
Abstract
The development of effective anticancer drugs remains a central objective in pharmaceutical research. In recent years, topological indices (TIs) have gained considerable attention for their ability to numerically represent molecular structures and support predictive modeling in cheminformatics. This study aims to explore the potential of recently introduced Sombor topological indices and their entropy-based extensions within the framework of quantitative structure–property relationship (QSPR) modeling. The study will focus specifically on anticancer compounds, utilizing graph theory and edge partition approach. A comprehensive Python-based computational framework was developed to compute the relevant topological descriptors and entropy measures. The calculated indices were then integrated with statistical regression and machine learning techniques to construct and evaluate QSPR models to predict characteristics such as boiling point, molar refractivity, heavy atom count, exact mass, flash point, and polarizability. A curated dataset of anticancer agents was employed to ensure data reliability and chemical diversity. Comparative regression analyses indicate that Sombor indices exhibit stronger predictive performance and higher statistical significance than their entropy-based counterparts. These findings highlight the promise of Sombor indices as reliable molecular descriptors for QSPR modeling and powerful tools in the cheminformatics-guided drug discovery process.
Similar content being viewed by others
Introduction
The development of effective anticancer drugs remains an important goal in the field of medicinal chemistry and pharmaceutical sciences. In recent years, the integration of chem-informatics and computational methods has significantly accelerated the drug discovery process, in particular by enabling the development of predictive modeling techniques1. In this context, quantitative structure-property relationship (QSPR) modeling has emerged as a powerful tool for establishing mathematical relationships between the chemical structures of compounds and their biological or physicochemical properties2.
Topological indices (TIs), which play a central role in QSPR modeling, are numerical descriptors derived from the molecular graph of a compound. As they encapsulate fundamental structural information, these indices enable the prediction of molecular properties without the need for experimental procedures. The first theoretical QSPR approaches can be traced back to the late 1940s2. These approaches correlated biological activities and physicochemical properties with theoretical numerical indices derived from molecular structure. Many TIs, ranging from classical degree-based indices such as the Randić index3 to the distance-based Wiener index4 and Zagreb indices5, have been successfully applied to the modeling of various molecular properties over the years6,7,8,9,10. The main advantage of these indices is their ability to establish strong correlations with both structural and physico-biological properties. In recent years, considerable research has been devoted to exploring and refining TIs due to their efficacy in QSPR analysis. The authors analyzed the molecular structures of drugs related to lung cancer treatment by computing various topological indices, including degree11, neighborhood12, and reverse-degree based indices13. The study in14 aimed to construct a QSPR model for 14 tuberculosis drugs by employing Revan degree–based topological indices to predict key physicochemical properties. Similarly, the work in15 applied QSPR analysis to 19 prostate cancer drugs, computed various topological indices, and compared them across 13 physicochemical properties to assess their predictive performance. In16,17, computational chemistry approaches were integrated with machine-learning techniques to investigate the relationships between diverse topological descriptors and the physicochemical characteristics of the examined compounds. Furthermore18, reported the computation of topological descriptors for several colorectal cancer drugs and evaluated their utility in predicting four physicochemical properties through QSPR modelling.
Recently, a new generation of topological descriptors has been developed beyond these classical indices. In particular, the Sombor topological index, proposed by Gutman in 2021 19, is defined based on the vertex degrees and provides a more balanced structural representation. The Sombor index provides a richer description in terms of topological information, especially when considering the edge structure of the molecular graph. The QSPR models based on ve-degree Sombor indices for predicting key properties of aromatic heterocyclic compounds were developed in20. The molecular structures of antiviral drugs were examined using graph theory and the edge-partition approach in21. The predictive performance of the Sombor index and its variants was evaluated using regression models developed for key PAHs in22. A theoretical investigation of Sombor indices is provided within the framework of chemical graph theory in23.
Shannon entropy quantifies the degree of unpredictability or uncertainty within a data set, where higher entropy indicates greater complexity and randomness, while lower entropy reflects more order and predictability24,25. A comparative study of the two versatile framework topologies, BCT and DFT, is presented and an entropy-based structural characterization is provided using bond-wise scaled comparison in26. Entropy-based descriptors and degree-based topological indices were generated from molecular graph structures using edge partitioning and computed for anticancer drugs with a Python-based algorithm in27. Entropy-based measures, when combined with TIs, offer a powerful framework for quantifying the structural complexity, diversity, and information content of molecular graphs28. The incorporation of principles from information theory enables these descriptors to provide a probabilistic perspective on molecular symmetry and irregularity features that are frequently critical in determining chemical behavior and biological activity. This is particularly evident in the context of pharmaceutical compounds, where entropy measures complement traditional topological indices by capturing subtle variations in molecular structure that influence drug performance and efficacy29.
