
Abstract
Mechanisms by which prior tuberculosis (TB) increases long-term risk for cancer, cardiovascular, and neurological disorders remain unclear, particularly in people with HIV (PWH). This study investigated DNA methylation (DNAm) patterns and associated pathways in PWH with and without prior TB infection. DNAm was analyzed in blood samples from 30 PWH (10 with prior latent TB infection [LTBI], 10 with previous successfully treated active TB, and 10 with no TB) using the Illumina MethylationEPIC BeadChip covering over 850,000 CpG sites. Epigenetic age was estimated, and age acceleration was calculated. Differentially methylated CpGs (dmCpGs) and regions (DMRs) were identified, and functional enrichment analyses for Gene Ontology, KEGG pathways, PANTHER database, and gene set enrichment analysis (DisGeNET, dbGaP) were performed. Statistical significance was set at a false discovery rate (FDR) of < 0.05. PWH exhibited significant epigenetic age acceleration, with a mean of 19.32 ± 10.82 years greater than chronological age. This accelerated aging was more pronounced in individuals with any prior TB infection (21.60 ± 12.03 years) compared to those without TB (17.42 ± 9.38 years). In the prior active TB vs. no TB comparison, 7461 dmCpGs were identified, corresponding to 150 DMRs (p < 0.05), with top associated genes including GRAMD1C (hypomethylation), DPP6 (hypermethylation), and HDAC4 (hypomethylation). In the LTBI vs. no TB comparison, 8598 dmCpGs were observed, corresponding to 39 DMRs (p < 0.05), associated with genes such as PLEKHG5 (hypermethylation), STK32C (hypermethylation), and SPATC1L. When comparing any prior TB (active or latent) to no TB, 71,774 dmCpGs and 14 DMRs were identified, including genes like PLEKHG5, KCNN3, and BRSK2. Pathway analyses of prior TB (active or latent) vs. no TB revealed enrichment in neurogenesis, neuron differentiation, axon guidance, and neuroactive ligand signaling. Additional enriched pathways included those related to platelet activation, vascular muscle contraction, and chemokine signaling. Cancer-related pathways such as proteoglycans in cancer, small cell lung cancer, prostate cancer, breast cancer, hepatocellular carcinoma, and thyroid cancer were also enriched. PANTHER analysis showed consistent enrichment in the Wnt signaling pathway and inflammation-mediated pathways across compared groups. DisGeNET analysis linked prior TB DNAm patterns to lymphoid leukemia, while dbGaP analysis identified associations with phenotypes like asthma, body mass index, tunica media, and lymphocyte count. Prior TB infection in PWH is associated with distinct DNAm changes in pathways related to neural function, cardiovascular health, and cancer risk, and is linked to more pronounced epigenetic age acceleration, suggesting epigenetic mechanisms for TB-related long-term complications.
Similar content being viewed by others
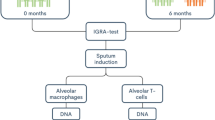

Introduction
Despite the improvement in diagnostic modalities and discovery of effective chemotherapeutic agents, tuberculosis (TB) is the leading cause of death from a single infectious agent, contributing 1.25 million deaths in 20231. Although the global TB treatment success for drug sensitive TB is high, long-term complications of TB contribute to significant disability and mortality even after TB treatment success2,3. After completion of TB treatment, previously treated patients are 3 times more likely to die than individuals without TB, and this is independent of age, sex, country income and type of TB4 or drug resistance profile5. For this reason, there are growing calls for programmatic follow up of TB patients after TB cure or treatment completion6,7. While these calls draw attention to the several post TB pulmonary complications resulting from structural changes in the lung (such as chronic obstructive airway disease, bronchiectasis, and fibrosis) 7, cardiovascular disease (CVD) and cancer account for almost 40% of deaths after TB treatment completion, while respiratory causes contribute only 14%4. Evidence from cohort studies shows that pulmonary TB (PTB) increases the risk for ischemic stroke8, acute coronary syndrome9, myocardial infarction10, and chronic kidney disease11. Additionally, PTB has been reported to increase the incidence of neurologic complications even in the absence of central nervous system tuberculosis. Dementia12, parkinsonism13 and depression14 are reported in population-wide cohort studies. Finally, TB is associated with an increased risk for cancer at ten sites15. Latent TB infection (LTBI) has been associated with similar risks for cancer, CVD and mental health problems16,17,18. There is an urgent need to understand the mechanisms underlying long term complications of active and latent TB.
