
Abstract
Benzimidazole derivatives are privileged heterocyclic scaffolds with broad-spectrum pharmacological activities, notably antitubercular and antibacterial. In particular, 1,2-disubstituted benzimidazoles have emerged as potent bioactive candidates due to their unique structural features and target specificity. In this study, fifty novel 1,2-disubstituted benzimidazole derivatives were computationally screened against Mycobacterium tuberculosis cell division protein FtsZ (PDB ID: 2Q1Y, GTP-γ-S complex) using AutoDock Vina v1.5.6. Docking poses were analyzed via PyMOL and Discovery Studio Visualizer to elucidate key binding interactions. Pharmacokinetic evaluation through SwissADME was performed to predict drug-likeness and ADME profiles. ADME analysis revealed that several lead candidates possessed favorable absorption, distribution, metabolism, and excretion properties, underscoring their suitability as orally bioavailable agents. Docking scores ranged from − 6.8 to − 9.6 kcal/mol, with multiple derivatives surpassing the binding affinity of standard antitubercular drugs, including isoniazid and para-aminosalicylic acid. The integration of structure-based molecular docking with in silico pharmacokinetic profiling highlights substituted benzimidazole scaffolds as promising next-generation FtsZ inhibitors for antitubercular drug development.
Similar content being viewed by others
Introduction
Tuberculosis (TB) remains one of the world’s deadliest infectious diseases, causing ~ 10 million new cases and 1.5 million deaths annually1. The emergence of multidrug-resistant (MDR) and extensively drug-resistant (XDR) strains has reduced the effectiveness of the standard Directly Observed Treatment, Short-Course (DOTS) regimen—isoniazid, rifampicin, pyrazinamide, and ethambutol—highlighting the need for new drugs2,3,4,5.
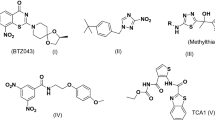
Benzimidazole derivatives exhibit diverse pharmacological activities, including antitubercular, antiviral, antifungal, and anti-inflammatory effects6,7,8,9,10,11,12,13. Their presence in marketed drugs underscores their versatility, and some have been reported to inhibit Mycobacterium tuberculosis enzymes such as InhA and KatG14. Figure 1 illustrates representative biologically active compounds containing the benzimidazole moiety, highlighting its significance as a privileged scaffold in drug design.

Biologically active compounds having Benzimidazole Moiety.
Previous studies have explored benzimidazoles as FtsZ inhibitors. Dhameliya et al. (2022) performed docking and MD simulations on benzimidazole-2-carboxamides targeting FtsZ, but without ADME profiling. Rode et al. (2020) synthesized 2,5- and 1,2,5-trisubstituted benzimidazoles, docking them against FtsZ with limited compound sets and no systematic ADME filtration. Thapa et al. (2024) examined KasA inhibitors with similar computational approaches but minimal ADME consideration. However, no study has conducted a large-scale, ADME-guided virtual screening of 1,2-disubstituted benzimidazoles against FtsZ (PDB 2Q1Y).
To address this gap, we docked a 50-compound library of 1,2-disubstituted benzimidazoles using AutoDock Vina, analyzed poses with PyMOL and Discovery Studio, prioritized leads via SwissADME, and benchmarked affinities against standard antitubercular drugs. This combined docking–ADME strategy aims to identify candidates with both strong target binding and favorable pharmacokinetic profiles for further development15.
Materials and methods
Software required
All molecular docking studies were performed on a Windows 10-based system with Intel Core i5 (8th Gen) processor, 16 GB RAM, using AutoDock Vina v1.5.6., PyMOL, and BIOVIA Discovery Studio Visualizer 2021.
Target protein preparation
The crystal structure of Mycobacterium tuberculosis FtsZ protein in complex with GTP-γ-S (PDB ID: 2Q1Y) was downloaded from the RCSB Protein Data Bank16. Water molecules and the native ligand were removed using Discovery Studio Visualizer. Hydrogen atoms were added, and energy minimization was performed to stabilize the structure. The binding site was defined based on the co-crystallized ligand’s coordinates, and the final structure was saved in PDB format. The crystal structure of M. tuberculosis FtsZ in association with GTP-γ-S (PDB ID: 2Q1Y) is shown in Fig. 2, providing insights into its functional conformation.

Crystal structure of the cell division protein FtsZ from Mycobacterium TB in association with GTP-gamma-S (PDB ID: 2Q1Y).