The identification and development of anticancer pharmaceuticals remains a key challenge in pharmaceutical research due to the heterogeneity and complexity of cancer. Computational techniques such as QSPR modeling have become widespread in experimental drug screening. This is driven by the dual objectives of enhancing efficiency and reducing the time and costs associated with the process. These approaches depend on molecular descriptors, with TIs derived from graph theory being particularly significant.
The 30 pharmaceutical compounds examined in this study represent a broad therapeutic spectrum within the field of oncology. These pharmaceuticals encompass agents employed in the management of diverse solid tumors, including those affecting the breast, lung, prostate, and bladder. Moreover, they are extensively utilized in the management of various hematological malignancies (blood cancers), such as leukemia, T-cell lymphoma, and multiple myeloma. The portfolio under consideration includes a variety of pharmacological approaches to cancer treatment, including targeted therapies, alkylating agents, and chemotherapy-supportive agents. These medications constitute the foundation of personalized therapeutic strategies guided by cancer type, disease stage, and individual genetic variability. To quantitatively elucidate the relationship between the structural characteristics of these compounds and their anticancer activities, this study introduces a systematic, Python-based computational framework for QSPR modeling. Within this framework, Sombor topological indices and their entropy-based extensions are employed to capture the underlying molecular information. A dedicated Python program has been specifically developed to automate the computation of these indices and facilitate the modeling process. The statistical significance of the data is ascertained through the implementation of Python during the analytical and modeling procedures. This multifaceted approach has been demonstrated to enhance the accuracy of modeling, whilst also enabling a comparative analysis of the performance of different regression models. This work provides a novel comparative evaluation of Sombor and entropy-based topological indices, demonstrating their potential as reliable predictors in anticancer drug modeling.
Motivation and methodology
Cancer continues to represent a significant global health challenge, thus necessitating the development of efficient and cost-effective methodologies to facilitate a comprehensive understanding and optimization of the physicochemical properties of anticancer compounds. The employment of experimental techniques for the characterization of these properties is frequently accompanied by significant expenses and extended periods of time. This underscores the necessity for reliable computational alternatives. In this study, graph-theoretical modeling is employed within a QSPR framework to analyze molecular structures. This modeling is based on degree-dependent and entropy-related topological indices. This approach provides an interpretable and low-cost method for assessing drug properties and offers valuable insights that may facilitate the rational design and discovery of more effective anticancer agents. The structural framework of the methodology, along with the tools employed throughout the study, is illustrated in the flowchart provided in Figure 1.

Flow chart of methodology.
In chemistry, TIs play a crucial role in the study of the structure and properties of chemical molecules. These indices are derived from the underlying molecular structure of the chemical molecule, which is represented as a graph. In this graphical representation, the atoms of a molecule are represented as vertices, while the chemical bonds connecting them are represented as edges. In this article, \(\mathscr {G}\) is denoted as a molecular graph, with \(\mathscr {G} = (V(\mathscr {G}), E(\mathscr {G}))\), where \(V(\mathscr {G})\) represents the set of vertices (atoms) and \(E(\mathscr {G})\) represents the set of edges (chemical bonds) in the graph. Any two vertices u and v of a graph \(\mathscr {G}\) are said to be adjacent or neighboring vertices if there exists an edge \(uv \in E(\mathscr {G})\) connecting them. The degree of a vertex \(u \in V(\mathscr {G})\) is defined as the number of edges that are connected to vertex u, denoted by d(u).
The general mathematical form of degree based topological index (\(\mathcal{T}\mathcal{I}\)) with function \(\psi\) is defined as
where \(\psi (d(u),d(v))\) is a real function of d(u) and d(v) with \(\psi (d(u),d(v))\ge 0\). The entropy measure based on the topological index function \(\psi\) is given by
The formulations of \(\psi (d(u),d(v))\) for the degree-based TIs and entropies of a graph \(\mathscr {G}\) are given in Table 1.
The Sombor index, a geometric approach to degree-based TIs, was introduced by Gutman19. The fundamental concept of this innovative index is to associate a pair \((X, \Upsilon )\) with an edge uv, where \(X = max \{d(u), d(v)\}\) and \(\Upsilon = min \{d(u), d(v)\}\). This is to say that each edge uv is mapped to a specific point \((X, \Upsilon )\) in a two-dimensional metric space \(R^2\), where the distance from the origin is defined as \(d(X, \Upsilon ) =\sqrt{X^2+ \Upsilon ^2}\). The Sombor index \(SO(\mathscr {G})\) is obtained by summing up the values of \(d(X, \Upsilon )\) for all edges in a graph \(\mathscr {G}\). In the aforementioned paper, Gutman proposed a reduced version of the Sombor index, which was termed the reduced Sombor index. In consideration of the Sombor and reduced Sombor indices, Kulli and Gutman30 proposed a modified version of these indices. Motivated by the work of Sombor indices, Kulli commenced the first and second Banhatti–Sombor indices31,32.
