
Abstract
Metformin, widely used for the treatment of type 2 diabetes, has recently gained attention for its potential anticancer properties. Several studies have shown that metformin treatment inhibits cell viability in colon adenocarcinoma (COAD); however, the research related to the tumor-node-metastasis (TNM) stage is limited. As COAD is frequently diagnosed at an advanced stage, understanding the genetic factors that regulate the pathogenesis of COAD at each TNM stage and the effects of metformin for potential treatment. Therefore, we identified differentially expressed factors at the TNM stage in metformin-treated COAD cells and investigated their regulatory mechanisms using microRNAs (miRNAs). Through bioinformatics analyses, four-jointed box kinase 1 (FJX1) and hsa-miR-1306-3p were identified as differentially expressed in COAD upon metformin treatment. Metformin treatment significantly reduced cell viability, with an observed decrease of approximately 50%. Analysis using quantitative real-time PCR showed an increase in hsa-miR-1306-3p and a decrease in FJX1 expression upon metformin treatment compared to untreated cells. Luciferase assay confirmed the sequence-specific binding of hsa-miR-1306-3p to FJX1. These findings highlight the potential of metformin as a therapeutic agent for COAD by modulating FJX1 expression via upregulation of hsa-miR-1306-3p, revealing novel avenues for COAD treatment.
Similar content being viewed by others


Introduction
Metformin is a biguanide used as a first-line treatment for type 2 diabetes and has a hypoglycemic effect by activating 5′-adenosine monophosphate-activated protein kinase (AMPK)1,2,3. Additionally, it inhibits cell viability, and recent studies have explored its therapeutic potential in various cancers including liver, colon, pancreatic, lung, breast, and prostate cancers4. Notably, metformin reduces the development and metastasis of colon adenocarcinoma (COAD), the second leading cause of cancer-related deaths worldwide5. One study found that COAD patients who take metformin had a higher survival rate compared to those who do not6. This was further supported by in vitro studies demonstrating that metformin modulates the AMPK-mTOR signaling pathway, significantly reducing the viability, migration, and invasion of COAD cell lines7. Additionally, studies suggested that metformin may improve the longevity of diabetic patients while reducing the incidence of COAD8. However, COAD is typically characterized by an absence of early symptoms and limited treatment options9,10. Therefore, understanding the molecular mechanisms of therapeutic agents at different stages of COAD is crucial11,12. However, no studies have investigated the effects of metformin across stages of COAD8,13,14.
The Dukes’ staging system is a highly effective method for classifying COAD15. However, it does not consider lymph node involvement, tumor grade, and other pathological characteristics, leading to variability in the assessments by different clinicians16. Consequently, the Dukes’ staging system is seldom used, and current cancer research predominantly employs the tumor-node-metastasis (TNM) staging system17,18,19,20. T stands for tumor and ranges from T1 to T4, reflecting the size of the primary tumor and its invasion into nearby tissues. ‘Node’ is represented by N and subdivided into N1, N2, and N3, based on the number, size, and location of the involved lymph nodes. ‘Metastasis’ is denoted by M and indicates whether the cancer has spread to other parts of the body, classified as M0 or M1, depending on the presence of distant metastasis21,22. This study used the TNM staging system to classify COAD cell lines, whereby Dukes’ A and B were categorized as T stage, Dukes’ C as N stage, and Dukes’ D as M stage23.
MicroRNAs (miRNAs) are small non-coding RNA, typically comprising 22 nucleotides24. They regulate gene expression by binding complementarily to the 3′ untranslated region (UTR) of target messenger RNAs (mRNAs)25. This regulation of miRNAs plays a central role in the pathophysiology of several diseases, including cancer26. In COAD, specific miRNAs such as miR-17-5p, miR-1224-5p, and miR-214-3p have been shown to suppress metastasis by targeting pathways involved in epithelial-mesenchymal transition (EMT) and other cancer-promoting mechanisms27,28,29. In addition to their role in cancer progression, miRNAs mediate the therapeutic effects of metformin on COAD30. Metformin has been demonstrated to modulate miRNAs, such as miR-342-3p and miR-361-5p, thereby enhancing the suppression of COAD31,32. MiR-342-3p has been shown to promote CD133 expression, increasing apoptosis and reducing cell viability31. In addition, metformin-regulated miR-361-5p has been observed to inhibit the SHH signaling pathway, a process that contributes to overcoming chemoresistance32. These findings highlight the potential of metformin as a therapeutic strategy through miRNA modulation in COAD.
Therefore, this study aimed to investigate the anticancer effects of metformin according to TNM staging in COAD. TNM-staged COAD cell lines were treated with metformin to identify changes in the expression levels of miRNAs and target genes, and to observe alterations in tumor characteristics. Thus, this study elucidated the mechanisms of action of metformin in COAD across TNM stages, with interactions between four-jointed box kinase 1 (FJX1) and hsa-miR-1306-3p.
Results
Inhibiting cell viability using metformin
WST assay assessed changes in COAD cell viability after metformin (10 mM) treatment using EZ-Cytox. This was accomplished by quantifying the absorbance at 450 nm, which is the wavelength used to detect live cell activity. The viability of SW480, SW620, and HCT116 cells, representing the T, N, and M stages, respectively, was significantly reduced by approximately 50% in metformin-treated cells compared to untreated cells (Fig. 1).

Inhibiting the viability of COAD cells using metformin. To evaluate the effects of metformin on COAD cells depending on their tumor node metastasis (TNM) stage, cell viability was assessed in three cell lines treated with metformin (10 mM) and the control group (0 mM metformin). (A) SW480, T stage; (B) SW620, N stage; and (C) HCT 116, M stage, respectively. *p-value < 0.05; **p-value < 0.01.
