
Abstract
Intervertebral Disc degeneration (IDD) is one of the leading causes of disability, and current therapies are ineffective. Phosphodiesterase 4B (PDE4B) plays an essential role in regulating the activation of nuclear factor E2-related factor 2 (Nrf2), while Nrf2 regulates ferroptosis. However, it is still unknown whether PDE4B is involved in the development of IDD. In this study, we explored the role of PDE4B on ferroptosis and Nrf2 in IDD pathogenesis by in vivo and in vitro experiments. The findings suggested that the expressions of PDE4B, ASCL4, and TRFC were significantly upregulated, and the expression of Nrf2 was significantly downregulated in nucleus pulposus (NP) tissues from human IDD patients dependent on IDD degeneration. Overexpression of PDE4B (PDE4B-OE) in NP cells upregulated the expression of ASCL4 and TRFC, and downregulated the expression of Nrf2. Meanwhile, the level of cytokine and oxidative stress were upregulated. Ferroptosis inhibitor Fer-1 or Nrf2 activator dimethyl fumarate (DMF) suppressed the effect of PDE4B-OE, while ferroptosis inducer elastin enhanced the effect of PDE4B-OE. In the IDD rat model, PDE4 inhibitor roflumilast, ferroptosis inhibitor Fer-1, or Nrf2 activator dimethyl fumarate (DMF) delayed IDD pathogenesis. While administration of ferroptosis inducer elastin enhanced IDD pathogenesis. Combination with PDE4B inhibitor and ferroptosis inhibitor Fer-1 significantly synergistic reversed IDD pathogenesis. While combination with PDE4B inhibitor or Nrf2 activator and elastin also decreased the degree of the IDD. The IHC suggested PDE4 inhibitor downregulated the expression of ASCL4 and TRFC. However, the combination effect of the Nrf2 activator was not obvious. Our study suggested that aberrant PDE4B activation in NP tissues induces pathological changes in IDD mediated by ferroptosis, and PDE4 inhibitor reveres the process of IDD by suppressing ferroptosis, and has a synergic effect with ferroptosis inhibitor. So PDE4B inhibition may be a potential therapeutic strategy for IDD.
Similar content being viewed by others


Introduction
Intervertebral Disc degeneration (IDD) is one of the leading causes of lower back pain, which is the fifth most common cause of medical treatment for adults, leading to a high disability rate. In severe cases, this may lead to labor loss and cause significant economic burdens on families and society. According to global disease survey data from 2016 to 2019, low back pain is the leading cause of years of life lost due to disability1,2. At present, the techniques available for treating IDD in clinical practice have limitations. Conservative treatments, such as traction, physical therapy, anti-inflammatory and analgesic methods, and surgical procedures mainly focus on relieving pain and current symptoms and cannot fundamentally block the pathological process of IDD3. Therefore, a deep understanding of the pathological mechanism of this disease, and the identification of new diagnostic and therapeutic targets are important ways to prevent or alleviate IDD and improve the quality of life of patients.
In terms of anatomical structure, the lumbar intervertebral disc is composed of three parts: the fibrous ring, the nucleus pulposus (NP), and the cartilage endplate. NP are located in the central posterior region of the intervertebral disc and are rich in proteins, polysaccharides, and water. The NP is the largest avascular tissue in the body, and its supply of nutrients and excretion of metabolites mainly rely on the permeation of cartilage4. In addition, degeneration of the NP is also affected by many other physiological and pathological factors, such as trauma, emergency braking, excessive exercise, smoking, diabetes, and vascular diseases5,6.
Many types of cell death mediate the pathological mechanism of IDD, such as apoptosis, pyroptosis, necrosis, etc. The role of ferroptosis in IDD has not been explored in depth. Ferroptosis was first proposed in 2012, and is a type of cell death caused by the small molecule compound erastin in the body. Moreover, erastin inhibits the transport of cysteine into cells, leading to depletion of glutathione and inactivation of glutathione peroxidase 4 (GPX4). This results in a large accumulation of lipid peroxidation products in cells, ultimately inducing ferroptosis7. In 2020, Zhang et al.8 reported the in vitro studies of rat NP cells, and found that homocysteine can induce an oxidative stress response and further activate ferroptosis, leading to the loss of NP cells. In 2021, Zhang et al.9 used single-cell RNA sequencing to analyze the NP tissues of degenerative lumbar intervertebral discs and normal lumbar intervertebral discs, and identified 7 major subgroups of chondrocytes. After gene enrichment analysis (from the GO and KEGG databases), it was found that iron metabolism pathways play a dominant role in the functional regulation of these chondrocytes. In the same year, Yang et al.10 conducted in vitro experiments using rat-derived annulus fibrosus cells and NP cells, and noted that oxidative stress-induced ferroptosis is involved in the mechanism of lumbar disc degeneration. In animal model studies, several researchers have noted that blocking iron-mediated cell death in the nucleus pulposus can provide limited relief from IDD11,12. These studies indicated that ferroptosis may play an important role in the lumbar disc degeneration, especially in nucleus pulposus.
Phosphodiesterase 4 (PDE4) family is the specific enzyme to hydrolyze cAMP. Based on the differences in coding genes, PDE4s can be further divided into four subtypes: PDE4A, PDE4B, PDE4C, and PDE4D. PDE4B is expressed mainly in the cytoplasm of inflammatory cells and is widely present in monocytes and neutrophils13. Current research has confirmed its important role in the pathological regulation of inflammatory and fibrotic damage14,15. Deletion of PDE4B protected spinal cord injury induced gut dysbiosis, bacterial overgrowth and endotoxemia16. At present, there are no reports on the correlation between PDE4B and IDD.
