
Abstract
Lung squamous cell carcinoma (LUSC) is one of the most common types of non-small cell lung cancer with poor prognosis. Druggable genome-wide Mendelian randomization (MR) was conducted to discover LUSC-related targets using expression quantitative trait loci (eQTL) and protein QTL (pQTL) in the ieu_b_4953 dataset and finngen dataset. Bayesian co-localization analysis, summary‑data‑based MR (SMR) analysis, and HEIDI test were conducted to verify the causal associations between genes and LUSC risk. Prediction of prognosis and immune infiltration was performed at the transcriptomic level, and expression patterns of genes were analyzed at the single-cell level. We identified DNMT1, ACSS2, YBX1, SELENOS, PPARA, MST1, CPA4, and MPO as LUSC-related genes based on MR analysis. Although Bayesian co-localization analysis showed negative co-localization results (PPH3 + PPH4 < 0.8), positive SMR (p < 0.05) and HEIDI (p > 0.05) were found in the ieu_b_4953 dataset and finngen dataset. Blood CPA4 and DNMT1 might be protective factors for bladder cancer and allergic rhinitis, respectively. Patients with low expression of CPA4, DNMT1 and YBX1 had better prognosis, while patients with high expression of MST1 had better prognosis. Tumor samples exhibited reduced infiltration of CD4 cells, CD8 cells, activated dendritic cells, eosinophils, myeloid-derived suppressor cells (MDSCs), macrophages, and mast cells. At the single-cell level, DNMT1, SELENOS and YBX1 were highly expressed in endothelial cells, epithelial cells, fibroblasts, hepatocytes, mast cells, and monocytes/macrophages. MST1 was overexpressed in hepatocytes. This study might deepen the understanding of the LUSC pathogenesis and identify potential targets for the management of LUSC.
Similar content being viewed by others
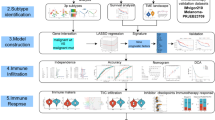

Introduction
Lung cancer accounts for 11.4% of global cancer incidence and 18% of cancer-related mortality1. In the USA, it is estimated that there will be 236,740 new cases of lung cancer and 130,180 deaths from the disease in 20222. Small cell lung cancer (SCLC) and non-small cell lung cancer (NSCLC) are two main types of lung cancer. NSCLC mainly includes lung adenocarcinoma and lung squamous cell carcinoma (LUSC), and LUSC is a common subtype of NSCLC, representing approximately 30% of all cases3. The prognosis and treatment decisions for LUSC patients are primarily based on the American Joint Commission on Cancer staging system4. Currently, the commonly used treatments for LUSC include surgery, chemotherapy, radiotherapy and targeted therapies5. However, due to the lack of clear driver mutations and poor response to targeted therapies, the overall survival rate for LUSC patients is about 30% shorter than for other NSCLC subtypes6,7. Therefore, it is necessary to develop potential targets for LUSC management.
The druggable genome is a subset of the human genome and it contains approximately 4500 genes. The druggable genome expresses proteins that bind with drug-like molecules8. The target-disease associations based on druggable genome may facilitate developing and validating drug targets for human disease9. Mendelian randomization (MR) is an impartial approach revealing causality that relies on genetic variation as instrumental variables (IVs) to assess whether an exposure leads to the corresponding outcome. MR analysis avoids confounding factors and reverse causality, and has stronger evidence than retrospective studies. Recently, MR analysis using the druggable genome has been applied to reveal genetically supported drug targets for human diseases such as nervous system disease, pulmonary disease, cardiovascular disease, and cancers10,11,12,13. Although HYKK (AGPHD1) has been identified as an increased risk of lung cancer using eQTL10, its predictive role in prognosis of LUSC as well as the association between HYKK and immune infiltration remains unclear. Therefore, the novel targets still require further development.
In this study, we conducted a systematic Mendelian randomization (MR) analysis to evaluate the causal effects of blood expression quantitative trait loci (eQTL) and protein quantitative trait loci (pQTL) on LUSC. Co-localization analysis and summary‑data‑based MR (SMR) with the heterogeneity in dependent instruments (HEIDI) test were conducted to verify the pleotropic associations between gene expression levels and LUSC. Repeated studies using different LUSC GWAS data sources were carried out to validate the identified genes. Subsequently, we assessed the causal relationship between identified genes and LUSC risk factors to explore the possible mechanisms by which these genes involved in the pathogenesis of LUSC. Next, we evaluated the potential adverse effects of the targets on LUSC treatment using a phenotype-wide MR analysis. Also, the associations between identified genes and immune infiltration were assessed to reveal the relationship between LUSC-related genes and tumor immune microenvironment at the transcriptomic level and single-cell level.
Results
Identifying druggable genes
Based on the data available in DGIdb v4.2.0, we identified 3952 genes as potential druggable genes (Supplementary Table 2). Additionally, we extracted 4479 druggable genes from the article by Finan et al. (Supplementary Table 3). Finally, 5859 unique druggable genes with Human Genome Organization (HUGO) names (Supplementary Table 4) were obtained from the aforementioned sources for further analysis.
Screening five LUSC-related genes using cis-eQTL
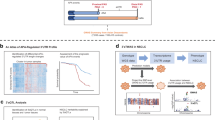
After LD analysis using blood eQTL, we identified 3724 genes for MR analysis. Based on the Wald ratio or IVW method, we identified 176 LUSC-related genes in the discovery cohort (p < 0.05, Supplementary Table 5 and Supplementary Fig. 1A), of which 95 genes were considered as risk factors for LUSC (OR > 1), and 81 genes were considered as protective factors for LUSC (OR < 1) (Fig. 1A,B). In the replication cohort, 216 LUSC-related genes were identified (p < 0.05, Supplementary Table 6 and Supplementary Fig. 1B). Among them, 103 genes were considered as protective factors for LUSC (OR < 1), while 113 genes were identified as risk factors for LUSC (OR > 1) (Fig. 1A,B). At the intersection between the discovery and replication cohorts, we found five LUSC-related genes including DNMT1, ACSS2, YBX1, SELENOS, and PPARA (Fig. 1C,D).

