
Abstract
Oral fungal microbiota plays an important role in many diseases, however, the role of oral fungal microorganisms in the development of patients infected with Omicron has not been reported. A total of 963 tongue coating samples were prospectively included in this study, and finally 336 samples from patients infected Omicron variant (PIOV), 234 samples from recovered patients infected with Omicron (RP), 71 samples from patients infected original strain of Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) (PIOS), 299 samples from healthy controls (HC) completed internal transcribed spacer (ITS) sequencing after screening and quality control. By comparing the difference of oral fungal microorganisms between PIOV, RP and HC, we found that with the recovery of PIOV, their oral fungal microecological diversity increased gradually. Besides, at the species level, there were 24 oral fungal species such as Zanclospora_jonesii increased gradually, while there were 24 oral fungal species such as Saccharomyces_cerevisiae decreased gradually. In addition, by comparing PIOS and PIOV, we found that the alpha diversity of oral fungal microorganisms in PIOV was significantly lower than PIOS and the main species of the two groups were different. At the same time, we randomly divided PIOV and HC into training and validation set. Based on random forest model and five-fold cross-validation, we identified three optimal microbial markers of oral fungi and constructed a diagnostic model of PIOV. The area under the curve (AUC) value of PIOV group was 99.01% in discovery phase and 97.84% in verification phase. In summary, based on large-scale samples, this study is the first to elucidate the characteristics of oral fungal microbiota changes during PIOV recovery and establish a supplemental non-invasive diagnostic model for PIOV based on the oral fungal microbiome.
Similar content being viewed by others


Introduction
Microecosystems include fungal microecology, bacterial microecology, viral microecology, archaea and small animals (including single-celled protozoa and worms). Fungal microecology refers to the interaction and dynamic relationship between fungi, their hosts and the microorganisms around them in a specific environment. Although the abundance of fungi is much lower than that of bacteria in the human body, studies have shown that it plays an important role in the occurrence and development of human diseases.
Studies show that the structural components of fungi can affect viral infections1. For example, Candida albicans can form biofilms that reduce viral sensitivity and enhance the survival of viral particles by protecting the virus from interference by the host’s immune system and other environmental factors, thereby maintaining the infectivity of viral particles for a long time2. Besides, a previous study on mosquitoes has shown that Talaromyces modifies the gut physiology, increasing susceptibility to Dengue virus (DENV) and amplifying viral infection3. Malassezia_globosa can reduce the overall survival of breast cancer patients and promote the occurrence and development of pancreatic cancer4,5. Based on another study, it also plays an important role in proinflammation by stimulating keratinocyte secretion of IL-23 and promoting the differentiation of pathogenic Th17 cells, which is related to the occurrence of a variety of immune-related skin diseases closely6,7. Besides, Lian Narunsky Haziza and his colleagues found that the relative abundance of fungi in tumor tissues was significantly higher than in healthy tissues, and with significant differences in the relative abundance and structural composition of fungi in different tumor types8. The team’s another study found that the inflammatory response can promote candida colonization in gastrointestinal tumors with a high concentration of candida flora, and candida can maintain a pro-inflammatory environment, which is related to the up-regulation of inflammatory signaling pathways mediated by the pro-inflammatory factor IL-1 and neutrophilic leukocytosis, thus affecting the survival outcomes and cancer metastasis of gastrointestinal patients9. Our previous research found significant associations between oral fungi and bacteria, fecal fungi and bacteria, cytokines and clinical markers during recovery from Coronavirus Disease 2019 (COVID-19)10. In summary, fungal microbiota plays an important role in the occurrence and development of diseases.
The COVID-19 is still circulating around the world at a low level and continues to have an ongoing impact on the physical and mental health of people around the world, which is inseparable from its strong ability to mutate. At present, Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) has appeared Alpha, Beta, Gamma, Delta, Omicron and other mutated strains, among which Omicron has the largest number of mutation sites11. Some of its mutation sites can enhance the affinity between the viral receptor binding domain (RBD) and the receptor of human angiotensin-converting enzyme 2 (ACE2)12which enhanced the virus’ ability to spread13. In addition, Omicron continues to evolve and new subtypes are emerging, and the immune escape ability of new variants is also increasing during evolution, resulting in the faster transmission14. Although there is evidence to prove that the relatively mild symptoms of patients infected Omicron variant (PIOV) are caused by its diminished ability to replicate compared with previous SARS-CoV-2 variants15. However, a proportion person of those who have recovered from Omicron continue to have physical, psychological or cognitive symptoms that affect their quality of life. Up to now, the number of cases of Omicron infections continues to grow worldwide, placing enormous pressure on global health systems.
In this study, we were the first to characterize the oral fungal microflora of PIOV, recovered patients infected Omicron (RP), patients infected original strain SARS-CoV-2 (PIOS), healthy controls (HC) and focused on describing the changes of oral fungal microecology during rehabilitation of PIOV. Meanwhile, based on oral fungal microbiome, we established a non-invasive diagnostic model of patients infected with omicron, which could accurately distinguish healthy controls from patients infected with Omicron. It provides a new basis for targeting and regulating fungal microecology to prevent and control new outbreak of infectious diseases.
Results
Study design and characteristics of the participants
We collected 963 tongue coating samples from Henan Province, China. After a rigorous screening and exclusion process, 940 tongue coating samples were sequenced by internal transcribed spacer (ITS). Including 336 samples from PIOV, 234 samples from RP, 71 samples from PIOS and 299 samples from HC (Details can be seen on the Fig. 1). The clinical features of PIOV and HC are shown in Table 1, in the discovery cohort, the mean age of PIOV was 28.4 years old, and the ratio of males to females was 125 : 99. While the mean age of HC was 28.6 years old, and the ratio of males to females was 105 : 95. In the validation cohort, the mean age of PIOV was 29.0 years old, and the ratio of males to females was 66 : 46. The mean age of HC was 30.4 years old, and the ratio of males to females was 54 : 45. Meanwhile, we analyzed the blood routine and biochemical indexes of the two groups of patients, there were significant differences between the two groups in neutrophils (P < 0.05), platelets (P < 0.05), hemoglobin (P < 0.05) and albumin (P < 0.05) in both cohorts.