Bioinformatic analyses of FJX1
Genes that exhibited increased expression in cancer tissues relative to normal tissues were identified in The Cancer Genome Atlas (TCGA). Conversely, genes that demonstrated decreased expression in metformin-treated cancer cells compared with untreated cells were selected from the Gene Expression Omnibus (GEO) database. A group of RNAs with a log2 fold change (log2FC) > 2 and p-value < 0.05 was established by comparing tumor tissues with normal tissues using RNA-sequencing (RNA-seq) data from TCGA (Fig. 2A). Additionally, the GSE67342 dataset was analyzed to detect RNAs with a log2FC < − 2 and p-value < 0.05, compared between metformin-treated and untreated cancer cells (Fig. 2B).

Bioinformatic analyses identified differentially expressed genes in COAD. Volcano plots were visualized to verify differentially expressed genes in (A) COAD tissues compared to normal tissues based on TCGA RNA-seq and (B) metformin-treated COAD cells in contrast to untreated COAD based on GSE67342 (GEO database). (C) Venn diagram was used to identify significant target genes, and there were 6 common genes between TCGA RNA-seq data and GSE67342. Among them, FJX1, which plays an important role in COAD, was selected as the target gene and highlighted in red. (D) Biological process data from gene ontology analysis using ShinyGO demonstrated that FJX1 is involved in organ development and cell signaling. (E) The construction of survival curves was based on the trisection of FJX1 expression levels for patients with COAD, utilizing the OncoLnc. The patients with high expression levels of FJX1 were depicted in a red line and those with low expression levels in a black line. (F) Expression of FJX1 in several cancers from TCGA was analyzed using the TIMER2.0 database, which revealed that FJX1 was upregulated in most cancers.
Six candidate genes were identified through the intersection of the differentially expressed genes (DEGs) from TCGA RNA-seq and GSE67342, including HILPDA, EPHB3, INHBA, SHH, FJX1, and RGS16. FJX1 was selected for further investigation owing to its consistently high |log2FC| values. It was ranked as the second most DEG in both TCGA and GEO datasets (Fig. 2C). Gene Ontology (GO) analysis indicated that FJX1 is involved in the development of tissue and cell signaling pathways (Fig. 2D). To assess the correlation between FJX1 expression and survival in patients with COAD, FJX1 expression groups were categorized into high (33%) and low (33%) and analyzed overall survival (OS) using the OncoLnc tool. High FJX1 expression was significantly associated with worse OS (p-value = 0.01), indicating a strong link between elevated FJX1 levels and poor prognosis in COAD (Fig. 2E). TIMER2.0 analysis revealed that FJX1 expression is elevated in most cancer types, including COAD (Fig. 2F).
Target miRNA prediction and bioinformatic analyses of hsa-miR-1306-3p
The miRNAs from the TCGA small RNA-seq data were analyzed to identify tumor suppressive miRNAs with a log2FC < − 2 and a p-value < 0.05 (Fig. 3A). miRNAs that were predicted to bind FJX1 complementarily were selected using the miRNA target prediction tools TargetScanHuman and miRDB. A combination of TCGA small RNA-seq data and two miRNA target prediction sites identified eight potential miRNAs: hsa-miR-7110-3p, hsa-miR-6716-5p, hsa-miR-1226-3p, hsa-miR-6756-5p, hsa-miR-3614-3p, hsa-miR-1229-3p, hsa-miR-4685-3p, and hsa-miR-1306-3p. Among these, hsa-miR-1306-3p was selected because of its markedly pronounced differential expression, exhibiting the lowest log2FC and p-value (Fig. 3B). Hsa-miR-1306-3p expression was screened across various cancer types using the dbDEMC database. Within the green squares, hsa-miR-1306-3p was significantly downregulated in breast cancer and COAD cells (Fig. 3C). The expression of hsa-miR-1306-3p was analyzed in normal colon and COAD tissues at different stages using the UALCAN database. The results indicated that hsa-miR-1306-3p levels were significantly decreased in COAD tissues across all stages compared to those in normal colon tissues (Fig. 3D). The relationship between hsa-miR-1306-3p and OS in patients with COAD was analyzed using the OncoLnc. Two groups of hsa-miR-1306-3p expression were categorized as high (33%) and low (33%). These results indicated that low hsa-miR-1306-3p expression was associated with lower OS rates (p-value = 0.01) (Fig. 3E).

Differential expression and prognostic impact of hsa-miR-1306-3p in COAD using bioinformatic analyses. (A) A volcano plot represented an analysis of differentially expressed miRNAs in COAD tissues compared to normal tissues from patients with COAD based on TCGA small RNA-seq data. (B) Two miRNA prediction databases, TargetScanHuman and miRDB, and TCGA miR-seq data that demonstrated significantly reduced miRNA expression in COAD tissues were used to find common miRNAs targeting FJX1. The list indicated common miRNAs targeting FJX1 based on log2FC < − 2, p-value < 0.05, and the lowest expressed miRNA (hsa-miR-1306-3p) was selected. (C) The expression of hsa-miR-1306-3p in several cancers from TCGA was analyzed using the dbDEMC. (D) The relative expression of hsa-miR-1306-3p in COAD stages was compared to normal colon data. The results demonstrated a significant down-regulation of expression in all COAD stages compared to normal. (E) The survival plot of hsa-miR-1306-3p showed low expression of hsa-miR-1306-3p resembling a low survival rate. **p-value < 0.01.
Sequence alignment between hsa-miR-1306-3p and FJX1
The complementarity of the interactions between hsa-miR-1306-3p and FJX1 was validated through RNA hybridization analyses using BioEdit and Bielefeld University Bioinformatics Server (BiBiServ). The 3′ UTR of FJX1 and hsa-miR-1306-3p were aligned, and primers were designed to amplify the region containing the complementary sequence to the seed region of hsa-miR-1306-3p (5′-CGUUGGC-3′) (Fig. 4A). The structure of the miRNA-mRNA hybrid was demonstrated using the BiBiServ RNA hybrid tool, and the minimum free energy (MFE) value was calculated to be − 25.3 kcal/mol, indicating a high degree of stability (Fig. 4B).

