
Abstract
Ionotropic glycine receptors (GlyRs) are chloride-permeable ligand-gated ion channels expressed in the nervous system. Alterations to glycinergic inhibition and the generation of dysfunctional GlyRs have been linked to chronic pain, a widely prevalent disease. Positive allosteric modulators (PAMs) targeting GlyRs exerted analgesic effects, motivating research on glycinergic PAMs as potential pain therapies. Rationally designed tricyclic sulfonamides are novel glycinergic PAMs with analgesic activity. However, detailed electrophysiological studies on these PAMs are still limited, and the GlyR binding site structural data has not been yet validated by mutational studies. Here, we combined electrophysiology and bioinformatics to systematically study the AM-1488 actions, a prototypical tricyclic sulfonamide, on recombinant GlyRs. We determined that AM-1488 is a potent, non-selective PAM of mammalian GlyR subtypes. In addition, the compound displayed agonistic activity, with partial preference for α1GlyRs. Single channel assays revealed that the compound increased the channel open probability without changing conductance. Mutational analyses on the tricyclic sulfonamide site confirm the molecular determinants contributing to functional activity. Our findings further define the mechanistic framework underlying the GlyR modulation by this PAM class, suggesting that further structure-driven exploration within the tricyclic sulfonamide site may originate novel glycinergic modulators for future development.
Similar content being viewed by others
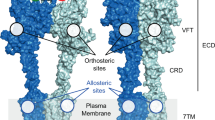


Introduction
Strychnine-sensitive glycine receptors (GlyRs) are pentameric ligand-gated ion channels (pLGICs) expressed along the mammalian central nervous system (CNS)1,2. GlyRs are integral membrane protein complexes formed by five subunits ordered around a central pore3. The activation of GlyR by its main physiological agonist glycine generates the opening of an anion-permeable pore, allowing the fast influx of chloride into neurons and the subsequent hyperpolarization of the membrane potential. Up to now, four α subunits (α1, α2, α3 and α4) and one β subunit of GlyRs have been identified4. The assembly of five α subunits can form functional homopentameric channels5,6,7,8, while the combination of β subunits do not form functional GlyRs. On the other hand, the assembly of 4α and 1β subunits configure functional heteropentameric channels that are the main GlyR conformation involved in synaptic neurotransmission3,9. Each GlyR subunit share the characteristic tertiary structure of the pLGICs, which include an extracellular domain (ECD), four transmembrane domains (TM1-4) and a variable intracellular domain (ICD) between TM3 and TM43,5,6,7,9. GlyR α subunits are highly homologous proteins, sharing a large degree of similarity on their primary sequence identity, especially on their ECDs and TM domains.
GlyRs are expressed throughout the entire central nervous system, including spinal and supraspinal structures1,10,11. GlyRs play a critical role in the inhibitory control of the spinal cord and brainstem12. While the expression pattern of GlyR α subunits vary along the CNS, β subunits are widely expressed. Compelling evidence has determined that α1 is widely expressed at the spinal cord and brainstem, while α3 is expressed at the spinal dorsal horn only after birth1. Conversely, the spinal expression of α2 is restricted to immature states but appears to be basally expressed in selected brain areas13. Nevertheless, more recent studies have pointed out that α1 and α3 subunits are also expressed in specific brain zones, possibly in combination with α211,14.
The GlyR activation is necessary to control key neurophysiological functions, such as respiratory rhythm, muscle tone, motor coordination and pain processing4. Cumulative evidence from human genetic studies have established that alterations in α1 and/or β GlyR genes lead to hyperekplexia, which is a disorder characterized by an elevated muscle tone and exaggerated responses to a sudden stimulus15. Further studies, mainly coming from genetically modified mouse models, have established that GlyR subunits contribute to several physiological processes and pathological conditions, such as respiratory control, memory, autism, epilepsy, alcohol addiction and chronic pain16,17,18,19,20,21. Many of the mentioned pathological states involve alterations in the glycinergic inhibition. Interestingly, diverse research groups have found that spinal glycinergic activity is impaired in chronic pain states. In this context, it has been reported that transient post-translational modifications on specific GlyR subunits are linked to the generation of dysfunctional receptors, leading to an altered inhibitory tone and pain hypersensitivity22. Thus, the pharmacological restoration of disrupted glycinergic activity has been established as a rational strategy against chronic pain.
The relevance of glycinergic dysfunction in chronic pain, at least in part, has motivated novel research on GlyR pharmacology4,12,23. The cumulative progress on compounds modulating GlyR activity has been well summarized in recent reviews4,12,23,24,25,26,27. Although these studies have highlighted that novel positive allosteric modulators (PAMs) of GlyRs have been discovered and characterized, whether these compounds are specific for GlyRs is still undefined. In addition, only several of these molecules have reported subunit-specific effects. Compounds with agonistic actions on GlyRs have been less explored. Despite these limitations, the available evidence supports the view that glycinergic PAMs displayed analgesic effects on chronic pain rodent models, at least partly due to the potentiation of α3-containing GlyRs23,28,29,30,31.
Within the set of GlyR modulators that displayed analgesic activity, the tricyclic sulfonamides AM-1488 and AM-3607 have been highlighted as a paramount example of rational design of novel glycinergic PAMs through a compelling process of identification, optimization, and biological testing8,29. Interestingly, it has been reported that the compound AM-3607 binds to α3GlyRs in a novel allosteric site located at the interface between two alpha subunits, near the top of the ECD and close to the orthosteric site where glycine binds8. Despite these advances, a detailed electrophysiological characterization of the actions of this new class of glycinergic PAMs is missing. In addition, whether these compounds can exert direct agonist effects or subunit-selective actions is not yet defined. Furthermore, the structural data of the binding site has not been validated by mutational and functional studies. Here, we use electrophysiological techniques and bioinformatics to provide a systematic study of the AM-1488 actions on recombinant GlyRs. We determined that AM-1488 is a potent, non-selective PAM of all mammalian GlyR configurations. In addition, AM-1488 displayed agonistic activity on GlyRs, with a partial preference for α1GlyRs. Single channel assays revealed that the compound increased the open probability of the ion channel without changing the channel conductance of wild-type receptors or phospho-mimetic α3GlyRs. Mutational analyses combined with molecular modelling uncover molecular determinants that contribute to the PAM activity of AM-1488. Altogether, our findings contribute to expand the knowledge about the biophysical and biochemical framework underlying the allosteric potentiation and direct activation of GlyRs by this class of PAMs. In a broader context, our data suggest that further structure-driven search within the tricyclic sulfonamide site may originate novel glycinergic PAMs and agonists for future drug development.
