A19 using siRNA, respectively, in RKO and HCT-116 cell lines (Fig. 10C). Subsequently, CCK-8 showed that silencing of MMRN1 promoted RKO and HCT-116 cell proliferation, while silencing of SLC6A19 significantly inhibited cell growth (Fig. 10D). Correspondingly, colony-forming ability was significantly increased after silencing MMRN1 in RKO (P = 0.0141) and HCT-116 (P < 0.001), but significantly decreased when silencing SLC6A19 expression (RKO: P = 0.0034, HCT-116: P = 0.0047) (Fig. 10E). Moreover, transwell assay confirmed that silencing of MMRN1 significantly promoted migration of RKO (P < 0.001) and HCT-116 (P = 0.0050) cell lines. Conversely, silencing of SLC6A19 significantly inhibited migration of these cell lines (RKO: P = 0.0056, HCT-116: P = 0.0064) (Fig. 10F).
Discussion
In cancer research, MR mitigates the errors and impacts associated with traditional randomized control trials and alleviates issues with reverse causality and confounding variables12. In medical research, observational studies are susceptible to confounding factors, and other variables may affect the results, making the results less reliable. Observational studies may also have problems with inverse causality, and the results may in turn affect exposure21,22. Compared with observational studies, MR uses genetic variation as an IV to infer causality, overcoming the limitations of observational studies because the exposure studied is random and without confounding factors, which can provide more reliable causal inference results23. Studies aiming to confirm and elucidate the causal relationships of known risk factors relevant to clinical settings, discover drug targets, and predict cancer risks are preferred24. For instance, an MR study by Yuan and colleagues provided evidence for a causal link between smoking and multiple gastrointestinal diseases, whereas alcohol consumption was linked to only a few gastrointestinal disorders25. Su and others employed MR in conjunction with the eQTL database to identify three promising genes for potential targeted therapies in Alzheimer’s disease26. Yang and others used two-sample MR to reveal the absence of a linear causal relationship between telomere length and liver cancer27. Thus, by employing univariate Cox analysis and integrating MR with the eQTL database, two crucial prognostic genes for CRC, MMRN1 and SLC6A19, were successfully identified in our study (Fig. 4A,B). Additionally, the nomogram model revealed that key genes accurately predict the survival of CRC patients. Using multiple datasets based on the two key genes, we investigated their potential molecular mechanisms via analyses such as immune infiltration, GSEA/GSVA pathway enrichment, drug sensitivity, and transcriptional regulation. Subsequently, the expression of both key genes was also observed in the scRNA-seq data of CRC. Finally, qRT-PCR confirmed the significant downregulation of MMRN1 and upregulation of SLC6A19 in CRC. Lower expression of MMRN1 and higher expression of SLC6A19 significantly promoted the proliferation and metastasis of colorectal cancer cells. Based on our findings, MMRN1 and SLC6A19 are promising candidates as key genes for predicting the prognosis of CRC.
Our research identified two crucial prognostic genes for CRC, MMRN1 and SLC6A19. MMRN1 and SLC6A19 contribute to tumor development through multiple mechanisms. MMRN1, which encodes a soluble adhesion glycoprotein, is regulated in cancer through methylation, protein interactions, and noncoding RNAs28. It is mainly distributed in platelets, megakaryocytes, endothelial cells (EC) and extracellular matrix (ECM), have significant effect on the tumor microenvironment28. The interaction between cancer cells and platelets provides the possibility of immune escape for cancer cells, allowing them to escape attack by natural killer cells (NK cells) and adhere to the blood vessel wall29. Subsequently, cancer cells cross the endothelium of blood vessels and leave the blood circulation, thus promoting metastasis29. Activated platelets can also recruit extracellular matrix components and granulocytes, promote the formation of early metastatic niches and promote the proliferation of cancer cells28,29. Additionally, activated platelets release various pro-angiogenic factors, such as vascular endothelial growth factor (VEGF), which promotes the migration and proliferation of endothelial cells and promotes tumor angiogenesis28. The dynamic changes of EC and cancer cells in the tumor microenvironment provide a rich blood supply for tumor growth and metastasis28. The ECM also plays a crucial role in tumor progression30. Tumor-associated ECM is synthesized mainly by tumor-associated fibroblasts (CAFs), and in CRC, ECM stiffness increases and has a specific molecular expression profile that enables cancer cell proliferation and metastasis30. These changes in the tumor microenvironment may be closely related to the immune escape mechanism of CRC, and the downregulation of MMRN1 may enhance the interaction between platelets and cancer cells and promote the immune escape mechanism of tumors, thereby promoting tumor progression by regulating tumor angiogenesis and extracellular matrix remodeling. What’s more, it serves as a novel adverse biomarker for pediatric acute myeloid leukemia and as a biomarker for the early detection of ovarian cancer31,32.