Participant flow diagram. We collected 963 tongue coating samples in this study, 336 samples from PIOV, 234 samples from RP, 71 samples from PIOS, 299 samples from HC. After the quality control, a total of 940 tongue samples (336 PIOV, 234 RP, 71 PIOS, 299 HC) completed ITS sequencing. We compared the difference of oral fungal microorganisms between PIOV, RP and HC, to analyze the dynamic characteristics of oral fungi in omicron infected patients during their recovery. And compared the oral fungal characteristics of PIOV with PIOS. Finally, we randomly divided PIOV and HC into training set and validation set at the ratio of 2:1. The best oral fungal microbial markers were identified in the training cohort, and the diagnostic model of PIOV was constructed for verification in the test cohort. Meanwhile, we collected blood samples from HC and PIOV to compare their liver and kidney function and routine blood. PIOV, patients infected Omicron variant; RP, recovered patients infected Omicron; PIOS, patients infected original strain of Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2); HC, healthy control; ITS, internal transcribed spacer.
Microbial characterization of oral fungi among PIOV, RP and HC
To explore the microecological changes of oral fungi during the rehabilitation of PIOV, we compared the characteristics of oral fungal microflora between 336 PIOV, 234 RP and 299 HC. Ace and chao indices are important indicators of species richness and microbial α diversity, the results showed that the α-diversity of oral fungal microorganisms in RP and PIOV groups were lower than HC group, and RP group was between them (P < 0.05) (Fig. 2A-B and Supplementary Table S1). The Venn diagram described that a total number of operational taxonomy units (OTUs) were shared between PIOV, RP and HC, 17,154 OTUs belonging to the PIOV group alone, 19,588 OTUs belonging to the RP group alone, and 40,243 OTUs belonging to the HC group alone (Fig. 2C). The results of nonmetric multidimensional scaling (NMDS) and principal coordinate analysis (PCoA) analysis showed that the distribution of oral fungal microbial community in RP group was significantly different from that in PIOV group and HC group, and the difference was statistically significant (Fig. 2D-F).

Oral fungal microbial diversity among PIOV group, RP group and HC group. (A,B) ace and chao maps showed that the α-diversity of oral fungal microorganisms in RP and PIOV groups was lower than that in HC group, and RP group was between the two. (C) Venn diagram displaying the overlaps between groups showed that 7361 of the total richness of 101401 OTUs were shared among the three groups, while the HC group had 40243 unique OTUs, significantly higher than the PIOV and RP groups. (D,E) he NMDS (D) and PCoA (E) analysis indicated the oral fungal taxonomic composition was conspicuously different in the three groups, and RP gradually approached HC. F) The ANOSIM analysis in the form of boxplot shows that the difference between PIOV group, RP group and HC group (R = 0.187, P < 0.05). PIOV, patients infected Omicron variant; RP, recovered patients infected Omicron; HC, healthy control; OTUs, operational taxonomy units; PCoA, principal coordinate analysis; NMDS, nonmetric multidimensional scaling; centerline, median; box limits, upper and lower quartiles; error bars, 95% CI.
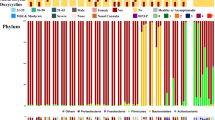
To compare the differences in the mean composition and structure of the oral fungal microbiota of PIOV, RP and HC, we calculated the relative abundance of the three groups at different levels. The results showed that the relative abundance of six phyla was significantly different between three groups (PIOV, RP, HC). To be specific, the relative abundance of Fungi_phy_Incertae_sedis and Zoopagomycota were lower in PIOV group, higher in HC group, and intermediate in RP group. However, the relative abundance of Ascomycota was higher in PIOV group and lower in HC group. The relative abundance of phylum Mucoromycota was significantly reduced in the PIOV group compared to the other two groups (Fig. 3A-B and Supplementary Table S2). We performed a differential analysis of the average composition and relative abundance of oral fungal microbiota among the three groups (PIOV, RP, HC) at the species level. (Fig. 3C-D). A total of 297 species were significantly different in relative abundance among the three groups (P < 0.05) (Supplementary Table S3). Among them, the relative abundances of 24 species, such as Zanclospora_jonesii and Cryptococcus_longus, were the lowest in PIOV group and the highest in HC group, while the relative abundances of RP group were between the two. The relative abundances of 24 species, including Saccharomyces_cerevisiae and Yarrowia_lipolytica, were the highest in PIOV group, the lowest in HC group, and the middle in RP group.

Comparison of mean composition and relative abundance of PIOV, RP and HC groups at phylum and species levels. The average composition of oral fungal flora in the three groups was compared at phylum (A) and species (C) levels. Comparison of the relative abundance of oral fungal flora in the three groups at phylum (B) and species (D) levels. *P < 0.05, **P < 0.01, ***P < 0.001. PIOV, patients infected Omicron variant; RP, recovered patients infected Omicron; HC, healthy control; OTUs, operational taxonomy units; centerline, median; box limits, upper and lower quartiles; circle or square symbol, mean; error bars, 95% CI.
