
Abstract
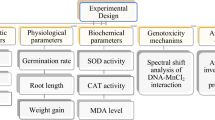
In this study, the biochemical and cytogenetic toxicity induced by the antibiotic active ingredient ornidazole in the non-target organism Allium cepa L. was investigated. In the toxicity assessment, the level of malondialdehyde (MDA), a biochemical marker of lipid peroxidation; genotoxicity indicators such as micronucleus (MN) frequency and mitotic index (MI); the incidence of chromosomal abnormalities (CAs); activities of antioxidant defense enzymes catalase (CAT) and superoxide dismutase (SOD); and the levels of chlorophyll a and b pigments reflecting photosynthetic capacity were analyzed. Additionally, DNA damage was assessed using the Comet test method, and the interaction of ornidazole with macromolecules—particularly DNA—was examined using the molecular docking approach. Four groups of A. cepa bulbs—one control and three treatments—were created. Three distinct dosages (0.0179, 0.0357 and 0.0714 mg/L) of ornidazole were used to germinate the bulbs in the treatment group, while tap water was used to germinate the bulbs in the control group. Following germination, samples from the roots and leaves were gathered and ready for examination. As a result, there was no cytogenetic damage or biochemical alteration that was statistically significant (p > 0.05) in the control group (Group I). MI value, DNA, and chlorophyll levels significantly (p < 0.05) decreased with ornidazole treatment, while MN frequency, CAs, MDA levels, SOD, and CAT activities significantly (p < 0.05) increased. At the ornidazole dosage of 0.0714 mg/L, these rises and declines were shown to be more noticeable. Ornidazole promoted several CAs in root meristem cells, the most common being the sticky chromosome. DNA damage was highlighted by the comet assay results, which indicated a drop in head DNA and an increase in tail DNA. In the control group, the Tail DNA was 1.00 ± 1.05 (%), whereas in the group treated with 0.0714 mg/L ornidazole, it increased to 72.0 ± 1.63 (5), indicating high DNA fragmentation. Molecular docking results showed ornidazole-DNA, ornidazole-tubulin, ornidazole-topoisomerase, ornidazole-glutamate-1-semialdehyde aminotransferase and ornidazole-protochlorophyllide reductase interaction supporting the biochemical and cytogenetic toxicity results. In conclusion, ornidazole exposure induced significant toxic effects in the non-target organism Allium cepa. The study further validated the efficacy of the Allium test as a reliable bioassay for detecting such toxicity. These findings underscore the urgent need for implementing appropriate environmental management strategies to mitigate pharmaceutical contamination and protect non-target organisms from drug-induced toxicity.
Similar content being viewed by others
Introduction
In today’s world, the widespread availability and consumption of pharmaceutical compounds have become an integral part of modern healthcare, yet their pervasive presence in the environment raises growing concerns about unintended toxic effects on non-target organisms and ecosystem health1. The worldwide pharmaceutical spectrum includes around 12.000 human and 2.500 veterinary medicines. Both the human health field and animal agriculture make extensive use of these pharmacological medicines. After use, pharmaceutical products are dispersed into the water environment or directly into the environment. Recent studies have shown that pharmaceuticals can be detected in wastewater, surface water, groundwater and even drinking water. Pharmaceuticals and their metabolites that cannot be metabolized in the body are excreted through feces or urine, and after passing through the sewage treatment system, they reach the soil through discharge or the water environment through drainage. Drugs commonly used in the veterinary field, especially as therapeutic and growth promoters, can be emitted into surface waters or directly into the soil through manure when animals are kept on pasture. Other major causes of potential drug contamination are the improper disposal of expired drugs in the environment and landfill, spillage into sinks and toilets, or drug residues from production accidents2. On the other hand, various production facilities have been found to cause much more environmental pharmaceutical pollution than that caused by the use of pharmaceuticals. Pharmaceutical factories generate large amounts of waste during production, cleaning and maintenance processes. Different groups of drugs such as antibiotics, analgesics, antihypertensives, antiepileptics, antiseptics, contraceptives, beta-blocker heart drugs, hormones and psychotherapeutics are recognized as environmental pollutants. In particular, 95% of antibiotic compounds have been demonstrated to be able to enter the sewage system unaltered3.
Although pharmaceuticals are essential for human and animal health, their release to soil, water, and air is a growing concern. The continued entry and persistence of pharmaceuticals and their active ingredients in the environment can have significant ecological impacts, and these substances can also have toxic effects on non-target species4. For example, they can accumulate in plant and animal tissues, increasing toxicity through the food chain. Furthermore, the spread of antibiotics in the environment can lead to the development of resistant bacterial strains, i.e., antimicrobial resistance. The average daily intake of antibacterial drugs for systemic human use in European Union (EU) and European Economic Area countries is set at 19.4 defined daily doses per 1000 population. However, antimicrobials are also consumed at high rates by animals. In this context, antimicrobial consumption in the global livestock sector is projected to increase by more than 60% between 2010 and 20305,6. One of the most widely used antimicrobial agents globally is ornidazole. Ornidazole is the active ingredient of antibiotics such as albitrol, avrazor, bitazol and biteral, with the chemical formula 1-(3-chloro-2-hydroxy)propyl-2-ethyl-5-nitroimidazole7. Due to its antibacterial and anti-protozoal properties, it is used in the treatment of anaerobic enteric protozoa. It is used in the treatment of amoebic liver abscesses, duodenal ulcers, giardiasis, trichomoniasis, intestinal lambliasis and vaginitis8,9. It is also used in the treatment of anaerobic infections susceptible to prophylaxis in dental and gastrointestinal surgery10. Conversely, it is employed as a sensitiser in the context of hypoxic tumours, when administered in conjunction with radiotherapy. Recent clinical applications have included the use of this treatment for active Crohn’s disease. Ornidazole is also preferred for surgical prophylaxis due to its long half-life compared to other nitroimidazole derivatives and its excellent penetration into lipidic tissues. The most commonly observed side effects are nausea, dizziness, abdominal pain, headache, bloating, diarrhea and skin rash. Ornidazole and its metabolites are primarily excreted in the urine. The amount excreted in urine constitutes 43%-63% of the ornidazole dose8.