Results
Allosteric and agonistic actions of AM-1488 on glycine receptors
The allosteric modulation of AM-1488 was previously characterized mainly using membrane potential dye assays in HEK293 cells expressing GlyRs of defined composition8. These experiments showed that AM-1488 was a PAM for GlyRs composed by α1 or α3 subunits, in both homomeric or heteromeric configurations. Therefore, we first examined the sensitivity to AM-1488 of recombinant GlyRs composed of α1, α2 and α3 subunits expressed in HEK293 cells using regular whole-cell electrophysiology. To define whether the compound show allosteric effects in all the GlyR subtypes expressed in the mammalian CNS, we performed these experiments in both homomeric and heteromeric GlyRs (Fig. 1). Using a sub-saturating concentration of glycine (EC10) co-applied with increasing concentrations of AM-1488 (0.01–100 µM), our assays defined that this compound is a potent, non-selective PAM for all subtypes of GlyRs (Fig. 1A-B; Table 1). A significant potentiation of the glycine-activated current through GlyRs was noticeable from ~ 0.1 µM of AM-1488, reaching plateau values at concentrations around 10-50 µM (Fig. 1A-B). This profile was not influenced by the expression of the β subunit. The concentration response curves for AM-1488 displayed a sigmoidal shape and showed similar EC50 values between the GlyR subtypes (Table 1; Fig. 1B). Although the maximal potentiation elicited by the compound on α1 and α3-containing GlyRs was higher than on α2-containing receptors, these differences were not significant (Table 1; Fig. 1C). The similar AM-1488 sensitivity displayed by GlyRs of different composition suggests that the structural features required for the tricyclic sulfonamide binding are also conserved. To explore this idea, we study the interaction of AM-1488 with GlyRs composed of α1, α2 and α3 subunits by using molecular docking procedures. We developed these models using the information coming from the crystal structure of α3GlyRs bound to AM-36078 combined with coordinates from other GlyRs structures as templates3,5,6,7,8,9. Our analyses showed that AM-1488 was able to interact with different GlyRs in the presence of glycine (Fig. 1D). The interaction of the tricyclic sulfonamide with its binding site was similar between the GlyR α subunits. Further analysis of the putative binding site for AM-1488 showed that the amino acids P10, F13, L14, R27, I28, R29, F32, Y78, L83, D84, L85, D86, G160 and Y161 are conserved between the α subunits. A comparison of the docking scores and the theoretical free energy involved in the compound-receptor interaction suggest a favourable binding of AM-1488 to the GlyR structures (Fig. 1D and Supplementary Table 1). Similar values were obtained with AM-3607 (Figure S1 and Supplementary Table 1).

Allosteric potentiation of recombinant GlyRs by AM-1488. (A) Representative whole-cell currents showing the potentiation generated by the application of 0.5 µM of AM-1488 to HEK293 cells transiently expressing homo and heteromeric GlyRs. The currents were evoked using 15 µM (α1), 30 µM (α2), 40–60 µM (α3), 15 µM (α1β), 25–50 µM (α2β), or 30–40 µM (α3β) of glycine. (B) Concentration response curves of homo and heteromeric GlyRs in the presence of AM-1488. Data are mean ± SEM. (C) The plot summarizes the maximal potentiation of the glycine-evoked currents elicited by the compound. Differences were not significant. (D) The left panel show the AM-1488 binding to homopentameric α3 GlyRs in the presence of glycine. The right panels summarized enhanced views of the predicted binding of AM-1488 to homologous sites of α1, α2 and α3GlyRs. Glycine is shown in purple. AM-1488 is depicted in red. The models showing the AM-1488-α3GlyR complex, in the presence of glycine, were created using the structural information available (PDB:5TIN)8.
Previous studies have shown that several compounds modulating GlyRs as PAMs are also acting as direct agonists at high concentrations32,33. Whether AM-1488 may exert agonistic actions on GlyRs is still undetermined. To explore this issue, we applied the compound alone, in the absence of glycine, to HEK293 cells expressing GlyRs (Fig. 2). To compare the potential agonist activity of AM-1488 on diverse GlyRs, we normalize the extent of receptor activation as percentage of the maximal current elicited by a saturating glycine concentration (i.e. 2 mM). We found that concentrations lower than 10 µM of AM-1488 evoked currents through homomeric GlyRs composed by α1 subunits, but not through α2 or α3GlyRs (Fig. 2B). Concentrations higher than 30 µM of AM-1488 evoked chloride currents on all the GlyRs studied (Fig. 2A-B). However, the GlyR activation elicited by concentrations > 30 µM was significantly higher on α1GlyRs, reaching more than 25% of the maximal current. At 100 µM, the highest concentration of compound tested, homomeric GlyRs composed of α1, α2 and α3 subunits were activated to values higher than the 20% of the maximal glycine evoked current. To evaluate whether the incorporation of the β subunits influence the direct actions of this compound, we performed recordings of heteromeric α1β GlyRs. These assays show that α1β GlyRs were activated by AM-1488 in a similar extent in comparison with homomeric α1GlyR (Supplementary Table 2). To define the nature of these currents, we next study the sensitivity of the AM-1488 evoked currents to strychnine and picrotoxin, two classical glycinergic inhibitors having different mechanisms of action34,35. The application of the competitive antagonist strychnine fully inhibited the compound evoked currents through homomeric α1, α2 and α3 GlyRs (Fig. 2C-D). On the other hand, the pore blocker picrotoxin completely reduced the AM-1488 evoked currents on α2 and α3 GlyRs, but only partially reduced the currents activated on α1GlyRs (Fig. 2C-D). These observations confirm the glycinergic nature of the sulfonamide-evoked currents but suggest that pore-opening events triggered by compound may differ between GlyR subunits. While we currently lack a definitive mechanistic explanation, we hypothesize that variations in pore-lining residues (particularly those TM residues involved in PTX blockade) could underlie this divergence. To further study the molecular features of the agonistic activity, we explore the putative interaction of AM-1488 with GlyRs by molecular modelling, in the absence of glycine. As structural information of tricyclic sulfonamides bound to GlyRs in the absence of glycine is missing, we performed molecular docking assays of AM-1488 in the glycine binding site3,5,6,8,9. Our results showed that AM-1488 likely interact with the orthosteric site of homomeric GlyRs (Fig. 2E). The tricyclic sulfonamide binding was stabilized by the F159, Y202, T204, F207 of the principal subunit, together with F63, R65, S129 of the complementary subunit. These residues also participate in the glycine and strychnine binding6,8. Calculation of the docking scores and the theoretical free energy involved in these interactions suggest a stable binding of AM-1488 to GlyRs in the closed state (Fig. 2E and Supplementary Table 1). Like the allosteric modulation, further analyses showed that binding of AM-3607 to the orthosteric site is likely (Figure S1 and Supplementary Table 1).

Direct activation of recombinant GlyRs by AM-1488. (A) Representative traces of the activation of α1, α2 and α3 GlyRs by 50 µM of AM-1488. The maximal glycine current (evoked using 2 mM glycine) is also shown. (B) Summary graph of the percentage of the maximal glycine evoked current activated by different concentrations of AM-1488 (1, 3, 10, 20, 50 and 100 µM) on α1 (red), α2 (green) or α3GlyRs (blue) expressed in HEK293 cells. AM-1488 was applied to cells in the absence of glycine, without pre-application. Each circle represents the mean ± SEM of 6–8 cells. ANOVA followed by Bonferroni post-hoc test; **, p < 0.01; ***: p < 0.001. (C) The representative traces show the blockade of AM-1488 (100 µM) activated currents by the competitive antagonist strychnine (STN, 2 µM, top) or the pore-blocker picrotoxin (PTX, 30 µM, bottom) in α1 (left), α2 (middle) and α3GlyRs (right). (D) Summary graph of the percentage of decrease of AM-1488 gated currents in the presence of glycinergic inhibitors. Each bar represents the mean ± SEM of 3–5 recorded cells. Statistical analysis, ANOVA followed by Bonferroni post-hoc test. α1GlyRs: AM-1488 vs. + STN, p < 0.01; AM-1488 vs. PTX, p = 0.2317. α2GlyRs: AM-1488 vs. + STN, p < 0.05; AM-1488 vs. PTX, p < 0.05; α2GlyRs: AM-1488 vs. STN, p < 0.05; AM-1488 vs. PTX, p < 0.05. (E) The left panel displays the AM-1488 binding to the orthosteric site of homopentameric α3GlyRs. The right panels are enriched outlooks of the predicted binding of AM-1488 to the orthosteric sites of α1, α2 and α3GlyRs. AM-1488 is shown in red. The models were generated from the structural coordinates available (α1, PDB:7TU9; α2, PDB:7KUY and α3, PDB:5CFB).