Furthermore, we used Linear discriminant analysis (LDA) Effect Size (LEfSe) to find microorganisms with significant differences between three groups, and the influence of the abundance of each different species on the overall difference effect was estimated by LDA score (LDA score (log10) > 3). Cladogram diagram can show the comparison of oral fungal microorganisms in different group and select oral fungal microorganisms with significant differences among the three groups, and the abundance of these microorganisms is higher in the group corresponding to the corresponding color. (Fig. 4A). Based on the Metabolic Pathways From all Domains of Life (MetaCyc) database, we identified the metabolic pathways of microbial communities through ITS sequencing technology and found 80 enriched metabolic pathways with the most significant differences between the three groups. Among them, 22 metabolic pathways including colanic_acid_building_blocks_biosynthesis and diacylglycerol_biosynthesis_I were significantly increased in HC group. 11 metabolic pathways, such as ornithine_biosynthesis and cycle_I_prokaryotic were significantly elevated in RP group. 32 metabolic pathways such as glyoxylate_cycle, glycogen_degradation_I_bacterial were significantly increased in PIOV group. In addition, we identified 80 enriched pathways with the most significant differences. Among them, 22 metabolic pathways, including colanic_acid_building_blocks_biosynthesis and diacylglycerol_biosynthesis_I, were significantly increased in the HC group. 11 metabolic pathways, such as ornithine_biosynthesis, cycle_I_prokaryotic, were significantly increased in the RP group. 32 metabolic pathways, including glyoxylate_cycle and glycogen_degradation_I_bacterial, were significantly increased in the PIOV group (Fig. 4B and Supplementary Table S4). Furthermore, heatmap shows the difference in the relative abundance of OTUs for each sample between the PIOV, RP and HC group. Overall, there were significant differences in the composition and relative abundance of oral fungi among the three groups (details can be seen in Supplementary Fig. S1 and Supplementary Table S5).

LEfSe and LDA analyses based on OTU characterization of microbiota in PIOV (n = 336), RP (n = 234), and HC (n = 299). (A Cladogram generated by Lefse method showing the phylogenetic distribution of oropharyngeal fungal microbiota associated with PIOV, RP, and HC. Each filled circle represents one phylotype. phylum and class are represented by name on the cladogram. Order, family, genus, and species are listed on the bottom panel. Red circles in the branches represent fungal microbial communities that playing pivotal roles in PIOV; Blue circles represent fungal microbiota that playing important role in RP; Green circles represent fungal microbiota that playing important role in HC. Yellow circles represent fungal microbiome of little significance in either group. (P < 0.05, LDA > 3). (B Differentially abundant pathways across PIOV (n = 336), RP (n = 234) and HC (n = 299) identified by LEfSe. Histogram of LDA scores calculated for selected pathways showing significant difference in gene functions between PIOV (red), RP (blue) and HC (green). LDA score on log10 scale indicated at bottom. Significance of the microbial marker increases with LDA score. Based on LDA selection, the study found 32 metabolic pathways were significantly enhanced in PIOV group, 11 metabolic pathways in RP group, and 22 metabolic pathways in the HC group (P < 0.05, LDA > 3). *P < 0.05, **P < 0.01, ***P < 0.001. PIOV, patients infected Omicron variant; RP, recovered patients infected Omicron; HC, healthy control; LEfSe, Linear discriminant analysis effect size; LDA, Linear discriminant analysis. OTUs, operational taxonomy units; centerline, median; box limits, upper and lower quartiles; error bars, 95% CI.
Microbial characterization of oral fungi between PIOV and PIOS
To explore whether there were differences in the characteristics of oral fungal microbiota among different strains, we conducted ITS sequencing analysis on the tongue coating samples of PIOS (n = 71) and compared the sequencing results with PIOV group (n = 336). Ace and chao results showed that the \(\:\alpha\:\:\)diversity of oral fungal microorganisms in PIOV group was significantly lower than PIOS group (P < 0.05) (Fig. 5A-B and Supplementary Table S6). Venn diagram shows that there was a total of 3532 OTUs between PIOV group and PIOS group, while 30,453 OTUs are unique to PIOV, but only 9736 OTUs are unique to the PIOS group (Fig. 5C). The results of NMDS and PCoA analysis showed that there were significant differences in the distribution of oral fungal microbial communities between two groups, and the differences were statistically significant (P < 0.05) (Fig. 5D- F).

Oral fungal microbial diversity between PIOV group and PIOS group. (A,B) The results of ace and chao showed that the α-diversity of oral fungal microorganisms in PIOV group was significantly lower than PIOS group. (C) Venn diagram showing overlap between groups shows that the PIOV and PIOS groups have a total of 3,532 OTUs, the PIOV group has 30,453 OTUs, and the PIOS group has 9,736 OTUs. (D,E) PCoA and NMDS based on OTU distribution showed that there were significant differences in the classification composition of oral fungal microorganisms between the two groups, and the differences were statistically significant (F). PIOV, patients infected Omicron variant; PIOS, patients infected original strain of Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2); OTUs, operational taxonomy units; PCoA, principal coordinate analysis; NMDS, nonmetric multidimensional scaling; centerline, median; box limits, upper and lower quartiles; error bars, 95% CI.
In addition, we further identified differences in the mean composition and structure of the oral fungal microbiota between PIOV group and PIOS group. Firstly, we calculated the relative abundance of two groups at different species levels, and the results showed that, at the phylum level. The fungal phyla with high abundance in the two groups were the same. namely Ascomycota, Fungi_phy_Incertae_sedis, Basidiomycota and Mucoromycota, which accounted for 99.98% and 99.94% of the total microbiome abundance, respectively (Fig. 6A and Supplementary Table S7). At the species level, the main species that account for more than 71% of the total abundance of fungi in the two groups were different, In PIOV group, the higher oral fungal abundance were Fungi_spe_Incertae_sedis, Saccharomyces_cerevisiae, Yarrowia_lipolytica and so on. In PIOS group, the higher oral fungal abundance were Fungi_spe_Incertae_sedis, Zanclospora_jonesii, Aspergillus_subflavus and so on (Fig. 6C and Supplementary Table S8). Then, we compared the relative abundance of oral flora in two groups by Wilcoxon rank sum test to find the microorganisms with significant differences. At the phylum level, phylum Basidiomycota and Mucoromycota were significantly increased in PIOV group. While phylum Chytridiomycota and Zoopagomycota were significantly increased in PIOS group (Fig. 6B and Supplementary Table S9). At the species level, in PIOV group, 53 species, including Saccharomyces_cerevisiae and Yarrowia_lipolytica were significantly increased, while 77 species, including Aspergillus_subflavus, were increased in PIOS group (Fig. 6D and Supplementary Table S10).