The mechanisms underlying the long-term complications of TB are not well elucidated. Chronic inflammation and traditional CVD risk factors such as diabetes mellitus, smoking, and age have been implicated in increasing CVD risk19,20. Active TB and LTBI could contribute to cancer development by inducing chronic inflammation, genomic instability, and modulation of host cell signaling pathways21. Further, DNA methylation is thought to occur in genes and pathways involved in immune-regulation even after TB treatment. For example, this has been observed in genes involved in cytokine production (Interleukin (IL)-6), toll-like receptor signaling (TLR2), and other immune-related pathways (PI3K-AKT, MAPK, mTOR)22. Moreover, people with prior active TB have been shown to have an increase in the DNA methylation age by 13 years above the chronological age while cellular aging is increased by 14 years among people with LTBI23. However, these mechanisms have been demonstrated in predominantly HIV negative cohorts despite evidence that people with HIV (PWH) already experience ongoing accelerated aging driven by persistent inflammation, immune senescence, mitochondrial dysfunction, epigenetic alterations, and long term toxicities attributed to anti-retroviral therapy (ART) 24. There is a paucity of studies that have evaluated DNA methylation patterns associated with prior TB in PWH.
Our study aim was to determine whether prior TB infection is associated with DNA methylation (DNAm) patterns and pathways that could confer future risk for long term complications among PWH. By elucidating these mechanisms, the findings could inform targeted interventions to mitigate long-term complications, offering new avenues for improving the care of individuals with TB-HIV coinfection.
Methods
Study design and population
In this cross-sectional study, 30 adult PWH on ART were randomly selected from three HIV clinics in Uganda. Detailed study methods are described elsewhere25. Participants were stratified into three groups: 10 with prior LTBI, 10 with previously treated active TB, and 10 with no history of TB infection. Previous treatment for active TB (diagnosed either by sputum GeneXpert, microscopy or urine lipoarabinomannan) was ascertained from the HIV care records and the unit TB register at the respective clinics. LTBI was defined as a positive Quantiferon (QFT)-plus assay, according to manufacturer’s instructions, in an individual without previous treatment for active TB26. PWH with LTBI had all completed a course of TB preventive therapy according to national guidelines prior to enrollment27. Prior TB was defined as either LTBI or previous treated active TB. PWH with no TB had never been treated for active TB and had a negative QFT-plus assay.
Sample collection and processing
Whole blood samples (5 ml) were collected from each participant using EDTA vacutainers. Genomic DNA was extracted from the blood using the QIAamp DNA Blood Mini Kit (QIAGEN, Hilden, Germany) according to the manufacturer’s instructions. DNA quantity was measured using Qubit 4 fluorometer (ThermoFisher Scientific, USA), following the manufacturer’s instructions. For the methylation assay, 500 ng of DNA was used. DNA methylation (DNAm) levels were assayed using the Illumina Infinium MethylationEPIC v2.0 BeadChip array and the iScan system (Illumina, Inc., San Diego, CA, USA), in accordance with the manufacturer’s instructions, generating IDAT files for downstream bioinformatics analysis to assess methylation levels.
Data processing and analysis
The raw data files obtained from the Illumina MethylationEPIC BeadChip were preprocessed and analyzed using the minfi package in the R statistical software environment. The minfi package is a comprehensive toolkit specifically designed for the analysis of Illumina Infinium methylation arrays28. The preprocessing steps involved an initial quality assessment of the raw data using various metrics, including visualization of beta value distributions and evaluation of control probes to ensure optimal bisulfite conversion efficiency. Subsequently, background correction and quantile normalization were performed using the preprocess Quantile function to minimize technical variation and ensure consistent methylation value distributions across samples. To ensure data quality, probes with a detection p-value greater than 0.01 were removed, and probes located on sex chromosomes (X and Y) were excluded to avoid confounding due to sex differences. Additionally, probes containing single nucleotide polymorphisms (SNPs) were removed using the dropLociWithSnps function to prevent methylation differences caused by genetic variation.
Blood cell composition was estimated using the estimateCellCounts2 function, referencing the FlowSorted.CordBloodCombined.450 k dataset and utilizing the IDOLOptimizedCpGsCordBlood marker set. The cell types assessed included CD4+ T cells, CD8+ T cells, B cells, monocytes, natural killer (NK) cells, granulocytes, and nucleated red blood cells. The consistent proportion of blood cell types across samples, as detailed in Supplementary Table 1, indicated the absence of biological bias in the experimental setup. This consistency is important because the selected Horvath’s methylation clock, used for age estimation, incorporates various cell types, including blood cells.
Methylation beta values were also used to estimate epigenetic age for each individual using PC-PhenoAge, a second generation methylation clock able to predict mortality29. Age acceleration represented the extent to which the biological age of an individual exceeds their chronological age at the time of measurement. Age estimates were determined using dnaMethyAge package version 0.2.0 and the measure for age acceleration was defined as residuals yielded from regressing epigenetic age on chronological age from which root mean squared deviation (RMSD) and mean absolute deviation (MAD) were computable.