Sequence alignment of hsa-miR-1306-3p with FJX1 3′ UTR and localization of designed primers. (A) The sequence was aligned between hsa-miR-1306-3p and the 3′ UTR of FJX1 using the BioEdit program. The black arrows represent sense and antisense primers designed for PCR. The dotted boxes indicate the hsa-miR-1306-3p seed region and complementary site within FJX1. (B) RNA hybrid was illustrated, and its minimum free energy value was also calculated using BiBiServ. Green and red exhibit hsa-miR-1306-3p and the 3′ UTR of FJX1, respectively.
Relative expression of FJX1 and hsa-miR-1306-3p
Quantitative real-time PCR (qPCR) investigated the changes in the expression of FJX1 and hsa-miR-1306-3p induced by metformin. In cells treated with 10 mM metformin, the expression of FJX1 was significantly reduced across all cell lines (SW480, SW620, and HCT 116) compared to that in untreated cells (Fig. 5A). Conversely, the expression of hsa-miR-1306-3p was significantly increased in metformin-treated cell lines compared to that in untreated cells (Fig. 5B).

Relative expression analysis of FJX1 and hsa-miR-1306-3p. The expression patterns were examined in three cell lines, representing different TNM stages, treated with metformin (10 mM) and the control group (0 mM metformin) using qPCR. Experiments were performed in triplicate, and the results were presented as mean ± SD. (A) The expression of FJX1 was significantly decreased in all COAD cell lines treated with metformin, while (B) hsa-miR-1306-3p showed increased expression. SD, standard deviation; *p-value < 0.05; **p-value < 0.01.
Relationship between hsa-miR-1306-3p and FJX1
To verify the interaction between hsa-miR-1306-3p and FJX1, a dual-luciferase assay was conducted on SW480 cells co-transfected with the plasmid vector and hsa-mir-1306 mimics, including inhibitors, mimics, or negative controls (N.C.). The plasmid vector, psi-CHECK2, was cloned using the 3′ UTR of FJX1, and cells transfected with the plasmid vector alone were used as controls in the experiment. Cells co-transfected with the plasmid vector and hsa-mir-1306 mimic showed a significant decrease in luciferase expression compared to the control and N.C., whereas co-transfection of the plasmid vector and hsa-miR-1306-3p inhibitor restored luciferase activity to control levels (Fig. 6).

Revealing the interplay between the 3′ UTR of FJX1 and hsa-miR-1306-3p through dual luciferase activity assay. Luciferase activity was measured in SW480 cells co-transfected for 24 h with the Psi-CHECK2 vector cloned with 3′ UTR of FJX1 (plasmid), N.C., hsa-mir-1306 mimic, or hsa-miR-1306-3p inhibitor. The luciferase activity significantly declined in cells co-transfected with plasmid and hsa-mir-1306 mimic. However, the luciferase activity of cells co-transfected with plasmid and hsa-miR-1306-3p inhibitor was enhanced to a similar level as control or N.C. The triplicate experiments were conducted and were represented as mean ± SD. SD, standard deviation; ns, not significant; *p-value < 0.05; **p-value < 0.01.
Discussion
Metformin, a widely used antihyperglycemic agent for first-line treatment in type 2 diabetes, has garnered considerable attention in cancer research due to its anti-viability properties33. Many studies have explored its therapeutic potential, particularly in high-risk cancers such as COAD34. In COAD, metformin improves survival rates and reduces disease progression, potentially by modulating pathways involved in cell viability and metastasis13,35. In particular, miRNAs involved with metformin appear to play a crucial role in COAD36. miRNAs are small non-coding RNAs that regulate gene expression by binding to complementary sequences within the 3′ UTR of target mRNAs, leading to mRNA degradation or translational repression37. Through these mechanisms, miRNAs have emerged as key regulatory molecules in tumorigenesis, acting as both tumor suppressors and oncogenic drivers depending on their targets38. This role underscores the potential for metformin to influence specific miRNAs that could modulate COAD progression, a therapeutic avenue of great interest in current oncology research39,40. This study identified whether metformin could be a candidate treatment for COAD and which genes and miRNAs are involved in the regulation of cell viability (Fig. 7).

Schematic illustration of the proposed mechanism for modulation of hsa-miR-1306-3p and FJX1 using metformin. In COAD, the upregulation of FJX1 has been demonstrated to promote cell viability. Metformin treatment has been shown to increase the expression of tumor suppressor miRNAs, hsa-miR-1306-3p, which bind to FJX1 mRNA, leading to the inhibition of cell viability.
Consistent with previous studies, a reduction in cell viability was observed across various TNM stages of COAD after metformin treatment, as illustrated in Fig. 141,42. This finding demonstrated the efficacy of metformin in a wide range of TNM stages. However, the use of a more extensive range of COAD cell lines encompassing each TNM stage would enhance the reliability of our results.