SLC6A19 is a neutral amino acid transporter involved in the transmembrane transport of amino acids33. Upregulation of SLC6A19 may play a role in metabolic reprogramming of CRC. When cancer cells proliferate, they often need to change metabolic pathways to meet their energy needs, and this metabolic reprogramming contributes to tumor growth and metastasis34. SLC6A19 has a crucial effect on the metabolic regulation of cancer cells by regulating the transport of amino acids, especially neutral amino acids34. Amino acids are not only involved in protein synthesis, but also important regulatory factors in cell metabolism. Studies have found that amino acid metabolism plays an important role in the occurrence and development of CRC35,36. By regulating amino acid metabolism, cancer cells are able to activate metabolism-related signaling pathways such as mTOR and HIF-1α, which can promote cell proliferation and regulate immune responses in the tumor microenvironment37. For instance, amino acid metabolites can influence immune cell function to promote immune escape, inhibit T cell activity, enhance tumor immunosuppressive environment, and better help tumors evade immune system attack34. The up-regulation of SLC6A19 may enhance the uptake of amino acids, activate tumor-related metabolic pathways, increase the energy supply for cancer cells, and create conditions suitable for the proliferation of cancer cells. At the same time, the amino acid metabolic reprogramming involved in SLC6A19 may also alter immune cell function in the tumor microenvironment, then promoting cancer cells to escape immune surveillance. Thus, SLC6A19 plays a dual role in the metabolic reprogramming and immune escape of CRC. The identified critical prognostic genes provide potential targets for managing CRC prognosis, potentially enhancing the poor prognosis associated with CRC.
The nomogram model is widely used to evaluate disease prognoses by integrating various prognostic and determinant variables to produce a singular numerical probability for clinical events38. For example, Fu and colleagues developed a nomogram model based on body composition changes detected through CT scans and clinical prognostic factors that reliably predicts survival rates in patients with locally advanced cervical cancer39. Tao and his team constructed a nomogram model using body composition and prognostic nutritional indices, accurately predicting the postsurgical survival period for gastric cancer patients40. Consequently, we developed a nomogram model (Fig. 5A) based on diverse clinical indicators and the expression profiles of MMRN1 and SLC6A19 to predict 3-year and 5-year overall survival (OS) in CRC patients (Fig. 5B). The outcomes closely aligned with the observed OS, demonstrating the strong predictive ability of the nomogram model, further suggesting that MMRN1 and SLC6A19 are effective predictors of CRC prognosis. The accurate prognostic prediction of MMRN1 and SLC6A19 prompted further investigation into their underlying molecular mechanisms. Traditionally, CRC has been considered to have low immunogenicity41. However, recent studies have revealed heterogeneous immune responses in CRC, especially through microsatellite instability41. The tumor microenvironment plays a critical role in aspects such as immunosuppression, drug resistance, metastasis, and survival41. Investigating the levels of immune infiltration within this environment is crucial for uncovering potential molecular mechanisms and improving clinical prognosis42. Our research demonstrated that MMRN1 and SLC6A19 are closely linked to immune cell infiltration levels and significantly influence the immune microenvironment in CRC (Fig. 6A–E). Immune infiltration analysis of C1QB, GNG8, and SUCLG2 showed that the genes excluded in MR analysis had little distributional correlation with the distribution of immune cell populations in colorectal cancer (Supplementary Fig. 4A), indicating that MMRN1 and SLC6A19 were associated with immune cell populations and distribution specific to key genes. Consequently, the expression levels of these key genes serve as predictors of CRC prognosis.