The majority of studies on drug toxicity in the current literature have focused on the effects on target organisms, particularly humans and animals. However, the effects of these compounds on non-target organisms that play critical roles in ecosystems have not been sufficiently investigated, and this lack of knowledge makes it difficult to fully assess environmental risks. In this context, the present study aims to address a significant gap in the extant literature by investigating the cytogenetic and biochemical toxicity of ornidazole, one of the active components of widely used antibiotics, on Allium cepa, a model non-target eukaryotic organism. The identification of the genotoxic and biochemical changes caused by ornidazole in A. cepa not only expands the current understanding of the environmental toxicology of pharmaceuticals but also emphasises the necessity of considering non-target species in environmental risk assessments. The findings contribute valuable data to the existing literature and provide a foundation for future studies. These future studies will aim to develop effective intervention strategies to mitigate the adverse effects of pharmaceutical pollutants on ecosystem health.
Materials and methods
Experimental material, test chemicals, and doses
Allium cepa (2n = 16) bulbs sold commercially in Giresun (Turkey) were used as experimental material. The antibiotic active ingredient ornidazole (Merck CAS No: 16773-42-5) was preferred as the test chemical. Dosage levels were determined based on the highest marketed dose of ornidazole-containing antibiotics, typically available as 500 mg tablets. Test concentrations were prepared as descending series of 250 mg and 125 mg, reflecting the commonly used pharmaceutical formulations containing ornidazole as the active ingredient. The antibiotic dosages used in this study are based on the recommended therapeutic doses for humans; however, to apply these doses to plants, an appropriate equation was employed to account for differences in physiology and exposure routes between humans and the test organism11. As a result, based on the doses determined using Eq. (1), ornidazole concentrations of 0.0179 mg/L, 0.0357 mg/L, and 0.0714 mg/L were selected for Group II, Group III, and Group IV, respectively. Guidelines and laws at the institutional, national, and international levels have been followed when conducting experimental research on plant samples, including the provision of plant material.
Mi = Ideal mass of the drug (mg),
10.000 = Average mass of bulb (mg),
Mc = Mass of one tablet (mg),
70.000.000 = Average mass of a woman (mg).
Experimental process
Total of 40 bulbs were individually placed in sterile glass beakers. Bulbs in Groups II, III, and IV were germinated at room temperature using ornidazole concentrations of 0.0179, 0.0357, and 0.0714 mg/L, respectively, while tap water was used as the germination medium for Group I (control). 72 h of germination were required for root tip studies, while at least 144 h were needed for leaf development in order to assess chlorophyll. After the time frame, root and leaf tissues were collected, washed with distilled water, homogenization and crushing-preparation methods were applied and prepared for analysis, measurements and observations.
Chromosomal abnormality and MN test
Chromosome abnormalities and MN were detected using the acetocarmine crushing technique. The tips of the roots were trimmed to a length of about 1 cm and left in “Clarke” solution for two hours. After 15 min of washing in 96% ethanol, they were hydrolyzed for 17 min at 60 °C in 1 N HCl, and they were then stored for 30 min in 45% glacial acetic acid. After being crushed on a slide, coverslipped, and stained with acetocarmine for 24 h, the root tips were inspected under an Irmeco IM-450 TI model research microscope and captured on camera at ×400 magnification12.
For MN detection, the criteria of Fenech et al.13 were taken into consideration.
-
(a)
The MN should have a diameter that is around one-third that of the nucleus,
-
(b)
The nucleus and MN should have the same color,
-
(c)
When MN and nuclear membranes come into touch, their boundaries should be easily discernible.
MI was computed using Eq. (2).
10 preparations for CAs, MN, and MI counts were made for each group. 100 cells in each preparation and a total of 1.000 cells were examined for CAs and MN, and 10.000 cells in total were examined for MI. Cells at the interphase stage and four distinct stages of mitosis were examined for all anomalies and counts. For MN, MI and CAs analysis, 0.0714 mg/L sodium azide (NaN3) was used as positive control. NaN3 is used as a standard positive control in genotoxicity tests to verify the accuracy of the test system due to its strong and reliable genotoxic effects.
Comet assay
The technique suggested by Sharma et al.14 was used to isolate DNA from root cells. Using the approach suggested by Dikilitaş and Koçyiğit15 comet assay was carried out. After soaking in ethanol for twenty-four hours, the slides were dried in an oven. The slides were covered with coverslips after 100 µL of agarose was put onto them using a pipette. To enable the gel to freeze, the slides were refrigerated for five minutes. Freshly isolated cell solution and 1% low melting point (LMP) agarose were combined to create the second layer. 100 µL of the mixture was put on the first layer and covered with a coverslip after the cell suspension and LMP agarose were well combined in distilled water at 40 °C in a ratio of 1/8 on a slide. After 5 min of cooling, the preparations were taken out of the coverslips. To loosen the DNA, the preparations were put in the buffer solution in the electrophoresis tank and left for 40 min. After that, the preparations underwent electrophoresis. To counteract the alkaline environment created following electrophoresis and to improve the slides’ visibility under a microscope, the slides were cleaned three times for five minutes each at room temperature using 0.4 M Tris-HCl. Following neutralization, the slides were stained with 100 µL ethidium bromide, coverslipped, examined under a fluorescence microscope, counted and photographed. The “TriTek 2.0.0.0.38 Automatic Comet Assay” software was used to conduct comet assays. For every group, 1.000 cells in total were examined. DNA fragments were separated into two sections, the head and the tail, and the percentages of DNA in each section were determined. The scale proposed by Pereira et al.16 based on tail DNA percentages was used to assess the degree of DNA damage based on the numerical data derived from comet research.
Measurement of MDA level
The method suggested by Unyayar et al.17 was used to measure MDA levels. 1 mL of 5% trichloroacetic acid was used to homogenize 0.5 g of root tips. After being moved to a new tube, the homogenate was centrifuged for 10 min at 12.000 g. After being moved to a new tube, the supernatant and equal parts of 0.5% thiobarbituric acid were incubated for 30 min at 96 °C in a 20% trichloroacetic acid solution. After cooling, the tubes were centrifuged for 5 min at 10.000 g. At 532 nm, the supernatant’s absorbance was measured.