Modulation of glycine receptors by AM-1488 at the single-channel level
We next performed cell-attached single channel recordings of α1GlyRs expressed in HEK293 cells. As we expected, when the pipette was charged with a control concentration of glycine (15µM), we observed brief openings of the channel followed by long shut periods (Fig. 3A). At a clamping voltage of + 60mV, we observed opening events characterized by stable amplitude and conductance (Table 2; Fig. 3A-B). This data is consistent with previous studies36. To evaluate the allosteric effects of AM-1488 on the openings of α1GlyRs, the recording pipette was filled with glycine and AM-1488 at a concentration of 0.5µM or 10µM. Using 0.5µM of AM-1488, we detected an increase in the frequency of the transitions, which produces a significant increment in the NPo of α1GlyRs in comparison to control recordings with glycine alone (Fig. 3B; Table 2). We did not find significant differences in the conductance of the α1GlyRs in presence of AM-1488 0.5µM (Fig. 3B; Table 2). When we tested the effects of AM-1488 10µM, we find that the recorded patches show an increment in the frequency of the open-close transitions of the channel, with scarce periods of shutting of the channel and a significant increment in the NPo compared with the control (Fig. 3B; Table 2). The conductance of patches recorded with 10µM of AM-1488 were not significantly different in comparison with controls obtained with glycine alone (Fig. 3B).

Single channel analyses. (A) Representative current traces showing the effects of 0.5 µM of AM-1488 on glycine-activated currents in a cell-patch expressing homomeric α1GlyRs. The channels were activated with 15 µM of glycine. Calibration bar, 5 pA, 1 s. (B). Summary of the normalized open probability (NPo) and of the main conductance in the absence or presence of AM-1488 (0.5 and 10 µM). ANOVA followed by Bonferroni post-hoc test, *, p < 0.05; **, p < 0.01. (C,D) Single channel current traces obtained from cells expressing wild-type (C) or S346E mutated (D) α3GlyRs in the absence or presence of AM-1488 (10 µM). The channels were evoked with 90 µM of glycine. Calibration bar, 5 pA, 2 s. (E) The histograms show the unitary current amplitude distributions of wild-type receptors (left) and S346E mutant receptors before and during the application of AM-1488 (blue). The control condition (i.e. no allosteric modulator) is shown in grey. (F) The plots summarize the normalized open probability and the unitary conductance of wild-type and S346E α3GlyRs constructs in the absence or presence of AM-1488. NPo: ***p < 0.001, unpaired Student t-Test. In all the cases, the conductances were not significantly modified by the compound.
Previous reports indicate that PKA-mediated phosphorylation of serine 346 residue of α3GlyRs produces a reduction of glycinergic currents in the dorsal horn of the spinal cord, generating a loss of inhibitory control and promoting persistent pain16,22,23,27. The inhibition of the α3GlyR function by phosphorylation has been linked to a reduction of the main conductance of the channel37. To investigate whether AM-1488 can modify the properties of a hypofunctional α3GlyRs related to chronic pain, we performed cell-attached recordings of a phospho-mimetic α3GlyR (i.e. α3GlyR S346E) that displayed a reduced conductance28,37. Control recordings obtained with glycine alone (90 µM) produced similar periods of opening in both wild-type and S346E α3GlyRs (Fig. 3C; Table 2). The number of events and the NPo calculated for wild-type and S346E α3GlyRs were similar (Fig. 3E-F). As expected, the amplitudes of the events from α3GlyR S346E patches were smaller than the ones detected in wild-type α3GlyRs, consistent with a significant reduction on the main conductance37 (Fig. 3E-F; Table 2). The co-application of glycine and AM-1488 into the recording pipette produced a significant enhancement of the number of events in both wild-type and S346E α3GlyRs, which was translated in a significant increase of the NPo (Fig. 3E-F). Nevertheless, AM-1488 does not generate a significant change in the conductance of either wild-type of phospho-mimetic α3GlyRs (Fig. 3F).
Residues involved in the allosteric and agonist actions of AM-1488
The crystal structure of glycine and AM-3607 bound to α3GlyRs define the allosteric interactions of a prototypic tricyclic sulfonamide with this class of receptors8. These results revealed that AM-3607 interact with a network of residues within the extracellular domain, in close vicinity to the orthosteric glycine binding site. The residues participating in the binding of AM-3607 to α3GlyRs are P10, F13, L14, R27, R29, F32, L83, D84, L85, D86, Y78 and Y1618 (Fig. 4A). The physical proximity of the glycine and sulfonamide binding sites suggests that mutations on residues stabilizing the interaction of AM-3607 with the receptor may also alter the orthosteric site, generating non-functional receptors. Considering this possibility, we first performed in silico mutagenesis analyses on the AM-3607 binding site to assess potential alterations on the glycine binding site. These analyses suggest that alanine mutations on F13, L14, R27 and L83 will decrease the sulfonamide binding but partially conserving the properties of the orthosteric site. Consequently, we generated and studied F13A, L14A, R27A and L83A mutated α3GlyRs by whole-cell electrophysiology (Fig. 4B-E). Our results show that these mutated receptors displayed glycine-activated currents, generating concentration-dependent responses (Fig. 4B-C; Table 3). The mutations generated receptors with a decreased apparent affinity for glycine, showing higher EC50 values (Table 3). The maximal currents of these mutated GlyRs were not significantly different from those of wild-type receptors. However, activation of the F13A, L14A, and R27A α3 GlyRs with 5 mM glycine elicited a brief but noticeable rebound-type current at the end of glycine application in most recorded cells. While the precise mechanism underlying this phenomenon remains unclear, we suspect it may result from an artifact caused by incomplete glycine washout. Nonetheless, the mutated GlyRs exhibiting rebound currents also show an impaired apparent affinity for glycine, suggesting the presence of undetected alterations that may contribute to the generation of these rebound currents. We next analyzed the sensitivity of these receptors to AM-1488 using equipotent glycine concentrations (EC10) (Fig. 4D). The application of 0.5 µM AM-1488 significantly potentiated glycine-evoked currents in wild-type α3GlyRs (Fig. 4D, Supplementary Table 2). The F13A, L14A, and L83A substitutions significantly reduced potentiation, with the L83A mutant exhibiting pronounced insensitivity to AM-1488 (Fig. 4D, Supplementary Table 2). Despite these differences, the sulfonamide sensitivity remained comparable among the F13A, L14A, and L83A mutants (Supplementary Table 2). In contrast, the R27A mutation did not significantly impact allosteric potentiation (Fig. 4D). We then examined whether the direct activation of α3GlyRs by AM-1488 is also altered by the mutations. These assays were performed using 100µM of AM-1488, which generated a current equivalent nearly 15% of the maximal glycine current through wild-type α3GlyRs (Fig. 4E and Supplementary Table 2)). We found that F13A, L14A, R27A and L83A mutated GlyRs displayed significantly reduced AM-1488 evoked currents (Fig. 4E). The F13A and R27A mutants showed small responses, equivalent to less than 1% of the maximal glycine current (Fig. 4E and Supplementary Table 2). Conversely, the application of the compound to L14A and L83A mutated GlyRs did not elicit detectable currents (Fig. 4E). Altogether, these results confirm the functional relevance of several of the residues involved in the tricyclic sulfonamide binding8 and suggest that both the allosteric and direct activation of α3GlyRs by AM-1488 are mediated by common, but not identical, molecular determinants.