The bioinformatics was analyzed using the TCGA and GEO databases to elucidate the molecular mechanisms underlying metformin’s effects on COAD (Fig. 2A and B). TCGA RNA-seq data revealed DEGs between COAD and normal tissues, while GEO dataset GSE67342 provided DEG data for metformin-treated and untreated cell lines. Although direct comparisons between these datasets were challenging, integrating them enhanced the biological insights of our study43. FJX1 was selected as the target gene in this study presented in Fig. 2C. FJX1 influences cell adhesion structures and modulates the organization of microtubules and actin (Fig. 2D)44. FJX1 was highly expressed in COAD and inversely correlated with patient survival (Fig. 2E), highlighting its potential role in prognosis. Additionally, FJX1 was associated with oncogenic behaviors such as viability, metastasis, and treatment resistance across various cancers (Fig. 2F)45,46,47,48. MiRNAs like miR-532-3p and miR-1249 were found to regulate FJX1, suppressing COAD cell viability, migration, and invasion49,50. Despite extensive research on the role of FJX1 in COAD, our understanding of the genetic mechanisms underlying its involvement in tumorigenesis remains limited. The specific contributions of FJX1 to cancer development are not clearly defined in the GO and Kyoto Encyclopedia of Genes and Genomes pathway data, indicating the need for further investigation of its oncogenic pathways and molecular interactions.
Hsa-miR-1306-3p was selected for further analysis because of its significantly lower expression in COAD, as shown in Fig. 3B. Furthermore, its low expression correlated with poor clinical outcomes, indicating its potential role in the progression and aggressiveness of COAD across TNM stages as indicated in Fig. 3E. Hsa-miR-1306-5p, which is derived from the same precursor as hsa-miR-1306-3p, investigated the role of miR-1306-5p in various cancers, including COAD51,52,53. Notably, hsa-miR-1306-5p is typically downregulated in COAD, and its overexpression suppresses oncogenic behavior by inhibiting the PI3K/AKT/mTOR signaling pathway54. While the role of hsa-miR-1306-5p in various cancers has been extensively studied in terms of oncogenic pathways, investigations on hsa-miR-1306-3p remain limited. As illustrated in Fig. 3C, hsa-miR-1306-3p exhibited a significantly reduced expression in COAD. However, no study has investigated its role in COAD. This study provides novel insights into the association between hsa-miR-1306-3p and COAD progression, highlighting its potential significance.
Figure 4 showed that the binding interaction between hsa-miR-1306-3p and FJX1 had a calculated MFE of below − 25 kcal/mol, indicating a stable and favorable interaction55. The high complementary conservation in the seed region of hsa-miR-1306-3p suggests a strong binding capability. This could enhance its regulatory effect on FJX1 expression, highlighting its potential as a therapeutic target in COAD treatment.
In vitro studies have shown that FJX1 expression negatively correlates with hsa-miR-1306-3p levels. FJX1 levels were reduced in the metformin-treated samples, as shown in Fig. 5A. Conversely, the increase in hsa-miR-1306-3p levels illustrated in Fig. 5B suggests that both factors are influenced by metformin treatment and are negatively correlated. The dual-luciferase assay confirmed that hsa-miR-1306-3p suppressed FJX1 expression, as presented in Fig. 6. Co-transfection of cells with the plasmid vector and hsa-mir-1306 mimic resulted in decreased luciferase activity compared to that in the control group. Conversely, when cells were co-transfected with the plasmid vector and the hsa-miR-1306-3p inhibitor, luciferase activity was restored to levels comparable to those of the control. Furthermore, metformin directly regulates miRNA expression and highlights its relevance to cancer pathogenesis, thereby supporting the implications of our study56,57,58.
The findings of this study provided a foundation for the development of a therapeutic strategy for COAD that utilizes the interaction between metformin and the hsa-miR-1306-3p/FJX1 axis. The results of the study demonstrated metformin’s anti-cancer efficacy across TNM stages and identified hsa-miR-1306-3p as a novel candidate involved in this process. To enhance the benefits of metformin, further research is needed to explore how its effects can be modified to different TNM stages, providing insights into stage-specific mechanisms. A detailed understanding of the signaling pathways involving hsa-miR-1306-3p and FJX1 is essential for developing targeted therapies for COAD. Additionally, repurposing drugs and supplementing with vitamins like vitamin D could offer a complementary strategy for COAD treatment59. This approach will improve therapeutic outcomes by integrating metformin with other interventions. Further studies should investigate the roles of hsa-miR-1306-3p and FJX1 in vitro and in vivo, clarifying their contributions to tumor growth and metastasis. This will support the development of more effective treatment protocols, incorporating metformin in a stage-specific manner.
Conclusion
In summary, our findings demonstrated that metformin treatment increased hsa-miR-1306-3p expression in COAD cell lines, leading to the suppression of FJX1 and a subsequent reduction in cell viability. This suggests that metformin has potential as a therapeutic agent for COAD, and its efficacy is potentially influenced by the TNM stage. The interplay between hsa-miR-1306-3p and FJX1 has emerged as a crucial element in this process, warranting further research to clarify their roles as potential therapeutic targets.
Materials and methods
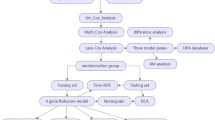
A summary of the materials and methods is presented as a flowchart in Fig. 8.

Process overview of the study’s methodology. Workflow for investigating the impact of metformin on COAD. Data collection was conducted using the GEO and TCGA databases. Differentially expressed genes and microRNAs were revealed using the DESeq2 package in R. The prediction of target microRNAs was performed with TargetScanHuman 8.0 and miRDB, and the identification of common genes and microRNAs was conducted using the Venny program. Bioinformatic analyses of the final target gene and miRNA were conducted using ShinyGO, TIMER2.0, OncoLnc, dbDEMC, UALCAN, and RNAfold. Sequence alignment of the target gene and miRNA was conducted using the RNA hybrid tool (BiBiServ and BioEdit). The experimental validation included cell viability assays, qPCR, and dual-luciferase assays with metformin treatment.