Furthermore, MMRN1 and SLC6A19 are involved in signaling pathways that are closely linked to the development and progression of CRC. MMRN1 was enriched in crucial pathways, such as the TGF-β signaling pathway and the Notch signaling pathway, which are essential for CRC (Fig. 8A,B,E). MMRN1 contains several domains, including the EMI domain, epidermal growth factor (EGF) -like domain, coiled coil and gC1q domain28. Our results suggest that MMRN1 is enriched in TGF-β and Notch signaling pathways, it may influence tumor immune escape and metastasis through these pathways. Previous studies have showed that the EMI domain regulates TGF-β signaling43 and enhances immune escape of cancer cells28. This is consistent with our results that MMRN1 plays an important role in immune escape of CRC by mediating activation of TGF-β pathway. The TGF-β signaling pathway has an important effect on the tumor microenvironment, influencing tumor progression44,45. By regulating the function of immune cells and their distribution in the tumor microenvironment, TGF-β can inhibit immune surveillance and promote tumor immune tolerance44. Downregulated MMRN1 enhances TGF-β signaling through the action of its EMI domain, thereby driving immune escape and helping cancer cells evade attack by the host immune system. In addition, the EMI and EGF-like domains of MMRN1 are closely related to the Notch signaling pathway. The Notch signaling pathway plays a key role in the proliferation, differentiation, and metastasis of cancer cells46. EGFL7 is a pro-angiogenic factor that exists in the endothelium and ECM47. Wang and his partners demonstrated the existence of EGFL7/NOTCH/c-Myc signaling pathway mediated by EMI and EGF-like domains in EGFL747. EMI and EGF-like domains also exist in MMRN128. Activation of this signaling pathway can promote cancer cells proliferation, metastasis, and enhance the invasive ability of cancer cells in CRC48. Dysfunctional Notch signaling in tumors also affects the function of immune cells, such as NK cells, T cells, thereby regulating the immune escape mechanism48. Thus, MMRN1 is involved in immune escape and metastasis in CRC through regulation of the TGF-β and Notch signaling pathways. SLC6A19 is associated with metabolic pathways, such as bile acid metabolism (Fig. 8C,D,F). Bile acids are a class of compounds synthesized by the liver that play a role in digestion and fat absorption49. Abnormal bile acid metabolism is closely related to the occurrence and development of CRC50. Bile acids regulate the proliferation, differentiation and apoptosis of intestinal epithelial cells by activating metabolist-related receptors such as the nuclear receptor farnesoid X nuclear receptor (FXR)51. These pathways affect immune cells in the gut, the integrity of the intestinal barrier, and the gut microbiome, which influence the onset and development of CRC51. In addition, bile acid metabolites, such as deoxycholic acid, can act on regulatory T cells and exert an immunosuppressive effect52. Arafath demonstrated that SLC7A5 maintains intracellular amino acid levels after KRAS activation through transcriptional and metabolic reprogramming, thereby maintaining effective growth of CRC53. SLC6A19 may be involved in amino acid metabolism and affect the progression of CRC, and its upregulation may affect intestinal flora composition and bile acid metabolism pathway by regulating intestinal amino acid metabolism. Gut microbiota plays an important role in bile acid metabolism, through which the immune environment in the gut is regulated51. Therefore, SLC6A19 may influence metabolic susceptibility to CRC through metabolic pathways. In view of the close correlation between SLC6A19 and metabolic pathways, we propose some possible therapeutic strategies, such as targeting SLC6A19 to inhibit the overuptake of amino acids by tumor cells by regulating the expression of SLC6A19 and destroying the favorable conditions for tumor growth54. This approach may help reduce the tumor’s adaptation to metabolic reprogramming, thereby inhibiting the proliferation of cancer cells. The intervention of intestinal microorganisms, bile acid metabolism and intestinal flora interaction play an important role. The use of probiotics to regulate the composition of intestinal microbiota may affect the metabolic pathway of bile acids, and thus affect the immune environment and metabolic adaptability of tumor54. These approaches may provide a new direction for the treatment of CRC. Chemoresistance poses a significant challenge in CRC treatment, substantially limiting the efficacy of therapies and adversely affecting prognosis55.
