
Abstract
Despite observational studies suggesting a link between chronic musculoskeletal pain (CMP) and increased risk of cognitive decline and dementia, the causal nature of this relationship remains uncertain due to potential confounding factors and reverse causality. We employed two-sample Mendelian Randomization (TSMR), bidirectional MR, mediation MR, drug-target MR, and colocalization analysis, along with gene set enrichment and protein–protein interaction (PPI) analyses. TSMR assessed the causal associations between CMP and the risk of dementia and its subtypes, including Alzheimer’s disease (AD), vascular dementia (VaD), Lewy body dementia (LBD), frontotemporal dementia (FTD), and Parkinson’s disease (PD). Bidirectional MR evaluated reverse causality, while mediation analyses identified potential mediators, focusing on neuroimaging and cognitive phenotypes. Drug-target MR investigated the role of the SLC39A8 gene, and colocalization analysis determined shared causal genetic variants. Gene set enrichment and PPI analyses elucidated the biological pathways implicated in the CMP-dementia relationship. Robust evidence established a causal relationship between chronic low back pain (LBP) and increased risk of PD, with knee osteoarthritis identified as a partial mediator, suggesting a pathway involving chronic inflammation. Bidirectional MR analysis revealed no evidence of reverse causality, further supporting the unidirectional causal link from LBP to PD. Colocalization analysis confirmed distinct genetic architectures for LBP and PD, while drug-target MR implicated the SLC39A8 gene as a potential mediator. Gene set enrichment and PPI analyses highlighted critical biological pathways, such as purine metabolism and glutamate receptor signaling. Suggestive evidence indicated potential causal links between limb pain and overall dementia, myalgia and VaD, as well as potential protective effects of Polymyalgia Rheumatica (PMR) against AD and rheumatism against PD. This study reveals a complex causal relationship between CMP and neurodegenerative diseases, particularly the robust link between LBP and PD. The findings underscore the need for further research to elucidate the underlying mechanisms and inform targeted prevention and treatment strategies.
Similar content being viewed by others


Introduction
Dementia, a syndrome characterized by the progressive decline in cognitive function, poses a significant challenge to global health and social care systems. Alzheimer’s disease (AD) is the most common cause of dementia, accounting for 60–80% of cases1. With the global prevalence of dementia projected to double every 20 years due to the aging population2,3, there is an urgent need to identify modifiable risk factors and develop effective prevention and treatment strategies.
Despite extensive research, the pathophysiological mechanisms underlying dementia are not fully elucidated, involving a complex interplay of genetic, environmental, and lifestyle factors4,5. While several well-established risk factors, such as advanced age, genetic predisposition, and vascular conditions, have been identified6, recent epidemiological studies have highlighted the potential role of chronic musculoskeletal pain (CMP) in the development of cognitive impairment and dementia7,8,9.
Multiple observational studies have reported associations between CMP and increased risk of cognitive decline, hippocampal atrophy, and dementia10,11. For instance, a study utilizing the UK Biobank cohort found that CMP was associated with significantly higher dementia risk, broader cognitive impairment, and greater hippocampal atrophy compared to those with single-site pain or pain-free individuals11. Furthermore, chronic low back pain (LBP) has been specifically linked to impairments in problem-solving, memory, and information processing12.
However, the evidence varies across different pain conditions, and the causal nature of these associations remains unclear. Observational studies are limited by confounding and reverse causality, challenging the establishment of a causal CMP-dementia link. Furthermore, the long-term impact of CMP on cognitive health remains elusive, as most studies have been limited by relatively short follow-up periods.
To address these limitations and provide robust evidence for a causal relationship, we employed Mendelian randomization (MR), a method that uses single nucleotide polymorphisms (SNPs) as instrumental variables (IVs) to mimic the randomization process of controlled trials. By leveraging genetic variants associated with CMP from large-scale genome-wide association studies (GWAS), MR minimizes confounding and reverse causality, enabling a more accurate estimation of the causal effect of CMP on cognitive decline and dementia risk. Clarifying this causal relationship may have significant implications for developing targeted prevention and treatment strategies, potentially informing clinical practice and public health policies to reduce the burden of dementia.
Methods
Study design and assumptions
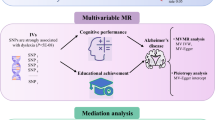
We utilized a comprehensive TSMR framework to investigate causal associations between CMP and dementia risk, including its subtypes. Additionally, we employed bidirectional MR, mediation MR, drug-target MR, colocalization analysis, and gene set enrichment and protein–protein interaction (PPI) analyses to thoroughly examine these relationships (Fig. 1).

The study design of this Mendelian randomization. Two-sample Mendelian randomization study design. This schematic illustrates the bidirectional MR framework assessing causal links between CMP and dementia subtypes, with arrows showing forward (CMP to dementia) and reverse (dementia to CMP) directions. MR assumptions ensure instrument validity: relevance, independence from confounders, and exclusion restriction. Mediation MR estimates direct (C') and indirect (A′ × B′) effects via mediators like hippocampus metrics (A′: CMP to mediator; B′: mediator to dementia). Additional analyses include colocalization using GWAS data and drug-target MR with cis-SNPs. The lower panel details 20 CMP conditions, hippocampus metrics, and dementia subtypes.
Bidirectional MR is particularly well-suited for studying CMP and dementia because it assesses both the causal impact of CMP on dementia and the reverse causality. This approach minimizes the risk of misleading results due to unclear causal direction, which is a common limitation in observational studies, and strengthens causal inferences by using separate sets of genetic instruments were used to assess the causal effects in each direction, ensuring the independence of the exposure and outcome.
To explore potential mediators, we performed Mediation MR analyses, focusing on neuroimaging and cognitive phenotypes, particularly hippocampus-related metrics. This focus is driven by recent literature suggesting that the hippocampus mediates the relationship between CMP and neurodegenerative diseases, a finding we aimed to validate in our analysis to elucidate the complex pathways through which CMP may influence dementia development.
We also conducted Drug-target MR analysis using SNPs associated with specific genes as IVs to investigate the role of potential genes in the CMP-dementia pathway and identify possible therapeutic targets. Recent studies have suggested that the SLC39A8 gene may mediate the relationship between CMP and neurodegenerative changes, prompting us to use drug-target MR to validate this potential mediation.
Colocalization analysis was conducted to evaluate whether CMP and dementia share causal genetic variants, thereby mitigating the potential confounding effects of genetic variants having different effects on each trait. Additionally, gene set enrichment analysis was performed to identify biological pathways and processes implicated in the CMP-dementia relationship, providing insights into the underlying molecular mechanisms. PPI analysis was also conducted to further elucidate the complex networks of proteins involved.
The validity of our MR analyses was ensured by rigorously evaluating three key assumptions: relevance (selecting genetic IVs strongly associated with CMP), independence (IVs independent of confounders influencing both CMP and dementia risk), and exclusion restriction (IVs influencing dementia solely through CMP).
All original GWAS included in our MR analyses were approved by the respective institutional review boards and ethical committees, with informed consent obtained from all participants. We strictly adhered to the Strengthening the Reporting of Observational Studies in Epidemiology Using Mendelian Randomization (STROBE-MR) guidelines to ensure the transparency, reproducibility, and overall quality of our MR study (Supplementary Table 1).
Data sources and populations
Genetic instrumental variables for CMP
This study utilized CMP data from the FinnGen study, specifically the Finnish R11 database (DF11, released June 24, 2024). DF11 is a comprehensive resource that includes 453,733 individuals (254,618 females, 199,115 males), providing substantial statistical power to investigate the associations between chronic pain conditions and dementia and its subtypes. We focused on 20 common chronic pain conditions, including LBP, joint pain, limb pain, fibromyalgia, and polyarthrosis, each systematically classified using International Classification of Diseases (ICD) codes.