Identification of differentially methylated CpGs and regions
Differentially methylated CpGs (dmCpGs) and regions (DMRs) were identified using the dmpFinder function in minfi, which applies a linear model to compare methylation levels while adjusting for age and sex, methylation batch effect and cell-type covariates. This function performed pairwise comparisons between the three groups (LTBI, active TB, and no TB) to identify CpG sites that exhibit significant differences in methylation levels. Significant regions were defined by β-value differences > 0.2 and permutation testing (B = 0). The dmpFinder function utilizes statistical tests to assess the significance of methylation differences, adjusting for multiple comparisons to control the false discovery rate (FDR). An FDR < 0.05 was considered statistically significant. The Comparisons included: 1. Previous Active TB vs. no TB; 2. Prior LTBI TB vs. no TB; 3. Prior TB infection (both LTBI and previous active TB) vs. no TB; and 4. Previous active TB vs. LTBI.
Pathway analysis
Pathway and gene ontology (GO) enrichment analyses were conducted using the missMethyl package to identify biological pathways associated with significant DMRs. These included GO enrichment analysis30 using the gometh function and pathway analysis through the gsameth function, leveraging gene sets from the Kyoto Encyclopedia of Genes and Genomes (KEGG)31, Molecular Signatures Database (MSigDB)32 and PANTHER version 16 database33. We further performed gene‐set enrichment analysis of our 120-gene signature using the Enrichr web server (http://amp.pharm.mssm.edu/Enrichr; accessed June 1, 2025)34. Up-regulated genes (FDR < 0.05, |log2FC|> 1) in prior-TB versus TB-naïve samples were submitted and tested against both the DisGeNET library of curated gene–disease associations (v7.0) and the dbGaP library of GWAS-derived phenotype associations. For each term, Enrichr computes a Fisher’s exact test p-value, Benjamini–Hochberg–adjusted p-value (FDR), odds ratio, and combined score (–log10 p × z-score of deviation from expected rank). We considered terms with FDR < 0.05 as significant and report, for all hits, the gene overlap, odds ratio, adjusted p-value, and combined score.
All statistical analyses were carried out using R version 4.3.1.
Results
Characteristics of study participants of PWH with and without prior TB
Of 30 participants, 17 (57%) were female and the median (interquartile range, IQR) age was 46.5 (40.0–50.0) years. All participants were on ART and had achieved viral load suppression (viral load < 1000 copies/ml). Table 1 shows characteristics of the participants.
Chronological and epigenetic age of in PWH with and without prior TB
In the combined 30 PWH (mean chronological age 43.6 ± 9.3 years), PC-PhenoAge estimates averaged 62.95 ± 10.65 years, yielding a mean epigenetic age acceleration of 19.32 ± 10.82 years. Overall, epigenetic age exceeded chronological age by a highly significant margin (paired t(29) = 9.77, p < 0.001, 95% CI 15.28–23.36).
Among the 10 TB–naïve individuals the mean chronological age was 43.8 ± 8.5 years while among the 20 TB survivors it was 44.3 ± 8.8 years. PC-PhenoAge estimates averaged 61.2 ± 10.7 years in the TB–naïve group and 65.9 ± 9.2 years in the prior-TB group. Within the TB–naïve cohort, epigenetic age significantly exceeded chronological age by a mean of 17.42 ± 9.38 years (paired t(9) = 5.87, p < 0.001; 95% CI 10.71–24.14). Similarly, among participants with a prior TB infection, PC-PhenoAge exceeded true age by 21.60 ± 12.03 years (paired t(19) = 8.03, p < 0.001; 95% CI 15.97–27.23). The mean epigenetic‐age acceleration was 4.18 years greater in TB survivors compared with TB–naïve individuals (95% CI: –4.13 to 12.49). However, this difference did not reach statistical significance (Welch’s t(22.6) = 1.04, p = 0.307), and the effect size (Cohen’s d ≈ 0.37) indicates only a small‐to‐moderate group separation, suggesting that a larger sample may be required to confirm this trend.
The comparison of epigenetic and chronological age between prior TB infection (both LTBI and active) and those without TB is shown in Fig. 1. The results demonstrate an overall trend toward accelerated aging among PWH that was more pronounced in people with prior TB infection (both LTBI and active TB).

Epigenetic Age (DNAm) Compared to Chronological Age in Individuals with and without Prior Tuberculosis Infection. (A) Scatter plot of DNAm age (y-axis) versus chronological age (x-axis) estimated using using PCPhenoAge methylation clock. The blue dashed line represents perfect agreement between DNAm age and chronological age, while the red dashed line represents the regression line derived from the actual data. Most samples are above the blue line, indicating an overestimation of DNAm age. The bottom panel shows age acceleration values (DNAm age minus chronological age residuals), with positive values indicating accelerated aging (DNAm age > chronological age) and negative values indicating decelerated aging (DNAm age < chronological age). The results demonstrate an overall trend toward accelerated aging. (B) Box plot comparing DNAm age (Epigenetic_Age) and chronological age (Age) between individuals with previous TB infection and those without TB infection. Both groups show epigenetic ages that exceed their true age, but this over‐estimation is more pronounced in those with prior TB infection.