Comparison of mean composition and relative abundance of PIOV group and PIOS group at phylum and species levels. At phylum (A) and species (C) levels, the average composition of oral fungal flora in the two groups. Comparison of the relative abundance of oral fungal flora between the two groups at phylum (B) and species (D) levels (We present the top 20 fungal species with significant differences, for complete information, please refer to Supplementary Fig. S3). *P < 0.05, **P < 0.01, ***P < 0.001. PIOV, patients infected Omicron variant; PIOS, patients infected original strain of Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2); OTUs, operational taxonomy units; centerline, median; box limits, upper and lower quartiles; circle or square symbol, mean; error bars, 95% CI.
Based on the above differences, we selected 49 OTUs with significant differences between the two groups, and the abundance content was represented by color in the heat map (Supplementary Fig. S2 and Supplementary Table S11). Finally, through LEfSe analysis, we found that there were significant differences in abundance between PIOV and PIOS from phylum to species, which we can see in the evolutionary branching diagram (Fig. 7A and Supplementary Table S12). In addition, 81 enrichment pathways with the most significant differences were identified between the two groups. There were 29 functions such as glyoxylate_cycle, heme_biosynthesis_from_glycine were significantly increased in PIOV group, and 20 functions, such as colanic_acid_building_blocks_biosynthesis and pyrimidine_ribonucleosides_degradation, were significantly increased in PIOS group (Fig. 7B and Supplementary Table S13).

Lefse and LDA analyses based on OTU characterization of fungal microbiota in PIOV (n = 336) and PIOS (n = 71). (A) Cladogram generated by Lefse method showing the phylogenetic distribution of oropharyngeal fungal microbiota associated with PIOV group and PIOS group. Each filled circle represents one phylotype. phylum and class are represented by name on the cladogram. Order, family, genus, and species are listed on the bottom panel. The size of the circle is proportional to phylotype abundance. By default, it is arranged outward from phylum to species. Red circles in the branches represent fungal microbial communities that playing pivotal roles in the PIOV group; Blue circles represent fungal microbiota that playing important role in PIOS group (P < 0.05, LDA > 3). (B) Differentially abundant pathways between PIOV (n = 336) and PIOS (n = 71) were identified by LEfSe. Histogram of LDA scores calculated for selected pathways showing significant difference in gene functions between PIOV (red) and PIOS (blue). LDA score on log10 scale indicated at bottom. Significance of the microbial marker increases with LDA score. Based on LDA selection, the study found 29 metabolic pathways were significantly enhanced in PIOV group and 20 metabolic pathways in the PIOS group (P < 0.05, LDA > 3). PIOV, patients infected Omicron variant; PIOS, patients infected original strain of Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2); LEfSe, Linear discriminant analysis effect size; LDA, Linear discriminant analysis. OTUs, operational taxonomy units; centerline, median; box limits, upper and lower quartiles; error bars, 95% CI.
Diagnostic model of PIOV based on oral fungal microorganism
We randomly divided 336 PIOV and 299 HC into discovery cohorts (224 PIOV and 200 HC) and validation cohorts (112 PIOV and 99 HC) in a ratio of 2:1. In the discovery phase, we characterized oral fungal microorganisms of 224 PIOV and 200 HC. Based on random forest model and five-fold cross-validation, three fungal markers including OTU4 (Cryptococcus_longus), OTU133954 (Raffaelea_lauricola) and OTU3 (Dioszegia_catarinonii) were identified as the best markers (Fig. 8A-B), then we constructed a diagnostic model of Omicron infection that could accurately distinguish between PIOV and HC. The probability of disease (POD) index was calculated according to the optimal OTUs. At the discovery phase, the POD value of PIOV group was significantly higher than HC group, and the area under the curve (AUC) value was 99.01% (95%CI 98.44%-99.59%, P < 0.05) (Fig. 8C-D). In the validation phase (112 PIOV and 99 HC), POD value in PIOV group was significantly higher than that in HC group, and AUC value was 97.84% (95% CI 96.38%-99.30%, P < 0.05) (Fig. 8E-F). The above results verified the accurate diagnosis effect of oral fungal microbial diagnosis model on Omicron infection. The study shows that microbial markers of oral fungi can be effective in diagnosing Omicron infection.

Identification of oral fungal classifier for PIOV. (A) Three oral fungal markers were selected as the best markers set by random forest model through five-fold cross-validation. (B) Importance distribution map of the selected microbial markers in the model. (C, D) In the train phase, the POD value in PIOV (n = 224) group was significantly higher compared with HC (n = 200) and achieved good diagnostic efficacy. Achieving an AUC of 99.01% (95%CI 98.44%-99.59%, P < 0.05). (E, F) In the validation phase, the POD value in PIOV (n = 112) was significantly higher than that in HC (n = 99) and achieved good diagnostic efficacy. Achieving an AUC of 97.84% (95% CI 96.38%-99.30%, P < 0.05). PIOV, patients infected Omicron variant; HC, healthy control; POD, probability of disease; AUC, area under the curve; centerline, median; box limits, upper and lower quartiles; circle or square symbol, mean; error bars, 95% CI.