Nuclear factor E2-related factor 2 (Nrf2) is an important transcription factor that regulates the cellular oxidative stress response and is also a central regulator maintaining intracellular redox homeostasis. Nrf2 can regulate the process of ferroptosis through the oxidative stress response17. GPX4 is the most important intracellular anti-lipid peroxidation enzyme and a core regulatory factor for ferroptosis. It synergistically exerts its anti-lipid peroxidation effect with GSH, thereby promoting the reduction of lipid peroxide levels on the cell membrane and inhibiting ferroptosis. Our previous study reported that PDE4B positively regulated the cytosolic Nrf2 by the NLRP3 complex18, and others reported that PDE4B negatively regulated the nucleus Nrf2 and the oxidative stress response mediated by heme oxygenase (HO-1)19. But whether PDE4B negatively correlates with Nrf2 in NP tissue is unclear. Based on the interaction between PDE4B and Nrf2, as well as its regulatory ability in inflammatory pathology, PDE4B may act on ferroptosis, but its existence in IDD is unknown.
In the current study, we supposed that PDE4B promotes ferroptosis related markers, such as acyl-CoA synthetase long chain family member 4 (ACSL4) and transferrin receptor (TFRC) in NP cells through the Nrf2 pathway and participates in IDD. We investigated the functions and molecular mechanisms of PDE4B in IDD by in vivo and in vitro study, and our study paves the way for future research targeting PDE4B as a promising therapeutic strategy for IDD.
Methods
Human NPC culture and treatment
NP cells were procured from Procell (Wuhan, Hubei, China). These cells were maintained at a concentration of 1 × 105 cells/ml in an environment consisting of 5% CO2 and 95% humidity at 37 °C using a complete human disc nucleus pulposus cell medium (#CM-H097, Procell, Wuhan, Hubei, China). Upon reaching 90% confluence, the cells were passaged using trypsin/ethylene diamine tetraacetic acid (EDTA) and then reseeded at a density of 104 cells/cm2. The cells were cultured continuously until passage no. 8, and for experiments, passage no. 3 to no. 8 were utilized. NPs were cultured at a seeding density of 2 × 105 cells per well in a 6-well plate. Subsequently, the cells were transfected and co-transfected with the vector control plasmid pIRES2-EGFP, the pIRES2-EGFP-PDE4B plasmid (PDE4B-OE)(# PPL00120-2a, Public Protein/Plasmid Library, Nanjing, Jiangsu, China) for 24 h. Following transfection of the PDE4B-OE plasmid, the cells were subjected to treatment with ferroptosis inducer erastin (HY-15763, MCE) at a concentration of 10 µM, ferroptosis inhibitor ferrostatin-1 (Fer-1, HY-100579, MCE) at 1 µM, or Nrf2 activator dimethyl fumarate (DMF, HY-17363, MCE) at 100 µM. Treatments were applied for 24 h for ELISA experiments and 6 h for qRT-PCR analysis. Following the treatment, both the cell supernatant and cell lysate were harvested and stored at a temperature of -80 °C, facilitating subsequent experimental analyses.
Animals and experiment design
Eight-week-old male Sprague‒Dawley rats were purchased from the Experimental Animal Center of Zhejiang University (Hangzhou, China) and housed at the Experimental Animal Center of Zhejiang University under standard conditions (23 ± 2 °C, 55 ± 5% humidity, and 12 –12 h light/dark cycle). All methods were carried out in accordance with relevant guidelines and regulations, and are reported in accordance with ARRIVE guidelines (https://arriveguidelines.org). All procedures involved with rats were approved by the Animal Care and Use Committee of Zhejiang University (No: ZJU20240617).
The rats were randomly divided into 9 groups (n = 5 per group) as follows: Normal control group; IDD model group; roflumilast (Rof, 5 mg/kg, NB1739-1, Meilune, Dalian, China. https://www.meilune.com/) + IDD model group; Fer-1 (5 mg/kg) + IDD model group; Eastin (5 mg/kg) + IDD model group; Fer-1(5 mg/kg) + Rof(5 mg/kg) + IDD model group; Eastin (5 mg/kg) + Rof(5 mg/kg) + IDD model group; Fer-1(5 mg/kg) + DMF(5 mg/kg) + IDD model group; Eastin (5 mg/kg) + DMF(5 mg/kg) + IDD model group.
Preparation of the IDD rat model
A rat IDD model was established in the coccygeal vertebra through a needle puncture. Briefly, before surgery, the extent of disc degeneration was measured using a magnetic resonance imaging (MRI) system (MAGNETOM Verio 3.0T, Siemens AG, Erlangen, Germany). After general anesthesia with 2.5% 2,2,2-tribromoethanol (20 ml/kg), the rat’s coccygeal discs between the 4th and 7th coccygeal vertebrae (Co4–Co7), were punctured using a 20-gauge sterile syringe needle from the dorsal side for 30 s. To ensure degeneration, the needle was passed through the nucleus pulposus (NP) tissue and the bilateral annulus fibrosus (AF), and the needle was rotated in the axial direction by 180° and held for 10 s. To fix the position of the puncture point, an X-ray system (Philips Allura Xper FD20, Amsterdam, Netherlands) was used to observe the process. Four weeks after surgery, according to the Pfimmann grading system, four blinded investigators used MRI results to classify the disc images into 5 grades20. Then, tail samples were harvested after the animals were sacrificed. The proximal and distal adjacent intervertebral discs were used as blank controls.
Hematoxylin-eosin staining and safranin O-fast green staining
Rat Co4-7 coccygeal discs were fixed in 4% paraformaldehyde for 48 h, decalcified in fast decalcifying solution (Preserve, Zhuoding, Beijing) for 72 h, embedded in paraffin and cut into 5 μm sections. Then, the sections were deparaffinized and dehydrated for histological analysis, and hematoxylin and eosin (H&E), and safranin O-fast green (SO&FG) staining were performed separately on consecutive tissue sections. Finally, the slices were dehydrated, cleared, and sealed. Images were obtained using a SLIDEVIEW VS200 workstation (Olympus, China). Histology degeneration score (HDS) was determined according to the histological characteristics of the nucleus pulposus following the standardized histopathology scoring system ranging from 0 for no degeneration to 2 for severe degeneration21.