In 2024, Gutman et al. introduced an alternative version of the Sombor index, termed the “elliptic Sombor index.” This index refers to the orbits of planets in the solar system and incorporates elliptic orbits with the Sun as the focus point. In the field of astronomy, the perimeter of an ellipse is of paramount importance. The elliptic Sombor index, a crucial metric in the study of celestial mechanics, was derived from this fundamental principle33. A new type of Sombor index, i.e, the Euler Sombor index was proposed and discussed by Gutman34and Tang et al35. This index in question is derived from an approximate expression of the circumference of an ellipse. Consequently, it can be regarded as a geometry-based invariant.
In order to assess the practical implications of the theoretically defined Sombor indices, drug molecules used in the treatment of various types of cancer have been included in the analysis. In this context, the pharmaceutical properties of the compounds examined in the study are presented below.
In this study, a total of 30 drugs have been discussed. Afatinib and Alpelisib have been shown to be effective in solid tumours, including those of the lung and breast cancer types36,37. Conversely, Anastrozole has been identified as particularly effective in cases of hormone receptor-positive breast cancer. Belinostat is a widely used drug in the treatment of haematological cancers, such as T-cell lymphoma. Bortezomib, Lenalidomide and Pomalidomide are used in the treatment of multiple myeloma38. Alkylating agents such as Busulfan, Carmustine and Lomustine are particularly used in the treatment of leukaemia and brain tumours. In the treatment of chronic myeloid leukemia (CML), Dasatinib and Nilotinib are commonly used, whereas Daunorubicin and Mitomycin C are employed against a range of tumors, including leukemia and bladder cancer. Erdafitinib is used to treat advanced bladder cancer, while Flutamide and Orgovyx are used to treat prostate cancer39. Futibatinib and Repotrectinib act as targeted therapy agents, while Granisetron is a pharmaceutical agent used to prevent chemotherapy-induced nausea and vomiting. Melphalan is used to treat epithelial ovarian cancer. Brutinib, Zanubrutinib, Prednisone, and Midostaurin have demonstrated efficacy in the treatment of lymphoma and certain types of leukemia. Olaparib is used for managing BRCA-mutated ovarian and breast cancers, while Olutasidenib is employed in treating IDH1 mutation–positive acute myeloid leukemia (AML). Plerixafor is indicated for stem cell mobilization, and the treatment of T-cell lymphoma involves the use of Pralatrexate40.

Chemical structures: (a) Afatinib, (b) Alpelisib, (c) Anastrozole, (d) Belinostat, (e) Bortezomib, (f) Busulfan, (g) Carmustine, (h) Dasatinib, (i) Daunorubicin, (j) Erdafitinib, (k) Flutamide, (l) Futibatinib, (m) Granisetron, (n) Ibrutinib, (o) Lenalidomide.

Chemical structures (continued): (a) Lomustine, (b) Melphalan, (c) Midostaurin, (d) Mitomycin C, (e) Nilotinib, (f) Olaparib, (g) Olutasidenib, (h) Orgovyx, (i) Plerixafor, (j) Pomalidomide, (k) Pralatrexate, (l) Prednisone, (m) Repotrectinib, (n) Ribociclib, (o) Zanubrutinib.
In the context of oncological treatments, Ribociclib has been utilized for the management of hormone receptor-positive and HER2-negative breast cancer. These medications form the basis of personalised treatment approaches that are informed by cancer type, disease stage, and individual genetic variations. It is imperative to acknowledge that the information pertaining to pharmaceuticals is intended solely for educational purposes and should not be construed as a substitute for professional medical counsel41. The molecular structures of the thirty cancer drugs discussed in this study, illustrated in Figures 2−3, were drawn using Chemcraft visualizer [Chemcraft - graphical software for visualization of quantum chemistry computations. Version 1.8, build 682. https://www.chemcraftprog.com].
