
Abstract
Hepatocellular carcinoma (HCC) is a predominant cause of cancer-related mortality globally, noted for its propensity towards late-stage diagnosis and scarcity of effective treatment modalities. The process of metabolic reprogramming, with a specific emphasis on lipid metabolism, is instrumental in the progression of HCC. Nevertheless, the precise mechanisms through which lipid metabolism impacts HCC and its viability as a therapeutic target have yet to be fully elucidated. In the current investigation, single-cell RNA sequencing in conjunction with weighted gene co-expression network analysis (WGCNA) was utilized to delineate lipid metabolism-related genes correlated with the prognostic outcomes of hepatocellular carcinoma (HCC). Data procurement encompassed transcriptomic and clinical datasets from HCC patients, sourced from The Cancer Genome Atlas (TCGA) and Gene Expression Omnibus (GEO) repositories. Subsequent to this, consensus clustering analysis was implemented to stratify patients into distinct subgroups, contingent upon the expression patterns of lipid metabolism genes. Further analytical procedures involved functional enrichment analysis, evaluation of immune infiltration, and examination of the mutation landscape.PTGES3 was identified as a pivotal gene associated with lipid metabolism. Subsequent to its identification, cellular communication analysis was employed to assess the immunological attributes of PTGES3 within the tumor microenvironment. The functional role of PTGES3 was further corroborated through molecular docking simulations and in vitro experimental assays. We identified 27 genes associated with lipid metabolism, 18 of which exhibited significant correlation with overall survival in HCC patients. PTGES3 emerged as a central gene, demonstrating a robust association with immune cell infiltration and unfavorable prognosis. Cellular communication analysis revealed that PTGES3 exhibits the highest communication intensity with T cells, modulating the tumor microenvironment by potentiating the FN1/CD44 + MDK/NCL signaling pathway. Elevated expression of PTGES3 was linked to immunosuppressive cascades, diminished responsiveness to immunotherapy, and inferior overall survival outcomes. Molecular docking analysis indicated that etoposide, methotrexate, and doxorubicin could effectively bind to PTGES3. In vitro experiments confirmed that PTGES3 knockdown significantly impaired the proliferation, invasion, and migration of HCC cells. This study highlights the pivotal role of lipid metabolism in HCC progression and identifies PTGES3 as a potential prognostic biomarker and therapeutic target. These findings offer new insights into the development of targeted therapies for HCC, particularly in patients with high PTGES3 expression.
Similar content being viewed by others


Introduction
Hepatocellular carcinoma (HCC) is the most common primary liver cancer worldwide, exhibiting rising incidence and mortality rates each year1,2. The etiology of HCC is intricate, involving a multifaceted progression. Owing to the scarcity of reliable biomarkers for early detection, a significant number of HCC cases are diagnosed at an advanced stage3,4,5. Presently, an integrative treatment strategy that combines surgical intervention with radiotherapy, chemotherapy, hormonal therapy, and targeted therapy is considered the optimal management for patients with liver cancer6,7. Recent breakthroughs indicate that the synergistic application of immunotherapy alongside targeted therapy significantly improves the efficacy of HCC treatment8. Nonetheless, long-term survival outcomes remain modest.
Metabolic reprogramming is a distinctive hallmark of cancer, catering to the biomass and energy demands of rapidly dividing cells during tumor growth9. In contrast to normal cells, tumor cells predominantly depend on glycolysis to meet their energy needs for proliferation, invasion, and metastasis10. However, the precise mechanisms behind metabolic reprogramming and how these alterations contribute to tumorigenicity are not yet fully elucidated. Emerging research underscores the pivotal role of lipid metabolism reprogramming in the proliferation and malignant transformation of hepatocytes during HCC progression. For instance, Cheng et al. demonstrated that a deficiency in the SLC25A47 gene results in the inactivation of AMPK α and disrupts lipid and cholesterol homeostasis, thereby promoting the development of liver cancer11. Hu et al. discovered that disruptions in fatty acid metabolism in hepatocytes lead to lipid accumulation within the tumor microenvironment (TME), which reduces mitochondrial activity and impairs the function of CD8 + T cells, facilitating immune evasion by tumor cells and exacerbating HCC progression12. Conversely, other studies suggest that modulating lipid metabolism can inhibit tumor cell proliferation and differentiation13. These findings imply that the role of metabolic reprogramming in tumorigenesis is more intricate than previously realized and merits further exploration as a potential therapeutic target for restraining tumor growth and overcoming drug resistance.
Currently, targeting fatty acid metabolism has emerged as a promising strategy in cancer treatment14. The development of drugs that modulate fatty acid levels or disrupt fatty acid metabolic signaling pathways has the potential to control tumor growth and enhance the sensitivity of cancer cells to treatment. In conclusion, lipid metabolism plays a multifaceted role in the tumor microenvironment, influencing various aspects of tumor biology, including cell survival, angiogenesis, immune regulation, migration, and therapeutic response15,16.However, the precise role of fatty acid metabolism in the liver cancer microenvironment remains uncertain. In our study, we employed single-cell sequencing technology and weighted correlation network analysis (WCGNA) for a comprehensive examination to identify characteristic genes related to fatty acid metabolism (FAM). These genes were classified into distinct subgroups using consensus clustering, and we further investigated the differences in clinicopathological characteristics and immune cell infiltration among these subgroups. Additionally, through cell communication analysis and receptor-ligand pair correlation analysis, it is possible to deeply elucidate the impact of PTGES3 on the communication and signaling mechanisms among different cells within the tumor.we investigated the heterogeneity of the hub gene PTGES3 in HCC and its impact on immunotherapy, screening relevant targeted drugs through molecular docking. Finally, through multi-level expression validation and a series of in vitro functional assays, we confirmed the crucial role of PTGES3 in hepatocellular carcinoma. Our work highlights the significant role of fatty acid metabolism (FAM) in the progression of HCC and offers hope for therapeutic strategies targeting the FAM pathway by elucidating its molecular characteristics, physiological functions, and clinical implications.
Methods
Data source
The Cancer Genome Atlas (TCGA) provides transcriptomic and clinical data for patients across up to 33 tumor types. In this study, we retrieved expression, mutation, and various clinical data from the TCGA database for 370 hepatocellular carcinoma (HCC) patients and 50 cases of paraneoplastic tissues. We also utilized the GSE149614 single-cell dataset from the GEO17 database(https://www.ncbi.nlm.nih.gov/geo/),which includes tissue samples from 10 HCC patients, encompassing primary tumor tissue, portal vein tumor thrombus (PVTT), metastatic lymph nodes, and non-tumor liver tissue.For single-cell analysis using Seurat 5.0.0, first perform quality control on the single-cell dataset. Before quality control, the cell numbers for the 10 samples are:
# HCC01 HCC02 HCC03 HCC04 HCC05 HCC06 HCC07 HCC08 HCC09 HCC10.
# 3368 4101 7426 6897 8009 8773 6079 13,770 4778 8714.
Filtering was performed using quality control criteria: 4000 > nFeature_RNA > 500 and percent. MT < 10, resulting in the following number of remaining cells:
# HCC01 HCC02 HCC03 HCC04 HCC05 HCC06 HCC07 HCC08 HCC09 HCC10.
# 2639 2568 6295 5038 6963 8241 5658 10,644 4041 7494.
The data was normalized and standardized using the NormalizeData and ScaleData functions, and the RunPCA function calculated the first 50 principal components (PCs). Based on the results of JackStraw and ElbowPlot, the first 30 PCs were used for subsequent analysis.
Identification and functional enrichment analysis of risk genes related to lipid metabolism
In this study, we employed the R package ‘clusterProfiler18’ for analysis, utilizing the Kyoto Encyclopedia of Genes and Genomes (KEGG)19and Gene Ontology (GO)20 databases to annotate the functions of 27 differentially expressed genes related to lipid metabolism. A P-value < 0.05 was considered statistically significant.
Weighted gene co-expression network analysis (WGCNA)
In this study, we applied weighted gene co-expression network analysis (WGCNA)21 to explore the gene sets most significantly associated with liver cancer. To determine an appropriate soft threshold, we used the “pickSoftThreshold” function in the “WGCNA” R package, considering a range from 1 to 10 (in increments of 1) and from 12 to 20 (in increments of 2).
Cell communication analysis
The CellChat “R” package (version 1.5.0) is utilized to uncover potential mechanisms of intercellular communication within the single-cell context. The createCellChat function is employed to construct a cellchat object, while the aggregateNet function is utilized to describe the signals emitted by each cell type. The netVisual_circle function is employed to visualize the quantity and weight of intercellular communication, while the netAnalysis_computeCentrality function infers the input and output weights of specific signaling pathways.