Screening of LUSC-related genes using cis-eQTL. (A) Three genes are identified as risk factors (OR > 1) at the intersection of the discovery cohort (ieu_b_4953 dataset) and replication cohort (finngen_R10_c3_HEPATOCELLU-CARC-EXALLC dataset). (B) Two genes are identified as protective factors (OR < 1) at the intersection of the discovery cohort (ieu_b_4953 dataset) and replication cohort (finngen_R10_c3_HEPATOCELLU-CARC-EXALLC dataset). (C) The MR analysis results of five intersecting genes in the discovery cohort. (D) The MR analysis results of five intersecting genes in the replication cohort.
Screening three LUSC-related genes using cis-pQTL
After LD analysis using pQTL, we identified 951 genes for MR analysis. A total of 47 LUSC-related genes were found in the discovery cohort (p < 0.05, Supplementary Table 7 and Supplementary Fig. 1C), with 19 as risk factors (OR > 1) and 28 as protective factors for LUSC (OR < 1) (Fig. 2A,B). In the replication cohort, 45 LUSC-related genes were identified (p < 0.05, Supplementary Table 8 and Supplementary Fig. 1D), containing 24 as risk factors for LUSC (OR > 1) and 21 as protective factors (OR < 1) (Fig. 2A,B). Next, we found MST1, CPA4, and MPO by intersecting the results of the discovery and replication cohorts (Fig. 2C,D).

Screening of LUSC-related genes using cis-pQTL. (A) Two genes are identified as risk factors (OR > 1) at the intersection of the discovery cohort (ieu_b_4953 dataset) and replication cohort (finngen_R10_c3_HEPATOCELLU-CARC-EXALLC dataset). (B) One gene is identified as protective factors (OR < 1) at the intersection of the discovery cohort (ieu_b_4953 dataset) and replication cohort (finngen_R10_c3_HEPATOCELLU-CARC-EXALLC dataset). (C) The MR analysis results of three intersecting genes in the discovery cohort. (D) The MR analysis results of three intersecting genes in the replication cohort.
Co-localization of LUSC-related genes
Subsequently, we conducted co-localization analysis using QTL data within 1 MB upstream and downstream of the chromosomal positions of eight genes, to assess whether identified genes and LUSC risk were consistent with a shared causal variant. The results were consistent for all genes in both the discovery and replication cohorts. The results showed that PPH3 + PPH4 < 0.8, indicating that there was not sufficient evidence to support the shared loci between these genes and LUSC (Table 1).
Summary‑data‑based MR (SMR) analysis and HEIDI test
The SMR analysis and HEIDI test provided further assessment for the causal associations between eight genes and LUSC risk. The p _SMR values for all genes passed the SMR test (p < 0.05), and all passed the HEIDI test (p > 0.05) both in the discovery cohort and the replication cohort (Table 2), which indicated that the genes associated with LUSC risk might be driven by a shared genetic variant.
Association between genes and risk factors for LUSC
Next, MR analysis was conducted to explore the associations between QTL data of eight genes and LUSC risk factors (Supplementary Table 9). Smoking was associated with several genes, such as DNMT1 (p = 0.035), ACSS2 (p = 0.006), PPARA (p = 0.041), and YBX1 (p = 0.024). Phthisis was notably associated with YBX1 (p = 0.032) and CPA4 (p = 0.031). Pulmonary fibrosis was significantly associated with SELENOS (p = 0.022), ACSS2 (p = 0.007), and PPARA (p = 0.042). Alcohol abuse was associated with YBX1 (p = 0.001) and MST1 (p = 0.037) (Fig. 3).

The associations between genes and risk factors for LUSC.
Phenome-wide association study (PheWAS)
Given that most drugs act through the bloodstream, we evaluated whether the expression of LUSC-related eight genes had beneficial or harmful effects on other indicators. Therefore, we conducted a comprehensive MR analysis screening 783 non-LUSC diseases or traits in the UK Biobank. Based on the IVW method, higher blood levels of CPA4 might serving as a protective factor for bladder cancer (Supplementary Fig. 2A). Higher blood levels of DNMT1 might be a protective factor for allergic rhinitis (Supplementary Fig. 2B).
Analyzing LUSC-related genes at the transcriptomic level
The results of the Wilcoxon test showed that the expression levels of DNMT1, CPA4, ACSS2, SELENOS, and YBX1 were significantly higher in the LUSC samples than that of the controls, while the expression of MST1 was significantly lower in the LUSC sample than that of the controls (Fig. 4A). SNV analysis indicated single nucleotide mutations in MPO, DNMT1, PPARA, MST1, and CPA4 in LUSC, with most of them being missense mutations (Fig. 4B). CNV analysis revealed that YBX1 and DNMT1 had extensive copy number deletions, while MPO had extensive copy number amplification in LUSC (Fig. 4C,D). Functional enrichment analysis was conducted to reveal the potential biological pathways for LUSC-related genes. The GO enrichment analysis indicated that these genes were mainly involved in biological processes such as lipoprotein metabolism, hexose biosynthesis, and glucose metabolism, with abundant cellular components including low-density lipoprotein particle and messenger ribonucleoprotein complex, as well as molecular functions including ubiquitin-coupled enzyme binding and antioxidant activity (Supplementary Fig. 3A–C). The results of KEGG enrichment analysis indicated that these genes were primarily enriched in glycolysis/gluconeogenesis and adipocytokine signaling pathways (Supplementary Fig. 3D).