Topological descriptors calculation
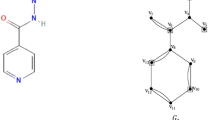
In this section, we present the mathematical calculations for the representative molecular graph \(\mathscr {G}\) of Belinostat, whose edge set is partitioned based on the vertex degrees of its 22 vertices, as shown in Figure 4. The subsets \(E_{i,j}=\{uv \in E(\mathscr {G}): d(u)=i,\ d(v)=j\}\) denote the equivalence classes of \(E(\mathscr {G})\) based on the terminal degrees. For Belinostat, we have \(\left| E_{1,2}\right| = 1\), \(\left| E_{1,3}\right| = 1\), \(\left| E_{1,4}\right| = 2\), \(\left| E_{2,2}\right| = 7\), \(\left| E_{2,3}\right| = 10\), \(\left| E_{2,4}\right| = 1\), and \(\left| E_{3,4}\right| = 1\). By applying equations in Table 1 for topological indices, the following results are obtained:

Graph for Belinostat.
Table 2 provides a concise overview of the edge partitions of the molecular graphs for the anticancer drugs under investigation. The edges are partitioned according to the degrees of their terminal vertices (e.g., edges of type (1,2), (2,3), etc.). This classification forms the basis for computing the Sombor-like topological indices and their entropy-related descriptors through mathematical simplification, as illustrated for the structure of Belinostat. Such calculations for the 30 structures considered in this study are both time- and space-consuming; however, within the Python framework, these partitions were automatically generated for each compound and subsequently utilized in all index and entropy calculations.
As illustrated in Algorithm 1, the degree-based topological indices are subsequently computed through the utilization of a Python script that has been developed for the purpose of this study. The program commences by importing the requisite libraries, including networkx and pandas for the purpose of numerical calculations. Please refer to Tables 3 and 4 for the calculated values of the topological indices and entropies, respectively.

Python code to compute several topological indices (TIs) and their entropies.
Modeling structure property through topological methods
QSPR analysis is a computational method for predicting a chemical compound’s properties based on its molecular structure. The selected drugs’ physicochemical properties and associated TIs have been elucidated using the QSPR modeling approach. This study investigated six physicochemical properties of the anticancer drugs in question: boiling point (BP), molar refractivity (MR), heavy atom count (HAC), exact mass (EM), flash point (FP), and polarizability (P). These properties play a crucial role in determining how a drug behaves, interacts, and performs within biological systems, influencing absorption, distribution, metabolism, and excretion, and ultimately affecting overall drug efficacy and safety. As shown in Table 5, the chemical and physical properties of the drugs under investigation have been obtained from the following sources: PubChem42 and ChemSpider43.
In this section, a rigorous evaluation of linear, quadratic and cubic regression models is conducted. In this analysis, the independent variables are the previously calculated TIs, and the dependent variables are the properties of the pharmaceutical compounds under investigation. \(\mathcal {Y}=\alpha +\beta \ \left( \mathcal{T}\mathcal{I}\right)\), \(\mathcal {Y}=\alpha +\beta \ \left( \mathcal{T}\mathcal{I}\right) +\gamma \ \left( \mathcal{T}\mathcal{I}\right) ^2\) and \(\mathcal {Y}=\alpha +\beta \ \left( \mathcal{T}\mathcal{I}\right) +\gamma \ \left( \mathcal{T}\mathcal{I}\right) ^2 +\delta \ \left( \mathcal{T}\mathcal{I}\right) ^3\) define the linear, quadratic and cubic regression relationship, respectively, with \(\mathcal {Y}\) describing the physical property of the drug, \(\mathcal{T}\mathcal{I}\) representing the topological index, and \(\alpha\), \(\beta\), \(\gamma\) and \(\delta\) serving as regression parameters.
A comprehensive array of statistical evaluations has been conducted to assess the models, incorporating correlation-based and error-based performance measures, including the correlation coefficient (R), the coefficient of determination (\(R^2\)), root mean square error (RMSE), mean absolute error (MAE), mean percentage error (MPE), the standard error (SE) of the predictions, the significance level (p-value), and Fisher’s statistical measure (F). The predictive capability of the model is evaluated using the RMSE metric, with the optimal model exhibiting the minimum error. RMSE is calculated as the square root of the mean of the squares of the differences between the predicted parameter value and the actual parameter value, divided by the sample size. The correlation coefficient gets closer to 1 when the results of experiments and theoretical predictions match each other. Furthermore, R-squared \((R^2)\), also referred to as the coefficient of determination, is a statistical metric that quantifies the extent to which the independent variables explain the variability in the dependent variable. It represents the extent to which the independent variables account for the variance in the dependent variable. The MAE is a metric that quantifies the average absolute deviation of the model’s predictions from the actual values, thereby indicating the overall magnitude of prediction errors. The MPE is a metric that calculates the average prediction bias in percentage terms, thus indicating whether the model systematically overestimates or underestimates the target values. The SE is a quantitative metric that quantifies the dispersion of the predictions around the mean, thereby providing an indication of the level of uncertainty associated with the model’s estimates.