Immunostaining assays
Deparaffinization and dehydration of the slice are described in the H&E staining part. After that, quenching the endogenous peroxidase activity was used by 0.3% H2O2 for 10 min; antigen retrieval was performed with 10 mM citrate buffer (pH 6.0) for efficient epitope exposure; and blocking buffer (5% BSA in PBS) was used to eliminate nonspecific binding of antibodies for 10 min. Afterwards, the sections were incubated with primary antibodies against ACSL4 (1:100) (#36176, signalway antibody), TFRC (1:200) (#33101, signalway antibody) at 4 °C overnight, followed by incubation with biotinylated IgG and streptavidin-horseradish peroxidase for 30 min at room temperature, then DAB (2 mg/ml) was used to visualize the immunoreactivity. Finally, the sections were counterstained with hematoxylin, and then mounted. (Fiji is just) ImageJ was used for semi-quantitative analysis of the average optical density of immunohistochemical staining, and intensity value was used to calculate and compare.
Enzyme-linked immunosorbent assay (ELISA)
The cultured cell supernatant was collected and centrifuged at 12,000 rpm and 4 °C for 10 min, after which the supernatant was harvested. The quantification of the inflammatory cytokines, including interleukin-1β (IL-1β), interleukin-6 (IL-6), and tumor necrosis factor-α (TNF-α), was carried out by using ELISAKit (R&D Systems, Minneapolis, MN, USA). Those analyses followed the manufacturer’s instructions.
Quantitative reverse transcription polymerase chain reaction (qRT–PCR)
Total RNA was isolated from the NP cells by using RNA iso Plus (9109, Takara Bio, Inc., Kusatsu, Shiga, Japan). RNA (1 µg) was collected and used to synthesize first-strand cDNA. SYBR Green Master Mix reagent was purchased from Takara Bio, Inc., and used for qRT‒PCR analysis, whereas GAPDH was used to normalize the data. All primers were obtained from Sangon Biotech (Shanghai, China). For each PCR, 4.5 µL of diluted cDNA, 5 µL of 2X SYBR Green mix, 0.25 µL of forward primer, and 0.25 µL of reverse primer were used. qPCR was performed under the following conditions: 95 °C for 10 min; 45 cycles of 95 °C for 10 s, 60 °C for 20 s, and 72 °C for 30 s. A quantitative reverse transcription PCR instrument was used for detection. The data were evaluated using the 2 − ΔΔCt formula22. The primer’s sequence and related information list in Table 1.
Detection of reactive oxygen species (ROS)
NP cells were seeded at 1 × 105 cells/ml density into 96-well plates. Following the transfection of the pIRES2-EGFP and pIRES2-EGFP-PDE4B plasmids, as well as the administration of erastin, Fer-1, or DMF, as described earlier in terms of time and concentration, intracellular ROS production was assessed using the fluorescent probe 2′,7′-dichlorofluorescein diacetate (DCFH-DA), which was obtained from Sigma Aldrich (St. Louis, United States). After treatment completion, the cells were incubated with 10 µM DCFH-DA at 37 °C for 30 min in a dark environment. The fluorescence intensity of DCF was measured using a Varioskan Flash microplate reader from Thermofisher Scientific (Vantaa, Finland) with an excitation wavelength of 485 nm and an emission wavelength of 530 nm.
Determination of superoxide dismutase (SOD) activity and GPX4 concentration
GPX4 and SOD activity in the cell were evaluated with a commercially available SOD determination kit (Nanjing Jiancheng Institute of Biological Engineering, Nanjing, China) and a GPX4 ELISA kit (Meimian, China) according to the manufacturer’s instructions. SOD activity can be calculated by the following formula (U/mg, the number of milligrams of protein): SOD activity in cells = (standard tube absorbance-measuring tube absorbance)/standard tube absorbance/50%× (total volume of reaction system/sampling volume)/protein content. For GPX4 calculation, creating a standard curve of GPX4 concentration and absorbance, the sample’s absorbance were determined, and the concentration were calculated from the standard curve.
Patient nucleus pulposus (NP) samples
Human NP samples were obtained from 14 male patients (mean age 54.4 ± 14.5 years) who were diagnosed with lumbar disc herniation and underwent nucleus pulposus resection. Ethics approval was obtained from the Ethics Committee of Tongde Hospital of Zhejiang Province (No. 202122-088). Informed consent was obtained from all participants enrolled in this study. The study was conducted according to the Code of Ethics of the World Medical Association (Declaration of Helsinki). The MRI-based Pfimmann grading system was used to assess the degenerative grade of the NP samples. Grade III (n = 3), Grade IV (n = 3), and Grade V (n = 3) NP samples were subjected to WB and ELISA.