Expression and mutation analysis of LUSC-related genes in TCGA dataset. (A) Expression of eight genes in LUSC and adjacent cancer samples. (B) Single nucleotide variants of eight genes. (C) Copy number variants of eight genes. (D) The positions of eight genes on chromosomes.
The Kaplan–Meier analysis showed that LUSC patients with low expression levels of CPA4 (p = 0.037), DNMT1 (p = 0.003), SELENOS (p = 0.035) and YBX1 (p < 0.001) had significant better survival probability than those with high levels, while patients with high expression levels of MST1 (p = 0.010) had better survival probability compared to those with low levels (Fig. 5A). The results from the protein–protein interaction (PPI) network indicated that these LUSC-related genes exhibited strong co-expression trends with ACOT1, ACOT2, and ENTPD5, suggesting that these genes might play a regulatory role in the occurrence and development of LUSC through synergistic effects (Fig. 5B). As shown in Fig. 5C, PPARA and DNMT1 had numerous clinical drugs, such as AZACITIDINE and DECITABINE, etc., suggesting that these drugs might be potential targets for LUSC management.

The Kaplan–Meier analysis of LUSC-related genes and PPI network analyses. (A) Kaplan–Meier analysis of eight genes in TCGA dataset. (B) Twenty genes that interact with eight LUSC-related genes. (C) Drugs in the DGIdb database that have regulatory effects on LUSC-related genes.
In the LUSC group, the infiltration scores of most immune cells were significantly lower than those in the control group, such as CD4 cells, CD8 cells, activated dendritic cells, eosinophils, myeloid-derived suppressor cells (MDSCs), macrophages, mast cells, and T cells (Fig. 6A). ESTIMATE algorithm also confirmed that the immune scores (StromalScore, ESTIMATEScore, and ImmuneScore) were lower in LUSC samples than that of the adjacent samples, while the tumor purity was higher in LUSC samples (Fig. 6B). The correlation analysis found that PPARA, MST1, and ACSS2 were negatively correlated with the infiltration of most immune cells, while DNMT1, CPA4, and YBX1 were significantly positively correlated with immune cell infiltration (Fig. 6C).

The relationship between eight LUSC-related genes and immune cell infiltration in TCGA dataset. (A) Differences in immune cell infiltration between LUSC and adjacent tissue samples. (B) ESTIMATE algorithm in LUSC and adjacent tissue samples. (C) The correlation between the expression of eight genes and immune cell infiltration in LUSC samples.
Analysis of LUSC-related genes at the single-cell level
The Vlnplots displayed the number of genes (nFeature), the total count (nCount), and the percentage of mitochondrial genes (percent.mt) in each cell before and after quality control (Supplementary Fig. 4A,B). After quality control, 33,694 genes were obtained from 20,895 cells. The correlation analysis showed that nCount was significantly positively correlated with nFeature (Supplementary Fig. 4C). Subsequently, 2000 highly variable genes were plotted in a scatter plot (Supplementary Fig. 4D). Principal components analysis (PCA) identified 20 principal components (PCs), and the top 20 PCs were selected for subsequent analysis (Supplementary Fig. 4E). Based on tSNE and cell type annotations, all cells were divided into 16,012 immune cells and 4,883 non-immune cells. The immune group consisted of B lymphocytes, mast cells, monocytes/macrophages, and T lymphocytes, while the non-immune group included endothelial cells, epithelial cells, hepatocytes, and fibroblasts (Fig. 7A–C). Cell markers used for annotation were shown in (Fig. 7D). Figure 7E displayed that T lymphocytes, monocytes/macrophages, epithelial cells, and hepatocyte accounted for the large number and proportion of various immune cells in each patient. Figure 7F displayed differentially expressed genes (DEGs) for each cell type. Furthermore, we examined the expression of genes in different cell types (Fig. 7G), and found DNMT1, SELENOS, and YBX1 were highly expressed in endothelial cells, epithelial cells, fibroblasts, hepatocytes, mast cells, and monocytes/macrophages. MST1 was specifically overexpressed in hepatocytes in the LUSC group.

The scRNA-seq data annotation. (A) t-SNE plots of the sample. (B) Clustering and grouping of cells. (C) Annotation results of cells. (D) Cell markers for annotation. (E) The number and proportion of each cell type in each patient. (F) Volcano plots of DEGs in different cells. (G) The expression of eight genes in different cell types.
Discussion
Integrating MR of GWAS and eQTL or pQTL data reveals genetic factors related to complex diseases, and may identify the genetic genes associated with the occurrence, development and treatment of diseases. In the present study, we identified DNMT1, ACSS2, YBX1, SELENOS, PPARA, MST1, CPA4, and MPO associated with LUSC through systematic druggable genome-wide MR analysis. Among them, high expression of genes such as DNMT1, YBX1, and CPA4 in LUSC was associated with poor prognosis, while high expression of the MST1 was associated with favorable prognosis. These findings may offer novel perspectives on potential targets and prognostic predictions for LUSC via genetic prediction.