The R11 database offers several advantages for our MR study. First, its large sample size and broad coverage of chronic pain conditions enable a thorough examination of the relationships between these exposures and the outcomes of interest. Second, the consistent use of ICD codes for classification minimizes the risk of misclassification bias and enhances the reliability of our findings. Finally, regular updates to the database ensure the quality and timeliness of the data, strengthening the validity of our results.
Outcome data for dementia with its subtypes
Outcome data for dementia and its subtypes were sourced from various studies, including the UK Biobank for overall dementia and vascular dementia (VaD), and specific GWAS meta-analyses for Lewy body dementia (LBD), frontotemporal dementia (FTD), AD, and Parkinson’s disease (PD).
For overall dementia and VaD, we extracted data from the UK Biobank using PheCodes 290.1 and 290.16, respectively. The UK Biobank provided 956 cases of overall dementia and 189 cases of VaD, with 410,833 controls13. The analysis was performed using SAIGE on 1,400 EHR-derived broad PheWAS codes for 20 million imputed variants in 400,000 white British individuals. To minimize potential confounding factors, phenotypes in the UK Biobank were weighted by age, age2, sex, age × sex, age2 × sex, principal components (PCs) 1–2014.
Data for LBD were obtained from a GWAS meta-analysis by Chia et al., which included 2591 LBD cases and 4027 neurologically healthy controls15. For FTD, we utilized data from a genome-wide association study by Van Deerlin et al., with a total sample size of 302416.
We got data from a genetic meta-analysis by Brian W. Kunkle et al. to investigate AD. This analysis identified new risk loci, highlighting key biological processes, and included data from 21,982 AD cases and 41,944 controls17. Lastly, for PD, we utilized data from the International PD Genomics Consortium, which included 33,674 PD cases and 449,056 controls18.
GWAS data sets for candidate mediators
The hippocampus may play a crucial mediating role in the relationship between CMP, particularly knee osteoarthritis (OA), and the increased risk of cognitive decline and dementia. Evidence suggests that individuals with knee OA experience accelerated brain aging, with the hippocampus being a primary contributor19. Moreover, the extent of accelerated brain aging predicts memory impairment and dementia risk in knee OA patients. To comprehensively investigate the potential mediating role of the hippocampus in the CMP-dementia relationship, we propose leveraging hippocampus-related GWAS data from multiple domains, including structural connectome, brain imaging phenotypes, white matter microstructure, and brain morphometry20,21,22,23. By integrating these diverse datasets, we aim to capture both structural and functional aspects of hippocampal involvement in the CMP-dementia pathway. Detailed data can be found in Supplementary Table 2.
Drug target MR is a novel extension of the MR paradigm that uses genetic variants associated with drug targets as IVs to infer the causal effects of these targets on disease outcomes .Previous research has shown that the SLC39A8 gene may mediate the relationship between knee OA and dementia19. To investigate whether this gene also mediates the association between other types of CMP and dementia, we plan to conduct a drug target MR study. We will select functional SNPs within or near the SLC39A8 gene as IVs. We will use GWAS data to assess the causal relationship between these SNPs and various types of CMP, such as chronic LBP and chronic neck pain, as well as the risk of dementia.
Statistical analysis
To ensure the robustness and reliability of our findings, we employed five complementary MR methods, each with its unique strengths in addressing different types of biases and violations of MR assumptions. First, the inverse variance weighted (IVW) method was used, as it provides efficient estimates under the assumption of no horizontal pleiotropy, making it ideal when all instruments are valid24. Next, to address the presence of outliers and potentially invalid instruments, we applied the contamination mixture (ConMix) method, which is robust to such violations of MR assumptions25. Additionally, we employed the robust adjusted profile score (RAPS) method to correct for weak instruments and account for both systematic and idiosyncratic pleiotropy, ensuring reliability in scenarios with pleiotropic effects26. To balance precision and robustness, particularly in heterogeneous datasets, the diversity-weighted IVW (DIVW) method was utilized, giving more weight to homogeneous causal effect estimates27. Finally, the constrained maximum likelihood (cML) method was applied to provide unbiased estimates even in the presence of systematic and idiosyncratic pleiotropy, further safeguarding against pleiotropic bias28.
The selection of genetic instruments was governed by stringent criteria to ensure the robustness of our analyses. SNPs were initially required to surpass a genome-wide significance threshold of P < 5 × 10−8 for the exposure of interest, ensuring that only the most strongly associated variants were included as instruments. Result categorization depended on the strength of the genetic instruments, with findings being considered robust causal evidence if at least 4 out of 5 MR methods yielded P < 0.05 using the SNPs meeting the P < 5 × 10−8 threshold. Such stringent criteria were necessary to ensure that the detected associations were not due to chance or weak instruments. Only these robust findings were subjected to a comprehensive series of follow-up analyses to deeply explore the underlying mechanisms.
In instances where fewer than 10 SNPs met the P < 5 × 10−8 threshold, the significance cut-off was relaxed to P < 1 × 10−5 to enhance the strength of the genetic instruments. This relaxation allowed for the inclusion of additional, albeit slightly weaker, instruments to maintain analytic power. If at this relaxed threshold, a minimum of 4 out of 5 MR methods still attained significance, the results were regarded as suggestive of a potential causal effect, though such findings were treated with greater caution and did not undergo the extensive follow-up analyses conducted for more robust associations.
Analyses lacking at least 10 SNPs at the 1 × 10−5 threshold were deemed underpowered and consequently excluded from the study. All retained results were reported as odds ratios (ORs) with accompanying 95% confidence intervals (CIs), providing a clear measure of the strength and precision of the observed associations. This approach balanced the need for strong instruments with the desire to explore multiple methodologies, ultimately enabling a thorough investigation of robust causal findings.
Bidirectional MR
To comprehensively assess the causal relationship between CMP and dementia, we employed bidirectional MR. This method allows us to evaluate the causal effects in both directions—specifically, from CMP to dementia and from dementia to CMP. First, we identified genetic IVs associated with CMP using GWAS data, which were then used to estimate the causal effect of CMP on dementia. Next, we selected a separate set of genetic IVs associated with dementia to evaluate its causal effect on CMP. Importantly, these dementia-associated IVs were chosen to be independent of those used in the CMP analysis. This methodological separation ensures that each causal pathway is evaluated independently, minimizing potential biases from reverse causation and allowing for a more precise assessment of the direction and magnitude of the causal effects.
Mediation MR
To explore how CMP influences dementia, including its subtypes, through potential mediating factors, we utilized Two-step MR. In the first step, we used genetic IVs associated with CMP to estimate its effect on potential mediators, yielding coefficient a. In the second step, we employed different genetic IVs, independent from those used initially, to estimate the mediator’s effect on dementia, resulting in coefficient b. The indirect effect of CMP on dementia through the mediator was calculated by multiplying these two coefficients (a × b). The total effect of CMP on dementia was then determined using a standard MR approach, and the direct effect was derived by subtracting the indirect effect from the total effect. This approach overcomes challenges such as confounding and measurement error present in traditional mediation analysis, ensuring more accurate and reliable estimates. Additionally, by using different IVs in each step, we avoid overlap and potential bias, which is crucial for the validity of our results29.