Measurement of chlorophyll content
The extraction and quantification of chlorophyll were done using the Kaydan et al.18 recommended method. After being weighed, 0.2 g sample of leaves was crushed in a mortar and refrigerated at + 4 °C. The samples were extracted by crushing them thoroughly with 5 mL of 80% acetone in a dark environment in the tube. The mixture was filtered, transferred to a new tube, topped with 5 mL of 80% acetone and centrifuged at 3.000 g. The sediment-free filtrate was moved to a fresh tube. This procedure was carried out again in a medium containing 80% acetone. Using a UV/VIS Spectrophotometer, the absorbance of the green chlorophyll solution was measured at 645 and 663 nm. Chlorophyll concentrations were computed using the Witham et al.19 equation.
Enzyme extraction
At + 4 °C, the extraction procedure was conducted. 5 mL of cold sodium phosphate buffer (50 mM, pH 7.8) was used in a mortar and pestle to crush and homogenize 0.5 g of root tip. After centrifuging the homogenate for 20 min at 10.500 g, the supernatant was kept at + 4 °C until analysis20.
Measurement of SOD activity
Beauchamp and Fridovich21 method was used to test the activity of the SOD enzyme. 3 mL of reaction solution were made in total (0.01 mL enzyme extract, 0.01 mL 4% insoluble polyvinylpyrrolidone, 0.28 mL de-ionized water, 0.3 mL 130 mM methionine, 0.3 mL 750 µM nitroblue tetrazolium chloride, 0.3 mL 0.1 mM EDTA-Na2, 0.3 mL 20 µM riboflavin and 1.5 mL 0.05 M sodium phosphate buffer). Sterile tubes were filled with the prepared solution. The tubes were exposed to a 15 W fluorescent lamp for 10 min to start the reaction, and then were left in the dark for 10 min to finish it. At 560 nm, absorbance was measured. One unit of SOD activity was defined as the amount of SOD required to produce 50% inhibition of nitroblue tetrazolium chloride reduction under experimental conditions22.
Measurement of CAT activity
Beers and Sizer23 method was used to assay the activity of the CAT enzyme. UV-VIS spectrophotometry was used to measure the CAT activity in a 2.8 mL reaction mixture (0.3 mL 0.1 M H2O2, 1.0 mL distilled water and 1.5 mL 200 mM monosodium phosphate buffer) at 25 °C. 0.2 mL of enzyme extract was added to start the reaction. By tracking the drop in absorbance at 240 nm brought on by H2O2 consumption, CAT activity was determined. Based on the fresh weight of the root tip, CAT activity was measured in units per minute22.
Molecular docking
Possible interactions of ornidazole with tubulin proteins, DNA topoisomerases, glutamate-1-semialdehyde aminotransferase and protochlorophyllide reductase enzymes and DNA molecules were analyzed by molecular docking method in computer environment. The 3D structure of tubulin (alfa-1B zinciri ve tübülin beta zinciri) (PDB ID: 6RZB)24 3D structures of DNA topoisomerase I (PDB ID: 1K4T) and II (PDB ID: 5GWK)25,26 3D structure of glutamate-1-semialdehyde aminotransferase (PDB ID: 2ZSL)27 3D structure of protochlorophyllide reductase (PDB ID: 6R48)28 3D structures of B-DNA dodecamer (PDB ID: 1bna)29 B-DNA dodecamer d (PDB ID: 195d)30 and (PDB ID: 1cp8)31 DNA were obtained from the protein database. The 3D structure of ornidazole (PubChem CID: 28061) was taken from PubChem. Proteins and enzymes were prepared for docking by identifying their active sites, removing water molecules and ligands, and adding polar hydrogen atoms. Energy minimization of proteins and enzymes was performed with Gromos 43B1 using Swiss-PdbViewer32 (v.4.1.0) software, while energy minimization of the 3D structure of ornidazole was performed with the universal force field (UFF) using Open Babel v.2.4.0 software33. Kollman charges were assigned to receptor molecules and Gasteiger charges to ornidazole. Molecular docking was performed with a grid box containing the active sites of proteins and enzymes and the entire structure of DNA molecules. Docking was then performed using Autodock 4.2.6 software34 based on the Lamarckian genetic algorithm. Docking analysis and 3D visualizations were performed with the Biovia Discovery Studio 2020 client.
Statistical analysis
Statistical analysis of the data was performed using one-way analysis of variance (One-way ANOVA) and Duncan tests using the SPSS Statistics 22 (IBM SPSS, Turkey) package program. Data are shown as mean ± standard deviation (SD). P value of less than 0.05 was considered statistically significant between means.
Results and discussion
Genotoxicity parameters
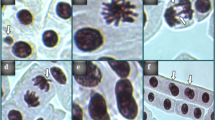
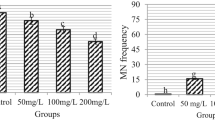
Figure 1; Tables 1 and 2 illustrate the genotoxicity induced by ornidazole treatment. The findings of the Comet test and chromosomal analysis revealed that the control (Group I) group had the lowest MN frequency, CAs numbers, and tail DNA amount, while the highest MI value and head DNA amount were found in this group. Treatment with ornidazole resulted in statistically significant (p < 0.05) increases in MN frequency, CAs numbers, and tail DNA quantity, and statistically significant (p < 0.05) decreases in MI value and head DNA amount. The most significant toxic effect was observed in the group administered 0.0714 mg/L ornidazole, and the number of dividing cells in this group decreased by 18.7% compared to the control group. MN frequency, which was observed at 0.20 ± 0.42 in the negative control group, increased significantly with ornidazole administration; this increase was recorded at 16.7 ± 1.77, 37.7 ± 2.98, and 69.3 ± 4.40 in Groups II, III, and IV, respectively. NaN₃, used as a positive control, showed a more pronounced genotoxic effect by increasing MN formation to 75.7 ± 8.27. Treatment with ornidazole promoted chromosomal abnormalities such as sticky chromosome > binuclear cell > vagrant chromosome > disorientation > unequal distribution of chromatin > bridge > nucleus bud > fragment in root meristem cells. Tail DNA ratio, a significant indicator of DNA damage, increased from 1.00 ± 1.05% in the negative control group to 75.0 ± 1.35% in the positive control group. Ornidazole applied at a dose of 0.0714 mg/L increased the tail DNA ratio to 72.0 ± 1.63%, causing DNA damage similar to that of NaN₃, a known genotoxic agent. This result demonstrates that ornidazole has a significant genotoxic effect at high doses.