Mutational analysis of AM-1488 binding site on α3GlyRs. (A) The images describe the AM-1488-α3GlyR complex in the presence of glycine. The center and right panels show enhanced views of the molecular interactions (4Å cutoff). The highlighted drops represent numbered residues of the AM-1488 binding pocket. The color code describes the amino acid biochemical features (light yellow, glycine; blue, positively charged residues; red, negatively charged residues; green, hydrophobic residues; cyan, polar residues). The green lines symbolize a pi–pi stacking. The models were generated from the structural coordinates of PDB:5TIN8. (B) Sample current traces obtained from cells expressing wild-type (WT) or mutated α3GlyRs. (C) Concentration response curves to glycine (1-5000 µM) of wild-type (WT) and alanine-mutated α3GlyRs. (D) The current traces and the bar graph describe the allosteric potentiation of AM-1488 (0.5 µM) on the glycine evoked current through wild-type and mutated α3GlyRs. Currents were evoked using 35–40 µM (WT), 700 µM (F13A), 250 µM (L14A), 500 µM (R27A), 250 µM (L83A) of glycine. F13A, L14A and L83A significantly reduced the allosteric potentiation, whereas R27A did not altered the AM-1488 actions. ANOVA followed by Bonferroni post-hoc test (***, p < 0.001; **: p < 0.01). (E) The current traces and the bar graph summarize the direct activation elicited by AM-1488 (100 µM) of wild-type and mutated α3GlyRs. All the mutants tested significantly reduced the agonistic actions of the compound. ANOVA followed by Bonferroni post-hoc test (*, p < 0.05).
Discussion
Allosteric modulation of glycine receptor subtypes by AM-1488
The tricyclic sulfonamides are possibly the only group of glycinergic PAMs with specificity for GlyRs8. To date, the available studies have defined the allosteric modulation of this class of molecules only in GlyRs composed of 1 and α3 GlyRs8. No studies have been reported for α2 GlyRs, which have also recently been associated with chronic pain and to other neurological disorders19,38,39,40. Our electrophysiological data indicate that both homo- and heteromeric GlyRs containing the α2 subunit are positively modulated by AM-1488, with a pharmacological profile similar to that of α1 and α3 GlyRs. This modulatory profile was similar in heteromeric GlyRs, regardless of the subunit. Thus, we conclude that AM-1488 is a potent glycinergic PAM with no preference for any specific GlyR subunit. Thus, our findings correlate with the previously reported data8 and expanded our knowledge to all the main GlyR subunits expressed in mammals.
The binding site of tricyclic sulfonamides was described in homomeric α3 GlyR structures. These studies indicate that AM-3607 binds at the interface of two α subunits8. Based on alanine substitutions within this site, our electrophysiological results confirm the relevance of several of these residues for the allosteric potentiation of α3 GlyRs by AM-1488. The residues involved in the tricyclic sulfonamides binding site are highly conserved between the α subunits and display similar binding properties according to our in silico docking studies8. This high degree of similarity correlates well with the comparable potency and efficacy of the PAM activity of AM-1488 on diverse GlyRs. In addition, our results showed that β subunit incorporation does not have a significant impact on the allosteric modulation of AM-1488. Previous data has suggested that the β subunit does not possess the key residues involved in the tricyclic sulfonamide binding8. Therefore, it is possible to suggest that the β subunit does not participate in the binding of AM-1488 or in the conformational changes that produce the PAM activity of the compound. The structural data available show that homomeric GlyRs possess five tricyclic sulfonamide binding sites8. On the other hand, high-resolution structures of heteropentameric GlyRs indicate a stoichiometry of 4α:1β3,9. Hence, we hypothesize that heteromeric GlyRs possess only three tricyclic sulfonamide binding sites. Since heteromeric GlyRs are positively modulated by AM-1488 with similar potency and efficacy in comparison to homomeric GlyRs, it is possible to suggest that three or even less AM-1488 molecules per receptor are necessary and sufficient to reach the maximal PAM effects detected in our whole-cell experiments. This modulatory profile diverges from previous reports showing that the presence of the β subunit produces functional and pharmacological changes compared with their homomeric counterparts. For example, all heteromeric GlyRs (α1β, α2β, and α3β) have lower main conductance values compared to homomeric receptors1. In addition, heteromeric GlyRs can have different sensitivities to modulatory compounds. For instance, α1GlyRs are positively modulated by the alkaloid gelsemine at concentrations up to 25 µM, but at higher concentrations, the compound behaves as an inhibitor. In contrast, gelsemine acts as an inhibitor of α1β GlyRs41. Other electrophysiological experiments showed that ethanol, propofol, and trichloroethanol have opposite modulatory effects on α2 and α2β GlyRs42. Similarly, whole-cell recordings showed that homomeric α1 and α3 GlyRs were positively modulated by the compound 2,6-di-tert-butylphenol, but heteromeric α1β and α3β GlyRs were largely insensitive28.
The initial characterization of the tricyclic sulfonamides indicated that this class of compounds binds to α3GlyRs in the desensitized state, increasing the affinity of the receptor for glycine by approximately 200 times8. The single-channel analysis conducted in this work showed that the PAM activity of AM-1488, and therefore the tricyclic sulfonamides, strongly increases the open probability of GlyRs without modifying the main conductance of the channel. The compound significantly increased the activity of hypofunctional phospho-mimetic α3 GlyRs but was unable to restore the channel conductance to wild-type values. These findings suggest that the mechanism of action of the tricyclic sulfonamides is related to an increase in the affinity of the receptor for its ligand, which likely produces modifications in the kinetic parameters of the channel, favoring transitions toward the open state and thus making their function more efficient. This mechanism is consistent with local structural rearrangements at the level of the ECD, precisely where the agonist glycine and the tricyclic sulfonamides are bound.
Activation of GlyRs by AM-1488
GlyRs are activated by total or partial agonists, such as glycine, β-alanine, and taurine4,26,43. Additionally, glycinergic currents can be evoked by exogenous compounds, such as ivermectin32. Our results show that a synthetic compound belonging to the tricyclic sulfonamide group can also act as a direct activator of GlyRs. The sulfonamide-evoked currents were inhibited by strychnine and picrotoxin, confirming its glycinergic nature. Our in silico studies suggest that AM-1488 may bind to GlyRs in the absence of glycine, while functional data with GlyRs containing mutations at the tricyclic sulfonamides binding site fully ablate the agonist activity of AM-1488. At present, we cannot discard that residual glycine could be present during our recordings, feasibly showing an allosteric potentiation instead of a direct activation of the receptors. Our experimental evidence is still inconclusive to define whether the supposed sulfonamide evoked currents were produced by trace quantities of glycine. Nevertheless, some of our findings support the idea that AM-1488 may have agonistic activity. The maximal allosteric potentiation effect of AM-1488 is reached at approximately 10 µM. In contrast, the experiments investigating direct activation showed a concentration-dependent increase in the amplitude of the currents evoked by AM-1488 from 10 µM. Additionally, our studies using mutated α3GlyRs showed that substitutions of on residues of the tricyclic sulfonamides site displayed similar, but not equal effects on the allosteric potentiation and on direct activation experiments. Since all the mutated α3GlyRs did not show any direct activation of AM-1488, but some of them still retain the allosteric potentiation, we suggest that both phenomena are associated to different mechanisms. Another element is that the putative direct activation of GlyRs by AM-1488 displayed partial but significant subunit selectivity. The GlyRs subunits have high sequence identity1. However, they exhibit, in some cases, subunit-dependent sensitivity to allosteric modulators16,26,36,44. Our data showed that the positive allosteric modulation of α1, α2, and α3GlyRs were similar in terms of potency and efficacy, but the putative agonistic activity of AM-1488 showed significant differences. GlyRs containing α1 subunits were activated by ≤ 10 µM of AM-1488, while current through homomeric α2 and α3 GlyR subunit were detected only with concentrations higher than 30 µM. The high degree of sequence identity in the tricyclic sulfonamide allosteric binding site between the α subunits suggests that AM-1488 has a similar binding affinity α1, α2, and α3GlyRs, in the presence of glycine. On the other hand, our in silico models show that AM-1488 can bind to the orthosteric site of all α GlyR subunits, with similar binding properties. Therefore, the binding of the compound to the orthosteric site or to the allosteric site (i.e. in the presence of glycine bound to the receptor) are divergent mechanisms that may explains the differences observed in the direct activation profile of homomeric GlyRs by AM-1488.