Tumor microenvironment correlation analysis
The CIBERSORT22 algorithm was utilized to evaluate the differences in immune cell infiltration among various subgroups related to lipid metabolism within the tumor immune microenvironment. The infiltration scores of immune cells and the activity of immune-related pathways were determined by employing the ‘GSVA’ R function and ‘ssGSEA’, respectively.
Consensus clustering analysis of lipid metabolism-related genes
Consensus unsupervised clustering analysis was performed using the R “ConsensusClusterPlus” package, and patients were divided into three different subtypes based on the CDF curve and Delta area. Subsequently, these molecular subtypes were compared with clinical pathological characteristics and prognosis. Principal Component Analysis (PCA) was conducted using the “ggplot2” R package. Furthermore, Kaplan-Meier analysis was used to compare the overall survival (OS) time among the three subtypes. The TIDE three-item scores were calculated among different subtypes using the TIDE website.
Mutation landscape of genes related to lipidmetabolism
The information on somatic mutations was obtained by downloading the TCGA database with maf format. This information was passed through the ‘maftools’ R package in R software and a waterfall plot was created in order to visualise the mutations in HCC patients. In parallel, we downloaded copy number variation (CNV) data from the UCSC Xena database and analysed the CNV frequencies of genes related to lipidmetabolism.
Comprehensive Analysis of Immunomic Molecular Characterization and immunotherapy response based on PTGES3
To understand the differences in tumor heterogeneity caused by PTGES3, we used the GSVA and GSEABase packages to analyze the immune processes of 29 immune cells. Additionally, based on the IOBR23software package, we used deconvolution-based CIBERSORT immune abundance assessment to evaluate the differences in immune cell infiltration between patients with high and low PTGES3 levels. We further assessed the delayed response survival rate of patients with different levels of PTGES3 to immunotherapy, and combined with the TIP algorithm, subtype mapping, and TIDE algorithm24 to estimate the response to immunotherapy.
Molecular docking and drug
To screen potential drugs for the treatment of PTGES3, we used the “pRRophetic” package in R software to calculate the half-maximal inhibitory concentration (IC50) values of the most commonly used chemotherapy drugs. Subsequently, the docking results were evaluated and analyzed using the PLIP system (https://plip-tool.biotec.tu-dresden.de/plip-web/plip/index). Finally, LIGPLOT software version 4.5.3 was used to visualize the molecular docking results in two-dimensional structures, and PyMOL was utilized to generate molecular docking diagrams. Protein structures were obtained from the PDB (https://www.pdb.org/).
Real-time quantitative qRT-PCR
Total RNA was extracted from cells using TRIzol reagent (Invitrogen, USA), and reverse transcription was performed using PrimeScript reverse transcriptase (Takara, Japan) prior to qRT-PCR. Quantitative PCR was conducted using SYBR Premix EX TaqTM II (Takara, Japan) and the LightCycler 480 Real-Time PCR System (Roche, Shanghai, China). The reaction conditions were as follows: maintaining at 95 °C for 10 s, at 60 °C for 30 s, and at 72 °C for 60 s, followed by 40 cycles of amplification. Relative expression levels were calculated using 2-ΔΔCt with GAPDH as an internal reference, and each sample was analyzed three times.
Cell culture and transfection
The hepatocellular carcinoma cell lines HCCLM3, Huh-7, and the normal hepatocyte cell line LO2 were all purchased from Shanghai FuHeng Biotech Co., Ltd. and cultured according to the standard procedures of the incubator. According to the siRNA product manual (Aiji Biotech), the liposome-mediated siRNA transfection was performed using the lipo3000 reagent (Invitrogen, USA). The siRNA sequence targeting PTGES3 is as follows:
Primer sequences | |
|---|---|
PTGES3-F | 5’-GATCGAAGGGACTATGTCTTCATT-3’ |
PTGES3-R | 5’-AAATTATCACTTCCTCCGAGACAAC3’ |
GAPDH-F | 5’-ATCCCTCCAAAATCAAGTGGGG-3’ |
GAPDH-R | 5’-GGGCAGAGATGATGACCCTTTT-3’ |
si-PTGES3#1-F | 5’-GGACUAUGUCUUCAUUGAAUUDTDT-3’ |
si-PTGES3#1-R | 5’-AAUUCAAUGAAGACAUAGUCCDTDT-3’ |
si-PTGES3#2-F | 5’-GGCUUAGUGUCGACUUCAAUADTDT-3’ |
si-PTGES3#2-R | 5’-UAUUGAAGUCGACACUAAGCCDTDT-3’ |
si-NC-F | 5’-UUCUCCGAACGUGUCACGUDTDT-3’ |
si-NC-R | 5’-ACGUGACACGUUCGGAGAADTDT-3’ |
Cell proliferation and Invasion Assay
After 48 h of siRNA transfection, 3 × 103 cells per well were seeded into a 96-well plate. Each well contained 100 µl of complete growth medium. To assess cell viability, 10µL of CCK-8 solution (Beyotime) was added to each well and mixed. After 2 hours, absorbance at 450 nm was measured. Transwell chamber was used for cell invasion test. In the invasion experiment, cells transfected with siRNA for 48 h (3 × 104) were inoculated into the upper chamber containing Transwell serum-free medium (Corning), and the lower chamber was coated with 1:8 diluted BD biosciences to simulate the extracellular matrix. After 48 h, the cells were fixed with 4% paraformaldehyde for 20 min, stained with 0.1% crystal violet for 20 min, and observed under an inverted microscope.
Cell scratch
The siRNA infected cells were evenly seeded in 6-well plates. And scrape three horizontal lines on the plate surface with a 10ul needle. After that, the cells were cultured in 2% serum medium for 24 h. Light microscopy was used to collect cell images after 0 and 24 h.
Sample collection and immunohistochemical staining(IHC)
Liver cancer samples were collected from 26 patients in the Department of Hepatobiliary Surgery at the Second Affiliated Hospital of Nanchang University between November 2022 and November 2023(Ethics No. o-2024147). All patients were diagnosed with hepatocellular carcinoma without distant metastasis. None of the patients had received radiotherapy or chemotherapy prior to surgery. The control group consisted of 26 cases of adjacent liver tissue. Experimental steps: this experiment used immunohistochemical SP staining, Santa Cruz kit, and strictly followed the instructions for dewaxing: put the prepared paraffin tissue Sect. (3 µ m) in a 60 ℃ incubator for about 2 h, then dewaxing in xylene, rehydrating with graded alcohol, and soaking in 3% hydrogen peroxide for 15 min to block endogenous peroxidase activity. The slides were pre incubated with 10% normal goat serum at room temperature for 30 min to reduce non-specific reactions. Subsequently, PBS was moistened for 3 min × 3, and then mouse anti human PTGES3 (Wuhan Sanying company, 1 ∶ 2000) was added dropwise at 4 ℃ overnight. After PBS was moistened for 3 min × 3, envision secondary antibody (Genetech) was added dropwise, and incubated at 37 ℃ for 30 min. finally, DAB (3,3 ‘- diaminobenzidine) staining was used, deionized water was used to stop the color development, hematoxylin was re stained for 20 s, and hydrochloric acid alcohol differentiation was used for 5 s; Rinse with tap water for 15 min and return to blue; Neutral resin seal. Known immunostaining positive slides were used as positive control. The staining intensity is usually divided into four grades: 0: no positive staining (negative) 1: light yellow (weak positive) 2: brownish yellow (positive) 3: the proportion of brownish (strong positive) positive cells is divided into four ranges: 1: cells less than 10% are positive 2: cells 11 −50% are positive 3: cells 51 −80% are positive 4: cells more than 80% are positive. Scoring formula: IHC Score = staining intensity × proportion of positive cells.
Statistical analyses
All statistical calculations and graphs were performed using R software version 4.2.0. Kaplan-Meier curves and log-rank tests were utilized to examine differences in survival among various risk groups. Additionally, the Wilcoxon test was employed to assess differences between two datasets. A two-sided p-value of < 0.05 was considered statistically significant.
Result
Research ideas
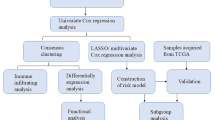
By reducing the dimensionality of single-cell sequencing samples from the GEO database (GSE149614), the authors conducted clustering and screening to identify differentially expressed genes in liver malignant parenchymal cells. They further utilized WGCNA analysis to pinpoint genes associated with lipid metabolism. Patients were categorized into distinct subgroups based on consistent clustering, and the clinical characteristics, survival times, and enrichment analyses were compared across these subgroups. Cellular communication analysis was employed to investigate the correlations between PTGES3 and other cells within the tumor microenvironment, as well as the underlying signaling pathways and ligand-receptor interactions. Patients were then stratified into two groups (high and low expression groups) based on the median expression level of PTGES3. Subsequent studies revealed distinct features of the immune microenvironment, immune checkpoint expression, and response to immunotherapy between the two groups. Ultimately, molecular docking was employed to predict potential targeted drugs for the PTGES3 high-expression group, and the efficacy of PTGES3 was validated in vitro (Fig. 1).