Studies that thoroughly examine the genotoxicity that ornidazole or other pharmaceutical compounds cause in plant cells are quite rare in the literature. In one of these studies, Drzymała and Kalka35 showed that diclofenac and sulfamethoxazole caused a decrease in MI and increases in MN and CAs rates in Vicia faba L. The number of studies addressing drug genotoxicity in animal organisms is higher. Lopez Nigro et al.36 reported that ornidazole treatment at four different concentrations (0.1, 1.0, 10 and 50 mg/mL) caused a significant decrease in the MI of peripheral blood lymphocyte cells in culture and an increase in the number of sister chromatid exchanges and CAs. Abrevaya et al.37 found that injection of metronidazole at a dose of 133 mg/kg c.a in mice (Mus musculus), NIH mice, Webster mice and Jackson (BALB/cJ) mice caused an increase in MN frequency in all mouse strains compared to the control group. In a similar study, Nigro and Carballo38 observed that tinidazole treatment at doses of 0.1, 1.0, 10 and 50 µg/mL caused a decrease in MI and an increase in sister chromatid exchanges and CAs in cultured human lymphocytes. Gül and Yılmaz39 found that tenoxicam and piroxicam caused a decrease in MI and an increase in MN frequency in human peripheral lymphocytes. Using the Comet test, Coşkun et al.40 demonstrated that metronidazole treatment at dosages of 125, 250 and 500 mg/kg ca. resulted in a dose-dependent decrease in head DNA and an increase in tail DNA in heart tissue.

Representative images showing various chromosome abnormalities induced by 0.0714 mg/L ornidazole treatment in Allium cepa root meristem cells. Bar = 10 μm.
The reduction in mitotic index observed following ornidazole treatment is thought to result from the disruption of microtubule organization, which is essential for spindle fiber formation during cell division, or from the inhibition of key enzymatic structures involved in DNA replication. This hypothesis is supported by previous studies demonstrating that certain pharmaceutical agents, can interfere with microtubule dynamics and inhibit enzymes. Mukhtar et al.41 reported that some drugs target microtubules and disrupt the normal function of the mitotic spindle thread, thereby halting cell cycle progression in mitosis. Similarly, Cheng et al.42 observed that some drugs can cause microtubule de-polymerization by binding to tubulin proteins, thereby interfering with mitosis of cells and preventing cell proliferation. Kohanski et al.43 have shown that some antibiotics reduce cell division by binding to topoisomerase II or IV, interfering with changes in DNA overcoiling. Shapiro44 showed that some antibiotics inhibit the ligase domain of topoisomerase enzymes, reducing mitotic activity. Van Ravenstein et al.45 determined that some drugs such as etoposide and doxorubicin decrease mitotic activity by trapping the enzyme topoisomerase II, which has an important role in DNA replication, behind the replication forks, causing topoisomerase II-dependent DNA breaks and fork stalling.
Mechanistic elucidation of ornidazole genotoxicity through molecular docking-based analyses
In this study, the effect of ornidazole on mitotic activity was not only explained with literature information, but also investigated using the molecular docking method in a computer environment. In this context, ornidazole-tubulin and ornidazole-topoisomerase interactions were examined and shown in Table 3; Fig. 2. Ornidazole exhibited a binding affinity of − 4.32 kcal/mol with the alpha-1B chain of tubulin. This indicates a moderate interaction. In other words, the inhibition constant of 682.06 µM indicates that ornidazole has a moderate inhibitory potential against this target. This interaction is stabilized by multiple hydrogen bonds with residues GLN11 (two bonds), ALA12, SER140, ASN101, ASP69 and GLU71. These hydrogen bonds probably contribute to the stability of the ornidazole-tubulin complex. However, the absence of hydrophobic interactions suggests that binding is primarily driven by polar interactions. The binding affinity of ornidazole with the beta chain of tubulin was − 4.30 kcal/mol, similar to its interaction with the alpha-1B chain. The inhibition constant of 703.55 µM also supports the moderate inhibitory potential of ornidazole. The interaction involves three hydrogen bonds with ARG276, indicating a strong polar interaction with this residue. In addition, hydrophobic interactions were observed with LEU215 (three bonds), LEU217 and ARG276. These hydrophobic interactions may increase the stability of the complex. Ornidazole binding to tubulin beta may disrupt microtubule dynamics. In conclusion, ornidazole-tubulin docking findings are consistent with the decrease in mitotic activity observed in our study, as tubulin protein is a critical component of microtubules and thus spindle threads.
Ornidazole showed a higher binding affinity (-6.27 kcal/mol) with DNA topoisomerase I than tubulin with an inhibition constant of 25.33 µM. This indicates that ornidazole has a stronger inhibitory potential against the DNA topoisomerase enzyme. The interaction is stabilized by two hydrogen bonds with ASP533 and one with ARG364. In addition, hydrophobic interactions with ARG364, HIS367 and LYS532 were observed. The binding affinity of ornidazole with DNA topoisomerase II is − 5.19 kcal/mol and the inhibition constant is 156.85 µM. This indicates a moderate inhibitory effect. The interaction involves two hydrogen bonds with ARG661 and two with GLY859, as well as one hydrogen bond with GLY857 and ALA652. In addition, hydrophobic interactions with THR858 and PHE653 were also observed. Since DNA topoisomerase enzymes play a critical role in DNA replication and chromosome segregation during cell division, their interaction with ornidazole may explain the decrease in mitotic activity observed in our study.