In general terms, the glycine binding stabilizes the extracellular GlyR domains generating a twisted conformation, which is crucial for the subsequent channel opening45. The efficiency of this process depends on the residues at the interface between the ECD and the TM domains, allowing the receptor to communicate the conformational changes produced by the agonist binding. The α1GlyRs possesses a more efficient gating process than the α2 or α3 GlyR subunits. For example, single-channel recordings showed that the maximal open probability of α2 and α3GlyRs is reached when the receptor interacts with 5 molecules of glycine44,46, while α1 GlyRs reach the maximal open probability with just 3 glycine molecules36. Differences in the gating pathways between the α subunits have been also used to explain the distinct intracellular modulation of GlyRs by the Gβγ dimer47,48. While α1 GlyRs were positively modulated by Gβγ, α2 and α3 GlyRs were unsensitive, despite all three subunits containing the basic intracellular residues needed for Gβγ-GlyR interaction47,48. These differences were explained by non-conserved residues in the TM domains of α subunits involved in the gating process47,49,50,51,52. A similar case was reported for the allosteric GlyR modulation by endocannabinoids. Acidic endocannabinoids, such as N-arachidonoyl glycine, act as PAMs for α1 GlyRs and NAMs for α2–α3 GlyRs53. Electrophysiological data using mutant GlyRs determined that this difference in sensitivity to the compound could be explained by non-conserved residues in TM2, close to the pore channel, that likely affect allosteric mechanisms rather than the binding of this class of compounds53. Thus, considering the results of this work and previous data, it could be suggested that α1 GlyRs possess structural features that make the agonist effect of AM-1488 more efficient than in the other α subunits.
The most representative case of a PAM that also possesses an agonist effect is ivermectin, which activates GlyRs at concentrations higher than 0.03 µM32. Ivermectin activates GlyRs through a binding site located in the TMD, at the interface of two adjacent α subunits5,8. Ivermectin-evoked currents possess different activation mechanisms than glycine-evoked currents. The classic antagonists of GlyRs, strychnine and picrotoxin, have no effect on ivermectin-evoked currents32. In contrast, our experiments show that the agonist activity of AM-1488 is blocked by classical GlyR antagonists, suggesting that the action mechanisms of AM-1488 are more similar to the agonist activation after binding to the orthosteric site. Given that the tricyclic sulfonamide binding site and glycine binding site are very close pockets in the ECD of GlyRs8 and considering that our in silico approaches suggest that AM-1488 interacts with the orthosteric binding site in the absence of glycine, we postulate that AM-1488 activates GlyRs primarily through interactions with the orthosteric site rather than with the allosteric binding site. This model fits well with our mutagenesis results, in which alanine substitutions of the sulfonamide allosteric site reduced the apparent affinity for glycine, the full GlyR agonist, and diminished the agonistic actions of AM-1488, a putative low agonist ligand. Nevertheless, these conclusions should be taken with care and further experiments are likely necessary to better understand these observations.
Implications for the neurophysiological mechanisms of tricyclic sulfonamide-mediated analgesic actions
Chronic pain is a significant public health issue, affecting around 30% of the global population54,55. Regardless of the underlying cause, chronic pain states are characterized by pathological alterations in the nociceptive pathways, leading to an imbalance between excitatory and inhibitory signals in the peripheral and central nervous system56,57,58. Current management strategies for chronic pain include opioids, NSAIDs, together with certain antidepressants and antiepileptics23,59. However, these treatments achieve only modest and highly variable efficacy in managing chronic pain and often come with intrinsic side effects60,61,62,63. This situation highlights the need for the development of novel, effective, and safe analgesics23,59. The advances in our understanding of the pain pathway neurophysiology have unveiled new protein targets for the development of such analgesics23,54,59. Previews reports have emphasized that pharmacological modulation of the glycinergic system could represent a promising strategy for developing novel analgesic drugs4,23,64,65.
Tricyclic sulfonamides, such as AM-1488, exemplify drugs targeted at GlyRs that produce analgesic effects in chronic pain models8. Distribution studies indicate that AM-1488 reaches a concentration of ≈ 1 µM in the brain. At this concentration, the compound effectively induces analgesic effects without significantly affecting locomotor activity in mice8. Our results suggest that concentrations of AM-1488 within this range are sufficient to allosterically potentiate glycine-evoked currents through all the mammalian GlyR subtypes. Since the actions of AM-1488 are most prominent using sub-saturating glycine concentrations, we hypothesize that the mechanism of action of AM-1488 is likely related to the modulation of extrasynaptic GlyRs, rather than synaptic GlyRs. While the activation of synaptic GlyRs by presynaptically released glycine is triggered by high agonist concentrations (≈ 1 mM), tonic glycinergic currents gated by the activation of extrasynaptic GlyRs are related to low glycine concentrations. Glycinergic tonic currents has been detected in various CNS regions and configure an effective mechanism of regulation on neuronal excitability11,13,14,66,67,68. Given that cerebrospinal fluid contains glycine at concentrations below 5–10 µM69, it is likely that AM-1488 will produce the allosteric potentiation of extrasynaptic GlyRs, which may either enhance the amplitude of detectable tonic currents or unmask unnoticeable glycinergic tonic currents gated from low glycine amounts conserved in nervous tissue preparations. Nevertheless, it is important to note that the composition of extrasynaptic GlyRs varies across different CNS regions11,14,21,66,67,68, indicating that the sensitivity of these currents to tricyclic sulfonamides may also differ.
A major proportion of chronic pain states are characterized by an unbalance between excitatory and inhibitory signals in the CNS58,65. We think that AM-1488 may counterbalance this alteration through the modulation of extrasynaptic GlyRs expressed in multiple sites within the pain pathway. For instance, a positive modulation of spinal GlyRs could effectively restore the inhibitory tone of the spinal cord, re-establishing a proper regulation of the nociceptive processing. Likewise, GlyRs expressed in supraspinal regions related with pain control, such as the anterior cingulate cortex, amygdala and periaqueductal grey11,66,70,71,72,73,74,75 may also be modulated by AM-1488, contributing to restore the inhibitory control. The modulation of presynaptic GlyRs by sulfonamides may provide an additional mechanism of neuronal regulation, dynamically shaping neurotransmitter release throughout the CNS4,76,77. Additional studies focused on these issues are necessary to reveal the neurophysiological mechanisms underlying the analgesic actions of tricyclic sulfonamides and its relationship with the glycinergic neurotransmission.
Methods
Chemicals
Glycine hydrochloride was obtained from Merck (Darmstadt, Germany). All other reagents used for electrophysiological recordings were purchased from Winkler (Santiago, Chile), Merck (Darmstadt, Germany) or Sigma-Aldrich (St. Louis, MO, USA). AM-1488 was a gift from Amgen (Thousand Oaks, CA, USA) under the RPA 2018773177.