Colocalization analysis
To explore whether the genetic variants associated with CMP also contribute to the risk of dementia and its subtypes, we conducted colocalization analysis. This analysis is crucial for determining whether the observed associations between CMP and dementia are driven by the same causal variant within a given genomic region, thereby reducing the likelihood that the associations are confounded by linkage disequilibrium or nearby genetic variants. We performed this analysis using approximate Bayes factor colocalization, implemented in the ‘coloc’ R package with default settings. This method calculates posterior probabilities for five possible hypotheses: H0 (no causal variants for either trait), H1 (a causal variant only for CMP), H2 (a causal variant only for dementia), H3 (distinct causal variants for both traits), and H4 (a shared causal variant for both traits). In the colocalization analysis, the lead SNP was selected as the variant with the smallest combined P-value across the two GWAS datasets (calculated as the sum of the P-values for the SNP in each dataset), a standard approach in the coloc package to anchor the comparison of genetic signals between traits. Summary statistics from our GWAS on CMP and publicly available GWAS data on dementia and its subtypes were utilized. A posterior probability for H4 (PP.H4) of at least 50% was considered indicative of possible colocalization, while a PP.H4 of at least 80% suggested a high likelihood of colocalization, confirming the presence of shared causal variants between CMP and dementia.
Drug-target MR
To explore the potential mediating role of the SLC39A8 gene in the relationship between CMP and dementia, including its subtypes, we conducted a Drug Target MR analysis. This analysis is crucial for understanding whether genetic variations in SLC39A8 could causally influence dementia risk through their impact on CMP. The Drug Target MR approach utilizes SNPs associated with a specific gene as IVs to infer the causal impact of that gene on disease risk. For our analysis, we focused on SNPs related to the SLC39A8 gene, applying stringent criteria to ensure the specificity and relevance of the selected IVs. We set a linkage disequilibrium (LD) threshold of r2 < 0.001 to avoid confounding from closely linked variants, a gene region window of 10,000 kb to capture relevant regulatory regions, and a minor effect allele frequency (EAF) threshold of 0.01 to ensure sufficient statistical power. By applying these criteria, we identified the appropriate SLC39A8-associated SNPs to be used as IVs, which will allow us to assess whether variations in this gene contribute to the causal pathway linking CMP to dementia.
Gene set enrichment PPI analysis
To elucidate the biological processes and pathways associated with the CMP and dementia-related genes identified from our MR analysis, we performed gene set enrichment analysis using the Metascape platform (https://metascape.org/). The analysis integrated gene sets derived from the robust evidence and suggestive evidence across multiple knowledgebases, including Gene Ontology (GO), Kyoto Encyclopedia of Genes and Genomes (KEGG), and Reactome. The Benjamini–Hochberg procedure was applied for false discovery rate (FDR) correction, with enriched terms considered significant at a q-value threshold of 0.05. Visualization of the results was achieved through bar plots highlighting the top enriched terms ranked by their − log10(q-values), as well as network diagrams depicting the relationships and gene overlaps among the enriched terms. This approach aimed to uncover potential mechanistic links between CMP and neurodegenerative outcomes, informing therapeutic target identification.
To investigate the molecular mechanisms underlying CMP and dementia, we constructed PPI networks using data from STRING, BioGrid, OmniPath, and InWeb_IM databases. Only high-confidence physical interactions (score > 0.132) from STRING and all physical interactions from BioGrid were included. The resulting subnetwork consisted of proteins encoded by the genes of interest and their direct physical interaction partners. We applied the Molecular Complex Detection (MCODE) algorithm to identify highly interconnected network components, representing protein complexes or signaling pathways with coordinated functions. Networks containing between 3 and 500 proteins were analyzed to ensure biological relevance while avoiding overly fragmented or large networks. To interpret the biological functions of the identified network components, we performed pathway and process enrichment analysis on each MCODE component. The three most significant functional terms based on P-values were selected as the functional annotation for each component.
Sensitivity analysis
To comprehensively evaluate the reliability and robustness of the MR study results, we conducted a series of sensitivity analyses.
First, we performed a pleiotropy test to assess whether the selected genetic IVs exhibited pleiotropic effects, ensuring that the key assumptions of the MR method were met. Specifically, we employed the MR-Egger regression, focusing on the Egger intercept to detect directional pleiotropy. A non-zero Egger intercept indicates the presence of pleiotropy, suggesting that some genetic variants may influence the outcome through pathways other than the exposure, potentially biasing the causal estimates30. Additionally, we used the MR-PRESSO (Mendelian Randomization Pleiotropy RESidual Sum and Outlier) analysis, which helped identify and remove any outlier genetic variants that could introduce bias into the causal effect estimates due to pleiotropic effects31.
Next, we assessed the consistency of the IVs using two methods: the MR-Egger regression and the IVW approach. These heterogeneity tests were used to detect any significant differences among the causal effect estimates derived from the individual genetic variants32.
To evaluate the strength and validity of the IV-exposure association, we calculated the F-statistic, statistical power, and R2. The F-statistic, given by (beta /SE)2, where beta is the effect estimate and SE is its standard error, indicates strong IVs when values exceed 1033. R2 was calculated with the formula [2 × EAF × (1 − EAF) × beta2]/(SE2 × N), where N is the sample size. Higher R2 values suggest the instruments strongly explain the exposure variability, crucial for reliable causal estimates34. Statistical power was assessed using an online tool (https://sb452.shinyapps.io/power/), which evaluates the study’s ability to detect causal effects based on sample size, effect size, and significance level35.
Furthermore, we employed the MR Steiger test to verify the directionality of the causal relationship between the IVs, the exposure, and the outcome, ensuring that the assumed causal pathway was correctly specified. This test can detect whether the correlation between the SNPs and the outcome is greater than the correlation with the exposure. SNPs that fail this test may be unrelated to the exposure and should be filtered out before the analysis36.
To improve the specificity of IVs in our MR analysis, we systematically excluded genetic variants associated with potential confounders, as identified through the GWAS Catalog. We conducted an extensive literature review to assess the associations between body mass index, smoking behavior, blood pressure, lipid levels, and related biomarkers with dementia and cognitive decline. SNPs that exhibited associations with these confounders were classified as confounding SNPs and subsequently removed from the final IV set (Listed in Supplementary Table 3).
Finally, various sensitivity analysis plots, including Leave-one-out, scatter plots, and funnel plots, were generated to visually assess the influence of individual genetic variants on the results. These plots are crucial for evaluating the consistency of causal effect estimates and identifying potential publication bias.
All analyses were conducted using the TwoSampleMR (version 0.6.6), MendelianRandomization (version 0.10.0), coloc (version 5.2.3), and MRPRESSO (version 1.0) packages in R Software (version 4.4.1).
Results
Robust evidence for the association between CMP and dementia with its subtypes
Our study provides robust evidence of a potential causal association between chronic LBP and an increased risk of PD, as demonstrated consistently across five MR methods (Fig. 2). The IVW method revealed a significant association [IVW: OR 1.329; 95% CI 1.007–1.752; p = 0.044], which was further supported by the ConMix approach [ConMix: OR 1.566; 95% CI 1.190–2.062; p = 0.009]. Consistent findings were also observed using the RAPS method [RAPS: OR 1.423; 95% CI 1.110–1.825; p = 0.005], the DIVW method [DIVW: OR 1.339; 95% CI 1.015–1.766; p = 0.039], and the cML method [cML: OR 1.390; 95% CI 1.049–1.841; p = 0.022], detailed in Supplementary Table 4.

Robust evidence from bidirectional MR between CMP and dementia with its subtypes. Forest plot shows causal effects of CMP on dementia subtypes using five MR methods: IVW, ConMix, RAPS, DIVW, and cML. ORs with 95% CIs are plotted, with P-values indicating significance. Methods are color-coded, and significance thresholds and N SNPs are listed.
Comprehensive sensitivity analyses strongly support the integrity and reliability of these findings. The F-statistic of 39.924 indicates robust IVs, and with 100% power and an R2 of 0.016, our study was well-powered to detect causal effects. Heterogeneity tests showed no significant heterogeneity among instruments [MR Egger Q = 0.657, p = 0.092; IVW Q = 18.498, p = 0.101], and the MR-Egger intercept of -0.026 [p = 0.465] suggests no pleiotropy. MR-PRESSO detected no outliers [p = 0.090], and the Steiger test affirmed the directional validity. Supplementary sensitivity analyses, including leave-one-out, funnel plots, and scatter plots, further corroborated the primary findings, showing no major issues that would undermine the reliability of the observed causal relationship between chronic LBP and PD risk (Supplementary Fig. 1).