Molecular docking interactions of ornidazole with key cellular targets. Ornidazole-tubulin alpha 1B chain (a), ornidazole-tubulin beta chain (b), ornidazole-DNA topoisomerase I (c), ornidazole-DNA topoisomerase II (d).
The reduction in DNA content, along with the increased frequency of MN and CAs observed following ornidazole treatment, may be attributed to two potential mechanisms. One possibility is that ornidazole interacts directly with DNA, leading to structural damage. Alternatively, it may enhance the generation of free radicals within the cell, which in turn could contribute to indirect genotoxic effects. These proposed mechanisms are supported by findings reported in previous studies. Shapiro44 reported that some antimicrobial drugs can bind directly to DNA and cause double-strand breaks. Similarly, Baquero and Levin46 observed that some antibiotics can directly cause single- or double-stranded DNA breaks. Arabski et al.47 showed that some antibiotics such as amoxicillin can cause DNA damage by increasing ROS production in cells. Dwyer et al.48 reported that antibiotics cause DNA double-strand breaks by causing nucleotide oxidation following ROS formation.
In this study, DNA and chromosomal damage induced by ornidazole was examined not only based on existing literature but also through molecular docking analyses to simulate the interaction between ornidazole and DNA. The results of these computational evaluations are presented in Table 4; Fig. 3. In this context, the binding interactions of ornidazole with three different DNA molecules, namely B-DNA dodecamer (1BNA), B-DNA dodecamer D (195D) and DNA (1CP8) were investigated. Ornidazole exhibits binding interactions with all three DNA molecules. However, the binding affinities differ. The free binding energy of ornidazole with different DNA molecules varies between − 4.68 kcal/mol and − 5.42 kcal/mol. This indicates a moderate interaction between ornidazole and DNA molecules. Among the DNA molecules examined, the B-DNA dodecamer (PDB ID: 1BNA) showed the strongest binding affinity with a free energy of -5.42 kcal/mol and an estimated inhibition constant of 107.07 µM. The primary interacting nucleotides in this sequence are A: G10, A: C11 and B: G16. This suggests that ornidazole associates with guanine and cytosine bases in a structural context. B-DNA dodecamer D (PDB ID: 195D) exhibited a slightly weaker binding affinity with a free energy of -4.68 kcal/mol and an inhibition constant of 369.12 µM. The key interacting nucleotides were A: G10, A: C11, B: G16 and B: T17. Thymine may play a role in stabilizing the interaction in this sequence. Finally, the third DNA structure (PDB ID: 1CP8) showed a binding energy of -5.15 kcal/mol with an inhibition constant of 169.02 µM. In particular, the interaction involved nucleotides A: G3, A: G4 and B: A7, emphasizing the potential importance of adenine and guanine residues in facilitating ornidazole binding. Overall, docking results showed that ornidazole can bind to guanine-rich nucleotide sites in DNA, especially guanine-cytosine (G-C), guanine-thymine (G-T) and guanine-guanine (G-G) pairs, and establish stable but moderate interactions with DNA types. This result implies that ornidazole can interact with cellular DNA, potentially affecting its structural stability and causing conformational changes. On the other hand, such interactions may also impair the ability of DNA to store genetic information and perform basic cellular functions. All these may explain the DNA damage and chromosomal abnormalities observed as a result of ornidazole treatment.

Visual representation of ornidazole binding to various DNA structures as determined by molecular docking. Ornidazole-1BNA (a), ornidazole-195D (b), ornidazole-1CP8 (c).
Biochemical toxicity parameters
Table 5 shows the biochemical changes caused by ornidazole treatment. The control (Group I) group had the lowest root MDA levels as well as the lowest SOD and CAT enzyme activity. This group also had the highest levels of chlorophyll a and chlorophyll b in their leaves. In addition to statistically substantial (p < 0.05) reductions in leaf chlorophyll a and b quantities, ornidazole treatment resulted in statistically significant (p < 0.05) increases in root MDA levels, SOD and CAT enzyme activities. These alterations were likewise found to be directly correlated with the ornidazole dosage. In this context, the greatest effect on biochemical toxicity was realized at 0.0714 mg/L dose of ornidazole. In group IV, which received the highest ornidazole, the MDA ratio increased by 70% compared to the control, chlorophyll a level decreased by 55.6%, and chlorophyll b decreased by 69.1%. In the control group, SOD activity increased from 69.3 ± 1.71 to 121 ± 3.84 U/mg, and CAT activity increased from 0.62 ± 0.08 to 3.19 ± 0.22 OD240nm min/g. The biochemical alterations caused by ornidazole in plant test materials have not been well examined in the literature. However, there are some studies on biochemical changes induced by other active pharmaceutical ingredients. Al-Mentafji et al.49 reported that 125 and 250 mg/L doses of chloroquine CQ used in the treatment of COVID-19 promoted H2O2 production in meristematic tissues of Mung bean (Vigna radiata), resulting in an increase in MDA levels and the activity of enzymatic antioxidants SOD and CAT. Li et al.50 determined that antibiotic applications of oxytetracycline and enrofloxacin at doses of 5, 10, 20, 40 and 80 mg/L caused an increase in CAT, SOD and peroxidase (POD) enzyme activities and MDA levels of wheat shoots and roots. Drzymała and Kalka35 showed that the application of diclofenac and sulfamethoxazole increased SOD activity in leaf tissues of Vicia faba L. Rydzynski et al.51 found that tetracycline hydrochloride reacts with the chlorophyll molecule and reduces the amount of chlorophyll.