The flowchart processing software utilized is Prism 9.
Analysis of single-cell datasets
Initial quality control of the single-cell dataset involved normalization and standardization of the data using the NormalizeData and ScaleData functions, respectively. Subsequently, principal component analysis (PCA) was executed via the RunPCA function, computing the first 50 principal components (PCs). The optimal number of PCs for further analysis was ascertained through the JackStraw and ElbowPlot procedures, which indicated the use of the first 30 PCs. The resolution of clustering was evaluated using the clustree function, with the most appropriate clustering identified at a resolution of 0.7. Dimensionality reduction and clustering were performed using the UMAP algorithm, which yielded 50 distinct clusters (designated as 0–49). These clusters were annotated based on specific cell type markers: B cells (MS4A1), dendritic cells (DCs, CD86), endothelial cells (PECAM1), fibroblasts (PDGFRB), liver parenchymal cells (CD24, MDK), macrophages (CD163, CD68), natural killer (NK) cells (NKG7, GNLY), plasma cells (JSRP1), and T cells (CD3D, CD3E) (Fig. 2A and B).The composition of cell types across various tissue types was subsequently quantified. The findings revealed that T cells constituted the highest proportion in normal tissues, whereas malignant liver parenchymal cells were most prevalent in parenchymal tumor tissues, lymph nodes, and portal vein tumor thrombi. Moreover, both parenchymal tumor tissues and portal vein tumor thrombi showed a significant presence of stromal cells (Fig. 2C). A comparison of cell type proportions across different clinical stages indicated that T cell infiltration increased with tumor progression, as all stages except Stage I displayed a high proportion of T cells. Dendritic cells were most prevalent in Stage IV, hinting at a potentially significant, yet understudied, role in advanced-stage tumors (Fig. 2D). To assess lipid metabolism in tumor tissues, the scMetabolism algorithm was utilized to evaluate the activity of various lipid metabolism pathways among different cell types. The analysis showed that malignant hepatic parenchymal cells exhibited the highest scores for multiple lipid metabolism pathways, suggesting that the enhanced lipid metabolism in these cells may be intricately associated with tumor initiation and progression (Fig. 2E).

Analysis of single-cell RNA sequencing data in hepatocellular carcinoma. (A) UMAP representations of nine distinct cell types, processed through filtration, normalization, and analysis with the Seurat package. (B) Bubble plots with annotations of cell type-specific markers, utilizing the SingeR package for annotation. (C-D) Utilization of the ggplot2 package to depict the distribution of cell types across various tissues and patient stages. (E) Bubble plots evaluating the enrichment of metabolic pathways, employing the scMetabolism package for analysis.
Acquisition of genes at the intersection of key lipid metabolism and HCC expression
For in-depth analysis, we extracted genes associated with the eight lipid metabolism-related pathways, resulting in a set of 165 unique genes after duplicate removal, which were then subjected to subsequent analysis. The FindAllmarker function was employed to identify differentially expressed genes within each cell subtype, with the screening criteria set at logFC > 0.25 and a corrected P-value < 0.05. This process yielded a total of 2,085 differentially expressed genes pertinent to liver parenchymal cells. To explore genes related to hepatocellular carcinoma (HCC), we conducted a Weighted Gene Co-expression Network Analysis (WGCNA) and constructed a scale-free network using the TCGA-LIHC gene set, achieving an R2 value of 0.9. This network conformed to a soft threshold of 12, indicating a satisfactory level of connectivity (Fig. 3A). Gene clustering resulted in the segregation of all genes into eight distinct modules (Fig. 3B). Evaluating the correlation between these modules and HCC, we found the turquoise module to have the strongest positive correlation, with a correlation coefficient of r = 0.52 and a p-value of 4e-31 (Fig. 3C). To pinpoint lipid metabolism-related genes in liver parenchymal cells that are associated with HCC, we intersected genes from the turquoise module with those that were differentially expressed in liver parenchymal cells and genes involved in lipid metabolism pathways as identified by WGCNA. This intersection, depicted in a Venn diagram, revealed 27 lipid metabolism-related genes that are specifically associated with liver parenchymal cells in the context of HCC (Fig. 3D).

Weighted gene coexpression network analysis (WGCNA). (A) Optimal soft threshold determination at β = 12, ensuring adherence to a power-law distribution and robust network connectivity. (B) Genes are partitioned into eight distinct modules. (C)The turquoise module exhibits the most pronounced positive correlation. (D) Identification of lipid metabolism-associated signature genes.
Identification and Mutational Landscape of Survival-related lipid metabolism differentially expressed genes in HCC patients
To delve into the metabolic genes linked to survival outcomes in hepatocellular carcinoma (HCC) patients, we conducted a one-way Cox regression analysis focusing on the 27 genes previously identified. The resulting forest plot indicated that 18 of these genes had a significant impact on overall survival (Fig. 4A). Analysis using GENEMANIA revealed complex protein interactions among the 18 lipid metabolism genes associated with survival in HCC (Fig. 4B). Correlation analysis of gene expression showed that AKR1C3, SQLE, HACD2, and PTGES3 exhibited negative correlations with the other genes, while the rest displayed positive correlations with one another. In the correlation heatmap, lime green signifies positive correlations, dark blue denotes negative correlations, ‘×’ marks non-significant correlations, and other colors represent significant correlations (Fig. 4C). Waterfall plot analysis uncovered prevalent mutation patterns among most genes, with missense mutations being the predominant type. Notably, CYP2C8 stood out with the highest mutation frequency (Fig. 4D).

Characteristics of prognostic genes associated with fatty acid metabolism. (A) Univariate Cox regression analysis was performed using the survival package, with forest plots visualized via ggplot2. (B) Protein interactions and pertinent pathways of 18 prognostic genes linked to fatty acid metabolism were investigated using the GENEMANIA platform, while ggcor and ggplot2 were utilized for correlation analysis and graphical representation. (C) Correlation analysis among the 18 prognostic genes related to fatty acid metabolism was conducted. (D) The mutational status of the 18 prognostic genes associated with fatty acid metabolism was examined, with waterfall plots generated using the maftools package. * p < 0.05; ** p < 0.01; *** p < 0.001; ns indicates no statistical significance.
Consistent clustering of HCC based on lipid metabolism-related genes
Employing consistent clustering to examine the expression profiles of the 18 survival-related lipid metabolism genes in hepatocellular carcinoma, we delineated three distinct subgroups within the HCC samples (Fig. 5A). Principal component analysis (PCA) revealed significant variations in the principal components among the subgroups, indicative of unique expression characteristics (Fig. 5B). Survival analyses showed that patients in subgroup three had the worst overall survival, whereas those in subgroup one had the best, with statistically significant differences observed across the three subgroups (p < 0.001, Fig. 5C). A comparison of clinical features across the subgroups revealed substantial differences in pathological stage, histological grade, T-stage, and survival outcomes. Subgroup three was characterized by a higher prevalence of patients with Stage III/IV, Grade 3/4, T3, T4, and higher mortality rates (Fig. 5D). TIDE scores suggested that Subgroup 1 had the lowest immune exclusion score, which may account for its superior overall survival (Fig. 5E). However, Subgroup 1 also displayed a relatively elevated score of immune dysfunction, as indicated by the TIDE score21, implying a general susceptibility to immunotherapy (Fig. 5F-G).

Differential Prognostic Responses and Immune Checkpoints Across Various Fatty Acid Metabolism (FAM) Subtypes. (A) Utilizing the ConsensusClusterPlus package with k = 3, consensus clustering analysis was conducted, segmenting patients into three distinct subgroups. (B) FactoMineR in conjunction with factoextra generated PCA vector plots, delineating the variance in principal components across different subgroups. (C) Survival analysis was performed using the survival package along with survminer, highlighting significant disparities in survival outcomes among the three subgroups. (D) Complex heat map was engaged to produce a heatmap of clinical correlations, evaluating the variations in clinical pathological features among the three subgroups. (E-G) The TIDE website was employed to compute the variations in immune function and TIDE scores across the three subgroups, with the gghalves package utilized for generating a cloud-and-rain plot for the respective subgroups. * p < 0.05; ** p < 0.01; *** p < 0.001; ns indicates no statistical significance.
