
Abstract
With human guidance, computers now use machine learning (ML) in artificial intelligence (AI) to learn from data, detect trends, and make predictions. Software can adapt and improve with new information. Imaging scans leverage pattern recognition to predict outcomes, diagnose disorders, and suggest treatments. Tuberculosis (TB) remains the most common bacterial disease affecting humans. The World Health Organisation reported that in 2022, 1.3 million people died from tuberculosis, with the death rate potentially reaching 66% if proper treatment isn’t provided. We trained ML-supervised algorithms like XG Boost, Logistic Regression, Random Forest Classifier, Ad- aBoost, and Support Vector Machine to help classify TB patients from large RNA-sequence count data. Such algorithms provided prediction accuracies of 0.963, 0.739, 0.773, 0.866, and 0.866 sequentially. This article highlights feature importance techniques using the ML model, XGBoost, with the highest prediction accuracy of 0.963, identifying significant genes in TB RNA sequence count data. Using key machine learning features, we here identified 20 pathways, 24 gene ontologies, 20 hub genes, and 22 drugs. Next, we applied advanced computational techniques, including pathway analysis, GO, hub-protein and protein–protein interactions (PPI), transcriptomic and miRNA interactions, and drug-protein interactions, to help analyze 100 highly expressed genes.
Similar content being viewed by others
Introduction
Machine Learning (ML) is being more widely utilized across various sectors including military, cybersecurity, healthcare, among many more. Additionally, ML algorithms are employed to address intricate and emerging challenges in biomedical research, encompassing tasks such as text mining, drug discovery, single-cell RNA sequencing, as well as early diagnosis and prognosis of diseases1. ML techniques are frequently used for analyzing and interpreting a variety of data types, including multi-omics, imaging, clinical records, medication details, and disease progression. Biomedical data science encompasses a variety of data formats, including genome sequences, omics data, medical imaging, and clinical records2. As time progresses, machine learning methods are increasingly gaining favor within the medical sector for their efficacy in aiding decision-making processes3. The study of RNA-sequence data reveals the effectiveness of machine learning algorithms in discovering splice variants within RNA-sequence data by utilizing a variety of ML methods for different detection objectives and to clarify sequence correlations4.
For thousands of years, tuberculosis (TB) has been a common infectious illness that has caused high rates of death in several vulnerable populations. Despite advances in medical science, this tendency continues into the modern period. According to the World Health Organization’s most current TB estimated 10.6 million new cases of TB and 1.3 million fatalities from the disease in 2022 (World Health - organization, 2023). Up to the advent of SARS-CoV-2, tuberculosis (TB) was the leading infectious cause of mortality in humans. Approximately 1.6 million people died from TB in 2021, mostly in low- and middle-income countries5. Early identification, drug resistance screening, and thorough treatment with short-course regimens can effectively cure tuberculosis6. Automated techniques for tuberculosis diagnosis and classification have improved disease identification precision, enabling healthcare professionals to make more informed decisions.
Additionally, innovators in advanced biotechnology have created a number of bioinformatics tools that have facilitated the investigation of illness studies. Many researchers have employed machine learning (ML) algorithms to predict chronic and viral diseases7,8 including the prevalence of tuberculosis based on associated risk factors9,10. Within these fields of artificial intelligence (AI), machine learning (ML) builds mathematical models using training data to make precise diagnoses and choices without the need for explicit manual programming for specific tasks.
We therefore used an XGBoost-based feature importance approach to help identify the precise highly expressed genes from RNA sequencing count data in this TB patient research. This approach was used instead of only relying on the values of Adjusted P-values and Log Fold Changes to identify important genes.
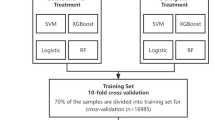
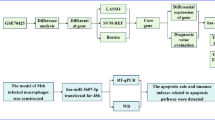
We then developed a classification pipeline for tuberculosis diagnosis using RNA-Seq microarray data, enabling rapid and reliable analysis of large transcriptome datasets for meaningful conclusions. Before proceeding with further analysis, the initial step involves assessing the quality of the unprocessed raw sequence data. Machine learning algorithms were used to identify significant genes, followed by bioinformatics analyses such as pathway enrichment, gene ontology, and drug prediction. The biological processes and roles associated with the discovered genes, as well as possible therapeutic possibilities for TB treatment, are clarified by these investigations. Our work’s visual depiction is seen in Fig. 1. Future efforts will use machine learning algorithms to quickly identify key genes and provide an instant prognosis estimate for TB patients based on gene sequence data. This will make it easier for medical personnel to handle the problem. In conclusion, these findings will be extremely helpful in controlling or reducing the hazards related to tuberculosis patients, providing invaluable assistance to researchers and medication makers.