Suggestive evidence for the association between CMP and dementia with its subtypes
Our analysis found suggestive evidence of causal links between limb pain and overall dementia, myalgia and VaD, polymyalgia rheumatica (PMR) and AD, and rheumatism and PD. All five MR methods showed a positive association between limb pain and overall dementia [ORs ranging from 1.390 to 1.491; all p-values < 0.05], and between myalgia and VaD [ORs ranging from 2.067 to 3.475; all p-values < 0.05], with the ConMix method indicating the strongest link for the latter [ConMix: OR 3.475; 95% CI 1.378–8.764; p = 0.007]. Sensitivity analyses confirmed the reliability of these results, with no significant heterogeneity or pleiotropy detected.
In contrast, our analysis suggested a potential protective effect of PMR against AD, as four MR methods indicated an inverse association [ORs around 0.93; all p-values < 0.05]. Similarly, all four MR methods indicated an inverse association between rheumatism and PD [ORs ranging from 0.895 to 0.925; all p-values < 0.05], suggesting a potential protective effect. Sensitivity analyses revealed no major issues, supporting the robustness of these results. These findings are detailed in Supplementary Table 5, and Supplementary Figs. 2–5, including leave-one-out, scatter, and funnel plots, further demonstrate the stability and reliability of our findings.
Bidirectional MR analysis results
Our reverse MR analysis revealed no evidence of a causal effect of PD on LBP, nor for other pain-dementia pairs (Fig. 3). For PD and LBP, all MR methods failed to show a significant causal effect in the reverse direction [IVW: OR 0.987; 95% CI 0.960–1.014; p = 0.339; ConMix: OR 0.946; 95% CI 0.904–0.990; p = 0.069; RAPS: OR 0.981; 95% CI 0.952–1.010; p = 0.195; cML: OR 0.986; 95% CI 0.961–1.011; p = 0.273]. Similarly, reverse analyses for limb pain and overall dementia, myalgia and VaD, PMR and AD, and rheumatism and PD also failed to demonstrate significant causal relationships. Sensitivity analyses further confirmed these findings, with no significant pleiotropy [MR-Egger intercept = 0.006; p = 0.236] and no heterogeneity [IVW Q = 29.107; p = 0.111], reinforcing the validity of the initial causal direction observed in our primary analyses.

Suggestive evidence from bidirectional MR for CMP and dementia subtypes. Forest plot shows reverse causal effects of dementia subtypes on chronic musculoskeletal pain using five MR methods: IVW, ConMix, RAPS, DIVW, and cML. ORs with 95% CIs are plotted, with P-values indicating significance. Methods are color-coded, with significance thresholds and N SNPs listed.
Mediation MR analysis results
We examined multiple hippocampus-related GWAS datasets across various domains, including structural connectome, brain imaging phenotypes, white matter microstructure, and brain morphometry. However, no evidence was found to support hippocampal mediation of the observed causal effect.
Subsequent mediation analysis identified knee OA as a potential mediator in the relationship between chronic LBP and PD. Specifically, the analysis revealed a significant indirect effect through knee OA [Indirect effect: OR 1.128; 95% CI 1.007–1.264; p = 0.034], accounting for approximately 42% of the total effect. Although the direct effect was not significant [Direct effect: OR 1.178; 95% CI 0.873–1.589; p > 0.05], the total effect estimate remained positive [Total effect: OR 1.329; 95% CI 1.007–1.752; p = 0.044]. A detailed description is provided in Supplementary Table 6.
Colocalization analysis results
Colocalization analysis was conducted to examine the potential shared genetic architecture between chronic LBP and PD, utilizing GWAS data (Supplementary Table 7). The analysis, spanning 1,350 SNPs, revealed a high posterior probability for the presence of genetic variants associated with LBP in the tested region [PP.H1 = 0.96]. Conversely, the probabilities for variants associated solely with PD [PP.H2 = 1.56 × 10−14] or distinct variants affecting both traits [PP.H3 = 0.024] were negligible. Crucially, the posterior probability for shared causal variants between LBP and PD [PP.H4 = 0.011] was very low, indicating a lack of common genetic determinants in this region for the two conditions. This finding suggests that the observed association between LBP and PD in MR analyses is likely driven by a direct causal effect of LBP on PD, rather than being influenced by horizontal pleiotropy. The locuscomparer plot (Fig. 4) visualizes the association signals of SNPs in the region for both LBP and PD, with −log10(P) values on the y-axis and LD (r2) with rs2523656 indicated by color. rs2523656, selected as the lead SNP due to its smallest combined P-value across the two GWAS datasets, shows a stronger association with LBP than with PD. This visualization aligns with the colocalization analysis result, which indicates distinct genetic architectures for LBP and PD, as evidenced by the low posterior probability for shared causal variants [PP.H4 = 0.011]37.

Colocalization analysis of chronic LBP and Parkinson’s disease. Regional association plots showing SNP association signals for LBP (left) and PD (right) on chromosome 6. The y-axis represents − log10(P) values, with rs2523656 marked as the lead SNP. Colors indicate LD (r2) with rs2523656, ranging from high to low linkage disequilibrium. Plots reveal distinct genetic architectures, supporting the colocalization result of low shared causal variant probability.
Drug target MR analysis results
Under the relaxed thresholds required to identify relevant SNPs for the SLC39A8 gene, our Drug Target MR analysis suggested a potential association between this gene and the outcomes under investigation, with odds ratios around 2.0. The CIs were generally supportive of a positive effect, although some intervals approached non-significance. While these findings provide an initial indication of a possible role for SLC39A8, the results should be interpreted cautiously due to the expanded criteria used. This exploratory analysis highlights a potential direction for future research to further investigate the gene’s involvement in the relationship between CMP and dementia.
To further illustrate the genetic architecture related to SLC39A8, Fig. 5 presents a fine-mapped scatter plot showcasing the reference SNP cluster ID (rsID) distribution of relevant SNPs within the genomic region of interest. This plot highlights the specific locations and significance levels of these SNPs, providing a visual representation of their potential involvement in the CMP—dementia relationship.

Association analysis of SNPs in the SLC39A8 gene region. Scatter plot showing SNP association signals within the SLC39A8 gene region on chromosome 4. The y-axis displays − log10(P) values, with rs1270 marked as the lead SNP. Colors indicate LD (r2) with rs1270, ranging from high to low linkage disequilibrium. Axes represent genomic positions and association strengths.
Gene set enrichment PPI analysis
The gene set enrichment and PPI analyses reveal significant pathways and protein interactions that may play critical roles in CMP and dementia. Figure 6A highlights the purine nucleoside metabolic process (GO: 0042278) as the most significantly enriched pathway, indicating its critical role in essential cellular functions such as DNA/RNA synthesis, energy transfer, and cell signalling. The enrichment of developmental pathways, including “positive regulation of developmental growth” and “cell morphogenesis,” underscores the gene set’s involvement in cellular growth and differentiation. Additionally, the glutamate receptor signalling pathway is notably enriched, suggesting its importance in neurotransmission, which is crucial for neurological disorders.

Analysis of gene enrichment and protein–protein interactions. Multi-panel visualization of pathways linking pain and dementia. (A) Bar graph depicts enriched terms colored by P-values. (B) Network displays enriched terms clustered by similarity using cluster IDs. (C) Network highlights term significance with P-value-based coloring. (D) Overall PPI network (left) and a key MCODE component (right) reveal interactions involving HLA-DPB1 and COL6A5, indicating shared inflammatory mechanisms.