DNA methylation patterns in PWH with and without prior TB
A total of 7461 differentially methylated CpGs (dmCpGs) were identified in the comparison between prior active TB vs. no TB groups. These dmCpGs corresponded to 150 differentially methylated regions (DMRs) with p < 0.05, associated with the following top genes: GRAMD1C (hypomethylation), DPP6 (hypermethylation), ANKRD53 (hypomethylation), HDAC4 (hypomethylation), ADAMTS16 (hypomethylation), FAM208B (hypomethylation), BFSP2(hypomethylation), ZNF665 (hypermethylation), DOCK1 (hypermethylation), and PRDM16 (hypermethylation) (Supplementary Table 2 and Fig. 2: A1&A2).

Heatmaps A1, B1, and C1 derived from beta values of the differently methylated CpG sites identified in pairwise comparison across different groups with stringency criteria of adjusted P < 0.01 and mean methylation difference > 0.2. Dendrogram shows separation based on groups. Enhanced Volcano plots A2, B2 and C2 of the differentially methylated CpG‐sites separating different groups. The x‐axis is the log fold change and the y‐axis is the negative log10 of p‐value with the cut off p‐value of 0.05 shown with dash‐dotted horizontal line. Blue CpGs indicate hypomethylated while red CpGs are hypermethylated.
In the comparison between LTBI vs. no TB, 8598 dmCpGs were observed. These were associated with 39 DMRs at p < 0.05, including genes such as PLEKHG5 (hypermethylation), STK32C (hypermethylation), MCF2L (hypermethylation), PTPN6 (hypermethylation), SAMD11 (hypermethylation), AP001468.58 (hypomethylation), EPCAM (hypermethylation), NEURL4 (hypomethylation), SLC25A31 (hypomethylation), DNMT3A (hypomethylation), TTC7B (hypomethylation), FAM208B (hypermethylation), BRSK2 (hypermethylation), as well as the SPATC1L gene region with both hypo and hypermethylated probes (Supplementary Table 2 and Fig. 2: B1&2).
Overall, 71,774 dmCpGs were identified when comparing individuals with any history of TB infection (both active and latent TB) to the control group. These dmCpGs corresponded to 14 DMRs associated with genes such as PLEKHG5 (hypomethylation), KCNN3 (hypermethylation), SLC16A9 (hypomethylation), BRSK2 (hypermethylation), MCF2L (hypermethylation), NUP98 (hypomethylation) and hypermethylated SH3GL2. (Supplementary Table 2 and Fig. 2: C1&2). Meanwhile even though only two DMRs (PLEKHG5 and KCNN3) in this comparison were active TB vs No TB and prior latent TB vs No TB comparisons.
Enriched pathways associated with DNAm patterns in PWH with and without prior TB
Gene ontology, KEEG and human C2 GSEA
In the prior TB (active or latent) vs. no TB comparison, DNAm changes were enriched in pathways related to neurogenesis, neuron differentiation, axon guidance, neuroactive ligand signaling, morphine addiction, and nervous system development. Additional pathways related to platelet activation, vascular muscle contraction, chemokine signaling, and cytokine-cytokine receptor interaction. Further, pathways relating to cancer, proteoglycans in cancer, small cell lung cancer, prostate cancer, lung cancer, breast cancer, hepatocellular carcinoma, and thyroid cancer were enriched (Fig. 3A).

Pathway enrichment analysis for association of prior TB and DNA methylation. In each panel, the x-axis represents the p-value for pathway enrichment (adjusted for false discovery rate [FDR]), while the y-axis lists the enriched pathways. The size of the dots indicates the number of genes involved in each pathway, and the color gradient represents the FDR, with red indicating higher significance (lower FDR).
The other pairwise comparisons (prior active TB vs. no TB and prior LTBI vs. no TB comparisons) are shown in Fig. 3B and C but they did not meet statistical significance.
PANTHER
Analysis using the PANTHER database consistently showed enrichment in the Wnt signaling pathway, with 38 associated genes in the active TB vs no TB comparison and 41 genes in the latent TB vs. no TB comparison. Pathways involved in inflammation mediated by chemokine and cytokine signaling, integrin signaling, angiogenesis, Cadherin signaling pathway, CCKR signaling pathway and Nicotinic Acetylcholine Receptor signaling pathway were also enriched in all compared groups (Table 2). De novo pyrimidine deoxyribonucleotide biosynthesis, Pyrimidine Metabolism, 2-arachidonoylglycerol biosynthesis, Cholesterol biosynthesis, Gamma-aminobutyric acid synthesis, Aminobutyrate degradation and Histamine synthesis pathways were observed in only active TB vs no TB comparison. Fructose galactose metabolism, Heme biosynthesis, Pentose phosphate pathway, Pyridoxal-5-phosphate biosynthesis, S-adenosylmethionine biosynthesis, Leucine biosynthesis, Isoleucine biosynthesis, Alanine biosynthesis, Valine biosynthesis, Anandamide and degradation were also entirely specific to the latent TB vs. no TB comparison.