Preparation of Ligand and macromolecule
Prior to initiating the molecular docking process, which involved removing the water molecules and native ligand linked to the target enzyme, ligand structures were sketched in ChemDraw Ultra 12.0 and saved in mol format. These were converted to 3D PDB structures using PyMOL, followed by addition of hydrogen atoms, assignment of rotatable bonds, and conversion to pdbqt format in AutoDock Tools (part of AutoDock Vina v1.5.6.). All ligands were energy minimized using MMFF94 force field to obtain low-energy conformations for docking.
Molecular docking was performed using AutoDock Vina v1.5.6. Blind docking was carried out by setting the grid box large enough to cover the entire protein surface (grid size: 40 Å × 40 Å × 40 Å; center coordinates: x = − 6.307, y = 53.272, z = 0.296). Docking exhaustiveness was set to 8. Binding poses were ranked by binding affinity (kcal/mol), and the best-ranked conformations were visualized using PyMOL and Discovery Studio Visualizer.
Docking of crystal structure of cell division protein FtsZ from Mycobacterium tuberculosis in complex with GTP-gamma-S (2Q1Y) with 1,2-disubstituted benzimidazole derivatives using AutoDock Vina v1.5.6., PyMol and discovery studio visualizer
To determine the compound's propensity for binding proteins, molecular docking tests were conducted17,18,19. PyMol and AutoDock Vina v1.5.6. were used to prepare the protein, remove water molecules, and add hydrogen. The target proteins in the Crystal Structure of cell division protein FtsZ from Mycobacterium tuberculosis in complex with GTP-gamma-S were retrieved from the RCSB PDB (protein data bank) in PDB format in 3D orientation. Prior to the docking process, energy minimization and conversion to PDBQT format were completed in the same software, and BIOVIA Discovery studio visualizer 2021 was used to build the 2D structure. Docking calculations were performed using AutoDock Vina v1.5.6.18
Ligand identification
The ligands were benzimidazole derivatives (Table 2). Currently available antitubercular medications include para-amino salicylic acid, ethambutol, and isoniazid. They participated in docking and comparison investigations after being obtained from the drug bank database (Table 1). Using PyMol, AutoDock Vina v1.5.6, and the Biovia Discovery Studio visualizer tools, the known structures were downloaded in SDF format and translated to PDB format before being used for docking experiments.
In silico ADME (absorption, distribution, metabolism and excretion studies)
The pharmacokinetics of chemicals in an organism's body are described by ADME. It evaluates the risk involved in giving people or other animals a medicinal drug. These pharmacokinetic characteristics are found In silico ADME predictions were performed using SwissADME 20,21,22,23and PreADMET web servers to evaluate the pharmacokinetic properties of the designed ligands. Parameters such as molecular weight, hydrogen bond donors/acceptors, logP, topological polar surface area (TPSA), and gastrointestinal absorption were assessed. Compounds violating more than one Lipinski’s rule were considered less drug-like. These properties help estimate a compound’s likelihood of being orally bioavailable and safely metabolized in the body.
Using the Biovia discovery studio tool, 50 distinct conformers were created in this manner (Fig. 3), and blind docking was used to determine whether these molecules bound in the active site or elsewhere in the target.

Synthetic Scheme.
Results and discussion
Swiss ADME results
In silico ADME tests were conducted on 1,2-disubstituted benzimidazole derivative analogues using a range of software programs, such as ChemDraw, Molinspiration, Swiss ADME, PyMol, and Discovery Studio Visualizer (DSV). Molinspiration software was used to calculate molecular descriptors and smiles notation. The Lipinski Rule of Five was examined using Molinspiration software, and it was found that nearly all of the analogues followed it. The drug likeness profile of analogues was anticipated by the methods developed by Molinspiration.
For a chemical to be a successful medicine, it must have low toxicity, strong biological activity at low effective dosages, and the ability to act continuously until the intended effect is achieved. Since the benzimidazole nucleus is well known for treating tubercular disease, new compounds containing it are created for their antitubercular properties. All of the proposed compounds were reported in Table 2. We found that suggested compound possesses antitubercular characteristics and a low frequency of toxicity potential.
According to the proposed compounds' structure–activity relationship (SAR) analysis, antitubercular action requires a 1,2-substituted benzimidazole core. Another important factor in determining the compounds' activity is the substitution pattern on the phenyl ring. If we substitute phenyl ring by electronegative group it will led to a better ADME/docking profile.