Figure 6B and C visualize a network of functionally related pathways, with nodes coloured by p-value significance. This network emphasizes key processes such as purine metabolism and glutamate signalling, which are relevant to CMP and dementia. These figures illustrate the interconnections between enriched terms, offering insights into potential molecular mechanisms underlying these conditions. Figure 6D shows the PPI network identified using the Molecular Complex Detection (MCODE) algorithm, highlighting significant clusters of protein interactions. Each node represents a protein involved in closely connected complexes, suggesting intricate interactions within biological processes such as “regulation of developmental growth” and “positive regulation of de-phosphorylation.” This network reveals the complex interplay among proteins related to CMP and dementia, identifying potential molecular complexes crucial for understanding disease pathology.
Discussion
Our study provides robust evidence for a causal association between chronic LBP and an increased risk of PD, consistently supported by multiple MR methods and corroborated by comprehensive sensitivity analyses. The unidirectional nature of this association, demonstrated by the lack of reverse causality in the bidirectional MR analysis, suggests that LBP may be a novel risk factor for PD, with important implications for understanding the etiology of this neurodegenerative disorder.
Epidemiological studies have consistently demonstrated an increased risk of PD in patients with OA. A recent meta-analysis found that individuals with OA had a 41% higher risk of developing PD compared to those without OA38. This association may be partly explained by the high prevalence of chronic LBP among patients with knee OA, which ranges from 54.6 to 58.1%39. The interaction between LBP and knee pain can exacerbate disabling effects, leading to impaired mobility and muscle strength, which are characteristic features of PD40,41. The increased risk of PD in patients with OA and LBP may be mediated by chronic inflammation and oxidative stress. Inflammatory mediators, such as IL-1β, IL-6, and TNF-α, play a crucial role in the progression of both LBP and knee OA, exacerbating local inflammation and potentially triggering neuroinflammation42,43. This sustained inflammatory state can damage cerebral vasculature and provoke glial-mediated responses, including increased expression of pro-inflammatory substances44,45. Peripheral inflammation may also activate microglia and astrocytes in the brain, particularly affecting midbrain dopaminergic neurons, which are highly sensitive to inflammatory stimuli. This cascade of events can lead to neurodegeneration and the development of the characteristic motor symptoms of PD38,46,47,48.Furthermore, degenerative joint disease, such as knee OA, can induce persistent oxidative stress, increasing the production of reactive oxygen species (ROS). Excessive ROS can damage neural cells, particularly impacting mitochondrial function. Chronic pain-induced oxidative stress and mitochondrial damage provide a potential molecular mechanism for PD’s neurodegenerative processes49,50,51,52. Addressing both LBP and knee OA could therefore offer a pathway to reducing PD risk.
Drug target MR analysis identified the SLC39A8 gene as a potential mediator of the LBP-PD relationship, providing a novel avenue for investigating the biological mechanisms underlying this association. SLC39A8 gene may mediate the progression from chronic LBP to PD through multiple mechanisms. This gene is involved in regulating inflammatory responses and ion balance, affecting neural system functions. Persistent pain stimuli may alter SLC39A8 expression, triggering cascade reactions including glial cell dysfunction, changes in synaptic plasticity, and structural alterations in specific brain regions. These factors collectively accelerate brain aging, increasing the risk of PD. Notably, the expression pattern of SLC39A8 in subcortical regions is closely related to brain aging, potentially explaining the characteristic pathological changes in PD19.
Gene set enrichment and PPI analyses identified pathways linking LBP and PD, including purine metabolism, glutamate receptor signaling, and developmental processes. Purine metabolism may increase oxidative stress, promoting dopaminergic neuron loss in PD51, while glutamate signaling disruptions may impair synaptic plasticity, contributing to cognitive decline in dementia53. The PPI network (Fig. 6D) highlights protein complexes like HLA-DPB1 and COL6A5, suggesting shared inflammatory mechanisms that may exacerbate neuro-inflammation in the brain. Our MR analysis supports a causal effect of musculoskeletal pain on PD (Fig. 2), with no reverse causality (Fig. 3), indicating a unidirectional relationship. However, these shared pathways likely mediate this effect by amplifying pain-induced neurodegeneration in the brain, enhancing dementia risk. Further studies are needed to clarify these mechanisms.
Beyond the robust evidence for the LBP-PD relationship, our study also provides suggestive evidence for associations between various chronic pain conditions and dementia subtypes. These findings underscore the broader implications of our research for understanding the complex connections between chronic pain and neurodegenerative disorders.
Chronic pain, particularly in the extremities, has been linked to altered cognitive function and an increased risk of dementia9,53,54. Neuroimaging studies have demonstrated that persistent peripheral neuropathic pain can lead to neuroanatomical and functional reorganization in the central nervous system, including reduced gray matter volume and disrupted connectivity in brain regions critical for cognition, such as the prefrontal cortex and hippocampus55,56. These changes may contribute to the observed cognitive decline and the development of dementia. The underlying mechanisms may involve neuro-inflammation, alterations in neurotransmitter systems, and impairment of synaptic plasticity, all of which affect both pain processing and cognitive function57.
The serotonergic and cholinergic systems have emerged as potential therapeutic targets for the management of chronic pain and cognitive disorders. The serotonergic system, which regulates pain, sleep–wake cycles, and emotional responses, is frequently disrupted in patients with chronic pain and may be associated with dementia58. Similarly, the cholinergic system, known for its effects on cognitive function, has been found to modulate pain59,60. Investigating these neurotransmitter systems may provide insights into the development of comprehensive therapeutic approaches addressing the comorbidity of chronic limb pain and cognitive decline.
Our finding that myalgia may contribute to an increased risk of VaD is consistent with the vascular hypothesis of cognitive decline, emphasizing the role of chronic inflammation in cerebrovascular disease61. The association between myalgia and statin use, a common preventive treatment for ischemic events, suggests a potential indirect link62,63,64. Furthermore, the overlapping impact of myalgia and COVID-19-related neurological complications highlights the need for further exploration into their combined effects on vascular health65,66.
Our reverse MR analysis (Fig. 3) supports the unidirectional nature of these associations, showing no robust or suggestive evidence of reverse causality, which aligns with literature suggesting chronic pain more commonly contributes to cognitive decline than vice versa9. However, some methods showed significant or near-significant results for certain comparisons, such as overall dementia with limb pain and AD with polymyalgia. These inconsistencies likely reflect method-specific sensitivities to biases like horizontal pleiotropy, with IVW being more prone to detecting associations compared to the more robust ConMix and cML methods. While these findings hint at potential bidirectional effects, possibly due to dementia-related changes in pain perception or shared inflammatory pathways67, their inconsistency suggests they are more likely statistical noise. Future studies with larger samples and stronger genetic instruments are needed to explore these potential effects.
The negative correlations observed between PMR and AD, as well as between rheumatic diseases and PD, are particularly intriguing. We hypothesize that the suggestive protective effect of PMR against AD may be linked to the anti-inflammatory effects of immunosuppressive therapies, such as corticosteroids, commonly used in PMR management. Chronic neuroinflammation is a key driver of AD pathology, promoting amyloid-beta accumulation and tau hyperphosphorylation67. By reducing systemic inflammation, these treatments might mitigate neuroinflammatory processes in the brain, potentially lowering AD risk. Indirect support for this hypothesis comes from studies on related conditions, such as Rheumatoid Arthritis (RA), where TNF-α inhibitors, also used in PMR, have been associated with a reduced AD risk68. However, direct evidence linking PMR or its treatments to AD risk reduction remains limited, and this potential mechanism requires further investigation. In contrast, the protective effect of rheumatism against PD is more strongly supported by evidence. Treatments for RA, such as disease-modifying antirheumatic drugs (DMARDs) and biologics like TNF-α inhibitors, likely reduce neuroinflammation—a key contributor to PD pathogenesis. Neuroinflammation, driven by cytokines like TNF-α, activates microglia and promotes dopaminergic neuron degeneration in PD69. By suppressing TNF-α and other pro-inflammatory pathways, RA treatments are associated with a reduced PD risk, as evidenced by MR studies and case–control studies showing that specific DMARDs, such as chloroquine/hydroxychloroquine, lower PD risk through modulation of inflammatory pathways70,71. Nevertheless, the relationship between rheumatic diseases and PD risk remains complex, with inconsistent findings across studies72,73. These discrepancies highlight the need for further research to elucidate the precise nature of these associations and the potential role of gender and other factors in modulating the relationship between rheumatic diseases and PD risk.