Bioinformatic analyses
RNA-seq and small RNA-seq data were obtained from TCGA (https://portal.gdc.cancer.gov/). RNA-seq data from 41 patients with COAD and small RNA-seq data from eight patients with COAD were randomly selected. Data from both primary tumor and normal tissues were extracted for further analyses. DEGs were identified by comparing tumor tissues with normal tissues from patients with cancer using the DESeq2 package in R. The resulting data were visualized as a volcano plot, using the ggplot2 package in R. Genes and miRNAs were selected based on a log2FC of ≥ 2 or ≤ -2 and a p-value of < 0.05. The GSE67342 dataset was obtained from the GEO (https://www.ncbi.nlm.nih.gov/geo/). DEGs were identified by comparing cells treated with metformin to untreated cells, selecting those with a log2FC ≤ − 2 and a p-value < 0.05. To gain further insights into the role of FJX1 in cancer, its expression levels in various cancer types were examined using the online tool TIMER2.0 (http://timer.cistrome.org/). Additionally, GO biological process analysis was performed on DEGs, including FJX1, using the online tool ShinyGO 0.80 (http://bioinformatics.sdstate.edu/go/). Lists of predicted target miRNAs were obtained from TargetScanHuman (https://www.targetscan.org/vert_80/) and miRDB (https://mirdb.org/). The Venny tool (https://bioinfogp.cnb.csic.es/tools/venny/) was used to identify target genes and miRNAs in TCGA RNA-seq and small RNA-seq data by creating visualized Venn diagrams. Differential expression of hsa-miR-1306-3p across various cancer types was analyzed using the dbDEMC database (https://www.biosino.org/dbDEMC/search). Differences in expression according to the stage of COAD were evaluated using UALCAN (https://ualcan.path.uab.edu/index.html). Survival curves were generated using OncoLnc (http://www.oncolnc.org/). The expression levels of FJX1 and hsa-miR-1306-3p were classified into two categories: the top 33% and the bottom 33%.
The sequence of the 3′ UTR of the selected target gene FJX1 was obtained from the National Center for Biotechnology Information (NCBI; https://www.ncbi.nlm.nih.gov/), and the sequence of hsa-miR-1306-3p was retrieved from the miRbase (https://www.mirbase.org/). The nucleotide sequences of hsa-miR-1306-3p and FJX1 were aligned using the BioEdit tool, revealing a binding region of hsa-miR-1306-3p within the 3′ UTR of FJX1. The binding interaction and MFE between hsa-miR-1306-3p and the 3′ UTR of FJX1 were confirmed using the RNA hybrid tool from BiBiServ (https://bibiserv.cebitec.uni-bielefeld.de/rnahybrid).
Cell culture and treatment
The COAD cell lines SW480, SW620, and HCT 116, representing the T, N, and M stages, respectively, were obtained from the Korean Cell Line Bank (Korea). All cell lines were grown in RPMI 1640 (Hyclone, USA) medium containing 10% inactivated fetal bovine serum (FBS; Gibco, USA) and 1% antibiotic–antimycotic (Anti-Anti; Gibco, USA) in a humidified atmosphere of 5% CO2 incubator at 37 °C. Metformin, obtained from Professor Ho Jeong Kwon at Yonsei University, was diluted in RPMI 1640 medium containing 10% FBS to a final concentration of 10 mM.
Viability test
All cell lines were seeded at a density of 2 × 103 cells/well in 96-well plates and incubated for 24 h to allow cell adhesion. Subsequently, cells were treated with either 10 mM metformin or FBS-only medium. Following an additional 24 h, 10 µL of EZ-Cytox (DoGenBio, Korea) was added to each well, and the cells were incubated at 37 °C with 5% CO₂ for a further 24 h. Absorbance was measured using a spectrophotometer, and cell viability was calculated according to the manufacturer’s protocol.
RNA extraction and complementary DNA (cDNA) synthesis
Hybrid-R™ (GeneAll, Korea) was used to extract total RNA from cell lines in accordance with the manufacturer’s instructions. An ND-1000 UV–Vis spectrophotometer (NanoDrop Technologies, Wilmington, NC, USA) was used to measure the RNA quantity and quality. The RNA samples were quantified to 500 ng and used for target mRNA and miRNA cDNA synthesis using the PrimeScript™ RT reagent kit with gDNA Eraser (TaKaRa, Japan) and HB miR Multi Assay Kit™ (SYSTEM I; HeimBiotek, Seoul, Republic of Korea), respectively. For reverse transcription of the target mRNA and miRNA, the following PCR settings were used: PrimeScript™ RT Reagent Kit with gDNA Eraser (TaKaRa, Japan) was used at 37 °C for 15 min and 85 °C for 5 s. The HB miR Multi Assay Kit™ (HeimBiotek, Korea) was used at 37 °C for 60 min and 95 °C for 5 min.
Gene cloning
Using synthesized cDNA from SW480 whole extracted RNA and the primers (sense primer: 5′-GCTGGGGTATGGATGATGGG-3′, antisense primer: 5′-TGAAAGAAAGGGGCAGGTGG-3′), the 3′ UTR of FJX1 was amplified using a PCR. The PCR was performed under the following conditions: an initial step of 5 min at 94 °C, followed by 45 cycles of 40 s at 94 °C for denaturation, 40 s at 59 °C for annealing, and 90 s at 72 °C for extension. The amplified DNA fragments were separated on a 1.5% agarose gel and purified using the Expin™ PCR SV kit (GeneAll, Korea) according to the manufacturer’s instructions. The purified PCR products were cloned into the dual-luciferase vector psi-CHECK2. The cloned plasmid was then extracted using the Exprep™ Plasmid SV, mini (GeneAll, Korea) according to the manufacturer’s instructions.