Western blot
The intervertebral disc tissues were lysed in cold radioimmunoprecipitation assay (RIPA) lysis buffer (containing 1% protease inhibitor cocktail (Roche Diagnostics, USA), 2% PMSF (Millipore Sigma) and 1% PhoSSTOP (Roche Diagnostics, USA). After centrifugation for 10 min at 12,000 × g, the protein concentration was determined using a Bradford assay (Thermo Fisher Scientific, USA). Fifty micrograms of protein were prepared for the western blot experiment. The protein sample was mixed with 5x loading buffer (Beyotime, Beijing, China) and denatured for 5 min at 100 °C. 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS‒PAGE) was used to separate the proteins, which were subsequently transferred to a nitrocellulose membrane (PALL BioTrace, #66485, USA) for 70 to 90 min at 300 mA using Mini-protein II System (Bio-Rad, USA). The membrane was blocked with Tris-buffered saline containing 5% BSA for 1 h at room temperature. The membranes were subsequently incubated with specific antibodies for PDE4B (# PD4B-201AP, 1:1000, FabGennix), ACSL4 (#36176, 1:1000, Signalway Antibody), TFRC (#33101) (1:1000, Signalway Antibody), Nrf2 (C-20) (SC722) (1:1000, Santa Cruz), β- tubulin (db3285) (1:5000, Diagbio) at 4 °C with gentle shaking overnight. After washing three times, the membranes were incubated with the appropriate secondary antibodies (IRDye 800CW goat anti-rabbit; IRDye 680CW goat anti-mouse; LI-COR Biosciences, 1:5000) for 2 h at room temperature. Immunoblots were scanned and quantified using an Odyssey imaging analysis system (Image Studio Ver 5.2, LI-COR Biosciences).
Quantification and statistical analysis
Data were presented as means ± SEM. All experiments were performed in at least three independent experiments. Data were analyzed with one-way ANOVA and Student’s t-test where appropriate. All statistical analyses were performed with GraphPad Prism 10.0 software. p < 0.05 was considered as a statistical difference.
Results
PDE4B suppresses Nrf2 and upregulates ACSL4 and TRFC in NP cells at the mRNA level
To study whether the PDE4B plays a role in activating ferroptosis signaling. NP cells were transfected with the vector control or PDE4B overexpressing plasmid (PDE4B-OE), and then treated with ferroptosis inducer erastin (10 µM), the ferroptosis inhibitor ferrostatin-1 (Fer-1) (1 µM), and the Nrf2 activator dimethyl fumarate (DMF) (100 µM). qRT‒PCR assay was performed to analyze the expression of ACSL4, TRFC, and Nrf2 in NP cells. PDE4B-OE significantly upregulated the mRNA expression of ACSL4, and TFRC, and suppressed Nrf2 expression(p < 0.05). Erastin further upregulated the mRNA expression of PDE4B, ACSL4, and TFRC, and suppressed Nrf2 expression; however, compared with the PDE4B-OE, the 1 µM Fer and DMF (100 µM) treatment largely suppressed the expression of PDE4B, ACSL4, and TFRC, and increased Nrf2 expression at the mRNA level(p < 0.05) (Fig. 1A). These data suggested that PDE4B overexpression in NP cells positively regulates ACSL4 and TRFC levels, and ferroptosis inhibitor and Nrf2 activator can downregulate PDE4B-induced ferroptosis.
PDE4B increases cytokine release and oxidative stress involved in ferroptosis and Nrf2 in human NP cells
To further determine whether PDE4B overexpression mediated cytokine release. IL-6, TNF-α, and IL-1β in NP cells were determined. The results revealed that pretreatment with the PDE4B-OE upregulated IL-6, TNF-α, and IL-1β secretion(p < 0.05), when compared to that in the control group. While erastin increased PDE4B-OE-induced cytokine release, there was no significant difference when compared to the PDE4B-OE group (Fig. 1B). In contrast, Fer-1 (1 µM) or DMF (100 µM) significantly suppressed the PDE4B-OE-induced secretion of IL-1β, TNF-α and IL-6 in NP cells(p < 0.05). These results suggested that PDE4B-OE-induced cytokine release is involved in ferroptosis and Nrf2. Ferroptosis can enhance PDE4B-OE-induced cytokine release, and Nrf2 can negatively regulate PDE4B-OE-induced cytokine release.

Effect of Fer-1, DMF and erastin on PDE4B-OE plasmid induced changes in NP cells. (A) PDE4B suppressed the mRNA expression of Nrf2, and upregulated mRNA expression of ACSL4 and TFRC. Cells were transfected with PDE4B-OE plasmid for 24 h, later, cells were with erastin (10 µM), Fer-1 (1 µM), and DMF (100 µM) for 6 h. mRNA level of PDE4B, ACSL4, Nrf2 and TFRC were determined by qRTPCR. (B) Effect of Fer-1, DMF and erastin on cytokines level against PDE4B-OE plasmid in NP cells. Cells were transfected with PDE4B-OE plasmid for 24 h, later, cells were with erastin (10 µM), Fer-1 (1 µM), and DMF (100 µM) for 24 h. Cells supernatant was analysed by ELISA. (C–E) Effect of Fer-1, DMF and erastin on SOD, ROS and GPX4 level against PDE4B-OE plasmid in NP cells. Cells were transfected with PDE4B-OE plasmid for 24 h later cells were with erastin (10 µM), Fer-1 (1 µM), and DMF (100 µM) for 6 h. (C) Percentage of inhibition of SOD. (D) Percentage of ROS generation; (E) GPX4 concentration; Data shown are means ± SEM of n = 3. * p < 0.05, ** p < 0.01, ***p < 0.001, ****p < 0.0001.
To evaluate the effect of PDE4B on oxidative stress, the levels of reactive oxygen species (ROS), superoxide dismutase (SOD), and glutathione peroxidase 4 (GPX4) were subsequently assessed. As anticipated, the overexpression of PDE4B increased ROS levels and decreased SOD levels in NP cells(p < 0.05), suggested that PDE4B induced oxidative stress. When PDE4B -OE combinate with erastin, they led to a further increase in ROS levels and a decrease in SOD levels in NP cells. In contrast, PDE4B -OE combination with Fer-1 (1 µM) and DMF (100 µM) had opposite effects, reducing ROS levels and promoting SOD levels in NP cells(p < 0.05) (Fig. 1C & D). These results suggested that PDE4B-OE induced oxidative stress mediated by ferroptosis and Nrf2.