Expression plasmids, cell culture, and transfection
HEK293 cells (CRL-1573; American Type Culture Collection, USA) were used as previously described41,78. The cells were transfected with plasmids encoding the following proteins: (i). rat GlyR α (1,2,3) subunits alone or combined with rat β subunits (Uniprot Accession Numbers: Q546L7, P22771, P24524, P20781). The EGFP expression was used as a marker of successful protein expression. The expression plasmids used has been previously characterized37,41,47,79,80. All experiments were conducted using the α3 GlyR long splice variant. Point-mutated α3GlyRs were obtained by site directed mutagenesis (Mutagenex Inc., USA). The amino acid numbering refers to the mature protein8. HEK293 cells were transiently transfected 0.5–3.0 µg of cDNA plasmids encoding GlyRs together with 0.5 µg of an EGFP-encoding plasmid. We used a ratio of 0.8 µL of lipofectamine per µg of transfected-DNA. For the experiments that required homomeric GlyRs expression, the cells were transfected with the αGlyR subunit of interest plus GFP in a 1:1 ratio. For the experiments that required the expression of heteromeric conformations of GlyRs, the cells were transfected with the αGlyR, EGFP and βGlyR in a ratio of 1:1:5 to avoid the formation of homomeric GlyRs28,41. The sequences of all constructs were verified by full-length sequencing (Plasmidsaurus, OR, USA). The electrophysiological recordings were made 16–48 h after the transfection.
Electrophysiology
The whole cell recordings were performed in voltage clamp configuration at room temperature (20–24 °C) using a holding potential of -60mV, following protocols previously published37,41,81. Patch pipettes (3–4 MΩ) were pulled from borosilicate glasses and were filled with internal solution, which contains (in mM): 120 KCl, 8 EGTA, 10 HEPES (pH 7.4), 4 MgCl2, 0.5 GTP and 2 ATP. The external solution contained (in mM) 140 NaCl, 5.4 KCl, 2.0 CaCl2, 1.0 MgCl2, 10 HEPES (pH 7.4) and 10 glucose. Whole-cell recordings were performed with an Axoclamp 200B amplifier (Molecular Devices, Sunnyvale, CA, USA) and were acquired using Clampex 10.1. Data analysis was performed off-line using Clampfit 10.1 (Axon Instruments, Sunnyvale, CA, USA). Exogenous glycine-evoked currents were obtained using a manually applied pulse of the agonist. Brief applications were obtained using outlet tube (200 μm ID) of a gravity-fed micro perfusion system, positioned 50–100 μm from the recorded cell. The concentration-response curves (CRC) were recorded and fitted to the Hill Equation: EC = (Emax*[C]nH)/([C]nH + [EC50]nH). In this equation, EC represents the evoked current at concentration C; Emax denotes the maximal evoked current; EC50 is the agonist concentration that produces 50% of Emax; and nH is the Hill coefficient80. In experiments involving allosteric modulation, paired recordings of glycine-evoked currents were made in the presence and absence of AM-1488. Based on preliminary experiments co-applying the compound and glycine for 10–20 s, we observed that the potentiation plateau was typically reached after 5–6 s. Consequently, we adopted this time frame along our experiments because balance glycine current desensitization with a rapid compound washout. The percentage change in the amplitude current induced by AM-1488 was calculated using the following equation: PC = ((EC-E0)/E0)*10028,41, where EC refers to the evoked current in presence of AM-1488 and E0 stand for the evoked current in absence of the PAM. All the experiments involving allosteric modulation were recorded using an EC5 − 10 value as control to ensure equipotent activation of receptors. We also examined the agonistic effect of AM-1488 by recording currents evoked solely by AM-1488 in the absence of glycine. The amplitude of these currents was then normalized by the maximum current triggered by this receptor. Reagents for both, allosteric and agonist studies were prepared in an external solution and then delivered to the recorded cell via perfusion system. The stock solution of AM-1488 was initially dissolved in DMSO and subsequently mixed with the external solution. In every instance, DMSO constituted no more than 1% of the solution volume. Prior studies have indicated that this concentration of DMSO does not affect the function of GlyRs41,81.
Single channel recordings have been previously described37,44,82. Recordings were acquired in cell-attached mode and digitized every 5–10 µs using an Axopatch 200B amplifier with a 2 kHz low-pass filter, a Digidata 13,222 A and the software pClamp (Molecular Devices, USA). Electrode pipettes of 7–12 MΩ were used and were manually fire-polished in a microforge (Narishige, Japan). The recording solution contained (in mM): Na-Gluconate 20, 102 NaCl, 2 KCl, 2 CaCl2, 1 MgCl2, 10 HEPES, 20 TEA-Cl, 15 sucrose y 14 glucose, pH 7.4. Recordings were performed at room temperature (20–24 °C) using a holding potential of + 60mV. To assess the allosteric modulation, the pipette was filled with extracellular solution containing glycine (EC5 − 10) alone or together with AM-1488 (0.5 or 10 µM). Single-channel conductances were determined from the relationship γ = I/(Vm − Vrev), in which I is the current amplitude of single channel events, Vm is the membrane potential and Vrev the reversal potential.
Molecular docking and bioinformatic procedures
Protein–ligand docking was performed using the structures of α1β, α2β, α3 GlyRs, and obtained from the Protein Data Bank (PDB ID: 7TU9, 7KUY, 5TIN, 5CFB)6,8,9,83. Before docking simulations, all protein structures were prepared using the Protein Preparation Workflow tool of the Maestro software (Schrödinger, LLC, NY, USA). This process included the addition of hydrogens, H-bond assignments optimization, the protonation states determination at pH 7 ± 0.2, and filling in missing side chains with Prime. Similarly, the AM-1488 structure was retrieved from the PubChem database (CID: 132547348) and prepared using LigPrep (Schrödinger, LLC, NY, USA) to generate ionization states at pH 7 ± 0.2 and possible conformations.
Site-directed docking calculations were performed using Glide (Schrödinger, LLC, New York, NY, USA, 2020) with a grid centered on the orthosteric binding site of the α/α, and the α/α interfaces of the AM-1488 binding site, respectively. Predictions were made employing the extra-precision (XP) configuration with a post-docking minimization that included 10 poses per ligand, from which the best pose was selected to represent each protein–ligand complex. Analysis of the complexes encompassed the structural and energetic parameters were summarized in the docking score values. In silico mutagenesis was conducted using the Residue and Loop Mutation module from Maestro. After substituting the selected amino acid, refinement was performed through implicit solvent minimization, including all residues within 5 Å around the mutation.
Statistical analyses
All results are presented as mean ± SEM. Statistical analysis and graph plotting were performed with Origin (version 6.0 or 8.0). Values of p < 0.05, p < 0.01, and p < 0.001 were considered statistically different. Statistical comparisons were performed using paired or unpaired Student’s t-tests. Multiple comparisons were analyzed with ANOVA followed by a Bonferroni post hoc test.
Data availability
The data supporting the findings of this study are provided within the manuscript and its Supplementary Information. Raw data can be made available upon request by contacting the corresponding authors.
References
Lynch, J. W. Native glycine receptor subtypes and their physiological roles. Neuropharmacology 56(1), 303–309 (2009).
Nys, M., Kesters, D. & Ulens, C. Structural insights into cys-loop receptor function and ligand recognition. Biochem. Pharmacol. 86(8), 1042–1053 (2013).
Zhu, H. & Gouaux, E. Architecture and assembly mechanism of native glycine receptors. Nature 599(7885), 513–517 (2021).
Lynch, J. W. et al. Glycine Receptor Drug Discovery Adv. Pharmacol., 79: 225–253. (2017).
Du, J. et al. Glycine receptor mechanism elucidated by electron cryo-microscopy. Nature 526(7572), 224–229 (2015).
Huang, X. et al. Crystal structure of human glycine receptor-alpha3 bound to antagonist strychnine. Nature 526(7572), 277–280 (2015).
Huang, X., Chen, H. & Shaffer, P. L. Crystal structures of human GlyRalpha3 bound to ivermectin. Structure 25(6), 945–950 (2017). e2.