DisGeNET (disease associations)
Out of 274 queried terms, only lymphoid leukemia remained significant after multiple-testing correction (Benjamini–Hochberg FDR = 0.0119) for the prior TB vs. no prior TB dmDNA patterns. Specifically, 139 of 213 DisGeNET-annotated lymphoid leukemia genes overlapped with our list (overlap = 139/213), yielding an odds ratio of 1.96 and a combined score of 26.7.
dbGaP (GWAS phenotypes)
In contrast, enrichment against the dbGaP library identified seven phenotype associations at FDR < 0.05 (out of ~ 300 tested) for the prior-TB vs. no-prior-TB DNAm patterns. These were lymphocytes (overlap = 36/44; OR = 4.69; FDR = 0.0025; combined score = 55.4), tunica media (71/104; OR = 1.58; FDR = 0.0095; combined score = 17.4), asthma (72/107; OR = 2.10; FDR = 0.0096; combined score = 18.6), body mass index (253/437; OR = 1.22; FDR = 0.0096; combined score = 16.8), leukocyte count (31/40; OR = 3.14; FDR = 0.0149; combined score = 15.2), body height (198/294; OR = 1.31; FDR = 0.0231; combined score = 12.5), body composition (154/241; OR = 1.28; FDR = 0.0314; combined score = 11.2), and cell adhesion molecules (89/135; OR = 1.45; FDR = 0.0427; combined score = 9.8).
Discussion
In this study, we investigated DNAm patterns associated with prior TB infection in individuals with HIV. Our findings reveal that both prior TB infection (LTBI or active TB) are associated with distinct DNAm changes, particularly in pathways related to neural function and development, cardiovascular health, and cancer risk. In addition, there was significant epigenetic age acceleration in PWH, which was notably more pronounced in individuals with a history of TB infection (21.60 years of acceleration) compared to TB-naïve individuals (17.42 years of acceleration). This suggests that prior TB may exacerbate the already accelerated aging processes driven by HIV, potentially contributing to an earlier onset of age-related comorbidities. Similar to our findings, Bobak et al. (2022) demonstrated that active tuberculosis in humans was associated with approximately 12.7 years of cellular aging, as measured by epigenetic clocks (Horvath clock), and 14.38 years using gene expression-based cellular clocks23.
The analysis of DNAm patterns identified a substantial number of dmCpGs and DMRs. Specifically, 7461 dmCpGs (150 DMRs) were found when comparing prior active TB to no TB, and 8598 dmCpGs (39 DMRs) when comparing LTBI to no TB. The combined group of any prior TB showed 71,774 dmCpGs (14 DMRs) compared to no TB. These findings highlight that both forms of prior TB exposure leave lasting epigenetic marks. Genes implicated in these DMRs, such as GRAMD1C and DPP6 in active TB history, and PLEKHG5 and STK32C in LTBI history, warrant further investigation for their roles in TB-related sequelae. For instance, genes like KCNN3 and BRSK2 were identified in the overall prior TB group, pointing to common epigenetic alterations irrespective of TB history type.
Functional enrichment analyses of these DNAm changes provide insights into the potential biological consequences. Across individuals with any prior TB exposure, pathways related to neural functions such as neurogenesis, neuron differentiation, axon guidance, and neuroactive ligand signaling were significantly enriched. This aligns with reports of increased risk for neurological and cognitive disorders post-TB and suggests an epigenetic basis for these observations in PWH12,13,35,36. The consistent enrichment of the Wnt signaling pathway across different TB comparison groups, as identified through PANTHER database analysis, further supports a role for altered neurodevelopmental and cell signaling processes37.
Cardiovascular health also appears to be epigenetically impacted by prior TB. Pathways including platelet activation and vascular smooth muscle contraction were enriched in the combined prior TB group. The dbGaP analysis further identified an association between prior TB DNAm patterns and “tunica media”, a component of blood vessels, suggesting structural or functional vascular changes that could engender atherosclerosis in the setting of hyperlipemia38. These findings support epidemiological evidence linking TB to increased cardiovascular disease risk19. The enrichment in pathways related to cholesterol biosynthesis and body mass index suggests epigenetic changes underlying the effect that prior TB migt have on cardiovascular disease risk factors as we have previously reported in our study setting39. Our previous work among PWH with prior active TB suggests that prior TB is associated with normal body mass index and a lower prevalence of dyslipidemia39.
The study also uncovered significant DNAm alterations in cancer-related pathways. In individuals with any prior TB, enrichment was seen for pathways associated with small cell lung cancer, prostate cancer, lung cancer, breast cancer, hepatocellular carcinoma, thyroid cancer, and proteoglycans in cancer. Furthermore, DisGeNET analysis directly linked DNAm patterns in the prior TB group to lymphoid leukemia. These epigenetic changes may contribute to the increased cancer incidence observed in TB survivors particularly for lung, hepatobiliary, and leukemia15,40. Moreover, the prominent enrichment in the gene sets associated with embryonic stem cell identity: BENPORATH_ES_WITH_H3K27ME3, BENPORATH_EED_TARGETS, and BENPORATH_SUZ12_TARGETS suggests potential risk for the development of aggressive, poorly differentiated, and estrogen receptor-negative tumors41. The association between prior TB and advanced cancer types needs to be demonstrated in epidemiological studies.