Co-transfection and luciferase assay
SW480 cells were seeded into 24-well plates at a density of 4 × 104 cells/well. Following 24 h of seeding, cells were co-transfected with the psi-CHECK2 vector containing the 3′ UTR of FJX1, an N.C., hsa-mir-1306 mimic, and hsa-miR-1306-3p inhibitor using jetPRIME® (Polyplus, France) as per the manufacturer’s instructions. The hsa-miR-1306-3p inhibitor is a chemically produced single-stranded RNA molecule that inhibits mature miRNA activity, whereas the hsa-mir-1306 mimic is an artificially created double-stranded RNA molecule that mimics mature miRNA duplexes. The experiment was designed using four-lane 24 well plates. Following a 24-h co-transfection period, 1 × passive buffer (Promega, USA) was used to lyse the cells following the manufacturer’s guidelines. Firefly and Renilla luciferase activities were measured after 24 h of cell lysis using a Dual Luciferase Assay Kit (Promega, USA) according to the manufacturer’s instructions.
qPCR amplification
Using the final cDNA products, qPCR was performed to examine the expression of hsa-miR-1306-3p. The following circumstances were met to analyze miRNA expression using the HB_I Real-Time PCR Master Mix Kit (HeimBiotek, Korea) under the manufacturer’s instructions (45 cycles of 95 °C for 10 s and 60 °C for 40 s; standard melting conditions at 55 °C for 90 s, then increasing by intervals of 5 s at 1 °C for each step up to 99 °C). The initialization phase lasted for 15 min. U6 was used as the reference gene for small nuclear RNA to standardize hsa-miR-1306-3p expression.
Target mRNA expression analysis was conducted using SYBR Green Q-PCR Master Mix with Low Rox (SmartGene, Korea) in accordance with the manufacturer’s instructions (hold at 95 °C for 2 min for initialization; 40 cycles of 95 °C for 2 s, 60 °C for 25 s, and 72 °C for 15 s; standard melting conditions are held at 55 °C for 90 s, and 5 s increase of 1 °C for each step up to 99 °C. GAPDH (sense primer: 5′-GAAATCCCATCACCATCTTCCAGG-3′, antisense primer: 5′-GAGCCCCAGCCTTCTCCATG-3′) was used as the reference gene for the target mRNA. The 2−ΔΔCt method was used to examine the data, with each sample being processed in triplicate.
Statistical analyses
All experiments were conducted in triplicate and error bars in the graphs represent the mean ± standard deviation (SD) of the data. Student’s t-tests were also performed for statistical analyses. P-values were used to indicate the statistical significance of the results, with a threshold of p-value < 0.05.
Data availability
Sequence data supporting the findings of this study have been deposited in GEO (URL: http://www.ncbi.nlm.nih.gov/geo/) and TCGA (URL: https://portal.gdc.cancer.gov/) databases. The datasets used in this study are available under the accession IDs: GSE67342 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE67342).
Change history
26 April 2025
The original online version of this Article was revised: The Funding section in the original version of this Article was incorrect. The Funding section now reads: “This work was supported in part by the 2024 BK21 FOUR Graduate School Innovation Support, funded by Pusan National University (PNU-Fellowship program), and by a grant from the National Research Foundation of Korea (MSIP; 2021R1A3B1077371) to HJK.”
Abbreviations
- COAD:
-
Colon adenocarcinoma
- TNM:
-
Tumor-node-metastasis
- miRNA, miR:
-
MicroRNA
- FJX1 :
-
Four-jointed box kinase 1
- UTR:
-
Untranslated region
- mRNA:
-
Messenger RNA
- AMPK:
-
Activating 5′-adenosine monophosphate-activated protein kinase
- WST assay:
-
Water soluble tetrazolium salt assay
- TCGA:
-
The cancer genome atlas
- GEO:
-
Gene expression omnibus
- log2FC:
-
Log2 fold change
- RNA-seq:
-
RNA-sequencing
- OS:
-
Overall survival
- GO:
-
Gene ontology
- HIPLDA :
-
Hypoxia-inducible lipid droplet-associated
- EPHB3 :
-
Ephrin type-B receptor 3
- INHBA :
-
Inhibin subunit beta A
- SHH :
-
Sonic hedgehog
- RGS16 :
-
Regulator of G-protein signaling 16
- BiBiServ:
-
The Bielefeld bioinformatics server
- MFE:
-
Minimum free energy
- qPCR:
-
Quantitative real-time polymerase chain reaction
- N.C.:
-
Negative control
- PI3K/AKT/mTOR:
-
Phosphatidylinositol-4,5-biphosphate 3-kinase/AKT/mechanistic target of rapamycin
- DEG:
-
Differential expression gene
- NCBI:
-
National center for biotechnology information
- RPMI:
-
Rosewell Park Memorial Institute
- Anti-Anti:
-
Antibiotic-antimycotic
- FBS:
-
Fetal bovine serum
- GAPDH :
-
Glyceraldehyde-3-phosphate dehydrogenase
- SD :
-
Standard deviation
References
Dong, Y. et al. The development and benefits of metformin in various diseases. Front. Med. 17, 388–431 (2023).
Lv, Z. & Guo, Y. Metformin and its benefits for various diseases. Front. Endocrinol. (Lausanne) 11, 191 (2020).
Kaneto, H., Kimura, T., Obata, A., Shimoda, M. & Kaku, K. Multifaceted mechanisms of action of metformin which have been unraveled one after another in the long history. Int. J. Mol. Sci. 22, 2596 (2021).
Skuli, S. J. et al. Metformin and cancer, an ambiguanidous relationship. Pharmaceuticals (Basel) 15, 626 (2022).
Bray, F. et al. Global cancer statistics 2022: GLOBOCAN Estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 74, 229–263 (2024).
Tarhini, Z. et al. The effect of metformin on the survival of colorectal cancer patients with type 2 diabetes mellitus. Sci. Rep. 12, 12374 (2022).
Sugiura, K. et al. Metformin inhibits the development and metastasis of colorectal cancer. Med. Oncol. 39, 136 (2022).