Discussion
Research has shown that the oral microbiome plays an important role in human health and disease16,17. A healthy oral microbiota is dynamic and complex microbial communities composed of bacteria, fungi, and viruses coexist closely18. The interactions between the host and the microbiota are complex, numerous, and bidirectional, as the microbiota affects many biological functions which are important for maintaining health and immune homeostasis19,20. Furthermore, there is a bidirectional interaction between intestinal fungi and viruses: The structural components and metabolites of fungi can significantly affect viral infections, and viral infections have also been proven to regulate fungal communities1. Our previous research has found that oral bacterial microbiota is strongly associated with recovery from viral infectious diseases21. For fungi, with the rich nutrients and gentle environment for fungal symbionts provided by the mucosal surfaces of humans, these commensal fungi establish stable colonization at mucosal interfaces, where they contribute to the maintenance of intestinal homeostasis and modulation of systemic immune responses22. Another study showed that there are 9 fungal species were overrepresented in the bronchoalveolar lavage of HIV-infected individuals compared to uninfected subjects (Pneumocystis jirovecii, Junghuhnia nitida and so on). Two of these species (Pneumocystis jirovecii and Ceriporia lacerata) are known lung pathogens that are associated with immunosuppression. Additionally, P. jirovecii is a leading cause of pneumonia in HIV-infected and other immunocompromised individuals23. Therefore, elucidation of the composition and functional changes of oral fungal microbiome after Omicron infection is of great clinical significance for disease prevention and control.
Firstly, this study was the first to elucidate the oral fungal microbial characteristics in the rehabilitation process of patients infected with omicron. We found 24 species of oral fungi such as Zanclospora_jonesii and Cryptococcus_longus in PIOV were significantly reduced compared with RP and HC (P < 0.05), while 24 species, including Saccharomyces_cerevisiae and Yarrowia_lipolytica, were significantly increased. Among the fungal species that gradually increased during the recovery of patients infected with omicron, many secondary metabolites of Chaetomium globosum have many biological activities such as anti-tumor, anti-malarial, anti-fungal, anti-bacterial, cytotoxic, enzyme inhibitor and so on 24. Another study showed that some compounds in the fermentation products of the fungus Talaromyces wortmannii can inhibit the activity of human breast cancer cells MCF-7 25. While among the fungus that gradually decreased in the recovery process, Saccharomyces_cerevisiae is closely relate to the occurrence of the gut fermentation syndrome (GFS)26. Candida albicans is an opportunistic pathogen that is widely distributed in the gastrointestinal tract and genitourinary tract2. Two other studies have found that the level of Candida albicans in COVID-19 patients is increased compared with healthy controls, which is consistent with the results of this study27,28. Aspergillus_restrictus is closely related to the progression of chronic respiratory diseases29. In addition, the study identified 80 metabolic pathways with the most significant differences between the three groups. Among them, pathways that promote energy metabolism and are closely related to biosynthesis, such as glyoxylate_cycle and glucose_and_glucose_1_phosphate_degradation, were significantly enhanced in the PIOV group30. However, pathways such as methyl_ketone_biosynthesis and diacylglycerol_biosynthesis_I, which promote the balance and coordination of metabolism, were significantly enhanced in HC group. In summary, on the one hand, oral fungi may affect the occurrence and development of diseases by altering the metabolic pathways of the host. On the other hand, it may affect the homeostasis of the bacteria through the interaction between bacteria and viruses, alter the immune response of the host, thereby influencing the occurrence and development of tumors and non-neoplastic diseases.
At present, although the pandemic of the COVID-19 has ended, it is still circulating around the world at a low level, mainly because the virus is constantly mutating. A large cohort study found that, compared with uninfected people or those who had been infected only once, reinfected people showed a higher risk of all-cause death, hospitalization, and multiple other sequelae, besides, the risk was increased regardless of whether or not reinfected people had been vaccinated and how many times they had been vaccinated31. Therefore, it is very important to study the characteristic changes of virus variant strains for disease treatment and prognosis. In this study, we compared the oral fungal microecology of PIOS and PIOV for the first time, the result showed that the diversity of oral fungal microbial in PIOV was significantly lower than PIOS. In addition, at the species level, the two groups had different microbial characteristics of oral fungi and different metabolic pathways, suggesting that the changes in oral fungal microecology caused by the mutation of SARS-CoV-2 may be closely related to the clinical symptoms and clinical typing after being infected.
In recent years, there are more and more reports about using fungus as markers to diagnose diseases. Xiaowen Huang et al. found models which based on fungal microbiome markers showed superior predictive power for disease32. Another study found that fungus were associated with clinical parameters, including survival index in patients with breast and ovarian cancer, and response to immunotherapy in patients with melanoma, and that there were significant associations between specific fungus and age, tumor subtypes, smoking status, immunotherapy response, and survival measures, supporting their clinical utility as potential biomarkers and therapeutic targets8. One of our previous studies showed that fungal microorganisms can be used as diagnostic markers for COVID-19 and successfully validated their effectiveness33. In this study, we further explored the diagnostic efficacy of oral fungal microbiome as patients infected with Omicron. In the discovery stage, the diagnostic AUC value of the diagnostic model based on oral fungal microbiome for PIOV was as high as 99.01%, and in the verification stage, the AUC value was 97.84%. The above results proved the high diagnostic efficiency of oral fungal microbial diagnosis model for PIOV. This study provides a new tool for the diagnosis of emerging infectious diseases based on oral fungal microecology.
However, the human microbiota is affected by many factors, and each person may have different oral fungal microbiota due to different physical conditions, which is our limitation. Therefore, the study may also be affected by the participants’ own diet and living habits, which may lead to certain bias in our research results.