Huang, X. et al. Crystal structures of human glycine receptor alpha3 bound to a novel class of analgesic potentiators. Nat. Struct. Mol. Biol. 24(2), 108–113 (2017).
Yu, H., Bai, X. C. & Wang, W. Characterization of the subunit composition and structure of adult human glycine receptors. Neuron 109(17), 2707–2716e6 (2021).
Aguayo, L. G. et al. Changes on the properties of glycine receptors during neuronal development. Brain Res. Brain Res. Rev. 47(1–3), 33–45 (2004).
McCracken, L. M. et al. Glycine receptor alpha3 and alpha2 subunits mediate tonic and exogenous agonist-induced currents in forebrain. Proc. Natl. Acad. Sci. U S A. 114(34), E7179–E7186 (2017).
Gallagher, C. I. et al. Positive allosteric modulators of Glycine receptors and their potential use in Pain therapies. Pharmacol. Rev. 74(4), 933–961 (2022).
Xu, T. L. & Gong, N. Glycine and glycine receptor signaling in hippocampal neurons: diversity, function and regulation. Prog Neurobiol. 91(4), 349–361 (2010).
Gallegos, S. et al. High ethanol sensitive glycine receptors regulate firing in D1 medium spiny neurons in the nucleus accumbens. Neuropharmacology 160, 107773 (2019).
Bode, A. & Lynch, J. W. The impact of human hyperekplexia mutations on glycine receptor structure and function. Mol. Brain 7, 2 (2014).
Harvey, R. J. et al. GlyR alpha3: an essential target for spinal PGE2-mediated inflammatory pain sensitization. Science 304(5672), 884–887 (2004).
Manzke, T. et al. Serotonin receptor 1A-modulated phosphorylation of glycine receptor alpha3 controls breathing in mice. J. Clin. Invest. 120(11), 4118–4128 (2010).
Winkelmann, A. et al. Changes in neural network homeostasis trigger neuropsychiatric symptoms. J. Clin. Invest. 124(2), 696–711 (2014).
Pilorge, M. et al. Genetic and functional analyses demonstrate a role for abnormal glycinergic signaling in autism. Mol. Psychiatry. 21(7), p936–p45 (2016).
Gallegos, S. et al. Reduced sedation and increased ethanol consumption in knock-in mice expressing an ethanol insensitive alpha 2 subunit of the glycine receptor. Neuropsychopharmacology 46(3), 528–536 (2021).
Munoz, B. et al. Influence of nonsynaptic alpha1 glycine receptors on ethanol consumption and place preference. Addict. Biol. 25(2), e12726 (2020).
San Martin, V. P. et al. Glycine receptor subtypes and their roles in nociception and chronic pain. Front. Mol. Neurosci. 15, 848642 (2022).
Lara, C. O. et al. Pentameric ligand-gated ion channels as pharmacological targets against chronic pain. Front. Pharmacol. 11, 167 (2020).
Cioffi, C. L. Modulation of glycine-mediated spinal neurotransmission for the treatment of chronic pain. J. Med. Chem. 61(7), 2652–2679 (2018).
Cerdan, A. H. et al. The glycine receptor allosteric ligands library (GRALL). Bioinformatics 36(11), 3379–3384 (2020).
Breitinger, U. & Breitinger, H. G. Modulators of the inhibitory glycine receptor. ACS Chem. Neurosci. 11(12), 1706–1725 (2020).
Zeilhofer, H. U. et al. Glycine receptors in spinal nociceptive control-an update. Biomolecules 11(6), 846 (2021).
Acuna, M. A. et al. Phosphorylation state-dependent modulation of spinal glycine receptors alleviates inflammatory pain. J. Clin. Invest. 126(7), 2547–2560 (2016).
Bregman, H. et al. The discovery and hit-to-lead optimization of tricyclic sulfonamides as potent and efficacious potentiators of glycine receptors. J. Med. Chem. 60(3), 1105–1125 (2017).
Hussein, R. A. et al. Modulation of glycine receptor-mediated pain signaling in vitro and in vivo by glucose. Front. Mol. Neurosci. 12, 280 (2019).
Argueta, D. A. et al. A balanced approach for cannabidiol use in chronic pain. Front. Pharmacol. 11, 561 (2020).
Shan, Q., Haddrill, J. L. & Lynch, J. W. Ivermectin, an unconventional agonist of the glycine receptor chloride channel. J. Biol. Chem. 276(16), 12556–12564 (2001).
Ahrens, J. et al. The nonpsychotropic cannabinoid cannabidiol modulates and directly activates alpha-1 and alpha-1-beta glycine receptor function. Pharmacology 83(4), p217–p22 (2009).
Curtis, D. R., Duggan, A. W. & Johnston, G. A. The specificity of strychnine as a glycine antagonist in the mammalian spinal cord. Exp. Brain Res. 12(5), 547–565 (1971).
Pribilla, I. et al. The atypical M2 segment of the beta subunit confers picrotoxinin resistance to inhibitory glycine receptor channels. EMBO J. 11(12), 4305–4311 (1992).
Beato, M. et al. The activation mechanism of alpha1 homomeric glycine receptors. J. Neurosci. 24(4), 895–906 (2004).
Moraga-Cid, G. et al. Modulation of glycine receptor single-channel conductance by intracellular phosphorylation. Sci. Rep. 10(1), 4804 (2020).
Kallenborn-Gerhardt, W. et al. Prolonged zymosan-induced inflammatory pain hypersensitivity in mice lacking glycine receptor alpha2. Behav. Brain Res. 226(1), p106–p111 (2012).
Imlach, W. L. et al. Glycinergic dysfunction in a subpopulation of dorsal horn interneurons in a rat model of neuropathic pain. Sci. Rep. 6, 37104 (2016).
Yu, H. et al. Temporal changes of spinal transcriptomic profiles in mice with spinal nerve ligation. Front. Neurosci. 13, 1357 (2019).
Lara, C. O. et al. Functional modulation of glycine receptors by the alkaloid gelsemine. Br. J. Pharmacol. 173(14), 2263–2277 (2016).
Munoz, B. et al. Modulatory actions of the glycine receptor beta subunit on the positive allosteric modulation of ethanol in alpha2 containing receptors. Front. Mol. Neurosci. 14, p763868 (2021).
Ivica, J. et al. The intracellular domain of homomeric glycine receptors modulates agonist efficacy. J. Biol. Chem. 296, 100387 (2020).
Marabelli, A. et al. The kinetic properties of the alpha3 rat glycine receptor make it suitable for mediating fast synaptic inhibition. J. Physiol. 591(13), p3289–p3308 (2013).
Gielen, M. & Corringer, P. J. The dual-gate model for pentameric ligand-gated ion channels activation and desensitization. J. Physiol. 596(10), 1873–1902 (2018).
Krashia, P. et al. The long activations of alpha2 glycine channels can be described by a mechanism with reaction intermediates (flip). J. Gen. Physiol. 137(2), 197–216 (2011).
Yevenes, G. E. et al. Molecular requirements for ethanol differential allosteric modulation of glycine receptors based on selective gbetagamma modulation. J. Biol. Chem. 285(39), 30203–30213 (2010).
Sanchez, A. et al. Control of ethanol sensitivity of the glycine receptor alpha3 subunit by transmembrane 2, the intracellular splice cassette and C-terminal domains. J. Pharmacol. Exp. Ther. 353(1), 80–90 (2015).
Crawford, D. K. et al. Roles for loop 2 residues of alpha1 glycine receptors in agonist activation. J. Biol. Chem. 283(41), 27698–27706 (2008).
Saul, B. et al. Point mutation of glycine receptor alpha 1 subunit in the spasmodic mouse affects agonist responses. FEBS Lett. 350(1), p71–p6 (1994).