Since all of the produced compounds follow the same criteria, we may conclude that the molecules are orally active based on absorption, distribution, metabolism, and excretion (ADME) findings. The results of ADME experiments are shown in Tables 3 and 4. The suggested compounds' octanol water partition coefficient (mol log P) cannot be greater than 5. Table 3 shows that the substances have an excellent oral bioavailability. The water solubility is represented by the logarithm of the molar concentration. Because the proposed compounds contain lipophilic functions meant to improve cell permeability, some of them are very soluble, soluble and most of them are moderately water soluble. The drug's distribution in the body was evaluated using a volume of distribution, % unbound, and blood–brain barrier permeability. A log volume of distributions value greater than 0.40 indicates a stronger tissue distribution, and a higher volume of distributions value indicate a better drug dispersion in tissues as opposed to plasma.
The in silico ADME evaluation (Table 4) of the designed 1,2-disubstituted benzimidazole derivatives (P1–P50) revealed favorable pharmacokinetic profiles supporting their potential as drug-like candidates.
The developed compounds have a good likelihood of reaching their intended targets. Using Swiss ADME techniques, the solubility and gastrointestinal absorption (percent absorbed) were calculated. Each molecule that is designed interacts with enzymes either as a substrate or as an inhibitor. Due to their good ADME and toxicity properties, all of the suggested compounds can be considered plausible lead candidates.
Molecular docking results
Molecular docking, which predicts the principal binding mechanism of a ligand with a target protein having a known three-dimensional structure, is an essential method in structure-based computer-assisted drug design (CADD). The suggested benzimidazole derivatives are successfully docked into the target protein's active site (PDB ID: 2Q1Y) using AutoDock Vina v1.5.6. software. The target protein exhibits appropriate hydrogen bonding with the targeted chemicals P9, P11, P12,P14, P20, P27, P30, P32, P34, P38 and P50 (Table 5). The GTP-gamma-S protein binding site and the cell division protein FtsZ from Mycobacterium tuberculosis were within five radii of the interactions that the active medications created. Since P32 has the lowest binding affinity ( − 9.6 kcal/mol), it was selected as a potent inhibitor even though the bulk of the compounds were active (PAS: binding affinity: − 4.7 and Isoniazid: binding affinity: − 5.5). The most effective compounds' mechanisms of binding with the target protein and the designer drug were revealed by docking experiments.
In this investigation, the binding interactions of P32 were examined in comparison to PAS and Isoniazid, which are clinically relevant compounds utilized as standards. PAS and Isoniazid were selected as standards because to their recognized modes of action against the target protein and their well-characterized binding patterns. Figure 4 illustrates P32's binding, Fig. 5 represents PAS binding, and Fig. 6 shows Isoniazid's binding. The established interactions between PAS and isoniazid were used as a standard to evaluate P32's potential.

Binding interaction of compound P32 with 2Q1Y protein.

Binding interaction of Para-amino salicylic acid (Standard) with 2Q1Y protein.

Binding interaction of Isoniazid (Standard) with 2Q1Y protein.
Figure 4 shows compound P32 bound within the active site of FtsZ (PDB ID: 2Q1Y). The ligand forms key interactions with Leu188(B), Gln192(B), Gly193(B), Arg304(B), and Ala262(B), contributing to stable binding. The 2D interaction diagram further reveals a combination of hydrophobic contacts and hydrogen bonding that enhance the ligand–protein affinity.
Figure 5 shows the standard drug PAS bound within the active site of FtsZ. The ligand engages in hydrogen bonding and polar interactions with Asp81(A), Asp43(B), Ala70(B), Thr106(B), Gly107(B), Gly17(B), Gly19(B), and Gly104(B). These multiple glycine interactions, along with acidic and polar residue contacts, help stabilize the complex and may contribute to its inhibitory activity.
Figure 6 shows the standard drug Isoniazid interacting within the FtsZ binding pocket. The ligand forms key contacts with Gly19(B), Asp43(B), and Ala70(B), indicating a relatively focused interaction profile compared to other ligands. These residues may play a role in stabilizing the ligand orientation and facilitating its known antitubercular activity.
Conclusion
In this study, a series of novel 1,2-disubstituted benzimidazole derivatives were designed and evaluated for their potential antitubercular activity using in silico methods. Most designed compounds showed favorable pharmacokinetic profiles based on SwissADME and PreADMET results and comply with major drug-likeness criteria, suggesting their potential as lead molecules for further development.
Molecular docking studies were performed against the cell division protein FtsZ from Mycobacterium tuberculosis in complex with GTP-gamma-S (2Q1Y), a critical target for bacterial proliferation. Among the tested derivatives, P32 demonstrated stronger binding affinity than standard TB drugs in docking simulations, through actual biological potency remains to be confirmed.
Overall, the combined ADME and docking results support that the designed benzimidazole derivatives, particularly P32, are promising candidates for the development of new antitubercular agents. These findings warrant further biological validation through in vitro and in vivo studies.