It is believed that ornidazole damages root cell membranes and accelerates lipid degradation. This may explain the increase in root MDA levels observed following treatment. This notion is supported by certain evidence in the literature. Lin and Zhang52 reported that antibiotics cause the production of free radicals in the cell, promoting oxidative damage and causing an increase in MDA. Naguib et al.53 showed that bactericidal antibiotics promote the formation of lipid peroxyl radicals in the cell, which causes an increase in membrane lipid peroxidation and increases MDA levels. Akhtara et al.54 found that exposure to antibiotics causes oxidative stress and increases MDA levels. One possible explanation for the rise in root SOD and CAT enzyme activity following ornidazole therapy is that the drug produces free radicals in the cell, which prompt the cell to produce more antioxidant enzymes to get rid of free radicals. This notion is supported by some information found in the literature. Nassar and Borjac55 demonstrated that plants can reduce oxidative stress induced by antibiotics through the activation of antioxidant enzymes such as SOD and CAT. Jamova et al.56 reported that drugs cause the formation of ROS such as superoxide radical, hydrogen peroxide, hydroxyl radical, peroxyl radical and increase the synthesis of SOD and CAT, which are first line antioxidant defense system enzymes, to balance the harmful effects of these ROS in cells. Topal et al.57 pointed out that metronidazole applications caused an increase in enzyme levels in non-target organisms and that the possible reason for this increase could be oxidative stress.
The decrease in leaf chlorophyll content as a result of ornidazole treatment may have been caused by damage to leaf structure, impaired chlorophyll biosynthesis and changes in Mg ion concentration. This notion is supported by some information found in the literature. Liu et al.58 reported that antibiotics significantly reduce chlorophyll content and photosynthetic rate by inhibiting enzymes involved in chlorophyll synthesis. Rydzyński et al.51 showed that antibiotics reduce the amount of chlorophyll and photosynthesis by causing the loss or removal of Mg2+ ions from the chlorophyll molecule. Similarly, many chemicals are reported in the literature to cause serious changes in chlorophyll levels in non-target organisms59,60,61,62,63.
Mechanistic elucidation of ornidazole toxicity on pigment levels with molecular docking
In this study, the decrease in chlorophyll content observed following ornidazole application was supported by the interaction of ornidazole with glutamate-1-semialdehyde aminotransferase and protochlorophyllide reductase, enzymes involved in chlorophyll synthesis. These interactions were analyzed in-silico using molecular docking, and the results are shown in Table 6; Fig. 4. Ornidazole showed a binding affinity of − 4.82 kcal/mol with glutamate-1-semialdehyde aminotransferase and the inhibition constant was 294.65 µM. The interaction involved hydrogen bonds with SER120 as well as with GLY276 and VAL279 (two bonds). Hydrophobic interactions were observed with PHE27, PRO278 (two bonds), PRO67 (two bonds) and LYS271. Ornidazole showed the highest binding affinity (− 6.96 kcal/mol) with protochlorophyllide reductase enzyme and exhibited a strong inhibitory potential with a very low inhibition constant of 7.94 μm. In this interaction, hydrophobic interactions were observed with TYR219 (two bonds), ILE153, CYS222, PHE229 (two bonds) and VAL142. Protochlorophyllide reductase and glutamate-1-semialdehyde aminotransferase enzyme play important roles in chlorophyll biosynthesis, depending on amino acid metabolism. Inhibition of these enzymes by ornidazole significantly reduces chlorophyll synthesis, which can lead to a decrease in chlorophyll amount and growth disorders.

Molecular docking interactions between ornidazole and key enzymes involved in chlorophyll biosynthesis. Ornidazole-glutamate-1-semialdehyde aminotransferase (a), ornidazole-protochlorophyllide reductase (b).
Conclusion
This comprehensive study examines the biochemical and genotoxic effects of ornidazole, a compound commonly used in medical applications, on non-target organisms. It combines modern computer-assisted methods, such as the classic Allium cepa test, with molecular binding. It demonstrated that exposure to ornidazole causes significant biochemical and cytogenetic toxicity in A. cepa, as evidenced by increased DNA damage, chromosomal aberrations and oxidative stress. Molecular docking analyses corroborated these findings by revealing robust interactions between ornidazole and pivotal cellular macromolecules. These results emphasise the importance of environmental measures aimed at preventing pharmaceutical pollution and protecting non-target species. The results are therefore expected to provide a foundation for a wide range of applications and policy development, not only in basic science, but also in disciplines such as environmental toxicology, drug safety and sustainable agricultural practices. By improving our understanding of the adverse effects of drugs on non-target organisms, this study makes a significant contribution to literature on environmental risk assessment and the development of preventive strategies.
Data availability
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Moreno-Barragán, A. S. & Benalcázar-Pozo, C. A. & Bermúdez-del sol, A. Contaminación ambiental Por productos farmacéuticos y Su impacto En La Salud humana. Rev. Cienc. Méd Pinar Río 27(S1), e5855 (2023).
Sayadi, M. H., Trivedy, R. K. & Pathak, R. K. Pollution of pharmaceuticals in environment. J. Ind. Pollut Cont. 26(1), 89–94 (2010).
Shalini, K., Anwer, Z., Sharma, P. K., Garg, V. K. & Kumar, N. A review on pharma pollution. Int. J. Pharmtech Res. 2(4), 2265–2270 (2010).
Ashiwaju, B. I., Uzougbo, C. G. & Orikpete, O. F. Environmental impact of pharmaceuticals: A comprehensive review. Matrix Sci. Pharm. 7(3), 85–94 (2024).
Malmqvist, E., Fumagalli, D., Munthe, C. & Larsson, D. J. Pharmaceutical pollution from human use and the polluter pays principle. Public. Health Ethics 16(2), 152–164 (2023).
Miettinen, M. & Khan, S. A. Pharmaceutical pollution: A weakly regulated global environmental risk. Reciel 31(1), 75–88 (2022).
Bone, W., Yeung, C. H., Skupin, R., Haufe, G. & Cooper, T. G. Toxicity of ornidazole and its analogues to rat spermatozoa as reflected in motility parameters. Int. J. Androl. 20(6), 347–355 (1998).
Hizarciolu, Y. S., Zeynep, A. Y. & Ozyazici, M. Bioavailability file: ornidazole. FABAD J Pharm. Sci. 29, 133–144 (2004).
Gopinath, K. G., Wilson, B. P., Viggeswarpu, S., Mathews, P. K. & Mani, S. Ornidazole-induced ataxia in an Indian woman: A case report. SAGE Open. Med. Case Rep. 3, 2050313X15621857 (2015).