These key prognostic genes are significantly correlated with chemotherapeutic drug sensitivity and present promising opportunities for CRC therapy, especially with drugs such as AICAR and BI.D1870, MS.275 and JNK.Inhibitor.VIII (Fig. 5E,F). To ensure that the associations were specific to key genes, we performed drug sensitivity analyses for C1QB, GNG8, and SUCLG2. C1QB was not associated with JNK.Inhibitor.VIII (Supplementary Fig. 4B), GNG8 was not associated with AICAR,BI.D1870 (Supplementary Fig. 4C), no correlation was found between C1QB and JNK. Inhibitor. SUCLG2 is not correlated with AICAR, MS.275 (Supplementary Fig. 4D), demonstrating that this correlation is specific to MMRN1 and SLC6A19. The four chemotherapeutic drugs mentioned above have significant potential in anti-CRC therapy and could serve as chemotherapeutic candidates for CRC prognostic treatment. Notably, transcription factors play a crucial role in signal transduction, and the loss of their activity can lead to tumor development, offering significant potential for targeted therapy56. Our research progressed from gene expression to transcriptional regulation, revealing that transcription factors such as TFAM and NRL coregulate the expression of MMRN1 and SLC6A19 (Supplementary Fig. 2A-B), potentially revealing the upstream regulatory mechanisms of these crucial prognostic genes. Transcriptional regulation analysis showed that cisbp_M2575 may be the common mechanism of regulating these two key genes, and the corresponding key gene transcription factor is TFAM. TFAM is a core factor for mitochondrial DNA (mtDNA) replication and transcription and plays a key role in the maintenance of mitochondrial function57. Wu found that TFAM was overexpressed in CRC58. Downregulation of TFAM can reduce the proliferation of CRC cells59, suggesting that TFAM have an important effect on the occurrence and development of CRC. Guo and his team proposed that frequent frameshift mutations occur in TFAM in primary CRC with microsatellite instability (MSI)60. A similar phenomenon was not observed in microsatellite stabilized (MSS) CRC60. In MSI-H CRC, the truncated mutation of TFAM leads to a significant reduction in its protein levels both in vivo and in vitro, is strongly associated with mitochondrial DNA depletion60. These results suggest that the expression and function of TFAM may be involved in metabolic regulation and drug resistance mechanisms in CRC by influencing mtDNA stability. In addition, by silencing TFAM in CRC cells, we found that the expression level of MMRN1 increased and that of SLC6A19 decreased (Supplementary Fig. 2C), indicating that TFAM can regulate the expression of MMRN1 and SLC6A19. Our study revealed the potential molecular mechanisms of MMRN1 and SLC6A19, providing new targets and strategies for the prognostic management of CRC.
Downregulation of MMRN1 is of great importance to the prognosis of CRC, especially as it is associated with immune escape and metastasis. MSI-H CRC is highly genetically unstable, and this subtype is often associated with an increased mutation load, which provides a more favorable condition for tumor immune escape61. Not only that, the tumor microenvironment of MSI-H CRC showed a more robust anti-tumor immune response, with cancer cells activating immunosuppression through multiple pathways to evade the surveillance of the immune system62. Downregulation of MMRN1 alters the susceptibility of cancer cells to immune responses by affecting TGF-β and Notch signaling pathways. In MSI-H CRC, it plays an important role in immune escape mechanism62. On the other hand, the role of up-regulation of SLC6A19 in the prognosis of CRC is also essential. In MSI-H CRC, reprogramming of amino acid metabolism plays an important role in cancer cell proliferation and immunosuppression34. SLC6A19 enhances the energy supply of cancer cells by promoting the uptake of neutral amino acids and activates metabolism-related signaling pathways. MSI-H CRC is generally associated with metabolic changes in the tumor microenvironment that may help cancer cells acquire a strong capacity for proliferation, may also promote tumor immune escape by regulating immune cells through amino acid metabolism34. In addition, SLC6A19 may affect the uptake of amino acids, affecting the interaction between cancer cells and immune cells, thereby enhancing the immune escape of tumors. In MSI-H CRC, the upregulation of SLC6A19 may promote the activation of these metabolic pathways and promote the proliferation and metastasis of cancer cells. Combining the findings of MMRN1 and SLC6A19 with the molecular characteristics of MSI-H CRC can provide a new perspective to explore the metabolic reprogramming and immune escape mechanism of CRC.
As sequencing technology has rapidly advanced in recent years, scRNA-seq has been developed for profiling tumor cells and their heterogeneity, introducing new methods for biodiversity and genomics research63. A crucial step in translating GWAS data to CRC molecular mechanisms is identifying the main cell types associated with CRC64. Through scRNA-seq, Wu and colleagues discovered numerous novel genes associated with schizophrenia risk, offering new insights into the biological mechanisms of schizophrenia64. To investigate whether critical genes demonstrate cell type-specific enrichment in CRC, we obtained single-cell data from GSE221575 for clustering and annotation analysis. The cells were categorized into 15 clusters and subsequently into seven cell types: CD8 + T cells, epithelial cells, B cells, monocytes, HSCs, fibroblasts, and endothelial cells (Fig. 9B). This finding suggests a strong association between these cell types and the molecular mechanisms of CRC, as indicated by the distinct expression levels of MMRN1 and SLC6A19 (Fig. 9C,D). The results of scRNA-seq revealed specific enrichment of MMRN1 and SLC6A19 in certain CRC cell types, providing compelling evidence for exploring CRC molecular mechanisms. Finally, the significant downregulation of MMRN1 and upregulation of SLC6A19 in CRC was confirmed by qRT-PCR (Fig. 10A,B), providing robust empirical evidence. Lower expression of MMRN1 and higher expression of SLC6A19 significantly promoted the proliferation and metastasis of colorectal cancer cells (Fig. 10D–F). It was further suggested that the key genes identified were closely related to the prognosis of CRC.