Enrichment analysis of lipid metabolism-related genes in three clustered clusters
The differences in biological processes and pathways among the three subclusters were further investigated. Cluster 1 was predominantly associated with the anabolic and catabolic metabolism of organic acids. Cluster 2 was primarily linked to steroid metabolism, while Subcluster 3 was enriched in amino acid metabolism and other related processes, highlighting distinct biological pathways (Fig. 6A-C). Subsequent KEGG enrichment analysis and score transformation revealed that Cluster 1 was characterized by upregulation of the complement cascade response and bile secretion (Fig. 6D). In contrast, both Clusters 2 and 3 exhibited downregulation in metabolic processes and the complement cascade response (Fig. 6E-F). Based on these findings, we hypothesize that the upregulation of immune and metabolic processes in patients may contribute to a better overall survival rate.

The FAM subgroups exhibited distinct enrichments in the Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) databases. Figures A and B illustrate the variations in GO enrichment across the three subgroups, while Figures D-F highlight the disparities in KEGG pathway enrichment among the respective subgroups.
Enrichment analysis of core genes and their association with tumor immune infiltration
The analysis further delineated a subset of core genes pertinent to lipid metabolic processes, with a focus on 18 genes for comparative examination between hepatocellular carcinoma (HCC) tissues and their corresponding controls via bulk RNA sequencing methodologies. This comparative analysis revealed that a mere quartet of genes, namely AKR1C3, HACD2, PTGES3, and SQLE, exhibited elevated expression levels within HCC tissues (Fig. 7A). Subsequent to categorizing the expression levels of these genes into high and low expression cohorts, survival curves were constructed based on median expression values. Statistical significance was discernible in the survival curves corresponding to AKR1C3, PTGES3, and SQLE ( Fig. 7B-E). The correlation analysis in conjunction with immune checkpoint molecules and chemokines elucidated that PTGES3 alone demonstrated a robust correlation with these immune-relevant molecules (Fig. 7F). In synergy with the immune process correlations indicated by the preceding enrichment analysis, this observation implies that PTGES3 is the predominant core hub gene among those investigated. Examination of radar correlation plots for PTGES3 revealed a significant positive correlation with dendritic cells, macrophages, Th1, and Th2 cells (as illustrated in Fig. 7G). Moreover, PTGES3 exhibited positive correlations with immune processes such as antigen-presenting cell (APC) co-stimulation and human leukocyte antigen (HLA), and conversely, negative correlations with interferon II processes (Fig. 7H). Consequently, PTGES3 was selected as the core gene for the lipid metabolism model for subsequent research endeavors.

The identification of a pivotal gene within the FAM gene family in hepatocellular carcinoma (HCC) is a crucial step towards elucidating its pathogenesis and identifying potential therapeutic targets. The research employed a comprehensive strategy to examine the expression profiles and clinical relevance of FAM risk genes in HCC.
Single-cell sequencing analysis and Bulk analysis reveal the immune landscape of PTGES3 gene and its clinical significance
To gain a more profound understanding of the role of PTGES3 in hepatocarcinogenesis, we initiated our investigation by assessing its expression profile within the single-cell dataset (GSE149614). Feature plot analysis disclosed that PTGES3 is predominantly expressed in hepatocellular carcinoma (HCC) cells and T lymphocytes, with the most negligible expression observed in plasma cells (refer to Fig. 8A-B). Notably, PTGES3 displays tissue-specific expression patterns. Specifically, in metastatic lymph nodes, PTGES3 is predominantly expressed in HCC cells. Conversely, in portal vein tumor thrombus (PVTT), the expression of PTGES3 is primarily localized in T lymphocytes. In the tumor microenvironment, PTGES3 demonstrates a dispersed pattern of high expression, devoid of discernible cellular aggregation. Conversely, the expression level of PTGES3 is notably reduced in normal tissue samples (refer to Fig. 8C). Regarding clinical correlations, elevated PTGES3 expression is associated with more advanced histopathological grades (e.g., G3/G4), higher clinical stages (e.g., Stage II, Stage III/IV), and is prevalent in patients who have succumbed to the disease. These observations imply that PTGES3 may serve as a prognostic indicator for clinical progression and staging (Fig. 8D-F). To delineate the underlying biological processes and signaling pathways modulated by this gene, we embarked on a two-tiered investigative approach.Initially, we stratified the samples into high and low expression groups utilizing the median expression level of PTGES3 in the TCGA-LICH cohort. Subsequently, we conducted a differential expression analysis to compare gene expression between these two groups. The accompanying figure illustrates the top 10 genes that were up-regulated and down-regulated in the high PTGES3 expression group (refer to Fig. 8G). In light of the outcomes from the differential expression analysis, we executed a HALLMARK Gene Set Enrichment Analysis (GSEA) and identified that biological processes pertinent to mitosis and DNA repair were markedly enriched in the high PTGES3 expression group, implying that PTGES3 may facilitate tumor proliferation (refer to Fig. 8H). Next, within the single-cell dataset, we similarly categorized cells into high and low expression groups based on PTGES3 expression levels and conducted GSEA analysis.We observed that in the high PTGES3 expression group, there was an upregulation of not only processes associated with lipid metabolism but also pathways related to reactive oxygen species (ROS) and mitotic activities (refer to Fig. 8I). Upon intersecting the up-regulated pathways identified in both bulk and single-cell RNA sequencing data, we pinpointed two common pathways: MYC_TARGETS_V1 and E2F_TARGETS, both of which are integral to signaling cascades governing proliferation and cell division (refer to Fig. 8J). These findings provide further evidence that the up-regulation of PTGES3 is significantly correlated with adverse patient prognoses and contributes to tumor progression.

Expression of PTGES3 at the single-cell level and its clinical significance.(A-B) Expression of PTGES3 in nine types of immune cells (C) Distribution of PTGES3 in four distinct tissues (D-F) Correlation between PTGES3 expression levels and clinical pathological characteristics (G) Top 10 genes with positive and negative correlations to PTGES3 (I-H) Enrichment analysis of PTGES3 in Bulk and scRNA-seq pathways (J) Venn diagram of the intersection between Bulk and scRNA-seq pathways for PTGES3.
Exploration of the impact of the PTGES3 gene in the tumor microenvironment through cellular communication
We performed cell communication analysis utilizing the single-cell sequencing dataset from GEO (GSE149614). Initial findings suggested that PTGES3 is predominantly involved in the lipid metabolism process of liver cancer cells. Consequently, we categorized liver cancer cells into Hepa_PTGES3_hi (high expression) and Hepa_PTGES3_lo (low expression) groups to examine their communication with other cell types. The analysis revealed that Hepa_PTGES3_hi cells significantly increased their communication with T cells (Fig. 9A). Further investigation into specific signaling pathways identified two key pathways: the FN1 pathway and the MK pathway, with Hepa_PTGES3_hi cells acting as the primary senders and T cells as the primary receivers (Fig. 9B).The communication heatmap revealed that the interaction between Hepa_PTGES3_hi cells and T cells was most intense in the FN1 and MK pathways, respectively (Fig. 9C-D). In comparison to the FN1 pathway, the most significant ligand-receptor pair was FN1-CD44, with FN1 being most highly expressed in Hepa_PTGES3_hi cells and CD44 being most highly expressed in T cells (Fig. 9E). For the MK pathway, the predominant ligand-receptor pair was MDK-NCL, with MDK showing the highest expression in Hepa_PTGES3_hi cells and NCL in T cells (Fig. 9F). Consequently, Hepa_PTGES3_hi cells may influence T cell functionality by regulating the FN1 and MK signaling pathways.

Analysis of cellular communication networks. (A) The CellChat “R” package (version 1.5.0) evaluates the interactions between Hepa_PTGES3_hi and other cell types. (B) The createCellChat function is utilized to construct a schematic diagram of incoming and outgoing signals in CellChat samples. (C-D) The netVisual_circle function is employed to visualize the communication intensity among different cell types in the FN1 and MK pathways. (E-F) The netAnalysis_computeCentrality function is used to infer and visualize the expression of key genes in the FN1 and MDK signaling pathways across 10 cell types.