Glutathione peroxidase 4 (GPX4) was subsequently assessed. The overexpression of PDE4B decreased GPX4 levels in NP cells(p < 0.05), suggested that PDE4B induced ferroptosis may be GPX4 -dependent. In contrast, PDE4B -OE combinate with Fer-1 (1 µM) or DMF (100 µM) had opposite effects, increasing GPX4 levels in NP cells (p < 0.05) (Fig. 1E). These results suggested that PDE4B-OE activates ferroptosis, but interestingly, when PDE4B-OE combinated with erastin, there was not a further decrease in GPX4, suggesting that PDE4B-induced GPX4 decrease is in the same pathway with erastin-induced GPX4 decrease. Meanwhile, Nrf2 can negatively regulate PDE4B-OE-induced GPX4 decrease.
PDE4B mediates ferroptosis in the nucleus pulposus in IDD rat model
To further confirm the relationship between PDE4B and ferroptosis, we conducted an IDD rat model. The MRI results revealed that PDE4 inhibitor roflumilast, and ferroptosis inhibitor Fer-1can significantly suppress the process of IDD, while administration of ferroptosis inducer elastin, IDD process unchanged. However, the combination of PDE4B inhibitor and Fer-1 significantly synergistic reverse IDD pathogenesis. While the combination of PDE4B inhibitor and elastin unchanged the degree of the IDD. Meantime, in combination with Nrf2 activator dimethyl fumarate (DMF) and ferroptosis inhibitor Fer-1 or ferroptosis inducer erastin unchanged the process of IDD in MRI image (Fig. 2). These results suggested that PDE4 inhibition has a protective effect on the IDD process, and has a synergic effect with ferroptosis inhibitor, and the effect is better than the Nrf2 activator.

MRI images and analysis of IDD model and drugs treated IDD model before and after modeling. PDE4 inhibitor reduces the progression of lesions in the IDD model. Orange arrow points out the needle puncture site; (A) Typical MRI representative images (B) MRI score diagram. Data shown are means ± SEM of n = 5. * p < 0.05, ** p < 0.01, ***p < 0.001.
The histological results suggested that PDE4 inhibitor roflumilast, ferroptosis inhibitor Fer-1, or Nrf2 activator dimethyl fumarate (DMF) can significantly reduce the injury of NP tissues, While administration of ferroptosis inducer elastin could not. Combination with PDE4B inhibitor and ferroptosis inhibitor Fer-1 significantly synergistically reduced injury of NP tissues. Combination with PDE4B inhibitor or Nrf2 activator and elastin also decreased injury of NP tissues (Fig. 3).

Pathological image and histologic degeneration score of IDD model and drugs treated IDD model. (A) Typical histological diagram of intervertebral disc tissue in control, IDD model and drugs treated IDD model, stained with H&E and SO&FG. Scale bar = 500 μm. (B) Histological score diagram. Data shown are means ± SEM of n = 5. *p < 0.05, **p < 0.01, ***p < 0.001.
And further, IHC results suggested PDE4 inhibitor downregulated ASCL4 and TRFC, and increased Nrf2, ferroptosis inhibitor Fer-1 or Nrf2 activator dimethyl fumarate (DMF) can significantly reduce the injury of NP tissues, While administration of ferroptosis inducer elastin could not. Combination with PDE4B inhibitor and ferroptosis inhibitor Fer-1 synergistic reduced injury of NP tissues. Combination with PDE4B inhibitor or Nrf2 activator and elastin also decreased injury of NP tissues (Fig. 4). Those results suggested PDE4 inhibitor can downregulate ASCL4 and TRFC.

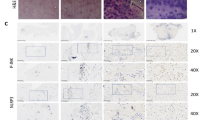
ASCL4 and TFRC expression in IDD model and drugs treated IDD model. Immunohistochemical staining of paraffin sections was performed with anti-ASCL4 antibody or anti-TFRC antibody, and (Fiji Is just) imageJ was used for average optical density analysis. (A) Representative IHC images of ASCL4 in control, IDD model and drugs treated IDD model; positive staining was brown color. (B) Quantitative analysis results of ASCL4 in NP cells. (C) Representative IHC images of TFRC in control, IDD model and drugs treated IDD model. Up line scale bar = 500 μm, down line scale bar = 50 μm. Data shown are means ± SEM of n = 3–6. *p < 0.05, **p < 0.01, ***p < 0.001.
Nrf2 negatively correlates with ferroptosis in the NP tissues of IDD patients
To further confirm the results, we investigated the effects of PDE4B, ferroptosis (ACSL4 and TFRC are known ferroptosis related hallmarkers), and Nrf2 activation in human nucleus pulposus samples from clinical IDD patients with different grades. Western blot analysis revealed that ACSL4 and TFRC protein expression increased with deteriorating NP, while the phosphorylation of Nrf2 decreased (Fig. 5). However, the PDE4B expression seems unchanged. The reason may be due to the grade of patients, in this study, we cannot collect the Grade II and Grade I NP tissue. Our previous data showed that PDE4B is lower in Grade II (data not shown). These observations confirmed that PDE4B plays an important role in NP tissues, and the levels of ACSL4 and TFRC in NP tissues from IDD patients were negatively correlated with phosphorylated Nrf2. These results demonstrated that the expression of ASCL4 and TFRC in human NP tissues is intricately regulated in response to the activation of the Nrf2 signaling pathway during IDD progression. Based on the above PDE4B overexpression induces ferroptosis, and PDE4B inhibition should be a promising approach for IDD treatment.

Effect of ferroptosis of nucleus pulposus through Nrf2 signalling pathway in IDD patients. The expression of ACSL4, TFRC, pNrf2 were determined by nucleus pulposus tissue of the IDD patients by western blotting. (A) Representative images of ACSL4, TFRC and pNrf2 (B) densitometric analysis in the IDD patients with different degrees of lesions. Data shown are means ± SEM of n = 3. *p < 0.05, **p < 0.01, ***p < 0.001.