Tables 6, 7 and 8 show the relationship between the experimental properties and different topological indices using a linear, quadratic and cubic regression models, respectively, where the highest correlation values are shown in bold. Notably, \(BSO_2\) exhibits a strong positive correlation with BP and FP. The correlation coefficients of 0.99 (linear\(\backslash\)quadratic) and 0.9904 (cubic) indicate a very strong positive relationship between mSO and both MR. The correlation coefficients of 0.9899, 0.99, 0.9903 indicate a very strong positive relationship between mSO and both P in linear, quadratic and cubic models, respectively. Similarly, \(BSO_1\) shows a significant positive correlation with HAC (R = 0.999). Furthermore, \(BSO_1\) shows a strong positive correlation with EM in all models, as evidenced by its high R-value of 0.989, 0.9911, and 0.9923, respectively. Subsequently, the utilization of linear, quadratic and cubic regressions facilitate the establishment of best mathematical models encompassing the physicochemical properties and topological indices. The best model was determined based on the \(R^{2}\), RMSE, and p-values. The performance metrics of best regression models, as detailed in Table 9, demonstrate their efficacy and accuracy in predicting molecular properties.
The linear, quadratic and cubic models explain 84.8% of the variation in BP and \(BSO_2\), as indicated by \(R^2= 0.848\). It accounts for 98% of the variation in MR and mSO (\(\hbox {R}^{2} = 0.980\)) in linear and quadratic models. The cubic model explains 98.1% of the variation in MR and mSO, as indicated by \(R^2= 0.981\). The \(R^2\) value of 0.998 demonstrates that the models provides an almost complete explanation (99.8%) of the variations in HAC and \(BSO_1\). Similarly, 97.8% of the variation in EM and \(BSO_1\) is explained by the linear model (\(R^2 = 0.978\)), while 98.2% and 98.5% of the variation are captured by the quadratic and cubic models (\(\hbox {R}^{2} = 0.982\) and \(R^2 = 0.985\), respectively). For all three models, 85.7% of the variation in FP and \(BSO_2\) is explained (\(R^2 = 0.857\)). Furthermore, the linear and quadratic models show a remarkable ability to explain 98% of the variation in P for mSO (\(R^2 = 0.980\)), while the cubic model accounts for 98.1% of the variation (\(R^2 = 0.981\)). As indicated by the statistical parameters displayed in Table 9, the ensuing conclusions can be derived concerning the linear, quadratic and cubic regression models for the indices in question. For all of the properties that were examined, the SE, MPE, and MAE values of the linear, quadratic, and cubic models were very close to each other. This indicates that the three regression forms demonstrate similar levels of predictive accuracy. For BP\(\sim BSO_2\), the cubic model provides slightly lower SE and MAE values, although the differences are minimal. A similar pattern is observed for MR\(\sim mSO\), HAC\(\sim BSO_1\), EM\(\sim BSO_1\), FP\(\sim BSO_2\), and P\(\sim mSO\), where the cubic model consistently yields the smallest SE and MAE values, but the improvements over the linear and quadratic models remain marginal. The error-based metrics demonstrate that all three models demonstrate high accuracy across all properties. The cubic models offer slightly better predictive stability and lower error levels, although the gains in performance are negligible.

The best predictors are based on the linear regression analyses.

The best predictors are based on the quadratic regression analyses.

The best predictors are based on the cubic regression analyses.

Collective comparison of linear, quadratic, and cubic regression models.
The best predictors estimated across all regression models for the considered physicochemical properties (BP, MR, HAC, EM, FP and P) are visualized in Figures 5, 6 and 7, while Figure 8 provides a collective comparison of linear, quadratic, and cubic regression curves to highlight differences in model behavior across polynomial degrees.
In the following part, the relationship between entropies and the physicochemical properties of the drugs under investigation is examined. In Tables 10, 11, and 12, where the highest correlation values are shown in bold, linear, quadratic, and cubic regression analyses have been used to calculate the correlation coefficients between the entropy values and the physicochemical characteristics of the drugs, respectively. In Table 10, a high R-value indicates a strong positive correlation between \(\mathcal {I}_{BSO_1}\) and MR, HAC, EM and P, with values of 0.9552, 0.9715, 0.9373 and 0.9550, respectively. Similarly, \(\mathcal {I}_{BSO_2}\) exhibits significant positive associations with FP, as reflected by its high R-value of 0.9550. Furthermore, \(\mathcal {I}_{ESO}\) shows a strong positive correlation with BP, with R-value of 0.9103. In Tables 11 and 12, a high R-value demonstrates a strong positive relationship between \(\mathcal {I}_{BSO_1}\) and MR as well as P. Likewise, \(\mathcal {I}_{ESO}\) displays a strong positive correlation with BP, HAC, and FP. Moreover, \(\mathcal {I}_{mRSO}\) reveals a significant positive association with EM.