Wang, Q. & Shi, M. Effect of metformin use on the risk and prognosis of colorectal cancer in diabetes mellitus: A meta-analysis. Anticancer Drugs 33, 191–199 (2022).
Sawicki, T. et al. A review of colorectal cancer in terms of epidemiology, risk factors, development, symptoms and diagnosis. Cancers (Basel) 13, 2025 (2021).
Taieb, J. et al. Deficient mismatch repair/microsatellite unstable colorectal cancer: Diagnosis, prognosis and treatment. Eur. J. Cancer 175, 136–157 (2022).
Hamdy, N. M. et al. Unraveling the ncRNA landscape that governs colorectal cancer: A roadmap to personalized therapeutics. Life Sci. 354, 122946 (2024).
Rizk, N. I. et al. Revealing the role of serum exosomal novel long non-coding RNA NAMPT-AS as a promising diagnostic/prognostic biomarker in colorectal cancer patients. Life Sci. 352, 122850 (2024).
Ng, C. W. et al. Metformin and colorectal cancer: A systematic review, meta-analysis and meta-regression. Int. J. Colorectal Dis. 35, 1501–1512 (2020).
Lee, J. W. et al. Metformin usage and the risk of colorectal cancer: A national cohort study. Int. J. Colorectal Dis. 36, 303–310 (2021).
Dukes, C. E. The classification of cancer of the rectum. J. Pathol. Bacteriol. 35, 323–332 (1932).
Puppa, G., Sonzogni, A., Colombari, R. & Pelosi, G. TNM staging system of colorectal carcinoma: A critical appraisal of challenging issues. Arch. Pathol. Lab. Med. 134, 837–852 (2010).
Geh, I. et al. Association of coloproctology of Great Britain & Ireland (ACPGBI): Guidelines for the management of cancer of the colon, rectum and anus (2017)—Anal cancer. Colorectal Dis. 19(Suppl 1), 82–97 (2017).
Amin, M. B. et al. The eighth edition AJCC cancer staging manual: Continuing to build a bridge from a population-based to a more “personalized” approach to cancer staging. CA Cancer J. Clin. 67, 93–99 (2017).
Argiles, G. et al. Localised colon cancer: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 31, 1291–1305 (2020).
Glynne-Jones, R. et al. Rectal cancer: ESMO Clinical practice guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 29, iv263 (2018).
Chen, K., Collins, G., Wang, H. & Toh, J. W. T. Pathological features and prognostication in colorectal cancer. Curr. Oncol. 28, 5356–5383 (2021).
Delattre, J. F. et al. A comprehensive overview of tumour deposits in colorectal cancer: Towards a next TNM classification. Cancer Treat. Rev. 103, 102325 (2022).
Akkoca, A. N. et al. TNM and modified dukes staging along with the demographic characteristics of patients with colorectal carcinoma. Int. J. Clin. Exp. Med. 7, 2828–2835 (2014).
Eldosoky, M. A. et al. Diagnostic significance of hsa-miR-21-5p, hsa-miR-192-5p, hsa-miR-155-5p, hsa-miR-199a-5p panel and ratios in hepatocellular carcinoma on top of liver cirrhosis in HCV-infected patients. Int. J. Mol. Sci. 24, 3157 (2023).
Macfarlane, L. A. & Murphy, P. R. MicroRNA: Biogenesis, function and role in cancer. Curr. Genomics 11, 537–561 (2010).
Hamdy, N. M. et al. Advancements in current one-size-fits-all therapies compared to future treatment innovations for better improved chemotherapeutic outcomes: A step-toward personalized medicine. Curr. Med. Res. Opin. 40, 1943–1961 (2024).
He, B. et al. miRNA-based biomarkers, therapies, and resistance in cancer. Int. J. Biol. Sci. 16, 2628–2647 (2020).
Hill, M. & Tran, N. miRNA interplay: Mechanisms and consequences in cancer. Dis. Model Mech. 14, dmm047662 (2021).
Segal, M. & Slack, F. J. Challenges identifying efficacious miRNA therapeutics for cancer. Expert. Opin. Drug. Discov. 15, 987–992 (2020).
Nangia-Makker, P. et al. Metformin: A potential therapeutic agent for recurrent colon cancer. PLoS One 9, e84369 (2014).
Zhang, Y., Chen, R., Deng, L., Shuai, Z. & Chen, M. The effect of metformin on the proliferation, apoptosis and CD133 mRNA expression of colon cancer stem cells by upregulation of miR 342–3p. Drug Des. Devel. Ther. 15, 4633–4647 (2021).
Hong, X. L. et al. Metformin abrogates FUSOBACTERIUM nucleatum-induced chemoresistance in colorectal cancer by inhibiting miR-361-5p/sonic hedgehog signaling-regulated stemness. Br. J. Cancer 128, 363–374 (2023).
Saini, N. & Yang, X. Metformin as an anti-cancer agent: Actions and mechanisms targeting cancer stem cells. Acta Biochim. Biophys. Sin. (Shanghai) 50, 133–143 (2018).
Saito, A. et al. Metformin changes the immune microenvironment of colorectal cancer in patients with type 2 diabetes mellitus. Cancer Sci. 111, 4012–4020 (2020).
Kamarudin, M. N. A., Sarker, M. M. R., Zhou, J. R. & Parhar, I. Metformin in colorectal cancer: Molecular mechanism, preclinical and clinical aspects. J. Exp. Clin. Cancer Res. 38, 491 (2019).
Lu, G. et al. The effects of metformin on autophagy. Biomed. Pharmacother. 137, 111286 (2021).
Niu, L. et al. Biological implications and clinical potential of metastasis-related miRNA in colorectal cancer. Mol. Ther. Nucleic Acids 23, 42–54 (2021).
Zhang, N., Hu, X., Du, Y. & Du, J. The role of miRNAs in colorectal cancer progression and chemoradiotherapy. Biomed. Pharmacother. 134, 111099 (2021).