The mechanisms underlying the observed DNAm changes and their specific contributions to long-term complications warrant further investigation by larger studies. Inflammation-related pathways, including chemokine and cytokine signaling and integrin signaling, were consistently enriched across comparisons, underscoring the role of persistent inflammation in driving these epigenetic modifications and subsequent long-term complications22. The association of prior TB DNAm with phenotypes such as asthma and altered leukocyte counts in the dbGaP analysis further supports ongoing immune dysregulation. Additionally, the interaction between TB infection, HIV infection, and antiretroviral therapy may further contribute to epigenetic changes and long-term complications.
Our findings of widespread and persistent DNA methylation changes in PWH with a history of prior TB complement and extend studies like DiNardo et al. (2020), which demonstrated DNA hypermethylation in critical immune pathways leading to dampened host immune responsiveness during active TB, with some changes persisting post-treatment42. While DiNardo et al. focused on hypermethylation as a mechanism for immediate immune evasion by M. tuberculosis, our study reveals a more complex, long-term epigenetic landscape in PWH following prior TB, characterized by both hyper- and hypomethylation across a broader range of pathways implicated in neural function, cardiovascular health, cancer, and accelerated aging. Thus, while active infection may induce specific hypermethylation to suppress acute immunity, our results suggest that prior TB exposure in PWH leads to a distinct and more diverse set of lasting epigenetic modifications that likely contribute to the increased risk of chronic comorbidities and accelerated biological aging observed in this population.
Our study has several limitations. The cross-sectional design limits our ability to establish causal relationships between prior TB infection and DNAm changes. The relatively small sample size may have limited our power to detect additional significant associations. Furthermore, our study focused on peripheral blood DNAm, which may not fully reflect epigenetic changes in other tissues relevant to long-term complications. The use of the IGRA to identify LTBI can misclassify PWH due to immune anergy. However, PWH in our study were mostly immune competent (median CD4 count of > 900 cells), making the risk of misclassification low.
Conclusion
In conclusion, prior TB infection in PWH is associated with extensive DNA methylation alterations and pronounced epigenetic age acceleration in pathways linked to neurocognitive health, cardiovascular function, and cancer susceptibility. These epigenetic signatures offer promising biomarkers for identifying PWH at highest risk of TB-related long-term comorbidities. Longitudinal studies are warranted to validate these methylation patterns as predictive markers and to explore targeted interventions—such as anti-inflammatory therapies or epigenetic modulators—to mitigate post-TB sequelae in this vulnerable population.
Data availability
Datasets used in this study are available from https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE304107.
Abbreviations
- ART:
-
Antiretroviral therapy
- CVD:
-
Cardiovascular disease
- DMR:
-
Differentially methylated region
- DNAm:
-
DNA methylation
- dmCpGs:
-
Differentially methylated CpG sites
- FDR:
-
False discovery rate
- HIV:
-
Human immunodeficiency virus
- KEGG:
-
Kyoto encyclopedia of genes and genomes
- LTBI:
-
Latent tuberculosis infection
- MsigDB:
-
Molecular signatures database
- NK:
-
Natural killer (cells)
- PANTHER:
-
Protein analysis through evolutionary relationships
- PWH:
-
People with HIV
- QFT:
-
QuantiFERON
- RMSD:
-
Root mean squared deviation
- TB:
-
Tuberculosis
References
World Health Organization. Global tuberculosis report 2024. World Health Organization (2024).
Shah, M. & Reed, C. Complications of tuberculosis. Curr. Opin. Infect. Dis. 27, 403–410 (2014).
Baluku, J.B., Namanda, B., Namiiro, S., Rwabwera, D.K., Mwesigwa, G., Namaara, C., et al. Death after cure: Mortality among pulmonary tuberculosis survivors in rural Uganda. Int. J. Infect. Dis. [Internet]. [cited 2024 Sep 16];144. Available from: https://www.ijidonline.com/article/S1201-9712(24)00140-1/fulltext (2024).
Romanowski, K. et al. Long-term all-cause mortality in people treated for tuberculosis: A systematic review and meta-analysis. Lancet Infect. Dis. 19, 1129–1137 (2019).
Blöndal, K., Rahu, K., Altraja, A., Viiklepp, P. & Rahu, M. Overall and cause-specific mortality among patients with tuberculosis and multidrug-resistant tuberculosis. Int. J. Tuberc. Lung Dis. 17, 961–968 (2013).