In addition, previous research has also been of great help to our research. Zhou and his team analysis using TCGA database and GTEs database showed that MMRN1 is downregulated in colon cancer and is closely related to the prognosis of cancer patients65. Li finds that MMRN1 was highly correlated with the clinical outcome of rectal cancer through the ESTIMATE algorithm and TCGA database66. These are consistent with our findings. However, the role of SLC6A19 in colorectal cancer has not been explored previously.
Certain limitations also existed in our study. Firstly, the data sources of MR analysis in this study are all individuals of European ancestry in the database, which can reduce the potential bias caused by demographic structure, but limits the generalization of MR Results to other populations. Second, the summary‐level data for these associations in the GWAS on the exposure could be inflated because of winner’s curse. Winner’s curse is a common problem in GWAS, Jiang conducted an empirical investigation on the impact of winner’s curse on MR estimates, and the research results show that winner’s curse may bias MR estimates, but its actual impact may not be large67. If avoiding sample overlap is not feasible, the analyst should consider performing a sensitivity analysis based on variants closely associated with exposure67. In this study, SNPs related to the genome-wide significance threshold of each gene (P < 1e−5) were selected as potential IVs, and the LD among SNPs was calculated. Sensitivity analysis was conducted using the leave-one method, and horizontal pleiotropy test was conducted using the MR-Egger intercept test (P < 0.05). No heterogeneity was observed among SNPs (Cochran’s Q < 0.05), suggesting high confidence in the results of our MR Analysis. In addition, in order to further study and identify the biological function of genes, due to nationality and legal restrictions, we chose China, a country with a large population, and collected patient samples from all regions of China to reduce population bias. While we attempted to validate findings in Chinese cohorts, two potential limitations warrant attention: differential LD patterns between Europeans and Chinese may alter instrument strength. Population-specific environmental factors, such as dietary patterns in lipid metabolism, could modify effect sizes. Finally, the sample size selected for this verification is small, so it is still necessary to expand the sample size for in-depth study, and it is best to select samples of European ancestry.
Conclusion
In conclusion, this study investigated the clinical significance and potential applications of key genes in CRC. We identified two key genes related to the prognosis of CRC, namely MMRN1 and SLC6A19, and analyzed their relationships with immune cell invasion, signaling pathways, chemotherapy drug sensitivity and single cell subpopulations. The biological functions of key genes and their effects on the prognosis of CRC were explored by cell experiments. These findings reveal the underlying molecular mechanisms of key genes and provide new targets and therapeutic approaches for the prognostic management of CRC.
Data availability
From the public database: the datasets analysed during the current study are available in the TCGA database, [https://portal.gdc.cancer.gov/], we downloaded raw mRNA expression data for colorectal cancer data; the GEO database, [https://www.ncbi.nlm.nih.gov/geo/info/datasets.html], we download the data file of single cell data GSE221575 from the NCBI GEO public database for single cell analysis; the eQTLGen consortium database, [https://www.eqtlgen.org]; the MR Base database (finngen_R10_C3_COLORECTAL_EXALLC), [http://app.mrbase.org/]; the GWAS summary data, [https://gwas.mrcieu.ac.uk/]; the Cancer Drug Sensitivity Genomics Database, [https://www.cancerrxgene.org/]. The original contributions presented in the study are included in the article or supplementary material. Further inquiries can be directed to the corresponding author.
References
Kastrinos, F., Kupfer, S. S. & Gupta, S. Colorectal cancer risk assessment and precision approaches to screening: Brave new world or worlds apart?. Gastroenterology 164, 812–827 (2023).
Dekker, E., Tanis, P. J., Vleugels, J. L. A., Kasi, P. M. & Wallace, M. B. Colorectal cancer. Lancet 394, 1467–1480 (2019).
Kotani, D. et al. Molecular residual disease and efficacy of adjuvant chemotherapy in patients with colorectal cancer. Nat. Med. 29, 127–134 (2023).
Ciardiello, F. et al. Clinical management of metastatic colorectal cancer in the era of precision medicine. CA Cancer J. Clin. 72, 372–401 (2022).