The link between rheumatic diseases, such as RA, and neurodegenerative conditions, including AD and PD, is rooted in chronic systemic inflammation and immune system dysfunction74,75. Persistent inflammation in RA can lead to cerebrovascular damage, reducing cerebral blood flow and increasing the risk of VaD76. The pathological similarities between RA and neurodegenerative diseases involve inflammatory markers like interleukin-6 and amyloid-related proteins, which contribute to neurodegeneration and cognitive decline67. In AD, for instance, IL-6 and TNF-α exacerbate amyloid-beta aggregation and tau pathology, while in PD, they contribute to microglial activation and dopaminergic neuron loss69. These shared inflammatory pathways suggest that systemic inflammation in rheumatic diseases may exacerbate neurodegenerative processes, highlighting the potential for anti-inflammatory therapies to mitigate such risks. However, the precise mechanisms linking rheumatic diseases and neurodegenerative disorders remain unclear, particularly for conditions like PMR and AD, where direct evidence of therapeutic impact is lacking. Further research is needed to fully understand the multiple pathways involved and to develop improved therapeutic strategies for both RA and neurodegenerative diseases.
In summary, our study highlights the complex associations between CMP conditions and dementia. The findings suggest causal relationships between chronic LBP and PD, as well as potential associations between limb pain, myalgia, polymyalgia, rheumatism, and various types of dementia. However, further research is needed to confirm these relationships and elucidate the underlying mechanisms. Given the high prevalence of both chronic pain and neurodegenerative disorders, our results have significant public health implications. Identifying chronic pain as a potentially modifiable risk factor for dementia underscores the importance of effective pain management strategies. Future research should focus on further elucidating the biological mechanisms, identifying novel therapeutic targets, and exploring lifestyle interventions to reduce the risk of both chronic pain and dementia. A better understanding of the link between chronic pain and neurodegenerative disorders can ultimately improve patient outcomes and reduce the overall burden of these conditions on individuals and society.
Conclusion
This study sheds new light on the complex relationship between CMP and dementia, providing robust evidence for a causal link and identifying potential biological pathways and mediators. These findings suggest that chronic pain may play a more significant role in the development of dementia than previously recognized, highlighting the importance of a holistic approach to understanding and managing these conditions.
Limitations
First, the primarily European study population may limit the generalizability of our findings; future research should include more diverse populations. Second, despite rigorous sensitivity analyses, unmeasured confounders like lifestyle factors or comorbidities may still affect causal estimates. Third, variability in dementia diagnosis across studies may impact comparability, highlighting the need for standardized diagnostic criteria. Fourth, reliance on self-reported pain measures could introduce bias; objective measures are recommended for future research. Fifth, while associations between chronic pain and dementia were observed, the exact biological mechanisms remain unclear, necessitating further experimental studies. Sixth, survival bias might underestimate associations due to early mortality in older populations. Lastly, our MR analysis focused on genetic variants linked to pain and dementia, which may not fully capture the genetic risks involved; comprehensive genetic risk scores or family-based studies are needed in future research.
Data availability
All data used in this study were obtained from IEU Open GWAS (https://gwas.mrcieu.ac.uk/).
References
Scheltens, P. et al. Alzheimer’s disease. Lancet 397, 1577–1590. https://doi.org/10.1016/S0140-6736(20)32205-4 (2021).
Gale, S. A., Acar, D. & Daffner, K. R. Dementia. Am. J. Med. 131, 1161–1169. https://doi.org/10.1016/j.amjmed.2018.01.022 (2018).
Hankey, G. J. Public health interventions for decreasing dementia risk. JAMA Neurol. 75, 11–12. https://doi.org/10.1001/jamaneurol.2017.3303 (2018).
Ferencz, B. & Gerritsen, L. Genetics and underlying pathology of dementia. Neuropsychol. Rev. 25, 113–124. https://doi.org/10.1007/s11065-014-9276-3 (2015).
Raz, L., Knoefel, J. & Bhaskar, K. The neuropathology and cerebrovascular mechanisms of dementia (2016). https://doi.org/10.1038/jcbfm.2015.164. Accessed August 23, 2024.
Ranson, J. M. et al. Modifiable risk factors for dementia and dementia risk profiling. A user manual for Brain Health Services—part 2 of 6. Alzheimer’s Res. Ther. 13, 169. https://doi.org/10.1186/s13195-021-00895-4 (2021).
Zhao, L. et al. Morphological and genetic decoding shows heterogeneous patterns of brain aging in chronic musculoskeletal pain. Nat. Mental Health 2, 435–449. https://doi.org/10.1038/s44220-024-00223-3 (2024).
Tian, J. et al. Association between chronic pain and risk of incident dementia: Findings from a prospective cohort. BMC Med. 21, 169. https://doi.org/10.1186/s12916-023-02875-x (2023).
Whitlock, E. L. et al. Association between persistent pain and memory decline and dementia in a longitudinal cohort of elders. JAMA Intern Med 177, 1146–1153. https://doi.org/10.1001/jamainternmed.2017.1622 (2017).
The potential contribution of chronic pain and common chronic pain conditions to subsequent cognitive decline, new onset cognitive impairment, and incident dementia: A systematic review and conceptual model for future research. J. Alzheimer’s Dis. 78:1177–1195. https://doi.org/10.3233/JAD-200960 (2020).
Elevated dementia risk, cognitive decline, and hippocampal atrophy in multisite chronic pain. Proc. Natl. Acad. Sci. U. S. A. 120(9), e2215192120–e2215192120 (2023). https://doi.org/10.1073/pnas.2215192120
Systematic review and meta- analysis on the association between chronic low back pain and cognitive function. Pain Pract 23, 399–408 (2022). https://doi.org/10.1111/papr.13194
Taliun, D. et al. Sequencing of 53,831 diverse genomes from the NHLBI TOPMed program. Nature 590, 290–299. https://doi.org/10.1038/s41586-021-03205-y (2021).
Sudlow, C. et al. UK biobank: An open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLos Med. 12, e1001779. https://doi.org/10.1371/journal.pmed.1001779 (2015).
Chia, R. et al. Genome sequencing analysis identifies new loci associated with Lewy body dementia and provides insights into its genetic architecture. Nat. Genet. 53, 294–303. https://doi.org/10.1038/s41588-021-00785-3 (2021).
Van Deerlin, V. M. et al. Common variants at 7p21 are associated with frontotemporal lobar degeneration with TDP-43 inclusions. Nat. Genet. 42, 234–239. https://doi.org/10.1038/ng.536 (2010).
Kunkle, B. W. et al. Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nat. Genet. 51, 414–430. https://doi.org/10.1038/s41588-019-0358-2 (2019).
Nalls, M. A. et al. Identification of novel risk loci, causal insights, and heritable risk for Parkinson’s disease: a meta-analysis of genome-wide association studies. Lancet Neurol 18, 1091–1102. https://doi.org/10.1016/S1474-4422(19)30320-5 (2019).
Zhao, L. et al. Morphological and genetic decoding shows heterogeneous patterns of brain aging in chronic musculoskeletal pain. Nat Mental Health 2, 435–449. https://doi.org/10.1038/s44220-024-00223-3 (2024).