Chakaya, J., Kirenga, B., Getahun, H. Long term complications after completion of pulmonary tuberculosis treatment: A quest for a public health approach (2016).
Visca, D., Centis, R., Munoz-Torrico, M. & Pontali, E. Post-tuberculosis sequelae: The need to look beyond treatment outcome. Int. J. Tuberc. Lung Dis. 24, 761–762 (2020).
Sheu, J.-J., Chiou, H.-Y., Kang, J.-H., Chen, Y.-H. & Lin, H.-C. Tuberculosis and the risk of ischemic stroke: A 3-year follow-up study. Stroke 41, 244–249 (2010).
Chung, W.-S. et al. Tuberculosis increases the subsequent risk of acute coronary syndrome: A nationwide population-based cohort study. Int. J. Tuberc. Lung Dis. 18, 79–83 (2014).
Huaman, M. A. et al. Tuberculosis and risk of acute myocardial infarction: A propensity score-matched analysis. Epidemiol. Infect. 145, 1363–1367 (2017).
Shen, T.-C. et al. The risk of chronic kidney disease in tuberculosis: A population-based cohort study. QJM: Int. J. Med. 108, 397–403 (2015).
Peng, Y.-H. et al. Increased risk of dementia among patients with pulmonary tuberculosis: A retrospective population-based cohort study. Am. J. Alzheimers Dis. Other Demen. 30, 629–634 (2015).
Shen, C.-H., Chou, C.-H., Liu, F.-C., Lin, T.-Y., Huang, W.-Y., Wang, Y.-C., et al. Association between tuberculosis and Parkinson disease. Medicine (Baltimore) [Internet]. [cited 2019 Sep 9];95. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4779022/ (2016).
Sweetland, A. C. et al. Addressing the tuberculosis–depression syndemic to end the tuberculosis epidemic. Int. J. Tuberc. Lung Dis. 21, 852–861 (2017).
Leung, C. Y. et al. Cancer incidence attributable to tuberculosis in 2015: Global, regional, and national estimates. BMC Cancer 20, 1–13 (2020).
Su, V.Y.-F. et al. Latent tuberculosis infection and the risk of subsequent cancer. Medicine (Baltimore) 95, 2352 (2016).
Hossain, M. B. et al. Role of latent tuberculosis infection on elevated risk of cardiovascular disease: A population-based cohort study of immigrants in British Columbia, Canada, 1985–2019. Epidemiol. Infect. 151, e68 (2023).
Wong, Y.J., Noordin, N.M., Keshavjee, S., Lee, S.W.H. Impact of latent tuberculosis infection on health and wellbeing: a systematic review and meta-analysis. European Respiratory Review [Internet]. [cited 2022 Aug 25];30. Available from: https://err.ersjournals.com/content/30/159/200260 (2021).
Yang, J. et al. Tuberculosis survivors and the risk of cardiovascular disease: Analysis using a nationwide survey in Korea. Front. Cardiovasc. Med. 11, 1364337 (2024).
Huaman, M.A., Henson, D., Ticona, E., Sterling, T.R., Garvy, B.A. Tuberculosis and cardiovascular disease: Linking the epidemics. Trop. Dis. Travel Med. Vaccines [Internet]. [cited 2021 Jan 3];1. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4729377/ (2015).
Malik, A. A., Sheikh, J. A., Ehtesham, N. Z., Hira, S. & Hasnain, S. E. Can Mycobacterium tuberculosis infection lead to cancer? Call for a paradigm shift in understanding TB and cancer. Int. J. Med. Microbiol. 312, 151558 (2022).
Qin, Y. et al. Important role of DNA methylation hints at significant potential in tuberculosis. Arch. Microbiol. 206, 177 (2024).
Bobak, C. A. et al. Increased DNA methylation, cellular senescence and premature epigenetic aging in guinea pigs and humans with tuberculosis. Aging 14, 2174–2193 (2022).
Rodés, B., Cadiñanos, J., Esteban-Cantos, A., Rodríguez-Centeno, J. & Arribas, J. R. Ageing with HIV: Challenges and biomarkers. EBioMedicine 77, 103896 (2022).
Baluku, J. B. et al. Mycobacterium tuberculosis infection and cytogenetic abnormalities among people with HIV. Mutat. Res./Genet. Toxicol. Environ. Mutagen. 888, 503640 (2023).
Qiagen. QuantiFERON-TB Gold Plus (QFT-Plus) ELISA package insert. (2017).
Musaazi, J. et al. Increased uptake of tuberculosis preventive therapy (TPT) among people living with HIV following the 100-days accelerated campaign: A retrospective review of routinely collected data at six urban public health facilities in Uganda. PLoS ONE 18, e0268935 (2023).
Aryee, M. J. et al. Minfi: A flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics 30, 1363–1369 (2014).
Levine, M. E. et al. An epigenetic biomarker of aging for lifespan and healthspan. Aging (Albany NY) 10, 573–591 (2018).