Bayesian co-localization analysis showed that there were no shared genetic variations between LUSC-related genes and LUSC. Sun has found a negative co-localization result (PPH4 < 0.75), and further enrichment analysis shows that the identified proteins are mainly enriched in several pathways, which suggests that the association between exposure and outcome is more likely to be driven by complex causal pathway14. It is necessary to verify the negative co-localization result using a more robust method. Sun and colleagues have performed Bayesian co-localization to determine whether protein and colorectal cancer risk are consistent with a shared causal variant. SMR analysis and HEIDI test are further conducted to verify the robustness of co-localization, and the HEIDI test (p > 0.05) indicates that the association of protein and colorectal cancer is not driven by LD15. Liu has identified dozens of DNA methylation sites and genes having pleiotropic associations with Alzheimer’s dementia based on SMR and HEIDI (p > 0.01)16. Only application of SMR and HEIDI also provides strong evidence supporting the pleiotropic association of exposure and outcome. In this study, after Bayesian co-localization analysis, SMR was conducted to verify the pleotropic associations between gene expression levels and LUSC. HEIDI test was used to distinguish genes associated with LUSC risk owing to a shared genetic variant rather than genetic linkage using multiple SNPs in a region. Hence, the integration of SMR and HEIDI results provided evidence supporting that the identified genes and LUSC risk were consistent with a shared causal variant.
Transcriptomic analysis revealed the characteristics of these genes in LUSC. SNV analysis indicated single nucleotide variants in MPO, DNMT1, PPARA, MST1, and CPA4 in LUSC, with most of them being missense mutations. CNV analysis revealed that YBX1 and DNMT1 had extensive copy number deletions, while MPO had extensive copy number amplification. The previous study has found genetic alterations of DNA methylation regulators in lung adenocarcinoma, with significant gene mutation relationships between DNMT1 and ZBTB4, MBD4, NSUN2, and UHRF117, suggesting the role of DNMT1 mutations in tumor microenvironment of lung adenocarcinoma. CPA4 belongs to the carboxypeptidase A/B subfamily and catalyzes the release of carboxy-terminal amino acids. It has been reported that CPA4 is implicated in the process of tumor microenvironment formation and distant metastasis in cancer. Wang and colleagues have demonstrated that CPA4 negatively regulates HGS-ETR1/2-induced pyroptosis through forming a positive feedback loop with the AKT pathway18, which reveals the important role of CPA4 in tumor immune response. YBX1 encodes a highly conserved cold shock domain protein that has broad nucleic acid binding properties. The abnormal YBX1 relates to cancer proliferation, and it may serve as a prognostic marker for poor outcome and drug resistance in certain cancers19,20. Additionally, previous study has found the deficiency of MST1 is associated with primary T-cell immunodeficiencies21, and this finding may emphasize the important role of MST1 in T-cell derived immunity. These findings from SNV analysis and CNV analysis suggest that genetic variation of these genes may be involved in the tumor microenvironment of LUSC.
Furthermore, we found some immune cells including CD4 cells, CD8 cells, activated dendritic cells, eosinophils, MDSCs, macrophages, mast cells, and T cells were decreased in LUSC. The tumor microenvironment is an entity in which tumor cells, inflammation and immune cells interact and evolve over time. Eosinophils are components of tumor microenvironment, and eosinophils play pivotal immunological role in innate immune and adaptive immune responses. Notably, eosinophils may exert pro-tumorigenic effects and anti-tumor effects22. The role of eosinophils is influenced by surrounding stimuli such as cytokines and exosomes, and different tumor microenvironment to a great extent22. Besides, the abnormal expression of chemokine (C–C motif) ligand 4 (CCL4) may affect the survival of CD8 + T cells in NSCLC23. Hence, the reduced CD8 + T cells may attribute to surrounding chemokine or chemokine ligand. Also, Leveque E and colleagues have demonstrated the heterogeneity of mast cells in NSCLC, where CD103 + tumor-associated mast cells are likely to interact with CD4 + T cells, and are closer to cancer cells than CD103- mast cells24. Dendritic cells are important immune cells in the tumor microenvironment and exert anti-tumor effects, whereas dendritic cells expressing PD-L1 may make itself keeping immature status, therefore inducing immune escape of tumor cells and progression of NSCLC25. The function of dendritic cells is inhibited in lung cancer patients, due to the inhibitory effects of lung tumors employing numerous strategies on dendritic cells-based immunity26. Collectively, infiltration of immune cells can be influenced by various factors, which may explain the lower expression levels of immune cells in LUSC in this study. Additionally, the characteristics of immune cell infiltration in LUSC may provide directions for immunotherapy in LUSC patients. Meanwhile, scRNA-seq analysis highlighted that DNMT1, SELENOS, and YBX1 were highly expressed in endothelial cells, epithelial cells, fibroblasts, hepatocytes, mast cells, and monocytes/macrophages, with the specific high expression of MST1 in hepatocytes. The results emphasized the relationship between LUSC-related genes and specific cell types in LUSC.
This study systematically identified multiple potential targets associated with LUSC through systematic druggable-wide MR analysis. Gene-drug interaction network analysis showed that PPARA and DNMT1 had multiple potential clinical drugs, such as AZACITIDINE and DECITABINE, suggesting that these drugs might serve as potential options for LUSC management. These findings not only provide new insights for the management of LUSC but also lay an important foundation for future research. However, further functional experiments and clinical studies are necessary to verify the actual efficacy and safety of these targets. With advances in technology and accumulation of data, future research can refine and expand this analytical method to discover more potential targets and treatment strategies.