Immunologic features associated with the hub gene PTGES3
The immunological profiling was conducted utilizing the IOBR20 toolkit, which incorporates a comprehensive array of over 200 genes with established immune associations. The functional status of these immunological markers was evaluated through the application of ssGSEA (single-sample Gene Set Enrichment Analysis) algorithms. Subsequent to analyzing the hepatocellular carcinoma (HCC) tumor microenvironment, we ascertained the prognostic significance of these immunological signatures within the TCGA-LIHC (The Cancer Genome Atlas-Liver Hepatocellular Carcinoma) cohort. Our findings culminated in the identification of the top 20 immunological signatures, which were prioritized based on their statistical significance as indicated by P-value rankings.Significantly, the MDSC pathway, hypoxia signatures, and DNA damage repair were all correlated with a hazard ratio (HR) exceeding 1 (Fig. 10A). Figure 10B displays a heatmap of the enrichment scores for the top 20 immune signatures in the high and low PTGES3 expression subgroups, suggesting that elevated PTGES3 expression is linked to the MDSC pathway, hypoxia signatures, and DNA damage repair, which implies a poorer overall survival outcome for patients with high PTGES3 expression. Figure 10C and D illustrate a comparison of the enrichment scores between immunosuppressive and immune rejection signatures, demonstrating that high PTGES3 expression is associated with a more pronounced immunosuppressive environment relative to immune rejection. Furthermore, an immune abundance assessment using CIBERSORT with inverse convolution revealed a significant enrichment of specific immune cell types in the low PTGES3 expression group, indicating a higher level of immune infiltration within this subgroup (Fig. 10E).

Characterization of the tumor microenvironment (TME) molecular features in patients with varying PTGES3 expression levels. (A) Analysis of the association between immune signaling pathways and patient survival rates, utilizing the IOBR package for evaluation. (B) Comparison of the top 20 immune signaling pathways between patients with high and low PTGES3 expression. (C) Representation of inhibitory signaling distribution in patients with high versus low PTGES3 expression. (D) Representation of immune rejection signaling distribution in patients with high versus low PTGES3 expression. (E) Profiling of immune cell abundance in patients with high and low PTGES3 expression, employing the CIBERSORT deconvolution method. *p < 0.05; **p < 0.01; ***p < 0.001; NS denotes not statistically significant.
PTGES3 highlights better predictive ability for immunotherapy response in HCC patients
To thoroughly evaluate the function of PTGES3 in hepatocellular carcinoma (HCC) immunotherapy, a systematic examination was performed. Initially, we scrutinized the correlation of PTGES3 expression with dysregulation scores, rejection scores, and microsatellite instability (MSI) scores, utilizing the TIDE algorithm. Our findings revealed a notable inverse correlation between PTGES3 expression and both dysregulation and MSI scores, coupled with a significant positive association with rejection scores. This suggests that patients exhibiting elevated PTGES3 levels are associated with higher rejection scores and diminished microsatellite instability, which may influence their responsiveness to immunotherapy (Fig. 11A-C). To corroborate these observations, we procured data from the immunotherapy cohort GSE91061 in the GEO database, encompassing patients who received PD1 and CTLA4 inhibitor therapies. Survival analysis indicated that patients with increased PTGES3 expression exhibited inferior overall survival (Fig. 11D), and non-responders to immunotherapy demonstrated heightened PTGES3 expression (Fig. 11E).The SUBMAP algorithm, which operates on the principle of bi-data clustering, suggested that patients with reduced PTGES3 expression exhibited greater responsiveness to PD1 inhibitors in both the GSE91061 and TCGA-LIHC cohorts (Fig. 11F). Furthermore, we conducted an ESTIMATE analysis and examined the correlation between PTGES3 expression and stromal scores, immune scores, and microenvironmental scores. Our findings revealed a notable inverse correlation specifically between PTGES3 expression and stromal scores, implying that PTGES3 may play a role in stromal remodeling (Fig. 11G-I).

Predictive Potential of Immunotherapy in Patients with High and Low PTGES3 Expression Levels. (A-C) Correlation analyses between the dysregulation score, exclusion score, microsatellite instability (MSI) score, and PTGES3 expression. (D) Survival analysis comparing patients with high and low PTGES3 expression within the GSE91061 dataset. (E) Distribution of patients with high and low PTGES3 expression across different immunotherapy response categories. (F) Application of the subclass mapping algorithm to forecast the immunotherapy response in patients with varying PTGES3 expression levels. (G-I) Associations between the stromal score, immune score, and estimated score, and their correlation with PTGES3 expression. *p < 0.05; **p < 0.01; ***p < 0.001; NS indicates the results are not statistically significant.
Selection of potential therapeutic drugs
Considering the unfavorable prognosis of immunotherapy in HCC patients with elevated PTGES3 expression, we systematically scrutinized the pertinent literature to identify potential therapeutic agents. Initially, we assessed the IC50 disparities of various pharmacological compounds between high and low PTGES3 expression groups utilizing the pRRophetic algorithm, with the objective of pinpointing agents that could effectively modulate PTGES3 expression. A correlation analysis of IC50 values in relation to PTGES3 expression yielded the top 10 drugs that exhibited a significant inverse and positive correlation with PTGES3 (Fig. 12A-B). The top three identified agents were etoposide, methotrexate, and doxorubicin.The AUC algorithm was employed to determine the density distribution of drug IC50 values within the two groups. This analysis indicated that the density distribution of these three drugs was reduced in the high PTGES3 group, implying that this cohort might derive greater benefit from these pharmacological agents (Fig. 12C-E). Subsequent investigation via molecular docking assessed the binding affinity of PTGES3 with the top three identified drugs (Fig. 12F-H). Molecular docking outcomes confirmed that the relative binding energies of PTGES3 with etoposide, methotrexate, and doxorubicin fell within the optimal range for molecular interaction, with the most favorable binding energies observed as −7.0 kcal/mol for etoposide, −6.8 kcal/mol for methotrexate, and − 6.1 kcal/mol for doxorubicin. Collectively, etoposide was identified as the most promising drug candidate for mitigating the role of PTGES3 in hepatocarcinogenesis.

Screening for potential therapeutic drugs targeting patients with high PTGES3 levels. (A-B) Calculation of the half-maximal inhibitory concentration (IC50) values for PR-deficient cells and presentation of the top 10 drugs with significant negative and positive correlations with PTGES3. (C-E) Distribution differences of three drugs in the high PTGES3 group and low PTGES3 group. (F-H) Evaluation and analysis of the docking results of the top three drugs with significant negative correlations with PTGES3 using the PLIP system (https://plip-tool.biotec.tu-dresden.de/plip-web/plip/index), visualization with LIGPLOT software version 4.5.3; and generation of molecular docking diagrams using PyMOL. * p < 0.05 **p < 0.01 ***p < 0.001; NS indicates no statistical.
Upregulation of PTGES3 in Liver Cancer and Knockdown of PTGES3 Significantly Suppresses the Proliferative, Invasive, and Migratory Abilities of HCC Cells
In synthesizing the aforementioned findings, we conclude that PTGES3 is a pivotal gene associated with lipid metabolism and displays significant immune relevance. Quantitative reverse transcription polymerase chain reaction (qRT-PCR) experiments revealed that, in comparison to normal liver cells, human HCC cell lines demonstrated elevated PTGES3 expression (Fig. 13A). Additionally, we transfected siRNA into HCCLM3 and Huh-7 cells, selecting the most potent siRNA-PTGES3#1 and #2 for subsequent experimentation (Fig. 13B-C). Immunohistochemistry (IHC) experiments indicated that PTGES3 expression was upregulated in liver cancer tissues, corroborating the cytological findings, and the IHC score for PTGES3 in liver cancer tissues was higher compared to adjacent non-cancerous tissues (Fig. 13F). Cell Counting Kit-8 (CCK-8) experiments demonstrated that PTGES3 knockdown suppressed the proliferative capacity of HCCLM3 and Huh-7 cells (Fig. 13D-E). Scratch assay and Transwell experiments further showed that PTGES3 gene silencing inhibited the migratory and invasive capabilities of HCCLM3 and Huh-7 cells. These data suggest that PTGES3 acts as a positive regulator in HCC cell biology (Fig. 13G-I).

Validation of PTGES3 Expression Levels and the Significant Inhibition of Proliferation, Invasion, and Migration in Liver Cancer Cells following PTGES3 Gene Knockout. (A) Assessment of PTGES3 mRNA expression in various liver cancer cell lines. (B-C) Evaluation of the knockout efficiency of PTGES3 mRNA in HCCLM3 and Huh-7 cells. (D-E) The CCK-8 assay reveals that the knockout of the PTGES3 gene significantly inhibits the proliferative capacity of HCCLM3 and Huh-7 cells. (F) Immunohistochemistry (IHC) experiments confirm elevated PTGES3 expression in liver cancer tissues, with correspondingly higher IHC scores. (G-H) Knockout of the PTGES3 gene significantly reduces the migratory and invasive capabilities of HCCLM3 and Huh-7 cells. (I) Statistical representation of the experimental data for HCCLM3 and Huh-7 cells; * p < 0.05; ** p < 0.01; *** p < 0.001; NS: not statistically significant.