Conclusion
This project prospectively collected 940 tongue coating samples. Through ITS sequencing, it was the first to clarify the characteristics of oral fungal microecological cohorts in PIOV, RP, PIOS and HC. We found that PIOV had different oral fungal microbial characteristics with PIOS. Besides, we also found that some fungus gradually increased or decreased with the recovery of PIOV. The results indicated that the oral fungal microbiota diversity of PIOV recovered after their recovery. However, the diversity of oral fungal microbiota in RP still did not reach the level of HC. Furthermore, our findings suggest that fungal microbiota may influence clinical outcomes via interactions with host metabolic pathway. Which offer a new strategy to target fungal microbiota for disease prevention and control. At the same time, based on oral fungal microbiome, we established a non-invasive diagnostic model for PIOV, which could accurately distinguish HC between PIOV. This provides a novel supplemental diagnostic tool for virus surveillance, particularly in scenarios where RT-PCR may be unavailable or yield inconclusive results (e.g., false negatives due to low viral load). In summary, first, this study represents the first longitudinal mapping of the dynamic changes in the oral fungi during the recovery of PIOV through large-scale analysis (n = 963). Second, it establishes a fungus-based supplemental diagnostic model validated by machine learning (AUC > 97%). Finally, it provides fundamental insights into host-fungal crosstalk that recalibrates the model of infection pathogenesis.
Methods
Study design
This study is a clinical practice guided by the Declaration of Helsinki and Good Rules, and its design is based on the principles of prospective specimen collection and retrospective blind evaluation. before collecting samples, all confirmed patients and healthy controls signed the written informed consent. This research has passed the Ethics Committee of the First Affiliated Hospital of Zhengzhou University (L2021-Y429-002).
We prospectively collected tongue swab samples from some hospitalized patients in designated hospital for COVID-19 in Henan Province during January-February 2022. All patients infected Omicron were confirmed to be infected with the BA.5.2 branch of Omicron variant. The diagnosis is based on the “COVID-19 diagnosis and treatment Program Trial V.9 Guidelines” issued by the National Health Commission of the people’s Republic of China (Supporting information). All patients were treated with standard guidelines. The healthy control samples came from the volunteers in the physical examination center of the First Affiliated Hospital of Zhengzhou University. At the same time, we collected basic information from all participants, including age and gender, as well as blood samples for liver and kidney function and routine blood tests. All tongue swab samples were sequenced and analyzed based on ITS. Following rigorous screening, we included 336 patients infected Omicron and 234 rehabilitation patients when they met the discharge criteria according to the guidelines, 299 healthy controls, 71 patients infected the original strain of SARS-CoV-2 and rehabilitation patients of infected Omicron. Finally, we randomly divided 336 PIOV and 299 HC into discovery cohorts (224 PIOV and 200 HC) and validation cohorts (112 PIOV and 99 HC) in a ratio of 2:1 for subsequent analysis.
Oropharyngeal specimen collection and DNA extraction
We collected two sets of tongue swab samples from each subject to ensure sufficient valid samples. Besides, to minimize the interference of factors such as diet and drugs with oral microecology, all the subjects were required to fasting at least 8 h and gargling with normal saline 2–3 times before collecting samples. Specially trained operators scrape the tongue coat with a throat swab 2–3 times, and then put the swab head into a test tube containing 2–3 ml of the virus preservation solution. All samples were stored in a refrigerator at -80℃ after being inactivated in a warm bath at 56℃ for more than 30 min.
DNA extraction, PCR amplification, and sequencing
All samples were subjected to the same procedures for DNA extraction and PCR amplification by the same laboratory staff. Samples were suspended in 790 µl of sterile lysis buffer (4 M guanidine thiocyanate; 10% N-lauroyl sarcosine; 5% N-lauroyl sarcosine-0.1 M phosphate buffer [pH 8.0]) in 2 ml screw-cap tube containing 1 g glass beads (0.1 mm BioSpec Products, Inc., USA). This mixture was vortexed vigorously then incubate at 70℃ for 1 h. After incubation by bead beating for 10 min at maximum speed. DNA was extracted by following the manufacturer’s instructions for fungal DNA extraction using The E.Z.N.A.® Stool DNA Kit (Omega Bio-Tek, Inc., GA), which excepted lysis steps and stored at -20 °C for further analysis.
The extracted DNA from each sample was used as the template to amplify the ITS2 region of Fungi by using the primer ITS3F (5’-GCATCGATGAAGAACGCAGC-3’) and ITS4R (5’-TCCTCCGCTTATTGATATGC-3’). PCR reactions were run in a EasyCycler 96 PCR system (Analytik Jena Corp., AG). The products from different samples were indexed and mixed at equal ratios for sequencing by Shanghai Mobio Biomedical Technology Co. Ltd. using the NextSeq 2000 platform (Illumina Inc., USA) according to the manufacturer’s instructions.
Bioinformatic analysis of ITS sequencing
The sequencing data were assembled and filtered to obtain high-quality, quality-controlled sequences. These sequences were then clustered into operational taxonomic units (OTUs) at a 97% similarity threshold using UPARSE (version 7.1, http://drive5.com/uparse/). The representative sequence of each OTU was taxonomically annotated against the UNITE reference database (https://unite.ut.ee/repository.php). Subsequently, taxonomic analysis was performed to determine the species composition within the samples. Alpha diversity metrics (ACE estimator, Chao 1 estimator) were assessed by using Mothur v1.42.1. The non-parametric Mann-Whitney U test was used to test for significant differences between two groups. Comparison of multiple groups was done using a nonparametric Kruskal-Wallis test. Both Bray-Curtis, weighted and unweighted UniFrac dissimilarity were calculated in QIIME. Principal coordinate analysis (PCoA) plots and PERMANOVA which was used to test for statistical significance between the groups using 10,000 permutations were generated in R (version 3.6.0) package vegan 2.5-7. The LEfSe was used to detect taxa with differential abundance among groups (lefse 1.1, https://github.com/SegataLab/lefse). A heatmap plot of the key OUTs identified by random forest models was generated through using the ‘pheatmap’ package of the R program. A receiver operating characteristics (ROC) analysis was performed to measure the quality of the classification models by the R software package pROC. PICRUSt2 v2.4.1 (https://github.com/picrust/picrust2/wiki) was used to predict functional abundances based on ITS gene sequences. Raw sequencing data and accompanying information are available in Sequence Read Archive database under accession number PRJNA1213158.