We now implement the linear, quadratic, and cubic regression models to establish the best mathematical relationships encompassing physicochemical properties and entropies. As demonstrated in Table 13, the performance metrics of the best regression models indicate their effectiveness and accuracy in predicting molecular properties. However, a comparison of Tables 9 and 13 shows that the correlations obtained using topological indices for each physicochemical property are more meaningful than those obtained using entropies. Therefore, the comparative analysis will be performed using topological indices in the following section.
A comparative analysis approach
In the case of linear regression models, the findings of this study indicate that BP and FP can be effectively predicted using \(BSO_2\). The mSO index successfully predicts P and MR, while the \(BSO_1\) index proves effective in predicting HAC and EM. We compared our models with existing models for drugs used in cancer treatment, cited in references44,45,46. The following observations are revealed: In the study referenced in44, it was proposed that, upon consideration of ten drugs and boiling point, the highest correlation for Detour index (D) was identified to be
In45, thirteen drugs are considered and the highest correlation for the harmonic index (H) was identified to be
As mentioned below, the highest correlation for boiling point is obtained in the linear model that we have suggested with thirty drugs and by involving the \(BSO_2\) index.
In the study referenced in45, it was proposed that, upon consideration of thirteen drugs and flash point, the highest correlation for the harmonic index (H) was identified to be
In the study referenced in46, it was proposed that, upon consideration of ten drugs and flash point, the highest correlation for Randic index (R) was identified to be
The highest correlation for flash point was obtained in the linear model that was suggested with thirty drugs and the \(BSO_2\) index, as mentioned below:
This study extends the dataset and the methodological advancements beyond previous studies in44,45,46. In44 and46, several physicochemical properties of ten anticancer drugs were predicted using topological indices through linear regression and its derivatives. Similarly, in45, employed nine indices to predict five physicochemical properties of thirteen anticancer compounds using a linear regression approach. In contrast, the present study extends these previous frameworks by incorporating a larger and more diverse dataset of anticancer agents and by employing higher-order regression models, including quadratic and cubic forms. This comprehensive framework enables more accurate and robust prediction of physicochemical properties and contributes to the development of more reliable QSPR models for anticancer compounds.
The comparisons of Tables 6, 7, 8 and 10, 11, 12 show that the correlations obtained using topological indices for each physicochemical property are higher than those obtained using entropies. Consequently, in the comparative analysis, the most effective predictive topological indices for each property are utilized in this section. Tables 14, 15, and 16 present comparisons of actual and predicted values for the most effective predictive models within the linear, quadratic, and cubic regression frameworks, respectively. The results further reinforce the robustness and predictive reliability of the QSPR approach proposed in this study.
Limitations and future work
The findings of this study emphasize the significance of Sombor TIs and their associated entropies in modeling the physicochemical properties of thirty selected anticancer drugs. Although the proposed models exhibit good predictive accuracy, this study is not without limitations. While a larger dataset could improve the generalizability of the findings, the primary objective was to investigate the correlations between drug properties and degree-based topological indices, along with their entropy extensions. The extension of this approach to predict biological activity or toxicity would require more comprehensive data and advanced descriptors. Despite the limited nature of the dataset, analogous sample sizes have been employed in prior QSPR studies, provided they are supported by rigorous statistical validation. The reliability of the model was confirmed through the utilization of internal validation metrics, including \(R^{2}\), RMSE, and p-values. The trends obtained are consistent with those reported in the extant literature, thereby further substantiating the robustness of the methodology.
Future research directions may include the incorporation of machine learning algorithms for the enhancement of predictive capabilities, extending beyond conventional regression models to capture more intricate correlations within the data, thereby facilitating a deeper and more sophisticated understanding of the complex interplay between cancer cells and their therapeutic response to novel pharmacological agents.
Conclusion
This research makes a substantial contribution to the fields of computational chemistry, QSPR modeling, and molecular design. The findings highlight the effectiveness of degree-based topological indices in elucidating the complex nature of molecular interactions. The developed models utilize topological indices derived from hydrogen-depleted molecular graphs of anticancer drugs to quantitatively characterize molecular structure and connectivity, as well as to predict key physicochemical properties of the compounds. These predicted properties are closely associated with a drug’s biological activity, stability, and interactions with cellular mechanisms, making them highly valuable for the early identification of promising drug candidates. Furthermore, the proposed models provide a scalable and generalizable framework for analyzing novel molecules, thereby laying a solid foundation for the discovery of more effective and personalized cancer therapies.