Discussion
The initial clinical manifestations of hepatocellular carcinoma (HCC) frequently lack specificity, resulting in delayed diagnosis and the missed opportunity for radical surgery, thereby enhancing the risk of postoperative metastasis and recurrence. Despite advancements in cancer prevention, early screening, and therapeutic strategies in recent years, the prognosis for HCC patients remains suboptimal, with low survival rates25.Zhu ZZ et al.26. have established that genetic amplification of MDM4 and BCL2L1 is a factor associated with copy number aberrations (CNAs) and is linked to extrahepatic metastasis-free survival, acting as novel prognostic indicators for liver cancer. Consequently, the identification of new diagnostic and prognostic biomarkers is vital to enhance the overall survival of HCC patients.In contemporary oncological research, metabolic reprogramming has emerged as a pivotal domain. Particularly, fatty acid metabolism, which serves as a critical energy source facilitating the rapid proliferation and metastasis of tumor cells, participates in numerous essential biological processes. An increasing body of evidence indicates that fatty acid metabolism is instrumental in the onset and progression of hepatocellular carcinoma (HCC)27. For instance, Ning Z et al.‘s research demonstrates that the deubiquitinating enzyme USP 22 stabilizes the ACLY/ACC axis by modulating the ubiquitination of PPARγ receptors, thereby altering the fatty acid metabolism in HCC cells and fostering the development and progression of liver cancer28. Consequently, the continued investigation of key molecular markers associated with fatty acid metabolism is anticipated to yield novel insights into the pathogenic mechanisms of HCC.
With the continuous application of bioinformatics techniques and other methodologies in clinical research, an increasing number of tumor prediction models have been developed by researchers to predict and evaluate the survival status and treatment outcomes of patients with malignant tumors. Compared to traditional classification methods, the characteristic genes obtained through various algorithmic combinations and validated screening exhibit superior predictive accuracy29,30.This investigation corroborated the pivotal role of fatty acid metabolism in the progression of hepatocellular carcinoma (HCC) utilizing single-cell RNA sequencing (scRNA-seq) and, in conjunction with weighted gene co-expression network analysis (WGCNA), discerned a high degree of correlation within the fatty acid metabolism module encompassing 27 key genes. To delineate diverse fatty acid metabolism patterns and their influence on the tumor microenvironment, the research team employed unsupervised clustering to classify patients into three distinct subgroups, subsequently comparing the variances in biological processes and metabolic pathways amongst these subgroups. Subgroup I was predominantly enriched in the anabolism and catabolism of organic acids, subgroup II was linked to steroid metabolism, and subgroup III was distinguished by amino acid metabolism.
These findings suggest that the regulation of distinct metabolic pathways plays a crucial role in the immune response within the tumor microenvironment. Notably, in subgroup III, patients exhibited higher immune rejection scores and TIDE scores, indicating compromised immune function due to immune rejection. This may contribute to their poorer clinical outcomes and survival rates. Univariate Cox regression analysis identified 18 prognostic genes related to fatty acid metabolism, of which only four genes, AKR1C3, HACD2, PTGES3, and SQLE, were significantly upregulated in HCC. Studies have demonstrated that AKR1C3 plays a positive role in the tumor growth and autophagy process of HCC, and its deletion can induce mitochondrial fission and apoptosis in sorafenib-resistant HCC cells31.
HACD2 is implicated in the DNA demethylation cascade and is significantly associated with the progression to advanced clinical stages in individuals diagnosed with liver cancer32. SQLE, emerging as a novel oncogenic determinant, facilitates the proliferation of pancreatic cancer cells by modulating the Src/PI3K/Akt signaling axis33. Furthermore, PTGES3 is identified as a high-risk indicator in breast cancer and is correlated with unfavorable prognostic outcomes. The suppression of its expression has been demonstrated to restrain the growth of breast cancer cells34. Correlative analysis of these four genes with immune checkpoint molecules and chemokines revealed a high degree of correlation between PTGES3 expression and the expression of these immune-relevant molecules, suggesting its potential role as a central gene in the fatty acid metabolism paradigm35,36. PTGES3 encodes prostaglandin E synthase 3 (PGES3), which catalyzes the conversion of prostaglandin H2 to prostaglandin E2 (PGE2) and modulates various pro-inflammatory signaling pathways, thereby influencing cellular proliferation, apoptosis, angiogenesis, and immune surveillance37.PGE2, acting as an inflammatory mediator, modulates the functionality of immune cells by interacting with EP receptors38, thereby repressing T cell activation and proliferation, facilitating the aggregation of immunosuppressive cell populations39,40(including regulatory T cells, Tregs, and M2 macrophages), and augmenting the capacity of tumor cells to evade the immune response. Moreover, PTGES3 potentially influences the dendritic cell (DC) function indirectly through its metabolic byproduct, PGE2. Dendritic cells are pivotal antigen-presenting cells that are crucial in initiating T cell-mediated immune responses41.Consequently, an elevation in PGE2 synthesis can compromise the functionality of dendritic cells, culminating in a reduction of their antigen-presenting capacity. This impairment subsequently affects T cell-mediated immune responses and tumor-specific immune surveillance. Although PTGES3 is recognized as a significant player in various carcinogenic processes, its precise role in hepatocellular carcinoma (HCC) necessitates further investigation. Tumor progression is intricately linked to the dynamic alterations in the extracellular matrix, with immune cells being pivotal constituents of the tumor microenvironment. Hence, the tumor microenvironment is critically implicated in patient prognosis. A notable characteristic of HCC is the presence of hypoxia and immunosuppression, which collectively diminish the body’s immune system’s anti-cancer defenses42. Furthermore, metabolic perturbations in tumor cells can instigate a metabolic reprogramming of the microenvironment, consequently impacting the biological behavior of the tumor. In this investigation, the examination of cell-to-cell communication via single-cell sequencing methodologies aids in elucidating alterations within the tumor immune microenvironment and the tumor itself43. We delineated PTGES3 expression in two distinct cell types: Hepa_PTGES3_hi and PTGES3_lo, and subsequently performed an intercellular communication analysis. Our findings revealed that both FN1 and MDK are markedly upregulated in Hepa_PTGES3_hi cells.Moreover, Hepa_PTGES3_hi cells predominantly engage in communication with T cells via the FN1/CD44 and MDK/NCL signaling pathways, suggesting their potential role in modulating the extracellular matrix remodeling and facilitating tumor cell evasion from T-cell mediated immune surveillance. Additionally, these pathways can also transmit signals to macrophages, thereby influencing the macrophage migration inhibitory factor (MIF) pathway. Prior research has indicated that MIF is implicated in diverse immune processes, mediating immune evasion that can lead to tumor metastasis. Consequently, Hepa_PTGES3_hi cells may exert a pivotal role in the tumor immune microenvironment by modulating the T-cell immune response through the FN1/CD44 and MDK/NCL pathways. We assessed the immune cell composition within the HCC microenvironment utilizing the ssGSEA algorithm in conjunction with the IOBR R software package. The findings indicated that immune cells, including dendritic cells, macrophages, Th1, and Th2 cells, exhibited a pronounced down-regulation trend in high-risk patients. Conversely, the expression of PTGES3 was positively associated with immune processes such as antigen-presenting cell (APC) costimulatory signals and HLA, and negatively associated with interferon gamma-related processes. These results align with extant literature, implying that patients in the high-risk group tend to exhibit immunosuppression44. Subsequent analysis utilizing the TIDE algorithm revealed that PTGES3 expression was significantly inversely correlated with the dysregulation score and microsatellite instability (MSI) score, yet positively correlated with the rejection score. Patients exhibiting elevated PTGES3 expression demonstrated higher rejection scores and diminished microsatellite instability (MSI), suggesting potential reduced responsiveness to immunotherapeutic interventions. To address the unfavorable prognosis associated with immunotherapy in patients with high PTGES3 expression, researchers employed a comprehensive screening framework to systematically identify potential therapeutic agents. Presently, the primary treatment modality for hepatocellular carcinoma (HCC) involves targeted therapies, such as Lenvatinib and Apatinib. These agents exert their efficacy by targeting vascular endothelial growth factor receptors (VEGFR), thereby inhibiting tumor angiogenesis, reducing tumor blood and nutrient supply, and subsequently suppressing tumor growth45. In recent years, the combination of targeted therapies with immune checkpoint inhibitors (e.g., PD-1/PD-L1 inhibitors) has emerged as a promising strategy in the treatment of HCC. For instance, the combination of lenvatinib and the PD-1 inhibitor pembrolizumab has demonstrated synergistic effects in clinical trials. Lenvatinib enhances the tumor microenvironment’s responsiveness to immunotherapy by mitigating tumor angiogenesis46. Molecular docking investigations revealed that etoposide exhibited an IC50 value of −7.0kcal/mol, suggesting its potential as a prime candidate for therapeutic intervention in the high PTGES3 expression group. Etoposide, a non-specific chemotherapeutic agent, induces apoptosis by causing DNA damage in tumor cells47. However, its variable efficacy poses challenges in achieving consistent therapeutic outcomes across diverse patient cohorts. Future therapeutic strategies may involve the combination of etoposide with PD-1 inhibitors to overcome drug resistance and to activate the immune system48. Additionally, immunohistochemistry (IHC) and reverse transcription polymerase chain reaction (qRT-PCR) experiments confirmed the significant upregulation of PTGES3 in hepatocellular carcinoma (HCC) tissues. Subsequent in vitro studies demonstrated that PTGES3 knockout significantly inhibits the proliferation, invasion, and migration of HCC cells, indicating the potential of PTGES3 as a crucial oncogenic factor in HCC.