Discussion
In present study, we first explored PDE4B mediated ferroptosis in the mechanism of IDD and identified PDE4B as a target in the nucleus pulposus. In vitro experiment results demonstrated the PDE4B mediated ferroptosis in NP cell, and Nrf2 can inhibit PDE4B induced ferroptosis. In vivo experiment results demonstrated that PDE4 inhibitor significantly suppress the ferroptosis of the NP cells, reduce the expression of ACSL4 and TFRC in NP cells, delay the IDD process. Meantime, PDE4 inhibitor have a synergic effect with ferroptosis inhibitor. Furthermore, we confirmed the upregulation of PDE4B and ferroptosis and downregulation of Nrf2 in the NP cells from IDD patients. So PDE4B inhibition supply a novel strategy to tamper the development of IDD.
Currently, lumbar disc degeneration is the main cause and important pathological basis of low back pain23. On the one hand, lumbar disc degeneration leads to spinal stenosis and nerve compression, directly causing lower back and leg pain. On the other hand, the decrease in intervertebral disc height after degeneration increases the biomechanical load on the spine, thereby accelerating adjacent segment damage24. The nucleus pulposus is located in the central posterior region of the intervertebral disc and is rich in proteins, polysaccharides, and water. Both the nucleus pulposus and the endplate are sensitive to age and are prone to degenerative changes with age. In childhood, the structural boundaries of various parts of the intervertebral disc are clear. As age increases, the protein polysaccharides and water inside the nucleus pulposus gradually decrease, collagen deposition increases, the texture becomes more difficult, and the nucleus pulposus becomes closely connected to the inner fibrous ring. Some inner fibrous rings extend into the nucleus pulposus, causing cracks in the nucleus pulposus, which is the process of nucleus pulposus degeneration. Thompson et al.25 classified the degeneration pathology of the nucleus pulposus into five levels: level 1: swelling of the nucleus pulposus but intact fibrous ring; level 2: Occasionally, fibrous bands extend into the nucleus pulposus, and cartilage-like substances appear between the fibrous annulus layers; level 3: An increase in fibrous tissue and cartilage-like substances between the layers within the nucleus pulposus; and level 4: Horizontal fissures appear in the nucleus pulposus and local collapse of the fibrous ring. Level 5: The fissure extends to the entire nucleus pulposus and annulus fibrosus. It is not difficult to see that degeneration of the nucleus pulposus has two key points: loss of proteoglycan components in the nucleus pulposus and abnormal proliferation of fibrotic structures, both of which ultimately lead to the death of functional cells.
Phosphodiesterase-4 (PDE4) hydrolyzes the second messenger cyclic adenosine monophosphate (cAMP) in the cell, thereby terminating the biochemical effects of cAMP. Four subtypes of PDE4 (A, B, C, and D) are encoded by four genes. The conserved sequence and structure of PDE4s indicate that they play a crucial role in the regulation of physiological functions26. PDE4B is the main subtype of PDE4 in immune cells, and plays an important role in inflammation, the immune response, and tumors27. Our previous work has reported that lipopolysaccharides (LPS) can specifically induce the expression of PDE4B, and PDE4B can mimic the LPS-induced inflammation. Meanwhile, deficiency of PDE4B inhibits the inflammasome activation and pyroptosis in LPS-stimulated lung injury model and macrophages by regulating ROS/Nrf2/NLRP3 activation18. But there is no report mentioning PDE4B and IDD.
Previous studies suggested circPDE4B downregulated in osteoarthritis tissue, and regulates chondrocyte cell viability and extracellular matrix metabolism. Moreover, circPDE4B adeno-associated virus inhibits the degradation of cartilage matrix28. Nucleus pulposus is composed of a crisscrossing fibrous network structure consisting of a proteoglycan mucinous matrix and chondroid-like cells, which are derived from notochord cells and with a phenotype similar to that of chondrocytes. Nucleus pulposus cells secreted type II collagen and proteoglycan which form the extracellular matrices. During intervertebral disc degeneration, the imbalance of extracellular matrix components is an important factor affecting intervertebral disc stability.
Ferroptosis is a new type of cell death caused by iron-dependent peroxidation of lipids29. Long-chain acyl-coenzyme A (CoA) synthase 4 (ACSL4) and glutathione peroxidase 4(GPX4) positively and negatively regulate ferroptosis, respectively30. Accumulated evidence suggested that the ACSL4 promotes the biosynthesis of specific polyunsaturated fatty acids, such as arachidonic acid and adrenic acid, and contributes to the execution of ferroptosis by triggering phospholipid peroxidation. So, ACSL4 facilitates the cross-talk between ferroptosis and FA metabolism31. Recently, ACSL4 Inhibitor might be a potential therapeutic target32. In the present study, PDE4B overexpression significantly increased ASCL4 expression and decreased GPX4 in NP cells, and PDE4 inhibitor suppressed the expression of ASCL4 in the NP tissue of the IDD rat model, those results suggested that PDE4B-induced phospholipid peroxidation. PDE4B-OE increased ROS, and decrease SOD activity in NP cells also supported this conclusion.
Transferrin receptor (TFRC), another specific ferroptosis marker, is a universal iron importer for all cells using extracellular transferrin. Transferrin is an iron-carrier protein that binds to Fe3 + to deliver iron to different tissues via TFRC-mediated endocytosis33. TFRC is a potential factor leading to abnormal iron accumulation and ferroptosis, and is involved in the process of numerous diseases, such as skeletal muscle regeneration34, acute liver injury35, and cancer36, indicating that TFRC will be a potential therapeutic target. In the present study, PDE4B overexpression significantly increased TFRC expression in NP cells, and PDE4 inhibitor suppressed the expression of TFRC in the NP tissue of IDD rat model, those results suggested that PDE4B promotes iron transportation.