Shin, A. E., Giancotti, F. G. & Rustgi, A. K. Metastatic colorectal cancer: Mechanisms and emerging therapeutics. Trends Pharmacol. Sci. 44, 222–236 (2023).
Evans, D. M. & Davey, S. G. Mendelian randomization: New applications in the coming age of hypothesis-free causality. Annu. Rev. Genom. Hum. Genet. 16, 327–350 (2015).
Tian, H., Tom, B. D. M. & Burgess, S. A data-adaptive method for investigating effect heterogeneity with high-dimensional covariates in Mendelian randomization. BMC Med. Res. Methodol. 24, 34 (2024).
Ference, B. A. How to use Mendelian randomization to anticipate the results of randomized trials. Eur. Heart J. 39, 360–362 (2018).
Mälarstig, A. et al. Evaluation of circulating plasma proteins in breast cancer using Mendelian randomisation. Nat. Commun. 14, 7680 (2023).
Zhang, J. & Zhao, H. eQTL studies: From bulk tissues to single cells. J. Genet. Genom. 50, 925–933 (2023).
Zhu, Z. et al. Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat. Genet. 48, 481–487 (2016).
Gala, H. & Tomlinson, I. The use of Mendelian randomisation to identify causal cancer risk factors: Promise and limitations. J Pathol. 250, 541–554 (2020).
Skrivankova, V. W. et al. Strengthening the reporting of observational studies in epidemiology using Mendelian randomization: The STROBE-MR statement. JAMA 326, 1614–1621 (2021).
Skrivankova, V. W. et al. Strengthening the reporting of observational studies in epidemiology using mendelian randomisation (STROBE-MR): Explanation and elaboration. BMJ 375, n2233 (2021).
Kanehisa, M., Furumichi, M., Sato, Y., Matsuura, Y. & Ishiguro-Watanabe, M. KEGG: Biological systems database as a model of the real world. Nucleic Acids Res. 53, D672–D677 (2025).
Kanehisa, M. Toward understanding the origin and evolution of cellular organisms. Protein Sci. 28, 1947–1951 (2019).
Kanehisa, M. & Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30 (2000).
Oliva, M. et al. DNA methylation QTL mapping across diverse human tissues provides molecular links between genetic variation and complex traits. Nat. Genet. 55, 112–122 (2023).
Dasgupta, D., Mahadev Bhat, S., Price, A. L., Delmotte, P. & Sieck, G. C. Molecular mechanisms underlying TNFα-Induced mitochondrial biogenesis in human airway smooth muscle. Int. J. Mol. Sci. 24, 5788 (2023).
Burgess, S. & Thompson, S. G. Interpreting findings from Mendelian randomization using the MR-Egger method. Eur. J. Epidemiol. 32, 377–389 (2017).
Zhang, Z. et al. Non-alcoholic fatty liver disease and vitamin D in the UK biobank: A two-sample bidirectional Mendelian randomisation study. Nutrients 15, 1442 (2023).
Kim, S. Overview of clinical study designs. Clin. Exp. Emerg. Med. 11, 33–42 (2024).
Liu, D. et al. Vitamin D and multiple health outcomes: An umbrella review of observational studies, randomized controlled trials, and Mendelian randomization studies. Adv. Nutr. 13, 1044–1062 (2022).
Richmond, R. C. & Davey, S. G. Mendelian randomization: Concepts and scope. Cold Spring Harb. Perspect. Med. 12, a040501 (2022).
Yuan, S. et al. Smoking, alcohol consumption, and 24 gastrointestinal diseases: Mendelian randomization analysis. Elife 12, e84051 (2023).
Su, W. M. et al. Systematic druggable genome-wide Mendelian randomisation identifies therapeutic targets for Alzheimer’s disease. J. Neurol. Neurosurg. Psychiatry. 94, 954–961 (2023).
Yang, C., Wu, X., Chen, S. & Xiang, B. Association between telomere length and hepatocellular carcinoma risk: A Mendelian randomization study. Cancer Med. 12, 9937–9944 (2023).
Posner, M. G. Multimerin-1 and cancer: A review. Biosci. Rep. 42, BSR20211248 (2022).
Bonfitto, P. H. L. et al. Involvement of platelet signaling pathways in colorectal cancer and new therapeutic targets. Am. J. Cancer Res. 14, 5133–5153 (2024).
Chen, E., Zeng, Z. & Zhou, W. The key role of matrix stiffness in colorectal cancer immunotherapy: Mechanisms and therapeutic strategies. Biochim. Biophys. Acta Rev. Cancer 1879, 189198 (2024).