In summary, this study distinguishes itself from previous research by several innovative approaches. Initially, we conducted an in-depth single-cell analysis of the cellular composition in hepatocellular carcinoma (HCC) patients, assessing the functionality of diverse lipid metabolism pathways across various cell types through the application of the single-cell metabolism algorithm. Furthermore, our study implemented a uniform clustering algorithm that not only maintains the informational integrity across each histological dimension but also curtails the potential bias arising from clustering methodological preferences. Additionally, the factors incorporated into our model were extracted from data spanning multiple HCC patient cohorts, thereby augmenting the model’s stability and the reliability of prognostic forecasts, and mitigating the risk of overfitting. Nonetheless, despite the significant advancements achieved in this study, certain limitations persist. Primarily, although we mitigated cohort discrepancies through the application of a correction algorithm, variations in dataset size and sequencing platforms may still exert some influence on the outcomes. Secondly, the precise mechanisms by which lipid metabolism genes specifically contribute to tumor initiation and progression require further experimental validation. Additionally, the clinical relevance of lipid metabolism-associated genes identified in this study should be corroborated in larger, prospective, multicenter cohorts. Future research endeavors should focus on the development of targeted therapies against PTGES3, accompanied by rigorous preclinical and clinical trials to assess the therapeutic potential of PTGES3 inhibitors. Identifying patient subgroups most likely to benefit from PTGES3 inhibition, addressing potential resistance mechanisms, developing combinatorial strategies targeting multiple oncogenic pathways, and implementing adaptive treatment strategies based on continuous patient response monitoring represent promising avenues for investigation. Collectively, this study elucidates a significant association between fatty acid metabolism-related genes and the survival outcomes of hepatocellular carcinoma (HCC) patients, suggesting a pivotal role for fatty acid metabolism in HCC pathogenesis. These findings offer novel perspectives for HCC treatment, positioning fatty acid metabolism as a potential therapeutic target.
Conclusion
Collectively, these findings corroborate the prognostic relevance of lipid metabolism genes in hepatocellular carcinoma (HCC) patients and highlight the correlation between dysregulated lipid metabolism and immune cell infiltration. Moreover, PTGES3 is identified as a promising therapeutic target for HCC. This research presents a robust prognostic risk stratification strategy, potentially offering invaluable perspectives for the development of targeted therapeutic approaches in HCC.
Data availability
The raw data utilized in this study was sourced from a publicly accessible data platform. The liver cancer sample dataset was obtained from TCGA (http://portal.gdc.cancer.gov). For the single-cell dataset, its accession number is GSE149614, and the pertinent data can be accessed through the following link: (https://www.ncbi.nlm.nih.gov/)
References
Rumgay, H. et al. Global, regional and national burden of primary liver cancer by subtype. Eur. J. Cancer. 161, 108–118. https://doi.org/10.1016/j.ejca.2021.11.023 (2022).
Vogel, A., Meyer, T., Sapisochin, G., Salem, R. & Saborowski, A. Hepatocellular carcinoma. Lancet 400 (10360), 1345–1362. https://doi.org/10.1016/S0140-6736(22)01200-4 (2022).
Sidaway, P. Adjuvant sintilimab effective in high-risk HCC. Nat. Rev. Clin. Oncol. 21 (3), 168. https://doi.org/10.1038/s41571-024-00865-3 (2024).
Yan, T. et al. The advanced development of molecular targeted therapy for hepatocellular carcinoma. Cancer Biol. Med. 19 (6), 802–817. https://doi.org/10.20892/j.issn.2095-3941.2021.0661 (2022).
Cho, Y. et al. Transarterial Chemoembolization for Hepatocellular Carcinoma: 2023 Expert Consensus-based practical recommendations of the Korean Liver Cancer Association. Korean J. Radiol. 24 (7), 606–625. https://doi.org/10.3348/kjr.2023.0385 (2023).
Zhou, Y. et al. Challenges facing percutaneous ablation in the Treatment of Hepatocellular Carcinoma: extension of ablation criteria. J. Hepatocell Carcinoma. 8, 625–644. https://doi.org/10.2147/JHC.S298709 (2021). Published 2021 Jun 21.
Scott, R. A. et al. Outcomes of National Survey of the Practice of Hepatocellular Carcinoma Surveillance. J. Hepatocell Carcinoma. 10, 725–731. https://doi.org/10.2147/JHC.S403702 (2023). Published 2023 Apr 29.
Gordan, J. D. et al. Systemic therapy for Advanced Hepatocellular Carcinoma: ASCO Guideline Update. J. Clin. Oncol. 42 (15), 1830–1850. https://doi.org/10.1200/JCO.23.02745 (2024).
Yang, K. et al. The role of lipid metabolic reprogramming in tumor microenvironment. Theranostics 13 (6), 1774–1808. https://doi.org/10.7150/thno.82920 (2023). Published 2023 Mar 13.
Liberti, M. V. & Locasale, J. W. The Warburg Effect: How Does it Benefit Cancer Cells? [published correction appears in Trends Biochem Sci. ;41(3):287] [published correction appears in Trends Biochem Sci. 2016;41(3):287. (2016). https://doi.org/10.1016/j.tibs.2016.01.004]. Trends Biochem Sci. 2016;41(3):211–218. doi:10.1016/j.tibs.2015.12.001.
Cheng, L. et al. Hepatic mitochondrial NAD + transporter SLC25A47 activates AMPKα mediating lipid metabolism and tumorigenesis. Hepatology 78 (6), 1828–1842. https://doi.org/10.1097/HEP.0000000000000314 (2023).
Hu, X. et al. Downregulation of 15-PGDH enhances MASH-HCC development via fatty acid-induced T-cell exhaustion. JHEP Rep. 5 (12), 100892. https://doi.org/10.1016/j.jhepr.2023.100892 (2023). Published 2023 Aug 23.
Vassiliou, E. & Farias-Pereira, R. Impact of lipid metabolism on macrophage polarization: implications for inflammation and tumor immunity. Int. J. Mol. Sci. 24 (15), 12032. https://doi.org/10.3390/ijms241512032 (2023). Published 2023 Jul 27.
Cheng, C., Geng, F., Cheng, X. & Guo, D. Lipid metabolism reprogramming and its potential targets in cancer. Cancer Commun. (Lond). 38 (1), 27. https://doi.org/10.1186/s40880-018-0301-4 (2018). Published 2018 May 21.
Yarla, N. S. et al. Targeting arachidonic acid pathway by natural products for cancer prevention and therapy. Semin Cancer Biol. 40–41, 48–81. https://doi.org/10.1016/j.semcancer.2016.02.001 (2016).
Harris, A. L. Development of cancer metabolism as a therapeutic target: new pathways, patient studies, stratification and combination therapy. Br. J. Cancer. 122 (1), 1–3. https://doi.org/10.1038/s41416-019-0666-4 (2020).
Barrett, T. et al. NCBI GEO: archive for functional genomics data sets–update. Nucleic Acids Res. 41 (Database issue), D991–D995. https://doi.org/10.1093/nar/gks1193 (2013).
Yu, G., Wang, L. G., Han, Y. & He, Q. Y. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16 (5), 284–287. https://doi.org/10.1089/omi.2011.0118 (2012).
Kanehisa, M. & Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucl. Acids Res. 28 (1), 27–30. https://doi.org/10.1093/nar/28.1.27 (2000).
Ashburner, M. et al. Gene ontology: Tool for the unification of biology the gene ontology consortium. Nat. Genet. 25 (1), 25–29. https://doi.org/10.1038/75556 (2000).