Limitations should be noticed in this study. The blood cis-eQTL and cis-pQTL, as well as the Summary GWAS data were from European, and this might induce data overlap and potential bias. Hence, further analyses should be performed to validate the results using datasets containing samples from other countries. Besides, this study was an exploratory work and multiple testing correction was not performed. Although LUSC-related genes have been identified in this study, the expression patterns of these genes, and their relationships with prognosis, mutational characteristics and immune landscape need further replication using experimental studies and clinical trials, with multiple testing correction. Additionally, we did not conduct reverse MR analysis of cis-eQTL and cis-pQTL. Cis-eQTL and cis-pQTL studies center on identifying genetic variants (SNPs) located in specific chromosomal regions near the target gene/protein-coding genes (usually within ~ 1 Mb upstream and downstream of the gene), and these variants have been shown to be significantly associated with the mRNA expression level or protein abundance of that particular gene. However, risk variants identified by GWAS in LUSC are typically dispersed throughout the genome, and there is a high probability that there is no strong association (i.e., no significant cis effect) between them and the expression of specific genes or the abundance of specific proteins within the narrow-localized regions defined by cis-eQTL/cis-pQTL studies. This might be the reason why reverse MR analysis was not performed in similar articles on drug-targeted MR analysis11,27,28.
Conclusion
This study identified eight LUSC-related genes based on systematic druggable-wide MR analysis, and provided evidence supporting the associations between these genes with LUSC prognosis, immune cell infiltration and potential drug interactions. The findings provide novel ideas for the potential mechanisms of LUSC pathogenesis and new directions for the management of LUSC.
Methods
eQTL and pQTL datasets
The peripheral blood eQTL dataset was obtained from the eQTLGen (https://www.eqtlgen.org/phase1.html). Among them, 16989 cis-eQTL of genes were obtained from 31684 blood samples of healthy individuals of European ancestry29. The peripheral blood pQTL dataset was obtained from the GWAS catalog (https://www.ebi.ac.uk/gwas). The cis-pQTL of 4782 proteins was obtained from 5368 blood samples of healthy individuals of European ancestry. We obtained completely significant cis-eQTL (FDR < 0.05) and cis-pQTL (p < 5e-08), as well as allele frequency information, as shown in (Supplementary Table 1).
Acquisition of GWAS summary data
The LUSC GWAS summary statistical data was downloaded from the IEU database (https://gwas.mrcieu.ac.uk/) and the finngen database (https://r10.finngen.fi/), namely ieu_b_4953 and finngen_R10_c3_HEPATOCELLU-CARC-EXALLC. The ieu_b_4953 dataset contained 168 LUSC patients and 372,016 control samples, while the finngen_R10_c3_HEPATOCELLU-CARC-EXALLC dataset included 500 LUSC patients and 314,193 control samples. In this study, the ieu_b_4953 dataset was used as the discovery cohort to find potential LUSC-related genes. The finngen_R10_c3_HEPATOCELLU-CARC-EXALLC dataset served as the replication cohort, to find LUSC-related genes and then to determine the genes that coexisted with the discovery cohort.
Acquisition of druggable genes
The druggable genes were downloaded from DGIdb (https://www.dgidb.org/) and from a recent review of the “drugability” of genes. DGIdb is a drug-gene interaction database for precision medicine and drug discovery30. DGIdb provides information on drug gene interactions and prodrug genes through publications, databases, and other online sources. We downloaded the “category data”, which included all genes in the druggable gene categories in DGIdb, from all sources mapped to Entrez genes.
We also obtained a list of druggable genes reported by Finan et al.9. Finan divided the druggable gene set into three tiers: 1) tier 1 incorporated the targets of approved drugs and drugs in clinical development, and proteins that are targets of approved small molecule and biotherapeutic drugs were identified using manually curated efficacy target information from release 17 of the ChEMBL database. 2) tier 2 incorporated proteins closely related to drug targets or with associated drug-like compounds. 3) tier 3 incorporated extracellular proteins and members of key drug-target families.
Criteria for inclusion/exclusion
Eligible cases were histologically confirmed cases of LUSC between 30 and 85 years of age and residents in the geographical area of each participating hospital. Controls were selected from those patients hospitalized with a diagnosis unrelated to the exposure of interest and matched for race, sex, age (± 5 years), and hospital. Individual epidemiologic information was collected during the first hospitalization by trained interviewers using a computer-assisted questionnaire. The structured questionnaire collected the sociodemographic characteristics, recent and previous smoking, environmental exposure (air pollution and passive smoking), diet, personal and family cancer history, and occupational history of each participant. Peripheral blood samples (or mouthwash samples when refusing to donate blood) were collected from all subjects. Histological code was based on who/iaslc in 2001.
MR analysis
We performed MR analysis using the R package TwoSampleMR and the cis-eQTL or cis-pQTL was used as exposure data. To generate IVs, SNPs in each eQTL (FDR < 0.05) or SNPs in each pQTL (p < 5e-08) within 1 MB upstream and downstream to the target gene transcription start site were selected for linkage disequilibrium (LD) analysis. LD analysis was conducted on the SNPs using European samples from the Thousand Genome Project (r2 < 0.001, kb = 10,000), and the resulting data was loaded and coordinated using built-in functions. The Wald ratio method was used to calculate the MR estimate for each SNP. When multiple SNPs were available, the weighted average of the ratio estimates and weighted by the inverse variance weighted (IVW). MR-Egger was applied to evaluate whether the MR Egger intercept significantly deviated from 0 to test for level pleiotropy when the number of SNPs was not less than 3. Additionally, Cochran’s Q method was used to test for the heterogeneity between Wald ratios.