Rajesh, P., Gunasekaran, S., Gnanasambandan, T. & Seshadri, S. Experimental and theoretical study of ornidazole. Spectrochim. Acta Mol. Biomol. Spectrosc. 153, 496–504 (2016).
Bohrer, J. K. et al. Cytotoxicity and mutagenicity of female contraceptives in Allium Cepa L. Res. Soc. Dev. 11(11), e393111133841–e393111133841 (2022).
Himtaş, D., Yalçın, E., Çavuşoğlu, K. & Acar, A. In-vivo and In-silico studies to identify toxicity mechanisms of permethrin with the toxicity-reducing role of ginger. Environ. Sci. Pollut Res. 31, 9272–9287 (2024).
Fenech, M. et al. HUMN project: detailed description of the scoring criteria for the cytokinesis-block micronucleus assay using isolated human lymphocyte cultures. Mutat. Res. 534(1), 65–75 (2003).
Sharma, A. D., Gill, P. K. & Singh, P. DNA isolation from dry and fresh samples of polysaccharide-rich plants. Plant. Mol. Biol. Rep. 20, 415 (2002).
Dikilitaş, M. & Koçyiğit, A. Analysis of DNA damage in organizms via single cell gel electrophoresis (technical note): Comet assay method. J. Fac. Agric. Harran Univ. 14(2), 77–89 (2010).
Pereira, C. S. A. et al. Evaluation of DNA damage induced by environmental exposure to mercury in Liza aurata using the comet assay. Arch. Environ. Contam. Toxicol. 58, 112–122 (2010).
Unyayar, S., Celik, A., Cekic, F. O. & Gozel, A. Cadmium-induced genotoxicity, cytotoxicity and lipid peroxidation in Allium sativum and Vicia Faba. Mutagenesis 21, 77–81 (2006).
Kaydan, D., Yagmur, M. & Okut, N. Effects of Salicylic acid on the growth and some physiological characters in salt stressed wheat (Triticum aestivum L). J. Agric. Sci. 13(2), 114–119 (2007).
Witham, F. H., Blaydes, D. R. & Devlin, R. M. Experiments in Plant Physiology 1–11 (Van Nostrand Reinhold, 1971).
Zou, J., Yue, J., Jiang, W. & Liu, D. Effects of cadmium stress on root tip cells and some physiological indexes in Allium Cepa var. Agrogarum L. Acta Biol. Cracov. Bot. 54(1), 129–141 (2012).
Beauchamp, C. & Fridovich, I. Superoxide dismutase: Improved assays and an assay applicable to acrylamide gels. Anal. Biochem. 44(1), 276–287 (1971).
Pehlivan, Ö. C., Çavuşoğlu, K., Yalçın, E. & Acar, A. In Silico interactions and deep neural network modeling for toxicity profile of methyl methanesulfonate. Environ. Sci. Pollut Res. 30, 117952–117969 (2023).
Beers, R. F. & Sizer, I. W. Colorimetric method for estimation of catalase. J. Biol. Chem. 195, 133–139 (1952).
Lacey, S. E., He, S., Scheres, S. H. & Carter, A. P. Cryo-EM of dynein microtubule-binding domains shows how an axonemal dynein distorts the microtubule. Elife 8, e47145 (2019).
Staker, B. L. et al. The mechanism of topoisomerase I poisoning by a camptothecin analog. Proc. Nat. Acad. Sci. 99(24), 15387–15392 (2002).
Wang, Y. R. et al. Producing irreversible topoisomerase II-mediated DNA breaks by site-specific Pt (II)-methionine coordination chemistry. Nucleic Acid Res. 45 (18), 10861–10871 (2017).
Mizutani, H. & Kunishima, N. Crystal structure of glutamate-1-semialdehyde 2,1-aminomutase from Aeropyrum pernix (2011). https://www.rcsb.org/structure/2EPJ
Zhang, S. et al. Structural basis for enzymatic photocatalysis in chlorophyll biosynthesis. Nature 574(7780), 722–725 (2019).
Drew, H. R. et al. Structure of a B-DNA dodecamer: conformation and dynamics. Proc. Nat. Acad. Sci. 78(4), 2179–2183 (1981).
Balendiran, K., Rao, S. T., Sekharudu, C. Y., Zon, G. & Sundaralingam, M. X-ray structures of the B-DNA dodecamer d (CGCGTTAACGCG) with an inverted central tetranucleotide and its netropsin complex. Acta Crystallogr. Sec D: Biol. Crystallogr. 51(2), 190–198 (1995).
Katahira, R. et al. Solution structure of the novel antitumor drug UCH9 complexed with d (TTGGCCAA) 2 as determined by NMR. Nucleic Acids Res. 26(3), 744–755 (1998).
Guex, N. & Peitsch, M. Cswiss-model and the Swiss‐Pdb viewer: An environment for comparative protein modeling. Electrophoresis 18, 2714–2723 (2005).
O’Boyle, N. M. et al. Open babel: An open chemical toolbox. J. Cheminform. 3, 33 (2011).
Morris, G. M. et al. AutoDock4 and AutoDockTools4: Automated Docking with selective receptor flexibility. J. Comput. Chem. 30, 2785–2791 (2009).
Drzymała, J. & Kalka, J. Assessment of genotoxicity, mutagenicity, and cytotoxicity of diclofenac and sulfamethoxazole at environmental concentrations on Vicia Faba. Int. J. Environ. Sci. Technol. 21(4), 3633–3648 (2024).
López Nigro, M. M., Palermo, A. M., Mudry, M. D. & Carballo, M. A. Cytogenetic evaluation of two nitroimidazole derivatives. Toxicol. In Vitro 17(1), 35–40 (2003).
Abrevaya, X. C., Carballo, M. A. & Mudry, M. D. The bone marrow micronucleus test and metronidazole genotoxicity in different strains of mice (Mus musculus). Genet. Mol. Biol. 30, 1139–1143 (2007).
Nigro, M. L. & Carballo, M. A. Genotoxicity and cell death induced by tinidazole (TNZ). Toxicol. Lett. 180(1), 46–52 (2008).