Langfelder, P. & Horvath, S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinform. 9, 559. https://doi.org/10.1186/1471-2105-9-559 (2008). Published 2008 Dec 29.
Chen, B., Khodadoust, M. S., Liu, C. L., Newman, A. M. & Alizadeh, A. A. Profiling Tumor infiltrating Immune cells with CIBERSORT. Methods Mol. Biol. 1711, 243–259. https://doi.org/10.1007/978-1-4939-7493-1_12 (2018).
Zeng, D. et al. IOBR: Multi-omics Immuno-Oncology Biological Research to Decode Tumor Microenvironment and signatures. Front. Immunol. 12, 687975. https://doi.org/10.3389/fimmu.2021.687975 (2021). Published 2021 Jul 2.
Brinkman, E. K. & van Steensel, B. Rapid quantitative evaluation of CRISPR Genome Editing by TIDE and TIDER. Methods Mol. Biol. 1961, 29–44. https://doi.org/10.1007/978-1-4939-9170-9_3 (2019).
Ioannou, G. N. HCC surveillance after SVR in patients with F3/F4 fibrosis. J. Hepatol. 74 (2), 458–465. https://doi.org/10.1016/j.jhep.2020.10.016 (2021).
Zhu, Z. Z. et al. Copy number aberrations of multiple genes identified as prognostic markers for Extrahepatic Metastasis-free survival of patients with Hepatocellular Carcinoma. Curr. Med. Sci. 39 (5), 759–765. https://doi.org/10.1007/s11596-019-2103-6 (2019).
Vallianou, N. et al. Understanding the role of the gut Microbiome and Microbial metabolites in non-alcoholic fatty liver disease: current evidence and perspectives. Biomolecules 12 (1), 56. https://doi.org/10.3390/biom12010056 (2021). Published 2021 Dec 31.
Ning, Z. et al. USP22 regulates lipidome accumulation by stabilizing PPARγ in hepatocellular carcinoma. Nat Commun. ;13(1):2187. Published 2022 Apr 21. (2022). https://doi.org/10.1038/s41467-022-29846-9
Bian, G. et al. Identification and validation of a Cancer-Testis Antigen-related signature to predict the prognosis in stomach adenocarcinoma. J. Cancer. 15 (11), 3596–3611. https://doi.org/10.7150/jca.91842 (2024). Published 2024 May 11.
Ye, Y. et al. Identification of a cancer associated fibroblasts-related index to predict prognosis and immune landscape in ovarian cancer. Sci. Rep. 13 (1), 21565. https://doi.org/10.1038/s41598-023-48653-w (2023). Published 2023 Dec 7.
Wu, C. et al. AKR1C3-dependent lipid droplet formation confers hepatocellular carcinoma cell adaptability to targeted therapy. Theranostics 12 (18), 7681–7698. https://doi.org/10.7150/thno.74974 (2022). Published 2022 Nov 7.
Wen, H. et al. High expression of Ten Eleven translocation 1 is Associated with Poor Prognosis in Hepatocellular Carcinoma. Mediators Inflamm. 2023, 2664370. https://doi.org/10.1155/2023/2664370 (2023). Published 2023 May 4.
Xu, R. et al. SQLE promotes pancreatic cancer growth by attenuating ER stress and activating lipid rafts-regulated Src/PI3K/Akt signaling pathway. Cell Death Dis. ;14(8):497. Published 2023 Aug 4. (2023). https://doi.org/10.1038/s41419-023-05987-7
Yin, Q. et al. The integration of multidisciplinary approaches revealed PTGES3 as a novel drug target for breast cancer treatment. J Transl Med. ;22(1):84. Published 2024 Jan 20. (2024). https://doi.org/10.1186/s12967-024-04899-0
González, L. M. et al. Influence of variability in the cyclooxygenase pathway on cardiovascular outcomes of nephrosclerosis patients. Sci Rep. ;13(1):1253. Published 2023 Jan 23. (2023). https://doi.org/10.1038/s41598-022-27343-z
Kopp, B. T. et al. Secondhand smoke alters arachidonic acid metabolism and inflammation in infants and children with cystic fibrosis. Thorax 74 (3), 237–246. https://doi.org/10.1136/thoraxjnl-2018-211845 (2019).
Chen, S. et al. Allosterically inhibited PFKL via prostaglandin E2 withholds glucose metabolism and ovarian cancer invasiveness. Cell. Rep. 42 (10), 113246. https://doi.org/10.1016/j.celrep.2023.113246 (2023).
Morotti, M. et al. PGE2 inhibits TIL expansion by disrupting IL-2 signalling and mitochondrial function. Nature 629 (8011), 426–434. https://doi.org/10.1038/s41586-024-07352-w (2024).
Schmidleithner, L. et al. Enzymatic activity of HPGD in Treg Cells suppresses Tconv cells to maintain adipose tissue homeostasis and prevent metabolic dysfunction. Immunity 50 (5), 1232–1248e14. https://doi.org/10.1016/j.immuni.2019.03.014 (2019).
Mazzoni, M. et al. Senescent thyrocytes and thyroid tumor cells induce M2-like macrophage polarization of human monocytes via a PGE2-dependent mechanism. J. Exp. Clin. Cancer Res. 38 (1), 208. https://doi.org/10.1186/s13046-019-1198-8 (2019). Published 2019 May 21.
Ielpo, S. et al. Crosstalk and communication of cancer-associated fibroblasts with natural killer and dendritic cells: New frontiers and unveiled opportunities for cancer immunotherapy. Cancer Treat. Rev. Published Online Oct. 15 https://doi.org/10.1016/j.ctrv.2024.102843 (2024).
Sun, Y. et al. Integrated multi-omics profiling to dissect the spatiotemporal evolution of metastatic hepatocellular carcinoma. Cancer Cell. 42 (1), 135–156e17. https://doi.org/10.1016/j.ccell.2023.11.010 (2024).
Cheng, Z. et al. Endoplasmic reticulum stress promotes hepatocellular carcinoma by modulating immunity: a study based on artificial neural networks and single-cell sequencing. J. Transl Med. 22 (1), 658. https://doi.org/10.1186/s12967-024-05460-9 (2024). Published 2024 Jul 15.
Chen, S. et al. Hypoxia-driven tumor stromal remodeling and immunosuppressive microenvironment in scirrhous HCC. Hepatology 79 (4), 780–797. https://doi.org/10.1097/HEP.0000000000000599 (2024).
Llovet, J. M., Montal, R., Sia, D. & Finn, R. S. Molecular therapies and precision medicine for hepatocellular carcinoma. Nat. Rev. Clin. Oncol. 15 (10), 599–616. https://doi.org/10.1038/s41571-018-0073-4 (2018).
Finn, R. S. et al. Phase ib study of Lenvatinib Plus Pembrolizumab in patients with Unresectable Hepatocellular Carcinoma. J. Clin. Oncol. 38 (26), 2960–2970. https://doi.org/10.1200/JCO.20.00808 (2020).
Yu, J. et al. Components of the JNK-MAPK pathway play distinct roles in hepatocellular carcinoma. J. Cancer Res. Clin. Oncol. 149 (19), 17495–17509. https://doi.org/10.1007/s00432-023-05473-9 (2023).
Luo, Y., Liu, H., Fu, H., Ding, G. S. & Teng, F. A cellular senescence-related classifier based on a tumorigenesis- and immune infiltration-guided strategy can predict prognosis, immunotherapy response, and candidate drugs in hepatocellular carcinoma. Front. Immunol. 13, 974377. https://doi.org/10.3389/fimmu.2022.974377 (2022). Published 2022 Nov 15.
Funding
None.
Author information
Authors and Affiliations
Contributions
ZR, XX, XS– designed study protocol, writing the manuscript; SB, HC – cell study in vitro; XT, DH– statistical data analysis; XP, XS– searching of world literature and discussion of the obtained results with the results of previous research; All the authors approved the final version of the article to be published.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Consent for publication
Not applicable.
Ethics aproval and consent to participate
This study was approved by the ethics committee of The Second Affiliated Hospital of Nanchang University(approval no.O-2024147). We certify that the study was performed in accorgence1964 declaration of HELSINKI and later amendments.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Dai, H., Tao, X., Shu, Y. et al. Integrating single-cell RNA-Seq and bulk RNA-Seq data to explore the key role of fatty acid metabolism in hepatocellular carcinoma. Sci Rep 15, 2077 (2025). https://doi.org/10.1038/s41598-025-85506-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-85506-0
Keywords
This article is cited by
-
Big data-driven machine learning: transforming multi-omics lung cancer research
Discover Oncology (2025)