Laszlo, G. S. et al. Multimerin-1 (MMRN1) as novel adverse marker in pediatric acute myeloid leukemia: A report from the children’s oncology group. Clin. Cancer Res. 21, 3187–3195 (2015).
Saini, A. et al. Analysis of Multimerin 1 (MMRN1) expression in ovarian cancer. Mol. Biol. Rep. 47, 9459–9468 (2020).
Bhutia, Y. D., Mathew, M., Sivaprakasam, S., Ramachandran, S. & Ganapathy, V. Unconventional functions of amino acid transporters: Role in macropinocytosis (SLC38A5/SLC38A3) and diet-induced obesity/metabolic syndrome (SLC6A19/SLC6A14/SLC6A6). Biomolecules 12, 235 (2022).
Zhu, M. et al. Role of amino acid metabolism in tumor immune microenvironment of colorectal cancer. Am. J. Cancer Res. 15, 233–247 (2025).
Solanki, S. et al. Dysregulated amino acid sensing drives colorectal cancer growth and metabolic reprogramming leading to chemoresistance. Gastroenterology 164, 376–391 (2023).
Liu, X. et al. CircMYH9 drives colorectal cancer growth by regulating serine metabolism and redox homeostasis in a p53-dependent manner. Mol. Cancer 20, 114 (2021).
Dong, S. et al. ROS/PI3K/Akt and Wnt/beta-catenin signalings activate HIF-1alpha-induced metabolic reprogramming to impart 5-fluorouracil resistance in colorectal cancer. J. Exp. Clin. Cancer Res. 41, 15 (2022).
Balachandran, V. P., Gonen, M., Smith, J. J. & DeMatteo, R. P. Nomograms in oncology: More than meets the eye. Lancet Oncol. 16, e173–e180 (2015).
Fu, B. et al. Nomograms combining computed tomography-based body composition changes with clinical prognostic factors to predict survival in locally advanced cervical cancer patients. J. Xray Sci. Technol. 32, 427–441 (2024).
Tao, C. et al. Nomogram based on body composition and prognostic nutritional index predicts survival after curative resection of gastric cancer. Acad. Radiol. 31, 1940–1949 (2024).
Mao, X. et al. Crosstalk between cancer-associated fibroblasts and immune cells in the tumor microenvironment: New findings and future perspectives. Mol. Cancer 20, 131 (2021).
Lei, X. et al. Immune cells within the tumor microenvironment: Biological functions and roles in cancer immunotherapy. Cancer Lett. 470, 126–133 (2020).
Zacchigna, L. et al. Emilin1 links TGF-beta maturation to blood pressure homeostasis. Cell 124, 929–942 (2006).
Peng, D., Fu, M., Wang, M., Wei, Y. & Wei, X. Targeting TGF-beta signal transduction for fibrosis and cancer therapy. Mol. Cancer 21, 104 (2022).
Wang, X., Eichhorn, P. J. A. & Thiery, J. P. TGF-beta, EMT, and resistance to anti-cancer treatment. Semin Cancer Biol. 97, 1–11 (2023).
Shi, Q. et al. Notch signaling pathway in cancer: From mechanistic insights to targeted therapies. Signal Transduct. Target Ther. 9, 128 (2024).
Wang, Y. et al. EGFL7 drives the evolution of resistance to EGFR inhibitors in lung cancer by activating NOTCH signaling. Cell Death Dis. 13, 910 (2022).
Li, X. et al. The Notch signaling pathway: A potential target for cancer immunotherapy. J. Hematol. Oncol. 16, 45 (2023).
Ocvirk, S. & O’Keefe, S. J. D. Dietary fat, bile acid metabolism and colorectal cancer. Semin. Cancer Biol. 73, 347–355 (2021).
Fu, T. et al. Paired microbiome and metabolome analyses associate bile acid changes with colorectal cancer progression. Cell Rep. 42, 112997 (2023).
Režen, T. et al. The role of bile acids in carcinogenesis. Cell Mol. Life Sci. 79, 243 (2022).
Zhang, Y. et al. Effect of gut flora mediated-bile acid metabolism on intestinal immune microenvironment. Immunology 170, 301–318 (2023).
Najumudeen, A. K. et al. The amino acid transporter SLC7A5 is required for efficient growth of KRAS-mutant colorectal cancer. Nat. Genet. 53, 16–26 (2021).