Bayesian co-localization analysis
Sometimes SNPs are located in two or more gene regions. If an SNP contains eQTL/pQTL information for two or more different genes, its impact on the disease will be mixed by different genes. Therefore, co-localization analysis was used to confirm that LUSC and eQTL/pQTL might share causal genetic variation. To obtain significant MR results, colocalization analysis of SNPs within 1 MB upstream and downstream in each eQTL/pQTL was performed using R “coloc” package31 with P1 = 1 × 10–4, P2 = 1 × 10–4, and P12 = 1 × 10–5. P1 was defined as the probability of association between a given SNP and LUSC; the probability that a given SNP was a significant eQTL/pQTL was denoted as P2, and the probability that a given SNP was both LUSC and eQTL/pQTL was denoted as P12. Then, the “COLOC” package was applied to test for five hypotheses. We used a posterior probability (PP) to quantify the support for all hypotheses, which were determined as PPH0- PPH4: PPH0, SNPs were not associated with any trait; PPH1, associated with gene expression but not with LUSC risk; PPH2, associated with LUSC risk, but not with the expression of gene; PPH3, associated with LUSC risk and gene expression, with significant causal variation; PPH4, associated with LUSC risk and gene expression, and has a common causal variation. Due to the limited ability of co-localization analysis, the threshold of significance for co-localization was set at PPH3 + PPH4 ≥ 0.8.
SMR analysis and HEIDI test
SMR was conducted to verify the pleotropic associations between gene expression levels and LUSC. The heterogeneity in dependent instruments (HEIDI) test was used to distinguish genes associated with LUSC risk owing to a shared genetic variant rather than genetic linkage using multiple SNPs in a region. The SMR and HEIDI analysis could determine whether the influence of genetic variations (SNPs) on phenotypes occurred through gene/protein expression or other biological pathways. The SMR and HEIDI tests were performed using SMR Linux (https://yanglab.westlake.edu.cn/software/smr, v1.3.1). p_SMR < 0.05 and p_HEIDI > 0.05 indicated the significance level.
Risk factors for LUSC
We obtained information related to LUSC risk factors from the GWAS catalog database (https://gwas.mrcieu.ac.uk/), including smoking (GCST90044186), pulmonary tuberculosis (GCST006356), pulmonary fibrosis (GCST90041714), and alcohol abuse (GCST90079789). Using the same parameters, MR analysis was conducted using eQTL/pQTL data of eight genes to explore potential risk factors for LUSC.
Phenome-wide association study (PheWAS)
To investigate the potential side effects of eight genes, the PheWAS was conducted using gene expression as exposure and summary statistics for diseases as outcomes in the UK Biobank cohort (n ≤ 408 961). The disease GWAS of the UK Biobank was analyzed using the Scalable and Accurate Implementation of GEneralized mixed model (SAIGE V.0.29) approach32 to address imbalanced case-control ratios. We selected 783 personality traits (diseases) and over 500 cases for phenotype MR analysis. Summary statistical data of disease-related SNPs were downloaded from SAIGE GWAS (https://www.leelabsg.org/resources). Next, Phe-MR analysis was conducted using the blood eQTL/pQTL with the same parameters based on IVW estimates. p < 0.05 was regarded as statistically significant for causal effects.
Transcriptomic analysis of LUSC
The gene expression profile of LUSC was downloaded from the The Cancer Genome Atlas (TCGA, https://www.genome.gov/Funded-Programs-Projects/Cancer-Genome-Atlas) using R package “TCGAbiolinks”33, and converted FPKM values to log2 (FPKM + 1) values. We also obtained the data of copy number variation (CNV) and somatic mutation data using “TCGAbiolinks”. The TCGA survival information (OS) and clinical information (including age, stage, gender, grade, etc.)34. The TCGA LUSC dataset included 369 LUSC patients and 50 paracarcinoma samples. The expression of eight genes in LUSC patients and control group was compared using Wilcoxon test. The Maftools package analyzed the single nucleotide mutations of eight genes in LUSC. Additionally, we also analyzed the copy number variation of eight genes. GO enrichment analysis is the main bioinformatics tool for gene and product annotation, which includes three categories: cellular components (CC), molecular functions (MF), and biological processes (BP). KEGG is a database resource for understanding high-level functions and utilities of the biological system, such as the cell, the organism and the ecosystem, from molecular-level information, especially large-scale molecular datasets generated by genome sequencing and other high-throughput experimental technologies35,36,37. GO and KEGG functional enrichment analysis were performed using the “clusterProfiler” package to predict the potential molecular functions. p < 0.05 was considered statistical significance. The survival curve was generated using the Kaplan–Meier. Furthermore, a protein–protein interaction (PPI) network was generated using the GeneMANIA database (https://genemania.org/). The single sample gene set enrichment analysis (ssGSEA) algorithm was used to quantify the relative abundance of cellular infiltrates in the immune microenvironment of LUSC patient and normal control samples38. Gene sets marking each TIME infiltrating immune cell type were obtained from Charoentong’s research39. This gene set is rich in multiple human immune cell subtypes, including activated CD8 T cells, activated dendritic cells, macrophages, and NK cells, regulatory T cells, etc. Spearman correlation analyzed the correlation between eight genes and immune cell infiltration scores. Next, we searched for drugs targeting eight genes in the DGIdb database (https://www.dgidb.org/).