In our prior research, we established that PDE4B can trigger pyroptosis in the MH-S cell line and LPS-injured lung tissue18. In our current investigation, the overexpression of PDE4B led to the increased expression of IL-1β, TNF-α, and IL-6 in NP cells. This compelling evidence strongly suggested that PDE4B activates the inflammasome in NP cells, demonstrating its ability to initiate at least two forms of cell death in NP cells. Moreover, further investigation is warranted to understand the molecular mechanism of PDE4B-induced ASCL4/TFRC activation in NP cells, such as E-cadherin-NF2-Hippo-YAP pathway37, which promotes ferroptosis by upregulating several ferroptosis modulators, including ACSL4 and TFRC.
The study has a few limitations. Firstly, due to the nature of pathological NP cells in rats, it was nearly impossible to obtain the pathological nucleus pulposus from the vertebral bodies of the rat. As a result, we only performed IHC to determine the molecular expression. Further insights could be gained from additional studies using large animals such as monkeys38 and baboons39. Secondly, GPX4 is a negative regulator of ferroptosis, making it difficult to determine its presence in the patient’s NP tissue. Therefore, we did not include GPX4 data in our study, GPX4 expression was undetectable in the clinical NP samples, indicating a decrease in GPX4. Thirdly, ferroptosis inducer erastin and ferroptosis inhibitor Fer-1 were used in the present study. For the low solubility and low metabolic stability, they may not be suitable for in vivo study30. Erastin indirectly deactivates GPX4 by inhibiting cysteine entry into cells, depleting intracellular cysteine levels. Cysteine deficiency (similar to erastin) induced ferroptosis depends on transferrin29. Fer-1 as a potent inhibitor of ferroptosis, attenuates hypoxic-ischemic brain damage40. However, in the present study, erastin and Fer-1 indeed affect the changes in NP tissue during the IDD process by altering TFRC and ASCL4 expression.
In summary, our study revealed a novel target and molecular mechanism of disc degeneration. Our results first demonstrated that PDE4B increases ferroptosis in NP cells by upregulating ASCL4 and TFRC, improving Nrf2 activation and contributing to the subsequent occurrence of IDD. And PDE4 inhibitor have a protective effect on IDD and have a synergic effect with ferroptosis inhibitors. Overall, our study provides new insights into the occurrence and development of IDD, and targeting PDE4B offers a promising approach to suppressing cell death through multiple pathways.
Data availability
The data used and/or analyzed in the current study are available from the corresponding author upon reasonable request.
Change history
26 September 2025
A Correction to this paper has been published: https://doi.org/10.1038/s41598-025-20243-y
References
Cieza, A. et al. Global estimates of the need for rehabilitation based on the Global Burden of Disease study 2019: a systematic analysis for the global burden of Disease Study 2019. Lancet 396, 2006–2017 (2021).
Chen, S. et al. Global, regional and national burden of low back pain 1990–2019: a systematic analysis of the Global Burden of Disease study 2019. J. Orthop. Translation. 32, 49–58 (2022).
Xin, J. et al. Treatment of intervertebral disc degeneration. Orthop. Surg. 14, 1271–1280 (2022).
Wang, F., Cai, F., Shi, R., Wang, X. H. & Wu, X. T. Aging and age related stresses: a senescence mechanism of intervertebral disc degeneration. Osteoarthr. Cartil. 24, 398–408 (2016).
Doraisamy, R., Ramaswami, K., Shanmugam, J., Subramanian, R. & Sivashankaran, B. Genetic risk factors for lumbar disc disease. Clin. Anat. 34, 51–56 (2021).
Shimony, N. et al. Adolescent disc disease: risk factors and treatment success-related factors. World Neurosurg. 148, e314–e20 (2021).
Wei, X., Yi, X., Zhu, X. H. & Jiang, D. S. Posttranslational modifications in ferroptosis. Oxid. Med. Cell Longev. 2020, 8832043 (2020).
Zhang, X. et al. Homocysteine induces oxidative stress and ferroptosis of nucleus pulposus via enhancing methylation of GPX4. Free Radic. Biol. Med. 160, 552–565 (2020).
Zhang, Y. et al. Single-cell RNA-seq analysis identifies unique chondrocyte subsets and reveals involvement of ferroptosis in human intervertebral disc degeneration. Osteoarthr. Cartil. 29, 1324–1334 (2021).
Yang, R. Z. et al. Involvement of oxidative stress-induced annulus fibrosus cell and nucleus pulposus cell ferroptosis in intervertebral disc degeneration pathogenesis. J. Cell. Physiol. 236, 2725–2739 (2021).
Yu, X. et al. circ_0072464 shuttled by bone mesenchymal stem cell-secreted extracellular vesicles inhibits nucleus pulposus cell ferroptosis to relieve intervertebral disc degeneration. Oxid. Med. Cell Longev. 2022, 2948090. (2022).
Zhang, Y., Liu, C., Li, Y. & Xu, H. Mechanism of the mitogen-activated protein kinases/mammalian target of rapamycin pathway in the process of cartilage endplate stem cell degeneration induced by tension load. Global Spine J. 21925682221085226. (2022).
Schick, M. A. & Schlegel, N. Clinical implication of phosphodiesterase-4-inhibition. Int. J. Mol. Sci. 23. (2022).
Abou Saleh, L. et al. Ablation of PDE4B protects from Pseudomonas aeruginosa-induced acute lung injury in mice by ameliorating the cytostorm and associated hypothermia. FASEB J. 35, e21797 (2021).
Karam, S. et al. Cardiac overexpression of PDE4B blunts β-adrenergic response and maladaptive remodeling in heart failure. Circulation 142, 161–174 (2020).