Data availability
All data used in the research are included in the manuscript.
References
Bajorath, J. Integration of virtual and high-throughput screening. Nat. Rev. Drug Discov. 3(2), 140–146. https://doi.org/10.1038/nrd1301 (2004).
Todeschini, R. & Consonni, V. Molecular Descriptors for Chemoinformatics (Vols. 1–2) (Wiley-VCH, 2009). https://doi.org/10.1002/9783527628766
Randic, M. On characterization of molecular branching. J. Am. Chem. Soc. 97(23), 6609–6615. https://doi.org/10.1021/ja00856a001 (1975).
Wiener, H. Structural determination of paraffin boiling points. J. Am. Chem. Soc. 69(1), 17–20. https://doi.org/10.1021/ja01193a005 (1947).
Gutman, N. & Trinajstič, N. Chem. Phys. Lett. 174, 535 (1972).
Trinajstič, N. Chemical Graph Theory 2nd edn. (CRC Press, 1992).
Kumar, V. & Das, S. On structure sensitivity and chemical applicability of some novel degree-based topological indices. MATCH Commun. Math. Comput. Chem. 92, 165–203 (2024).
Zaman, S. et al. QSPR analysis of some novel drugs used in blood cancer treatment via degree based topological indices and regression models. Polycycl. Aromat. Compd. 44(4), 2458–2474 (2024).
Arockiaraj, M. et al. QSPR analysis of distance-based structural indices for drugs in tuberculosis treatment. Heliyon 10, e23981 (2024).
Hayat, S. et al. Predictive potential of eigenvalues-based graphical indices for determining thermodynamic properties of polycyclic aromatic hydrocarbons with applications to polyacenes. Comput. Mater. Sci. 238, 112944 (2024).
Kara, Y. et al. Computational insights and predictive models for lung cancer molecular structures. Chem. Pap. 79, 1869–1878 (2025).
Saǧlam Özkan, Y. & Kara, Y. Topological coindices and QSPR analysis for some potential drugs used in lung cancer treatment via CoM and CoNM-polynomials. Phys. Scr. 99, 105058 (2024).
Arockiaraj, M. et al. Comparative study of degree, neighborhood and reverse degree based indices for drugs in lung cancer treatment through QSPR analysis. Scientific Reports 15(1), 3639 (2025).
Shenoy, B. G. et al. Statistical analysis of Revan topological indices for drugs used in treatment of tuberculosis. Physica Scripta 100(3), 035203 (2025).
Balasubramaniyan, D. et al. Estimating physico-chemical properties of drugs for prostate cancer using degree-based and neighbourhood degree-based topological descriptors. Physica Scripta 99(6), 065233 (2024).
Ahmed, W. et al. Topological and statistical regression study of chemical structures using graph-theoretic descriptors: applications to cancer therapeutics. Chemical Papers, 1-23 (2025).
Kara, Y. et al. Data-driven QSPR analysis of anti-cancer drugs using python-based topological techniques. Journal of the Indian Chemical Society, 101993 (2025).
Nasir, S. Topological descriptors of colorectal cancer drugs and characterizing physical properties via QSPR analysis. International Journal of Analytical Chemistry 1, 5512172 (2025).
Gutman, I. Geometric approach to degree-based topological indices: Sombor indices. MATCH Communications in Mathematical and in Computer Chemistry 85(1), 105–118 (2021).
Rauf, A. et al. Quantitative structure-property relationship (QSPR) modeling for evaluating fluorescence attributes across various aromatic heterocyclic compounds with ve-degree-based Sombor indices. Chem. Pap. 78(11), 6343–6354 (2024).
Samiei, Z. & Movahedi, F. Investigating graph invariants for predicting properties of chemical structures of antiviral drugs. Polycycl. Aromat. Compd. 44(10), 6696–6713 (2024).
Kirana, B. et al. Comparative study of Sombor index and its various versions using regression models for top priority polycyclic aromatic hydrocarbons. Scientific Reports 14(1), 19841 (2024).
Gutman, I. et al. On Sombor indices of chemical graphs. Symmetry 13(8), 1406 (2021).
Shannon, C. E. A mathematical theory of communication. Bell Syst. Tech. J. 27(3), 379–423 (1948).
Estrada, E. The Structure of Complex Networks: Theory and Applications (Oxford University Press, 2011).