Bormann, J. et al. Residues within transmembrane segment M2 determine chloride conductance of glycine receptor homo- and hetero-oligomers. EMBO J. 12(10), 3729–3737 (1993).
Lynch, J. W. Molecular structure and function of the glycine receptor chloride channel. Physiol. Rev. 84(4), 1051–1095 (2004).
Yevenes, G. E. & Zeilhofer, H. U. Molecular sites for the positive allosteric modulation of glycine receptors by endocannabinoids. PLoS One. 6(8), e23886 (2011).
Cohen, S. P., Vase, L. & Hooten, W. M. Chronic pain: an update on burden, best practices, and new advances. Lancet 397(10289), 2082–2097 (2021).
Duran, J. et al. Chronic pain in Chile: first prevalence report of noncancer chronic pain, fibromyalgia, and neuropathic pain and its associated factors. Pain 164(8), 1852–1859 (2023).
Zeilhofer, H. U., Benke, D. & Yevenes, G. E. Chronic pain states: pharmacological strategies to restore diminished inhibitory spinal pain control. Annu. Rev. Pharmacol. Toxicol. 52, 111–1 33 (2012).
Luo, C., Kuner, T. & Kuner, R. Synaptic plasticity in pathological pain. Trends Neurosci. 37(6), 343–355 (2014).
Kuner, R. & Kuner, T. Cellular circuits in the brain and their modulation in acute and chronic pain. Physiol. Rev. 101(1), 213–258 (2021).
Yekkirala, A. S. et al. Breaking barriers to novel analgesic drug development. Nat. Rev. Drug Discov. 16(8), 545–564 (2017).
Varrassi, G. et al. Pharmacological treatment of chronic pain - the need for CHANGE. Curr. Med. Res. Opin. 26(5), 1231–1245 (2010).
Labianca, R. et al. Adverse effects associated with non-opioid and opioid treatment in patients with chronic pain. Clin. Drug Investig. 32(Suppl 1), 53–63 (2012).
Finnerup, N. B. Advances in pain management and research presented at the 2015 scientific meeting of the Scandinavian Association for the study of Pain (SASP). Scand. J. Pain. 8(1), p27 (2015).
Finnerup, N. B. et al. Pharmacotherapy for neuropathic pain in adults: a systematic review and meta-analysis. Lancet Neurol. 14(2), 162–173 (2015).
Zeilhofer, H. U. et al. Glycine receptors and glycine transporters: targets for novel analgesics? Cell. Mol. Life Sci. 75(3), 447–465 (2018).
Zeilhofer, H. U., Wildner, H. & Yevenes, G. E. Fast synaptic inhibition in spinal sensory processing and pain control. Physiol. Rev. 92(1), 193–235 (2012).
Salling, M. C. & Harrison, N. L. Strychnine-sensitive glycine receptors on pyramidal neurons in layers II/III of the mouse prefrontal cortex are tonically activated. J. Neurophysiol. 112(5), 1169–1178 (2014).
McLaughlin, C. et al. The role of tonic glycinergic conductance in cerebellar granule cell signalling and the effect of gain-of-function mutation. J. Physiol. 597(9), 2457–2481 (2019).
Munoz, B. et al. Presence of inhibitory glycinergic transmission in medium spiny neurons in the nucleus accumbens. Front. Mol. Neurosci. 11, 228 (2018).
Wishart, D. S. et al. HMDB 5.0: the human metabolome database for 2022. Nucleic Acids Res. 50(D1), D622–D631 (2022).
Zhuo, M. Contribution of synaptic plasticity in the insular cortex to chronic pain. Neuroscience 338, p220–229 (2016).
Qiu, S. et al. An increase in synaptic NMDA receptors in the insular cortex contributes to neuropathic pain. Sci. Signal. 6(275), ra34 (2013).
McCool, B. A. & Farroni, J. S. Subunit composition of strychnine-sensitive glycine receptors expressed by adult rat basolateral amygdala neurons. Eur. J. Neurosci. 14(7), 1082–1090 (2001).
McCool, B. A. & Botting, S. K. Characterization of strychnine-sensitive glycine receptors in acutely isolated adult rat basolateral amygdala neurons. Brain Res. 859(2), 341–351 (2000).
Choi, K. H., Nakamura, M. & Jang, I. S. Presynaptic glycine receptors increase GABAergic neurotransmission in rat periaqueductal gray neurons. Neural Plast. 2013, 954302 (2013).
Jeong, H. J. et al. Role of protein kinase C in opioid modulation of glycine-gated Cl(-) current in rat periaqueductal gray neuron. Eur. J. Pharmacol. 431(2), 143–150 (2001).
Werynska, K. et al. A phospho-deficient α3 glycine receptor mutation alters synaptic glycine and GABA release in mouse spinal dorsal horn neurons. J. Physiol. 601(18), 4121–4133 (2023).
Hruskova, B. et al. Differential distribution of glycine receptor subtypes at the rat calyx of held synapse. J. Neurosci. 32(47), 17012–17024 (2012).
Marileo, A. M. et al. Modulation of GABA(A) receptors and of GABAergic synapses by the natural alkaloid gelsemine. Front. Mol. Neurosci. 15, 1083189 (2023).
Yevenes, G. E. et al. Molecular determinants for G protein betagamma modulation of ionotropic glycine receptors. J. Biol. Chem. 281(51), 39300–39307 (2006).
Moraga-Cid, G. et al. A single phenylalanine residue in the main intracellular loop of alpha1 gamma-aminobutyric acid type A and glycine receptors influences their sensitivity to propofol. Anesthesiology 115(3), 464–473 (2011).
Marileo, A. M. et al. Molecular pharmacology of gelsemium alkaloids on inhibitory receptors. Int. J. Mol. Sci. 25(6). (2024).
Lasala, M. et al. Molecular modulation of human alpha7 nicotinic receptor by amyloid-beta peptides. Front. Cell. Neurosci. 13, 37 (2019).
Gibbs, E. et al. Conformational transitions and allosteric modulation in a heteromeric glycine receptor. Nat. Commun. 14(1), 1363 (2023).
Acknowledgements
This work was supported by ANID-FONDECYT 1211082 (to G.E.Y.) as well as ANID-FONDECYT 1211095 (to G.M.-C), ANID-FONDECYT 1231038 (to P.A.C.) and ANID FONDECYT 11221211 (to C.F.B.). This work was also supported by the Millennium Nucleus for the Study of Pain (MiNuSPain) and by the “IBRO Collaborative Research grant” program of the International Brain Research Association (IBRO) (to G.E.Y.). The authors thank L. Aguayo, and I. Cid, for their outstanding technical assistance. K. F-O, P. S-O, A. S, and K. G-R were supported by the University of Concepcion through Graduate School Fellowships. C.O.L, V. P. S-M, D. F, and A.M.M. were supported by ANID doctoral fellowships.
Author information
Authors and Affiliations
Contributions
C.O.L., G.M-C, C.F.B., C.B., and G.E.Y. conceived, designed, and performed experiments, analyzed the data, and wrote the paper. K. F-O., P. S-O, V.P.SM, C.O.L, A.M.M., A.S., O.V.C., D. F., J.C. and C.M-M. performed experiments and analyzed the data. C.F.B., P. S-O, and D.F. performed bioinformatics analyses. C.B., J.F., P.A.C, and L.G.A., contribute with resources and equipment. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Lara, C.O., Burgos, C.F., Fariña-Oliva, K. et al. Allosteric modulation and direct activation of glycine receptors by a tricyclic sulfonamide. Sci Rep 15, 5515 (2025). https://doi.org/10.1038/s41598-025-90209-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-90209-7