Single-cell transcriptomic data processing
We downloaded the single-cell RNA-seq (scRNA-seq) data of LUSC from the GEO database (https://www.ncbi.nlm.nih.gov/geo/). The GSE166635 dataset contains two LUSC patients40. The scRNA-seq data were preprocessed using the R package “Seurat” (version 4.0.0)41. First, we excluded cells with less than 200 genes and excluded cells with a mitochondrial percentage higher than 25%. The scRNA-seq dataset was normalized using the “NormalizeData” function and 2000 highly variable genes were identified using the “FindVariableFeatures” function based on the “vst” algorithm. Next, dimensionality was reduced using the principal components analysis (PCA), and the first 20 principal components were selected for analysis. We used the tSNE algorithm to visualize and mine the data. Cell clusters were identified by using the k-nearest neighbor (KNN) algorithm and the “FindClusters” function with a resolution of 1. We annotated cell populations based on the studies of Lichun Ma and Yan Meng40,42. The “FindAllMarkers” function in Seurat was utilized to identify differentially expressed genes (DEGs) between different cell types based on min.pct = 0.25, logfc.threshold = 0.25, and p < 0.05. The expression of eight genes in different cell types was evaluated using the “FeaturePlot” and “VlnPlot” functions.
Data availability
The blood eQTL dataset was obtained from the eQTLGen (https://www.eqtlgen.org/phase1.html). The blood pQTL dataset was obtained from the GWAS catalog (https://www.ebi.ac.uk/gwas/home). The GWAS summary data are available in the IEU database (https://gwas.mrcieu.ac.uk/) and the finngen database (https://r10.finngen.fi/). The druggable genes were downloaded from DGIdb (https://www.dgidb.org/). The gene expression profile of LUSC was downloaded from the The Cancer Genome Atlas (TCGA, https://www.genome.gov/Funded-Programs-Projects/Cancer-Genome-Atlas), and the scRNA-seq data of LUSC was available in the GEO (https://www.ncbi.nlm.nih.gov/geo/). Further inquiries can be directed to the corresponding author.
References
Nasim, F., Sabath, B. F. & Eapen, G. A. Lung cancer. Med. Clin. North. Am. 103, 463–473. https://doi.org/10.1016/j.mcna.2018.12.006 (2019).
de Sousa, V. M. L. & Carvalho, L. Heterogeneity in lung cancer. Pathobiology 85, 96–107. https://doi.org/10.1159/000487440 (2018).
Wu, F., Wang, L. & Zhou, C. Lung cancer in China: current and prospect. Curr. Opin. Oncol. 33, 40–46. https://doi.org/10.1097/CCO.0000000000000703 (2021).
Cheung, C. H. Y. & Juan, H. F. Quantitative proteomics in lung cancer. J. Biomed. Sci. 24, 37. https://doi.org/10.1186/s12929-017-0343-y (2017).
Alexander, M., Kim, S. Y. & Cheng, H. Update 2020: Management of non-small cell lung cancer. Lung 198, 897–907. https://doi.org/10.1007/s00408-020-00407-5 (2020).
Abu Rous, F., Singhi, E. K., Sridhar, A., Faisal, M. S. & Desai, A. Lung cancer treatment advances in 2022. Cancer Invest. 41, 12–24. https://doi.org/10.1080/07357907.2022.2119479 (2023).
Li, Y., Wu, X., Yang, P., Jiang, G. & Luo, Y. Machine learning for lung cancer diagnosis, treatment, and prognosis. Genom. Proteom. Bioinformat. 20, 850–866. https://doi.org/10.1016/j.gpb.2022.11.003 (2022).
Sharma, K. R., Colvis, C. M., Rodgers, G. P. & Sheeley, D. M. Illuminating the druggable genome: Pathways to progress. Drug Discov. Today 29, 103805 (2023).
Finan, C. et al. The druggable genome and support for target identification and validation in drug development. Sci. Transl. Med. 9, 1166 (2017).
Song, W. et al. Systematic druggable genome-wide Mendelian randomization identifies therapeutic targets for lung cancer. BMC Cancer 24, 680. https://doi.org/10.1186/s12885-024-12449-6 (2024).
Su, W. M. et al. Systematic druggable genome-wide Mendelian randomisation identifies therapeutic targets for Alzheimer’s disease. J. Neurol. Neurosurg. Psychiatr. 94, 954–961. https://doi.org/10.1136/jnnp-2023-331142 (2023).
Yang, L. Z. et al. Systematic Mendelian randomization exploring druggable genes for hemorrhagic strokes. Mol. Neurobiol. https://doi.org/10.1007/s12035-024-04336-9 (2024).
Zhang, N., Li, Y., Sundquist, J., Sundquist, K. & Ji, J. Identifying actionable druggable targets for breast cancer: Mendelian randomization and population-based analyses. EBioMedicine 98, 104859. https://doi.org/10.1016/j.ebiom.2023.104859 (2023).
Song, M., Chen, F., Li, X. & Chen, L. Exploring causal correlations between plasma proteins and peripheral neuropathy: a Mendelian randomization. Front. Neurol. 15, 1431669 (2024).
Sun, J. et al. Identification of novel protein biomarkers and drug targets for colorectal cancer by integrating human plasma proteome with genome. Genome Med. 15, 75 (2023).
Liu, D., Wang, Y., Jing, H., Meng, Q. & Yang, J. Novel DNA methylation loci and genes showing pleiotropic association with Alzheimer’s dementia: a network Mendelian randomization analysis. Epigenetics 17, 746–758. https://doi.org/10.1080/15592294.2021.1959735 (2022).
Yuan, D. et al. DNA methylation regulator-meditated modification patterns define the distinct tumor microenvironment in lung adenocarcinoma. Front. Oncol. 11, 734873 (2021).
Wang, L. et al. Carboxypeptidase A4 negatively regulates HGS-ETR1/2-induced pyroptosis by forming a positive feedback loop with the AKT signalling pathway. Cell Death Dis. 14, 793 (2023).
Shibata, T. et al. Targeting phosphorylation of Y-box–binding protein YBX1 by TAS0612 and everolimus in overcoming antiestrogen resistance. Mol. Cancer Ther. 19, 882–894 (2020).