Saberinia, A., Alinezhad, A., Jafari, F., Soltany, S. & Akhavan Sigari, R. Oncogenic miRNAs and target therapies in colorectal cancer. Clin. Chim. Acta 508, 77–91 (2020).
Pichler, M. et al. MiR-200a regulates epithelial to mesenchymal transition-related gene expression and determines prognosis in colorectal cancer patients. Br. J. Cancer 110, 1614–1621 (2014).
Mogavero, A. et al. Metformin transiently inhibits colorectal cancer cell proliferation as a result of either AMPK activation or increased ROS production. Sci. Rep. 7, 15992 (2017).
Wang, Y., Wu, Z. & Hu, L. The regulatory effects of metformin on the [SNAIL/miR-34]:[ZEB/miR-200] System in the epithelial-mesenchymal transition (EMT) for colorectal cancer (CRC). Eur. J. Pharmacol. 834, 45–53 (2018).
Wang, Z. et al. Identification of potential diagnostic and prognostic biomarkers for colorectal cancer based on GEO and TCGA databases. Front. Genet. 11, 602922 (2020).
Bu, T. et al. Regulation of sertoli cell function by planar cell polarity (PCP) protein Fjx1. Mol. Cell. Endocrinol. 571, 111936 (2023).
Huang, M. et al. Comprehensive analysis of the prognosis and immune effect of the oncogenic protein four jointed box 1. Front. Oncol. 13, 1170482 (2023).
Al-Greene, N. T. et al. Four jointed box 1 promotes angiogenesis and is associated with poor patient survival in colorectal carcinoma. PLoS One 8, e69660 (2013).
Zou, J. et al. Multi-omics analysis of the tumor microenvironment in liver metastasis of colorectal cancer identified FJX1 as a novel biomarker. Front. Genet. 13, 960954 (2022).
Liu, L. et al. FJX1 as a candidate diagnostic and prognostic serum biomarker for colorectal cancer. Clin. Transl. Oncol. 24, 1964–1974 (2022).
Cheng, T., Zhu, X., Lu, J. & Teng, X. MiR-532-3p suppresses cell proliferation, migration and invasion of colon adenocarcinoma via targeting FJX1. Pathol. Res. Pract. 232, 153835 (2022).
Dang, W. & Zhu, Z. MicroRNA-1249 targets four-jointed box kinase 1 and reduces cell proliferation, migration and invasion of colon adenocarcinoma. J. Gene. Med. 22, e3183 (2020).
Pan, Y., Zhang, Y., Hu, X. & Li, S. Construction of a novel cuproptosis-related ceRNA network-SNHG3/miR-1306-5p/PDHA1 and identification of SNHG3 as a prognostic biomarker in hepatocellular carcinoma. ACS Omega 8, 38690–38703 (2023).
Dang, Y. et al. lncRNA AC007207.2 promotes malignant properties of osteosarcoma via the miR-1306–5p/SIRT7 axis. Cancer Manag. Res. 13, 7277–7288 (2021).
Liu, W. et al. Identifying a novel IRF3/circUHRF1/miR-1306-5p/ARL4C axis in pancreatic ductal adenocarcinoma progression. Cell Cycle 21, 392–405 (2022).
Wang, W., Zhang, J., Fan, Y. & Zhang, L. MiR-1306-5p predicts favorable prognosis and inhibits proliferation, migration, and invasion of colorectal cancer cells via PI3K/AKT/mTOR pathway. Cell Cycle 21, 1491–1501 (2022).
Trotta, E. On the normalization of the minimum free energy of RNAs by sequence length. PLoS One 9, e113380 (2014).
Coronel-Hernandez, J. et al. Combination of metformin, sodium oxamate and doxorubicin induces apoptosis and autophagy in colorectal cancer cells via downregulation HIF-1alpha. Front. Oncol. 11, 594200 (2021).
Orang, A. et al. A functional screen with metformin identifies MicroRNAs that regulate metabolism in colorectal cancer cells. Sci. Rep. 12, 2889 (2022).
Sevim, C. et al. Investigation of the effects of metformin on the miR-21/PTEN/Akt pathway in HT-29 human colorectal adenocarcinoma cell and huvec co-culture. Cancer 72, 1 (2024).
Chen, Y. C. et al. Effect of vitamin D supplementation on primary dysmenorrhea: A systematic review and meta-analysis of randomized clinical trials. Nutrients 15, 2830 (2023).
Acknowledgements
We would like to thank Editage (www.editage.co.kr) for English language editing.
Funding
This work was supported in part by the 2024 BK21 FOUR Graduate School Innovation Support, funded by Pusan National University (PNU-Fellowship program), and by a grant from the National Research Foundation of Korea (MSIP; 2021R1A3B1077371) to HJK.
Author information
Authors and Affiliations
Contributions
Conceptualization: J.-m.K., H.J.S., W.R.K., H.-S.K.; Methodology: J.-m.K., H.J.S., W.R.K., E.G.P., H.-S.K.; Formal analysis: J.-m.K., H.J.S., D.H.L.; Investigation: J.-m.K., H.J.S., Y.J.L.; Writing–original draft: J.-m.K.; Writing–review & editing: J.-m.K., H.J.S., W.R.K., E.G.P., D.H.L., Y.J.L., H.-s.J., H.-Y.R., H.-S.K.; Resources: H.J.K., Y.H.C., S.-H.L.; Project administration: H.-S.K. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Kim, Jm., Shin, H.J., Kim, W.R. et al. Metformin modulates FJX1 via upregulation of Hsa-miR-1306-3p to suppress colon adenocarcinoma viability. Sci Rep 15, 6658 (2025). https://doi.org/10.1038/s41598-025-91022-y
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-91022-y