Myers, S. A. et al. Following spinal cord injury, PDE4B drives an acute, local inflammatory response and a chronic, systemic response exacerbated by gut dysbiosis and endotoxemia. Neurobiol. Dis. 124, 353–363 (2019).
Dodson, M., Castro-Portuguez, R. & Zhang, D. D. NRF2 plays a critical role in mitigating lipid peroxidation and ferroptosis. Redox Biol. 23, 101107 (2019).
Dhar, R. et al. Phosphodiesterase 4B is required for NLRP3 inflammasome activation by positive feedback with Nrf2 in the early phase of LPS- induced acute lung injury. Free Radic. Biol. Med. 176, 378–391 (2021).
Xu, B. et al. Inhibition of PDE4 protects neurons against oxygen-glucose deprivation-induced endoplasmic reticulum stress through activation of the Nrf-2/HO-1 pathway. Redox Biol. 28, 101342 (2020).
Pfirrmann, C. W., Metzdorf, A., Zanetti, M., Hodler, J. & Boos, N. Magnetic resonance classification of lumbar intervertebral disc degeneration. Spine 26, 1873–1878 (2001).
Lai, A. et al. Development of a standardized histopathology scoring system for intervertebral disc degeneration in rat models: an initiative of the ORS spine section. JOR Spine. 4, e1150 (2021).
Livak, K. J. & Schmittgen, T. D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 25, 402–408 (2001).
Fraser, R. D., Osti, O. L. & Vernon-Roberts, B. Intervertebral disc degeneration. Eur. Spine J. 1, 205–213 (1993).
Kos, N., Gradisnik, L. & Velnar, T. A brief review of the degenerative intervertebral disc disease. Med. Arch. 73, 421–424. (2019).
Thompson, R. E. et al. Disc lesions and the mechanics of the intervertebral joint complex. Spine 25, 3026–3035 (2000).
Liu, Z., Liu, M., Cao, Z., Qiu, P. & Song, G. Phosphodiesterase4 inhibitors: a review of current developments (2013–2021). Expert Opin. Ther. Pat. 32, 261–278 (2022).
Bailly, C. The potential value of amlexanox in the treatment of cancer: molecular targets and therapeutic perspectives. Biochem. Pharmacol. 197, 114895 (2022).
Shen, S. et al. circPDE4B prevents articular cartilage degeneration and promotes repair by acting as a scaffold for RIC8A and MID1. Ann. Rheum. Dis. 80, 1209–1219 (2021).
Dixon III, S. J. et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149, 1060–1072 (2012).
Dixon, S. J. & Olzmann, J. A. The cell biology of ferroptosis. Nat. Rev. Mol. Cell. Biol. (2024).
Ding, K. et al. Acyl-CoA synthase ACSL4: an essential target in ferroptosis and fatty acid metabolism. Chin. Med. J. 136, 2521–2537 (2023).
Huang, Q. et al. Identification of a targeted ACSL4 inhibitor to treat ferroptosis-related diseases. Sci. Adv. 10, eadk1200 (2024).
Richardson, D. R. Mysteries of the transferrin-transferrin receptor 1 interaction uncovered. Cell 116, 483–485 (2004).
Ding, H. et al. Transferrin receptor 1 ablation in satellite cells impedes skeletal muscle regeneration through activation of ferroptosis. J. cachexia Sarcopenia Muscle. 12, 746–768 (2021).
Wu, Y. et al. Ubiquitin ligase E3 HUWE1/MULE targets transferrin receptor for degradation and suppresses ferroptosis in acute liver injury. Cell. Death Differ. 29, 1705–1718 (2022).
Cheng, E. L. et al. Discovery of a transferrin receptor 1-binding aptamer and its application in cancer cell depletion for adoptive T-cell therapy manufacturing. J. Am. Chem. Soc. 144, 13851–13864 (2022).
Wu, J. et al. Intercellular interaction dictates cancer cell ferroptosis via NF2-YAP signalling. Nature 572, 402–406 (2019).
Longo, U. G., Ripalda, P., Denaro, V. & Forriol, F. Morphologic comparison of cervical, thoracic, lumbar intervertebral discs of cynomolgus monkey (Macaca fascicularis). Eur. Spine J. 15, 1845–1851 (2006).
Platenberg, R. C., Hubbard, G. B., Ehler, W. J. & Hixson, C. J. Spontaneous disc degeneration in the baboon model: magnetic resonance imaging and histopathologic correlation. J. Med. Primatol. 30, 268–272 (2001).
Zhang, M. et al. Ferrostatin-1 attenuates hypoxic-ischemic brain damage in neonatal rats by inhibiting ferroptosis. Transl Pediatr. 12, 1944–1970 (2023).
Acknowledgements
We are thankful for the technical support of Li Liu and Guifeng Xiao in the Core Facilities, Zhejiang University School of Medicine.
Funding
This study was supported by the Basic Public Welfare Research Program of Zhejiang Province (China) (No. TGY24H060008), the Medical and Health Technology Plan of Zhejiang Province (China) (No. 2023KY078), and the Chinese Medicine Research Program of Zhejiang Province (China) (No: 2019ZB023).
Author information
Authors and Affiliations
Contributions
XWX contributed to the methodology, investigation, and resources. RD contributed to the methodology, investigation, writing, data analysis, and visualization. ZDY contributed to the clinical sample collection. PQ, LY, and MS contributed to the methodology and investigation. THF contributed to conceptualization, supervision, and revision.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original online version of this Article was revised: The original version of this Article contained an error in Figure 5, where the Grade in panel A did not match that in panel D.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Xu, W., Dhar, R., Zheng, D. et al. PDE4B promotes ferroptosis in nucleus pulposus cells and is involved in intervertebral disc degeneration. Sci Rep 15, 3984 (2025). https://doi.org/10.1038/s41598-025-87639-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-87639-8