Shibata, T. et al. Y-box binding protein YBX1 and its correlated genes as biomarkers for poor outcomes in patients with breast cancer. Oncotarget 9, 37216 (2018).
Nehme, N. T. et al. MST1 mutations in autosomal recessive primary immunodeficiency characterized by defective naive T-cell survival. Blood J. Am. Soc. Hematol. 119, 3458–3468 (2012).
Sibille, A. et al. Eosinophils and lung cancer: from bench to bedside. Int. J. Mol. Sci. 23, 5066 (2022).
Chen, R., Ma, L., Jiang, C. & Zhang, S. Expression and potential role of CCL4 in CD8+T cells in NSCLC. Clin. Transl. Oncol. Off. Public. Federat. Spanish Oncol. Soc. Natl. Cancer Inst. Mexico 24, 2420–2431. https://doi.org/10.1007/s12094-022-02913-9 (2022).
Leveque, E. et al. Phenotypic and histological distribution analysis identify mast cell heterogeneity in non-small cell lung cancer. Cancers 14, 1394 (2022).
Mu, C.-Y., Huang, J.-A., Chen, Y., Chen, C. & Zhang, X.-G. High expression of PD-L1 in lung cancer may contribute to poor prognosis and tumor cells immune escape through suppressing tumor infiltrating dendritic cells maturation. Med. Oncol. 28, 682–688 (2011).
Wang, J. B., Huang, X. & Li, F. R. Impaired dendritic cell functions in lung cancer: a review of recent advances and future perspectives. Cancer Commun. 39, 43 (2019).
Sun, X. et al. Multi-omics Mendelian randomization integrating GWAS, eQTL and pQTL data revealed GSTM4 as a potential drug target for migraine. J. Headache Pain 25, 117. https://doi.org/10.1186/s10194-024-01828-w (2024).
Cao, Y., Yang, Y., Hu, Q. & Wei, G. Identification of potential drug targets for rheumatoid arthritis from genetic insights: a Mendelian randomization study. J. Transl. Med. 21, 616. https://doi.org/10.1186/s12967-023-04474-z (2023).
Vosa, U. et al. Large-scale cis- and trans-eQTL analyses identify thousands of genetic loci and polygenic scores that regulate blood gene expression. Nat. Genet. 53, 1300–1310. https://doi.org/10.1038/s41588-021-00913-z (2021).
Griffith, M. et al. DGIdb: mining the druggable genome. Nat. Methods 10, 1209–1210. https://doi.org/10.1038/nmeth.2689 (2013).
Rasooly, D., Peloso, G. M. & Giambartolomei, C. Bayesian genetic colocalization test of two traits using coloc. Curr. Protocols 2, e627 (2022).
Zhou, W. et al. Efficiently controlling for case-control imbalance and sample relatedness in large-scale genetic association studies. Nat. Genet. 50, 1335–1341 (2018).
Colaprico, A. et al. TCGAbiolinks: an R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res. 44, e71–e71 (2016).
Liu, J. et al. An integrated TCGA pan-cancer clinical data resource to drive high-quality survival outcome analytics. Cell 173 (400–416), e411. https://doi.org/10.1016/j.cell.2018.02.052 (2018).
Kanehisa, M., Furumichi, M., Sato, Y., Matsuura, Y. & Ishiguro-Watanabe, M. KEGG: biological systems database as a model of the real world. Nucleic Acids Res. 53, D672-d677. https://doi.org/10.1093/nar/gkae909 (2025).
Kanehisa, M. Toward understanding the origin and evolution of cellular organisms. Protein Sci. Public. Protein Soc. 28, 1947–1951. https://doi.org/10.1002/pro.3715 (2019).
Kanehisa, M. & Goto, S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30. https://doi.org/10.1093/nar/28.1.27 (2000).
Hanzelmann, S., Castelo, R. & Guinney, J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinformat. 14, 7. https://doi.org/10.1186/1471-2105-14-7 (2013).
Charoentong, P. et al. Pan-cancer immunogenomic analyses reveal genotype-immunophenotype relationships and predictors of response to checkpoint blockade. Cell Rep. 18, 248–262 (2017).
Meng, Y. et al. A TNFR2-hnRNPK axis promotes primary liver cancer development via activation of YAP signaling in hepatic progenitor cells. Cancer Res. 81, 3036–3050. https://doi.org/10.1158/0008-5472.CAN-20-3175 (2021).
Gribov, A. et al. SEURAT: visual analytics for the integrated analysis of microarray data. BMC Med Genom. 3, 21. https://doi.org/10.1186/1755-8794-3-21 (2010).
Ma, L. et al. Single-cell atlas of tumor cell evolution in response to therapy in hepatocellular carcinoma and intrahepatic cholangiocarcinoma. J. Hepatol. 75, 1397–1408. https://doi.org/10.1016/j.jhep.2021.06.028 (2021).
Funding
This study was supported by Tianjin Key Medical Discipline (Specialty) Construction Project (TJYXZDXK-018A).
Author information
Authors and Affiliations
Contributions
Shizhao Cheng wrote the manuscript; Hao Zhang performed interpreted the results; Zhenliang Shi prepared figures; Daqiang Sun designed the research, revised the manuscript and gave the final approval of the version to be published. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Consent for publication
All authors have approved the manuscript for publication and that it has not been published elsewhere.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Cheng, S., Zhang, H., Shi, Z. et al. Druggable genome-wide Mendelian randomization integrating GWAS and eQTL/pQTL data identifies targets for lung squamous cell carcinoma. Sci Rep 15, 30116 (2025). https://doi.org/10.1038/s41598-025-15977-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-15977-8