Operational taxonomy unit (OTU) clustering and taxonomy annotation
We use abundance to obtain quantity-controlled sequence from all samples, bind OTUs using the UPARSE pipeline, and then identify representative sequences. 97% was set as the homology threshold, the gene sequences were collected into OTUs and discard the OTUs with low coverage. The total OTUs of phyla, class, order, family, genus, species levels were counted, and the phylogenetic relationships of each OTUS were marked by RDP classifier V.2.6 according to the developer’s documents.
Statistical analysis
The data was analyzed by SPSS v.20.0 for Windows (SPSS, Chicago, Illinois, USA). To compare the differences among the three groups, we use one-way analysis of variance to compare the continuous variables of normal distribution and Kruskal-Wallis test to compare the continuous variables of non-normal distribution. p < 0.05 (two-tailed) was defined as statistical significance. To compare the differences between the two groups, we use t-test to analyze continuous variables of normal distribution; use Mann Whitney U test to analyze continuous variables of non-normal distribution; and use chi-square test to analyze classified variables.
Data availability
The raw Illumina read data for all tongue coating samples have been deposited in the European Bioinformatics Institute European Nucleotide Archive database and are publicly available as of the data of publication(accession number: PRJNA1213158, https://dataview.ncbi.nlm.nih.gov/object/PRJNA1213158?reviewer=m24b5hd8chpbgnc447m6atu8u9). Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact: Zhigang Ren (fccrenzg@zzu.edu.cn).
References
Wang, Y., Liang, J., Huang, X. & Yu, X. Insights into gut fungi: fungal succession, viral interactions and clinical applications. Microb. Pathog. 206, 107775. https://doi.org/10.1016/j.micpath.2025.107775 (2025).
Nobile, C. J. & Johnson, A. D. Candida albicans biofilms and human disease. Annu. Rev. Microbiol. 69, 71–92. https://doi.org/10.1146/annurev-micro-091014-104330 (2015).
Angleró-Rodríguez, Y. I. et al. An Aedes aegypti-associated fungus increases susceptibility to dengue virus by modulating gut trypsin activity. Elife 6 https://doi.org/10.7554/eLife.28844 (2017).
Liu, M. M. et al. Breast cancer colonization by Malassezia globosa accelerates tumor growth. mBio 15, e0199324 https://doi.org/10.1128/mbio.01993-24 (2024).
Alam, A. et al. Fungal mycobiome drives IL-33 secretion and type 2 immunity in pancreatic cancer. Cancer Cell. 40, 153–167e111. https://doi.org/10.1016/j.ccell.2022.01.003 (2022).
Jia, Q. et al. Malassezia globosa induces differentiation of pathogenic Th17 cells by inducing IL-23 secretion by keratinocytes. Mycopathologia 189, 85. https://doi.org/10.1007/s11046-024-00890-x (2024).
Li, W. et al. Malassezia globosa Activates NLRP3 Inflammasome in Human Sebocytes. J Invest Dermatol 142, 2042–2046.e2011https://doi.org/10.1016/j.jid.2021.11.038 (2022).
Narunsky-Haziza, L. et al. Pan-cancer analyses reveal cancer-type-specific fungal ecologies and bacteriome interactions. Cell 185, 3789–3806e3717. https://doi.org/10.1016/j.cell.2022.09.005 (2022).
Dohlman, A. B. et al. A pan-cancer mycobiome analysis reveals fungal involvement in Gastrointestinal and lung tumors. Cell 185, 3807–3822e3812. https://doi.org/10.1016/j.cell.2022.09.015 (2022).
Ren, Z. et al. Alterations of the oral and gut mycobiome and cytokines during long-term follow-up of COVID-19 convalescents. Signal. Transduct. Target. Therapy. 8, 166. https://doi.org/10.1038/s41392-023-01417-4 (2023).
Ortega, J. T., Jastrzebska, B., Rangel, H. R. & Omicron SARS-CoV-2 variant Spike protein shows an increased affinity to the human ACE2 receptor: an in Silico analysis. Pathogens 11 https://doi.org/10.3390/pathogens11010045 (2021).
Hoffmann, M. et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 181, 271–280e278. https://doi.org/10.1016/j.cell.2020.02.052 (2020).
Shao, W., Zhang, W., Fang, X., Yu, D. & Wang, X. Challenges of SARS-CoV-2 Omicron variant and appropriate countermeasures. J. Microbiol. Immunol. Infect. 55, 387–394. https://doi.org/10.1016/j.jmii.2022.03.007 (2022).
Karim, S. S. A., Karim, Q. A. & Omicron SARS-CoV-2 variant: a new chapter in the COVID-19 pandemic. Lancet 398, 2126–2128. https://doi.org/10.1016/s0140-6736(21)02758-6 (2021).
Suzuki, R. et al. Attenuated fusogenicity and pathogenicity of SARS-CoV-2 Omicron variant. Nature 603, 700–705. https://doi.org/10.1038/s41586-022-04462-1 (2022).
Ghannoum, M. A. et al. Characterization of the oral fungal Microbiome (mycobiome) in healthy individuals. PLoS Pathog. 6, e1000713. https://doi.org/10.1371/journal.ppat.1000713 (2010).