Smith, S. M. et al. An expanded set of genome-wide association studies of brain imaging phenotypes in UK Biobank. Nat Neurosci 24, 737–745. https://doi.org/10.1038/s41593-021-00826-4 (2021).
Fürtjes, A. E. et al. General dimensions of human brain morphometry inferred from genome-wide association data. Hum Brain Mapp 44, 3311–3323. https://doi.org/10.1002/hbm.26283 (2023).
Zhao, B. et al. Common genetic variation influencing human white matter microstructure. Science 372, 3736. https://doi.org/10.1126/science.abf3736 (2021).
Wainberg, M. et al. Genetic architecture of the structural connectome. Nat. Commun. 15, 1962. https://doi.org/10.1038/s41467-024-46023-2 (2024).
Burgess S, Scott RA, Timpson NJ, Davey-Smith G, Thompson SG, EPIC-Interact Consortium. Using published data in Mendelian randomization: A blueprint for efficient identification of causal risk factors. Eur. J. Epidemiol. 30, 543–552 (2015) https://doi.org/10.1007/s10654-015-0011-z
Burgess, S., Foley, C. N., Allara, E., Staley, J. R. & Howson, J. M. M. A robust and efficient method for Mendelian randomization with hundreds of genetic variants. Nat. Commun. 11, 376. https://doi.org/10.1038/s41467-019-14156-4 (2020).
Yu, K. et al. Assessment of bidirectional relationships between brain imaging-derived phenotypes and stroke: a Mendelian randomization study. BMC Med. 21, 271. https://doi.org/10.1186/s12916-023-02982-9 (2023).
Ye T, Shao J, Kang H. Debiased inverse-variance weighted estimator in two-sample summary-data Mendelian randomization. Ann. Stat. 49 (2021). https://doi.org/10.1214/20-AOS2027
Yin, Q. & Zhu, L. Does co-localization analysis reinforce the results of Mendelian randomization?. Brain 147, e7–e8. https://doi.org/10.1093/brain/awad295 (2024).
Carter, A. R. et al. Mendelian randomisation for mediation analysis: Current methods and challenges for implementation. Eur. J. Epidemiol. 36, 465–478. https://doi.org/10.1007/s10654-021-00757-1 (2021).
Bowden, J., Davey Smith, G. & Burgess, S. Mendelian randomization with invalid instruments: Effect estimation and bias detection through Egger regression. Int. J. Epidemiol. 44, 512–525. https://doi.org/10.1093/ije/dyv080 (2015).
Verbanck, M., Chen, C.-Y., Neale, B. & Do, R. Detection of widespread horizontal pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and diseases. Nat. Genet. 50, 693–698. https://doi.org/10.1038/s41588-018-0099-7 (2018).
Burgess, S., Butterworth, A. & Thompson, S. G. Mendelian randomization analysis with multiple genetic variants using summarized data. Genet. Epidemiol. 37, 658–665. https://doi.org/10.1002/gepi.21758 (2013).
Bowden, J. et al. Improving the accuracy of two-sample summary-data Mendelian randomization: Moving beyond the NOME assumption. Int. J. Epidemiol. 48, 728–742. https://doi.org/10.1093/ije/dyy258 (2019).
Burgess, S., Thompson, S. G. & CRP CHD Genetics Collaboration. Avoiding bias from weak instruments in Mendelian randomization studies. Int. J. Epidemiol. 40, 755–764 (2011). https://doi.org/10.1093/ije/dyr036
Pierce, B. L., Ahsan, H. & Vanderweele, T. J. Power and instrument strength requirements for Mendelian randomization studies using multiple genetic variants. Int. J. Epidemiol. 40, 740–752. https://doi.org/10.1093/ije/dyq151 (2011).
Hemani, G., Tilling, K. & Davey, S. G. Orienting the causal relationship between imprecisely measured traits using GWAS summary data. PLoS Genet. 13, e1007081. https://doi.org/10.1371/journal.pgen.1007081 (2017).
Liu, B., Gloudemans, M. J., Rao, A. S., Ingelsson, E. & Montgomery, S. B. Abundant associations with gene expression complicate GWAS follow-up. Nat. Genet. 51, 768–769. https://doi.org/10.1038/s41588-019-0404-0 (2019).
Heng, H. et al. WDR43 is a potential diagnostic biomarker and therapeutic target for osteoarthritis complicated with Parkinson’s disease. Front. Cell. Neurosci. 16, 1013745. https://doi.org/10.3389/fncel.2022.1013745 (2022).
Iijima, H., Shimoura, K., Aoyama, T. & Takahashi, M. Low Back Pain as a Risk Factor for Recurrent Falls in People With Knee Osteoarthritis. Arthritis Care Res. 73, 328–335. https://doi.org/10.1002/acr.24136 (2021).
Iijima, H., Suzuki, Y., Aoyama, T. & Takahashi, M. Interaction between low back pain and knee pain contributes to disability level in individuals with knee osteoarthritis: A cross-sectional study. Osteoarthr. Cartil. 26, 1319–1325. https://doi.org/10.1016/j.joca.2018.06.012 (2018).
Teder-Braschinsky, A., Märtson, A., Rosenthal, M. & Taba, P. Parkinson’s disease and symptomatic osteoarthritis are independent risk factors of falls in the elderly. Clin. Med. Insights Arthritis Musculoskelet. Disord. 12, 1179544119884936. https://doi.org/10.1177/1179544119884936 (2019).
Risbud, M. V. & Shapiro, I. M. Role of cytokines in intervertebral disc degeneration: Pain and disc content. Nat. Rev. Rheumatol. 10, 44–56. https://doi.org/10.1038/nrrheum.2013.160 (2014).
Du, X. et al. Research progress on the pathogenesis of knee osteoarthritis. Orthop. Surg. 15, 2213–2224. https://doi.org/10.1111/os.13809 (2023).
Wang, Y. et al. The role of IL-1β and TNF-α in intervertebral disc degeneration. Biomed. Pharmacother. 131, 110660. https://doi.org/10.1016/j.biopha.2020.110660 (2020).
Cebrián, C., Loike, J. D. & Sulzer, D. Neuroinflammation in Parkinson’s disease animal models: A cell stress response or a step in neurodegeneration? In Behavioral Neurobiology of Huntington’s Disease and Parkinson’s Disease (eds Nguyen, H. H. P. & Cenci, M. A.) 237–270 (Springer, Berlin, 2015). https://doi.org/10.1007/7854_2014_356.
Wang, J., Song, Y., Chen, Z. & Leng, S. X. Connection between systemic inflammation and neuroinflammation underlies neuroprotective mechanism of several phytochemicals in neurodegenerative diseases. Oxid. Med. Cell. Longev. 2018, 1972714. https://doi.org/10.1155/2018/1972714 (2018).
Peripheral inflammation in neurodegeneration. J. Mol. Med. https://doi.org/10.1007/s00109-013-1026-0. Accessed August 8, 2024.
Muñoz-Delgado, L. et al. Peripheral inflammatory immune response differs among sporadic and familial Parkinson’s disease. NPJ Park. Dis. 9, 12. https://doi.org/10.1038/s41531-023-00457-5 (2023).
Han, S. Osteoarthritis year in review 2022: Biology. Osteoarthr. Cartil. 30, 1575–1582. https://doi.org/10.1016/j.joca.2022.09.003 (2022).
Wang, Y. et al. Oxidative stress in intervertebral disc degeneration: Molecular mechanisms, pathogenesis and treatment. Cell Prolif. 56, e13448. https://doi.org/10.1111/cpr.13448 (2023).
Tresse, E. et al. Mitochondrial DNA damage triggers spread of Parkinson’s disease-like pathology. Mol. Psychiatry 28, 4902–4914. https://doi.org/10.1038/s41380-023-02251-4 (2023).