Limitations of the study
“However, computational predictions may not always reflect real biological activity, so further lab validation is essential.”
Data availability
All data generated or analysed during this study are included in this published article.
References
World Health Organization. (2020). Global Tuberculosis Report 2020.
Dheda, K. et al. The emergence of extensively drug-resistant tuberculosis (XDR-TB): a global concern. J. Med. Microbiol. 66(11), 1371–1383 (2017).
Gandhi, N. et al. Multidrug-resistant and extensively drug-resistant tuberculosis: A threat to global control of tuberculosis. Lancet 375(9728), 1830–1843 (2010).
Zhang, Y. et al. Mechanisms of drug resistance in Mycobacterium tuberculosis. Int. J. Tubercul. Lung Disease 22(9), 973–983 (2018).
World Health Organization. Treatment of tuberculosis: Guidelines for treatment of drug-susceptible tuberculosis and patient care (2019).
Kumar, V. et al. Benzimidazole derivatives as antitubercular agents: a review. J. Pharm. Pharmacol. 71(8), 1048–1063 (2019).
Sharma, P. et al. Benzimidazole derivatives as potential antitubercular agents: A molecular docking and dynamics study. J. Mol. Graph. Model. 81, 237–246 (2018).
Patel, R. et al. Synthesis, biological evaluation, and molecular docking studies of benzimidazole derivatives as antitubercular agents. Eur. J. Med. Chem. 163, 111–123 (2019).
Kumar, P. et al. Benzimidazole derivatives as antitubercular agents: A computational study using molecular docking and SwissADME. J. Mol. Modell. 26(10), 251 (2020).
Chauhan, P. et al. Benzimidazole derivatives: A review of their pharmacological and biological activities. Eur. J. Med. Chem. 166, 241–255 (2019).
Kumar, V. et al. Synthesis and biological evaluation of benzimidazole derivatives as antitubercular agents. Bioorg. Med. Chem. Lett. 28(10), 1739–1744 (2018).
Patel, R. et al. Design, synthesis, and biological evaluation of benzimidazole derivatives as antitubercular agents. J. Enzyme Inhib. Med. Chem. 33(1), 1240–1248 (2018).
Moulishankar, A. et al. Pharmacophore, QSAR, molecular docking, molecular dynamics and ADMET study of trisubstituted benzimidazole derivatives as potent anti-tubercular agents. Chem. Phys. Impact. 8(1), 100512 (2024).
Singh, P. et al. Design, synthesis, and biological evaluation of benzimidazole derivatives as inhibitors of InhA and KatG enzymes. Bioorg. Med. Chem. Lett. 30(15), 127447 (2020).
Kapetanovic, I. M. Computer-aided drug discovery and development (CADDD): in silico-chemico-biological approach. Chem. Biol. Drug Des. 89(1), 44–54 (2017).
RCSB. Protein data bank; Available from: https://www.rcsb.org/
Trott, O. & Olson, A. J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J. Comput. Chem. 31, 455–461. https://doi.org/10.1002/jcc.21334 (2010).
Morris, G. et al. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 30(16), 2785–2791 (2009).
Dallakyan, S. et al. Small-molecule library screening by docking with Autodock. Methods Mol Biol. 1263, 243–250 (2015).
Daina, A. et al. Swiss ADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sc Rep. 7(1), 1–13 (2017).
Kumar, P. et al. SwissADME and molecular docking studies of benzimidazole derivatives as antitubercular agents. J. Mol. Graph. Model. 99, 107641 (2020).
Patel, R. et al. Molecular docking and SwissADME studies of benzimidazole derivatives as inhibitors of InhA enzyme. J. Mol. Modell. 26(5), 123 (2020).
Sharma, P. et al. Computational studies of benzimidazole derivatives as antitubercular agents using molecular docking and SwissADME. J. Comput. Chem. 41(2), 112 (2020).
Acknowledgements
The authors are thankful to SMBT College of Pharmacy, Nashik, Maharashtra, India for encouraging throughout the research work.
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Author information
Authors and Affiliations
Contributions
P. N. S. conceived and designed the study, collected and analyzed the data, and drafted the manuscript. M. R. K. provided critical revisions and editing of the manuscript, ensuring its clarity and scientific rigor. Both authors reviewed and approved the final draft of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Sonwane, P.N., Kumbhare, M.R. Molecular docking and pharmacokinetics of benzimidazole-based FtsZ inhibitors for tuberculosis. Sci Rep 15, 35270 (2025). https://doi.org/10.1038/s41598-025-18084-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-18084-w