Nenkov, M., Ma, Y., Gaßler, N. & Chen, Y. Metabolic reprogramming of colorectal cancer cells and the microenvironment: Implication for therapy. Int. J. Mol. Sci. 22, 6262 (2021).
Ma, S. C. et al. Novel strategies to reverse chemoresistance in colorectal cancer. Cancer Med. 12, 11073–11096 (2023).
Shiah, J. V., Johnson, D. E. & Grandis, J. R. Transcription factors and cancer: Approaches to targeting. Cancer J. 29, 38–46 (2023).
Song, Y., Wang, W., Wang, B. & Shi, Q. The protective mechanism of TFAM on mitochondrial DNA and its role in neurodegenerative diseases. Mol. Neurobiol. 61, 4381–4390 (2024).
Wu, K. et al. Down-Regulation of MicroRNA-214 contributed to the enhanced mitochondrial transcription factor A and inhibited proliferation of colorectal cancer cells. Cell Physiol. Biochem. 49, 545–554 (2018).
Buchwaldt, J. et al. Decreased mitochondrial transcription factor A and mitochondrial DNA copy number promote cyclin-dependent kinase inhibitor 1A expression and reduce tumorigenic properties of colorectal cancer cells. Discov. Oncol. 15, 701 (2024).
Guo, J. et al. Frequent truncating mutation of TFAM induces mitochondrial DNA depletion and apoptotic resistance in microsatellite-unstable colorectal cancer. Cancer Res. 71, 2978–2987 (2011).
Wilbur, H. C., Le, D. T. & Agarwal, P. Immunotherapy of MSI cancer: Facts and hopes. Clin. Cancer Res. 30, 1438–1447 (2024).
Wang, Q., Yu, M. & Zhang, S. The characteristics of the tumor immune microenvironment in colorectal cancer with different MSI status and current therapeutic strategies. Front. Immunol. 15, 1440830 (2024).
Liu, W. et al. Construction of an immune predictive model and identification of TRIP6 as a prognostic marker and therapeutic target of CRC by integration of single-cell and bulk RNA-seq data. Cancer Immunol. Immunother. 73, 69 (2024).
Wu, Y., Zhang, C. Y., Wang, L., Li, Y. & Xiao, X. Genetic insights of schizophrenia via single cell RNA-sequencing analyses. Schizophr Bull. 49, 914–922 (2023).
Zhou, Q., Liu, Y., Zhou, J. & Zhang, W. Prognostic value and immunological role of MMRN1: A rising star in cancer. Nucleosides Nucleotides Nucleic Acids. 44, 148–169 (2025).
Li, C., Liu, T., Liu, Y., Zhang, J. & Zuo, D. Prognostic value of tumour microenvironment-related genes by TCGA database in rectal cancer. J. Cell Mol. Med. 25, 5811–5822 (2021).
Jiang, T., Gill, D., Butterworth, A. S. & Burgess, S. An empirical investigation into the impact of winner’s curse on estimates from Mendelian randomization. Int. J. Epidemiol. 52, 1209–1219 (2023).
Acknowledgements
Thanks for the technical support by the Core Facilities, Health Science Center, Ningbo University. Thanks for the authors who shared the original dataset in this study.
Funding
This study was supported by grants from the Key Scientific and Technological Projects of Ningbo (No. 2021Z133, No.2022Z130), Ningbo Top Medical and Health Research Program (No. 2023020612), and the Youth Medical Backbone Talents Training Program of Ningbo.
Author information
Authors and Affiliations
Contributions
GL.Ye and YF.Shao made substantial contributions to conception and design of this manuscript. M.Hu and HT.Dong were involved in drafting the manuscript and revising it critically for important intellectual content. X.Yu, JN.Yan, CY.Ye and CJ.Chen drew the figure in this manuscript. GL.Ye and YF.Shao reviewed and revised the final manuscript. All authors contributed to the figure discussions and approved the final manuscript submitted.
Corresponding authors
Ethics declarations
Competing interest
The authors declare no competing interests.
Ethical approval and consent to participate
The Ethics Committee of the First Affiliated Hospital of Ningbo University granted approval for this project (No.KY20220101). All human studies were carried out in accordance with the principles of the Declaration of Helsinki. Written informed consent was obtained from the patients.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Hu, M., Dong, H., Chen, C. et al. Comprehensive analysis of Mendelian randomization and scRNA-seq identify key prognostic genes and relevant functional roles in colorectal cancer. Sci Rep 15, 25039 (2025). https://doi.org/10.1038/s41598-025-10354-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-10354-x