Garcia-Esparcia, P. et al. Mitochondrial activity in the frontal cortex area 8 and angular gyrus in Parkinson’s disease and Parkinson’s disease with dementia. Brain Pathol. 28, 43–57. https://doi.org/10.1111/bpa.12474 (2018).
Kuner, R. & Kuner, T. Cellular circuits in the brain and their modulation in acute and chronic pain. Physiol. Rev. 101, 213–258. https://doi.org/10.1152/physrev.00040.2019 (2021).
Li, G. et al. Association between pain interference and motoric cognitive risk syndrome in older adults: A population-based cohort study. BMC Geriatr. 24, 437. https://doi.org/10.1186/s12877-024-04974-7 (2024).
Wang, Z., Sun, Z. & Zheng, H. Association between chronic pain and dementia: A systematic review and meta-analysis. Eur. J. Ageing 21, 17. https://doi.org/10.1007/s10433-024-00812-2 (2024).
De Ridder, D., Adhia, D. & Vanneste, S. The anatomy of pain and suffering in the brain and its clinical implications. Neurosci. Biobehav. Rev. 130, 125–146. https://doi.org/10.1016/j.neubiorev.2021.08.013 (2021).
Ong, W.-Y., Stohler, C. S. & Herr, D. R. Role of the prefrontal cortex in pain processing. Mol. Neurobiol. 56, 1137–1166. https://doi.org/10.1007/s12035-018-1130-9 (2019).
McCarberg, B. & Peppin, J. Pain pathways and nervous system plasticity: Learning and memory in pain. Pain Med. 20, 2421–2437. https://doi.org/10.1093/pm/pnz017 (2019).
Naser, P. V. & Kuner, R. Molecular, cellular and circuit basis of cholinergic modulation of pain. Neuroscience 387, 135–148. https://doi.org/10.1016/j.neuroscience.2017.08.049 (2018).
Jiang, Y.-Y. et al. Neural pathways in medial septal cholinergic modulation of chronic pain: distinct contribution of the anterior cingulate cortex and ventral hippocampus. Pain 159, 1550–1561. https://doi.org/10.1097/j.pain.0000000000001240 (2018).
Inoue, Y., Shue, F., Bu, G. & Kanekiyo, T. Pathophysiology and probable etiology of cerebral small vessel disease in vascular dementia and Alzheimer’s disease. Mol. Neurodegener. 18, 46. https://doi.org/10.1186/s13024-023-00640-5 (2023).
Thompson, P. D., Panza, G., Zaleski, A. & Taylor, B. Statin-associated side effects. J. Am. Coll. Cardiol. 67, 2395–2410. https://doi.org/10.1016/j.jacc.2016.02.071 (2016).
Tramacere, I. et al. Comparison of statins for secondary prevention in patients with ischemic stroke or transient ischemic attack: A systematic review and network meta-analysis. BMC Med. 17, 67. https://doi.org/10.1186/s12916-019-1298-5 (2019).
Collins, R. et al. Interpretation of the evidence for the efficacy and safety of statin therapy. Lancet 388, 2532–2561. https://doi.org/10.1016/S0140-6736(16)31357-5 (2016).
He, Y., Bai, X., Zhu, T., Huang, J. & Zhang, H. What can the neurological manifestations of COVID-19 tell us: A meta-analysis. J. Transl. Med. 19, 363. https://doi.org/10.1186/s12967-021-03039-2 (2021).
Jasti, M., Nalleballe, K., Dandu, V. & Onteddu, S. A review of pathophysiology and neuropsychiatric manifestations of COVID-19. J. Neurol. 268, 2007–2012. https://doi.org/10.1007/s00415-020-09950-w (2021).
Su, F., Bai, F. & Zhang, Z. Inflammatory cytokines and Alzheimer’s disease: A review from the perspective of genetic polymorphisms. Neurosci. Bull. 32, 469–480. https://doi.org/10.1007/s12264-016-0055-4 (2016).
Zhou, M., Xu, R., Kaelber, D. C. & Gurney, M. E. Tumor Necrosis Factor (TNF) blocking agents are associated with lower risk for Alzheimer’s disease in patients with rheumatoid arthritis and psoriasis. PLoS ONE 15, e0229819. https://doi.org/10.1371/journal.pone.0229819 (2020).
Cebrián, C., Loike, J. D. & Sulzer, D. Neuroinflammation in Parkinson’s disease animal models: A cell stress response or a step in neurodegeneration? In Behavioral Neurobiology of Huntington’s Disease and Parkinson’s Disease (eds Nguyen, H. H. P. & Cenci, M. A.) 237–270 (Springer, Berlin, 2015).
Guo, X., Chong, L., Zhang, X. & Li, R. Immunosuppressants contribute to a reduced risk of Parkinson’s disease in rheumatoid arthritis. Int. J. Epidemiol. 51, 1328–1338. https://doi.org/10.1093/ije/dyac085 (2022).
Paakinaho, A. et al. Disease-modifying antirheumatic drugs and risk of Parkinson disease: Nested case-control study of people with rheumatoid arthritis. Neurology 98, e1273–e1281. https://doi.org/10.1212/WNL.0000000000013303 (2022).
He, L., Zhao, H., Wang, F. & Guo, X. Inflammatory rheumatic diseases and the risk of Parkinson’s disease: A systematic review and meta-analysis. Front. Neurol. 13, 999820. https://doi.org/10.3389/fneur.2022.999820 (2022).
Kang, J. et al. Rheumatoid arthritis and risk of Parkinson disease in Korea. JAMA Neurol. 80, 634–641. https://doi.org/10.1001/jamaneurol.2023.0932 (2023).
Vassilaki, M. et al. Rheumatoid arthritis, cognitive impairment, and neuroimaging biomarkers: Results from the mayo clinic study of aging. J. Alzheimers Dis. 89, 943–954. https://doi.org/10.3233/JAD-220368 (2022).
Mohapatra, L., Mishra, D., Shiomurti Tripathi, A. & Kumar, P. S. Immunosenescence as a convergence pathway in neurodegeneration. Int. Immunopharmacol. 121, 110521. https://doi.org/10.1016/j.intimp.2023.110521 (2023).
Trzeciak, P., Herbet, M. & Dudka, J. Common factors of Alzheimer’s disease and rheumatoid arthritis-pathomechanism and treatment. Molecules 26, 6038. https://doi.org/10.3390/molecules26196038 (2021).
Acknowledgements
We gratefully acknowledge the participants and investigators involved in the original genome-wide association studies and the developers of the GWAS Catalog (https://www.ebi.ac.uk/gwas/home) and IEU OpenGWAS project (https://gwas.mrcieu.ac.uk/). This study was supported by Capital’s Funds for Health Improvement and Research (No.2020-2-2231). The funder played no role in study design, data collection, analysis and interpretation of data, or the writing of this manuscript. Figure 1 was created with BioRender.com.
Author information
Authors and Affiliations
Contributions
K.D., SM.L. and YL.Z took on the task of conceptualization. They worked together to develop the framework and theoretical underpinnings of the study. K.D. and YL.Z also assumed the responsibility of drafting the original draft. CY.Z and A.L. were in charge of reviewing and editing the manuscript, refining the text to ensure clarity and coherence. All authors participated in the acquisition, analysis, or interpretation of data. Furthermore, all authors critically revised the manuscript for important intellectual content. SM.L. and R.G. were responsible for the overall supervision of the research, providing guidance and support throughout the entire research process. The final version of the manuscript was approved by all the authors before publication.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Du, K., Zuo, YL., Zhang, ZM. et al. The role of the hippocampus and SLC39A8 in chronic musculoskeletal pain-induced dementia: a Mendelian randomization study. Sci Rep 15, 13211 (2025). https://doi.org/10.1038/s41598-025-97428-y
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-97428-y