The Gene Ontology Consortium. The gene ontology resource: 20 years and still going strong. Nucleic Acids Res. 47, D330–D338 (2019).
Kanehisa, M. & Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30 (2000).
Liberzon, A. et al. Molecular signatures database (MSigDB) 3.0. Bioinformatics 27, 1739–1740 (2011).
Mi, H. et al. PANTHER version 16: A revised family classification, tree-based classification tool, enhancer regions and extensive API. Nucleic Acids Res. 49, D394-403 (2021).
Kuleshov, M. V. et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 44, W90–W97 (2016).
Nightingale, R. et al. Post-TB health and wellbeing. Int. J. Tuberc. Lung Dis. 27, 248–283 (2023).
Hestad, K. A. et al. Cognitive impairment in Zambians with HIV infection and pulmonary tuberculosis. J. Acquir. Immune Defic. Syndr. 80, 110 (2019).
Freese, J. L., Pino, D. & Pleasure, S. J. Wnt signaling in development and disease. Neurobiol. Dis. 38, 148–153 (2010).
Belhoul-Fakir, H. et al. Injury to the tunica media initiates atherogenesis in the presence of hyperlipidemia. Front. Cardiovasc. Med. https://doi.org/10.3389/fcvm.2023.1152124/full (2023).
Baluku, J. B. et al. Association of prior tuberculosis with altered cardiometabolic profiles of people with HIV: A comparative cross-sectional study in Uganda. J. Clin. Tuberc. Other Mycobact. Dis. 39, 100523 (2025).
Luczynski, P., Poulin, P., Romanowski, K. & Johnston, J. C. Tuberculosis and risk of cancer: A systematic review and meta-analysis. PLoS ONE 17, e0278661 (2022).
Ben-Porath, I. et al. An embryonic stem cell-like gene expression signature in poorly differentiated aggressive human tumors. Nat. Genet. 40, 499 (2008).
DiNardo, A. R. et al. DNA hypermethylation during tuberculosis dampens host immune responsiveness. J. Clin. Investig. 130, 3113–3123 (2020).
Acknowledgements
None
Funding
This project was supported by funding from the National Cancer Institute through the Case Comprehensive Cancer Center (Grant number U54CS254566). The funding source had no role in the study design; in the collection, analysis and interpretation of data; in the writing of the report; and in the decision to submit the article for publication.
Author information
Authors and Affiliations
Contributions
JBB—conceptualization, methodology, investigation, data accrual, formal analysis, interpretation of results, drafting manuscript, editing manuscript, final approval. SN—investigation, data accrual, interpretation of results, drafting manuscript, editing manuscript, final approval. DK—investigation, data accrual, interpretation of results, drafting manuscript, editing manuscript, final approval. BN—investigation, data accrual, interpretation of results, drafting manuscript, editing manuscript, final approval. HK—methodology, investigation, data accrual, formal analysis, interpretation of results, drafting manuscript, editing manuscript, final approval. IJ—interpretation of results, drafting manuscript, editing manuscript, final approval. WG—methodology, investigation, formal analysis, interpretation of results, drafting manuscript, editing manuscript, final approval. NN—investigation, interpretation of results, drafting manuscript, editing manuscript, final approval. NB—investigation, interpretation of results, drafting manuscript, editing manuscript, final approval. EW—investigation, data accrual, interpretation of results, drafting manuscript, editing manuscript, final approval. JR—investigation, interpretation of results, drafting manuscript, editing manuscript, final approval. NJ—investigation, interpretation of results, drafting manuscript, editing manuscript, final approval. CK—investigation, interpretation of results, drafting manuscript, editing manuscript, final approval. MS—investigation, interpretation of results, drafting manuscript, editing manuscript, final approval. AC—investigation, interpretation of results, drafting manuscript, editing manuscript, final approval. IN—methodology, investigation, data accrual, formal analysis, interpretation of results, drafting manuscript, editing manuscript, final approval. SM—investigation, interpretation of results, drafting manuscript, editing manuscript, final approval. SG—investigation, interpretation of results, drafting manuscript, editing manuscript, final approval. BK—investigation, dinterpretation of results, drafting manuscript, editing manuscript, final approval.
Corresponding author
Ethics declarations
Competing interests
The authors declare that they have no relevant competing interests.
Consent for publication
Not applicable.
Ethical approval
Study participants provided written informed consent before study measurements were undertaken. The study protocol was approved by the Mildmay Uganda Research and Ethics Committee (#REC REF MUREC-107-2022). Further, the Uganda National Council of Science and Technology provided additional approval as required by the guidelines for conducting research in Uganda (HS2328ES). All methods were performed in accordance with the relevant guidelines and regulations.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Baluku, J.B., Namiiro, S., Zawedde, D.K. et al. DNA methylation patterns associated with prior tuberculosis infection in people with HIV. Sci Rep 15, 30349 (2025). https://doi.org/10.1038/s41598-025-15532-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-15532-5