Arockiaraj, M. et al. Entropy structural characterization of zeolites BCT and DFT with bond-wise scaled comparison. Sci. Rep. 13, 10874 (2023).
Ashraf, T. et al. Molecular insights into anticancer drugs through predictive mathematical modelling: a QSPR perspective. Journal of Micromechanics and Molecular Physics 10(03), 35–52 (2025).
Dehmer, M. Information processing in complex networks: Graph entropy and information functionals. Appl. Math. Comput. 201(1–2), 82–94 (2008).
Bonchev, D. & Trinajstič, N. Information theory, distance matrix, and molecular branching. J. Chem. Phys. 67(10), 4517–4533. https://doi.org/10.1063/1.435385 (1977).
Kulli, V. R. & Gutman, I. Computation of Sombor indices of certain networks. SSRG Int. J. Appl. Chem. 8(1), 1–5 (2021).
Kulli, V. R. On Banhatti-Sombor indices. SSRG Int. J. Appl. Chem. 8(1), 21–25 (2021).
Kulli, V. R. On second Banhatti-Sombor indices. Int. J. Math. Arch. 12(5), 11–16 (2021).
Gutman, I. et al. Geometric approach to vertexdegree-based topological indices-Elliptic Sombor index, theory and application. Int. J. Quantum Chem. 124(2), e27346 (2024).
Gutman, I. Relating sombor and Euler indices. Vojnoteh. Glas. 72, 1–12 (2024).
Tang, Z. et al. The Euler Sombor index of a graph. Int. J. Quantum Chem. 124, e27387 (2024).
Zhang, Y. et al. Efficacy of afatinib in the treatment of patients with non-small cell lung cancer and head and neck squamous cell carcinoma: A systematic review and meta-analysis. Front. Oncol. 12, 803593. https://doi.org/10.3389/fonc.2022.803593 (2022).
Slamon, D. J. et al. Alpelisib monotherapy for PI3K-altered, pretreated advanced breast cancer: A phase II study. Cancer Discov. 12(9), 2058–2069. https://doi.org/10.1158/2159-8290.CD-21-1696 (2022).
Richardson, P. G. et al. Pomalidomide, bortezomib, and dexamethasone for patients with relapsed or refractory multiple myeloma previously treated with lenalidomide (OPTIMISMM): A randomised, open-label, phase 3 trial. Lancet Oncol. 20(6), 781–794 (2019).
Montazeri, K. & Bellmunt, J. Erdafitinib for the treatment of metastatic bladder cancer. Expert Rev. Clin. Pharmacol. 13(1), 1–6 (2020).
Hong, J. Y. et al. Pralatrexate in patients with recurrent or refractory peripheral T-cell lymphomas: A multicenter retrospective analysis. Scientific Reports 9(1), 20302 (2019).
National Cancer Institute. https://cancer.gov/about-cancer/treatment/drugs
PubChem. An open chemistry database at the National Institutes of Health (NIH). https://pubchem.ncbi.nlm.nih.gov
Chemspider. Search and share chemistry. http://www.chemspider.com
Kour, S. & Sankar J, R. Machine learning regression models for predicting anti-cancer drug properties: Insights from topological indices in QSPR analysis. Contemporary Mathematics, 6515–6526 (2024).
Nasir, S. et al. Topological indices of novel drugs used in blood cancer treatment and its QSPR modeling. AIMS Mathematics 7(7), 11829–11850 (2022).
Bokhary, S. A. U. H. et al. On topological indices and QSPR analysis of drugs used for the treatment of breast cancer. Polycycl. Aromat. Compd. 42(9), 6233–6253. https://doi.org/10.1080/10406638.2021.1977353 (2022).
Funding
This study was supported by Bursa Uludag University Research Projects Coordination Office under the Grant Number FGA-2025-2068. The authors extend their gratitude to the BUU BAP Unit for their invaluable support.
Author information
Authors and Affiliations
Contributions
Methodology, Y.K., Y.S.O., A.B.B., and M.A.; validation, Y.K., and Y.S.O.; formal analysis, Y.K., Y.S.O., A.B.B., and M.A.; investigation, Y.K., Y.S.O., A.B.B., and M.A.; resources, Y.K., Y.S.O., and A.B.B.; visualization, Y.K., Y.S.O., A.B.B., and M.A.; supervision, Y.K. All authors have read and agreed to the current version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Kara, Y., Sağlam Özkan, Y., Bektaş, A.B. et al. Applications of Sombor topological indices and entropy measures for QSPR modeling of anticancer drugs: a Python-based methodology. Sci Rep 16, 3005 (2026). https://doi.org/10.1038/s41598-025-32906-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-32906-x