Gül, H. & Yılmaz, E. A. Investigation of the genotoxic effects of the analgesic drugs piroxicam and tenoxicam in human lymphocytes. BSJ Eng. Sci. 6(4), 540–549 (2023).
Coşkun, F., Yalçın, E. & Çavuşoğlu, K. Metronidazole promotes oxidative stress and DNA fragmentation-mediated myocardial injury in albino mice. Chemosphere 352, 141382 (2024).
Mukhtar, E., Adhami, V. M. & Mukhtar, H. Targeting microtubules by natural agents for cancer therapy. Mol. Cancer Ther. 13 (2), 275–284 (2014).
Cheng, Z., Lu, X. & Feng, B. A review of research progress of antitumor drugs based on tubulin targets. Transl Cancer Res. 9(6), 4020 (2020).
Kohanski, M. A., Dwyer, D. J. & Collins, J. J. How antibiotics kill bacteria: From targets to networks. Nat. Rev. Microbiol. 8(6), 423–435 (2010).
Shapiro, R. S. Antimicrobial-induced DNA damage and genomic instability in microbial pathogens. Plos Pathog. 11(3), e1004678 (2015).
Van Ravenstein, S. X. et al. Topoisomerase II poisons inhibit vertebrate DNA replication through distinct mechanisms. EMBO J. 41(12), e110632 (2022).
Baquero, F. & Levin, B. R. Proximate and ultimate causes of the bactericidal action of antibiotics. Nat. Rev. Microbiol. 19(2), 123–132 (2021).
Arabski, M. et al. Interaction of amoxicillin with DNA in human lymphocytes and H. pylori-infected and non-infected gastric mucosa cells. Chem. Biol. Interac. 152 (1), 13–24 (2005).
Dwyer, D. J. et al. Antibiotics induce redox-related physiological alterations as part of their lethality. Proc. Nat. Acad. Sci. 111(20), E2100–E2109 (2014).
Al-Mentafji, H. N. H. Effect of chloroquine on antioxidant enzymes and oxidative system on (Vigna radiata) plant tissues exposed to water stress. J. Phys. Conf. Ser. 1879(2), 022030 (2021).
Li, L. et al. Effects of antibiotics stress on root development, seedling growth, antioxidant status and abscisic acid level in wheat (Triticum aestivum L). Ecotoxicol. Environ. Saf. 252, 114621 (2023).
Rydzyński, D., Piotrowicz-Cieślak, A. I., Grajek, H. & Wasilewski, J. Investigation of chlorophyll degradation by Tetracycline. Chemosphere 229, 409–417 (2019).
Lin, Y., Li, T. & Zhang, Y. Effects of two typical quinolone antibiotics in the marine environment on Skeletonema costatum. Front. Mar. Sci. 11, 1335582 (2024).
Naguib, M. et al. An evolutionary conserved detoxification system for membrane lipid–derived Peroxyl radicals in gram-negative bacteria. PLoS Biol. 20(5), e3001610 (2022).
Akhtara, N. & Bharali, M. K. Genotoxicity assessment after sub-chronic exposure to amoxicillin, azithromycin, and Ciprofloxacin in male albino mice. Mol Cell. Toxicol. 1–10 (2025).
Nassar, S. & Borjac, J. Effect of a commonly used veterinary antibiotic on oxidative stress and root transporters of edible legumes and leafy crops. BAU J. Sci. Technol. 3(2), 1–20 (2022).
Jomova, K. et al. Several lines of antioxidant defense against oxidative stress: antioxidant enzymes, nanomaterials with multiple enzyme-mimicking activities, and low-molecular-weight antioxidants. Arch. Toxicol. 98(5), 1323–1367 (2024).
Topal, T. R., Karan, M., Çavuşoğlu, K. & Yalçın, E. Metronidazole induces genotoxicity, hepato-and nephrotoxicity by triggering DNA fragmentation in Swiss albino mice (Balb/c). Sci. Rep. 15 (1), 26187 (2025).
Liu, B. et al. Growth response and toxic effects of three antibiotics on Selenastrum Capricornutum evaluated by photosynthetic rate and chlorophyll biosynthesis. J. Environ. Sci. 23 (9), 1558–1563 (2011).
Krupka, M., Piotrowicz-Cieślak, A. I. & Michalczyk, D. J. Effects of antibiotics on the photosynthetic apparatus of plants. J. Plant. Interact. 17 (1), 96–104 (2022).
Kutluer, F., Güç, İ., Yalçın, E. & Çavuşoğlu, K. Toxicity of environmentally relevant concentration of esfenvalerate and Taraxacum officinale application to overcome toxicity: A Multi-bioindicator ın-vivo study. Environ. Pollut. 373, 126111 (2025).
Kuloğlu, S. S., Çavuşoğlu, K. & Yalçın, E. LC–MS/MS phenolic profileand remedial role of urtica dioica extract against Li2CO3-induced toxicity. Environ. Sci. Pollut Res. 31(42), 54589–54602 (2024).
Annakkaya, N., Çavuşoğlu, K., Yalçin, E. & Özkan, B. Investigation of in-vivo and in-silico toxicity induced by environmental drug contamination in a non-target organism. Sci. Rep. 15(1), 21082 (2025).
Topatan, Z. Ş. et al. Alleviatory efficacy of Achillea millefolium L. in etoxazole-mediated toxicity in Allium Cepa L. Sci. Rep. 14(1), 31674 (2024).
Acknowledgements
This study has not been financially supported by any institution.
Author information
Authors and Affiliations
Contributions
M.T.G: investigation; methodology; visualization; writing-review and editing. K.Ç: conceptualization; data curation; investigation; methodology; visualization; writing-review. E.Y: conceptualization; methodology; data curation; software; visualization; writing-review and editing.A.A: methodology; data curation; software; visualization; writing-review and editing.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Gürsoy, M.T., Çavuşoğlu, K., Yalçin, E. et al. Toxicological insights into the non-target effects of ornidazole using the Allium cepa bioassay system. Sci Rep 15, 33015 (2025). https://doi.org/10.1038/s41598-025-18500-1
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-18500-1