Chen, B. Y. et al. Roles of oral microbiota and oral-gut microbial transmission in hypertension. J. Adv. Res. 43, 147–161. https://doi.org/10.1016/j.jare.2022.03.007 (2023).
Balskus, E. P. The human Microbiome. ACS Infect. Dis. 4, 1–2. https://doi.org/10.1021/acsinfecdis.7b00248 (2018).
Li, X., Liu, Y., Yang, X., Li, C. & Song, Z. The oral microbiota: community composition, influencing factors, pathogenesis, and interventions. Front. Microbiol. 13, 895537. https://doi.org/10.3389/fmicb.2022.895537 (2022).
Zheng, D., Liwinski, T. & Elinav, E. Interaction between microbiota and immunity in health and disease. Cell. Res. 30, 492–506. https://doi.org/10.1038/s41422-020-0332-7 (2020).
Ren, Z. et al. Alterations in the human oral and gut microbiomes and lipidomics in COVID-19. Gut 70, 1253–1265. https://doi.org/10.1136/gutjnl-2020-323826 (2021).
Iliev, I. D. & Leonardi, I. Fungal dysbiosis: immunity and interactions at mucosal barriers. Nat. Rev. Immunol. 17, 635–646. https://doi.org/10.1038/nri.2017.55 (2017).
Cui, L. et al. Topographic diversity of the respiratory tract mycobiome and alteration in HIV and lung disease. Am. J. Respir Crit. Care Med. 191, 932–942. https://doi.org/10.1164/rccm.201409-1583OC (2015).
Sarmales-Murga, C., Sato, M., Kosaka, M., Akaoka, F. & Watanabe, K. Mechanism of unexpected In-Trans Post-PKS polyketide reduction in cochliodone biosynthesis. J. Am. Chem. Soc. 147, 11555–11563. https://doi.org/10.1021/jacs.5c03717 (2025).
Li, X. F. et al. Wortmannine H, a phenylpentenol isolated from an endophytic fungus, talaromyces wortmannii LGT-4. Nat. Prod. Res. 35, 3204–3209. https://doi.org/10.1080/14786419.2019.1690488 (2021).
Bayoumy, A. B., Mulder, C. J. J., Mol, J. J. & Tushuizen, M. E. Gut fermentation syndrome: A systematic review of case reports. United Eur. Gastroenterol. J. 9, 332–342. https://doi.org/10.1002/ueg2.12062 (2021).
Talaga-Ćwiertnia, K. et al. Variations in oral health outcomes and mycobiome composition among COVID-19 convalescents. Sci. Rep. 15, 21638. https://doi.org/10.1038/s41598-025-05078-x (2025).
Alfaifi, A. A. et al. Oral microbiota alterations in subjects with SARS-CoV-2 displaying prevalence of the opportunistic fungal pathogen Candida albicans. Microorganisms 12 https://doi.org/10.3390/microorganisms12071356 (2024).
Arruda, L. K., Mann, B. J. & Chapman, M. D. Selective expression of a major allergen and cytotoxin, asp f I, in aspergillus fumigatus. Implications for the Immunopathogenesis of aspergillus-related diseases. J. Immunol. 149, 3354–3359 (1992).
Di, J. et al. Gut microbiota metabolic pathways: key players in knee osteoarthritis development. Exp. Gerontol. 196, 112566. https://doi.org/10.1016/j.exger.2024.112566 (2024).
Bowe, B., Xie, Y. & Al-Aly, Z. Acute and postacute sequelae associated with SARS-CoV-2 reinfection. Nat. Med. 28, 2398–2405. https://doi.org/10.1038/s41591-022-02051-3 (2022).
Huang, X. et al. Multi-kingdom gut microbiota analyses define bacterial-fungal interplay and microbial markers of pan-cancer immunotherapy across cohorts. Cell Host Microbe 31, 1930–1943.e1934 (2023). https://doi.org/10.1016/j.chom.2023.10.005
Hu, X. et al. Oral fungal alterations in patients with COVID-19 and recovered patients. Adv. Sci. (Weinh). 10, e2205058. https://doi.org/10.1002/advs.202205058 (2023).
Acknowledgements
We thank all the generous volunteer subjects who enrolled in the study. We are also very grateful to Hongyan Ren and Chao Liu (Shanghai Mobio Biomedical Technology Co., Ltd.) for their generous help in sequencing.
Funding
This work was supported by the National Key Research and Development Program of China (2023YFC3043514), Young and Middle-aged Academic Leaders of Henan Provincial Health Commission (HNSWJW-2022013), the Scientific Research and Innovation Team of The First Affiliated Hospital of Zhengzhou University (QNCXTD2023002 and ZYCXTD2023002), the Postdoctoral Fellowship Program of China Postdoctoral Science Foundation under Grant (GZC20232430).
Author information
Authors and Affiliations
Contributions
Z.R. and Z.Y. designed the study. B.R., H.W. and Z.R. collected clinical samples. Y.M., S.L., L.Liu., G.Z., J.S., Y.Z., Y.S. and L.Li. collected and analyzed the clinical data of the subjects. Y.M., and S.L. extracted the DNA and analyzed the data. Z.R. and Z.Y. performed MiSeq sequencing. Y.M. and S.L. wrote the manuscript, Z.R. and Z.Y. revised the manuscript. All authors reviewed and approved the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
This study was approved by the Institutional Review Board from the First Affiliated Hospital of Zhengzhou University (L2021-Y429-002). The study was performed in accordance with the Helsinki Declaration and Rules of Good Clinical Practice. All participants signed written informed consent after the study protocol was fully explained.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ma, Y., Liu, S., Zhang, G. et al. Dynamic alterations of oral fungal microbiota in Omicron infected patients. Sci Rep 15, 35831 (2025). https://doi.org/10.1038/s41598-025-19819-5
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-19819-5