The suggested approach and the workflow.
Methods and materials
Data sets and pre-processing
GEO, located at https://www.ncbi.nlm.nih.gov/geo/, is a global public archive for genetic and genomic data that enables access to large datasets created by scientific communities, such as data from microarrays, next-generation sequencing, and other analytical methods. GEO makes this essential information more easily shared and accessible to researchers around the world11. We retrieve our dataset from GEO with accession ID GSE10314712. Furthermore, we acquired this dataset by analyzing raw count data through “GREIN”, an alternative tool facilitating the exploration of publicly available gene expression data13.
The dataset was assembled utilizing the “Illumina HiSeq 2000 (Homo sapiens)” platforms. 6,363 healthy adolescents aged between 12 and 18 years, were enrolled in the study and followed for 24 months or longer. Six months was the period between the collection of blood. Among them, 41 were diagnosed with active TB, while 104 served as asymptomatic control12. The period between blood collection and the diagnosis of active TB referred to as “time to diagnosis”, varied from 1 to 894 days. Nevertheless, given that it is count data, we obtained 28,091 genes for each individual and the dataset has dimensions of 28,089 rows and 1,608 columns. The count data are presented in tabular form, illustrating the number of sequence fragments associated with each gene across every sample14. The overview of the dataset is provided in Table 1.
After acquiring the data, we conducted pre-processing by utilizing the functionalities provided by the “pandas” library. The “pandas” library offers built-in, user-friendly functions for handling routine data manipulations and conducting analyses on datasets like this one. Its objective is to serve as the fundamental framework for Python’s statistical computing endeavors in the foreseeable future15.
Machine learning algorithms
This section discusses the experimental use of numerous ML algorithms for the prediction of tuberculosis.
Random forest (RF) classifier
Random Forest, a machine learning algorithm developed by Breiman and Cutler, combines many decision trees for both regression and classification problems, offering versatility and user-friendliness16. It is a classifier that uses multiple decision trees on a dataset, based on the majority votes, to improve predictive accuracy and prevent overfitting, resulting in higher accuracy16.
The Random Forest algorithm is a low-training, high-accuracy prediction method that performs efficiently on large datasets. It uses a two-phase process, combining N decision trees to create a random forest17. The process involves choosing K data points at random, creating decision trees linked to these points, selecting N decision trees, and performing the first two steps. The predictions from each tree are then allocated to the group with the most votes.
AdaBoost classifier
AdaBoost is an ensemble boosting classifier, proposed by Freund and Schapire since 1996. It combines multiple weak classifiers to increase accuracy. AdaBoost is an iterative method that sets classifier weights and trains data samples for accurate predictions of unusual observations18. AdaBoost is a classifier that aims to reduce training errors in each iteration by increasing the weight of incorrectly classified instances using a Decision Tree method. It is recommended to train the classifier interactively with a range of weighted training samples. However, this technique cannot be parallelized as each predictor needs to be trained after the preceding one. The AdaBoost algorithm involves assigning each observation the same weight, creating a model using a subset of data, computing errors by contrasting predicted and actual values, assigning higher weights to mistaken data points, and repeating these steps until the error function remains unchanged or the maximum number of estimators is achieved19.
Logistic regression classifier
Logistic regression is a popular machine learning algorithm used to predict a categorical dependent variable using independent variables. It predicts probabilistic values between 0 and 1, instead of providing exact values like 0 and 120.
Procedure for logistic regression
The procedure for implementing Logistic Regression in Python is the same as it was in earlier regression topics. The steps are as follows:
-
1.
Pre-processing of the data
-
2.
Assessing the training set for logistic regression fit
-
3.
Projecting the exam outcome
-
4.
Test the correctness of the outcome (matrix creation)
-
5.
Showing the test set outcome visually.
Logistic regression equation
From the linear regression equation, one may get the logistic regression equation. The following lists the mathematical procedures to obtain equations for logistic regression.
Here the straight-line equation may be expressed as follows:
In Logistic Regression y can be between 0 and 1 only, so for this let’s divide the above equation by (1−y):
But we need a range between − [infinity] to +[infinity], then take the logarithm of the equation it will become:
This classification algorithm is used to classify linear logarithms. However, it can be defined in this way:
XGBoost classifier
XGBoost is a gradient-boosting machine learning algorithm, renowned for its computational efficiency, feature importance analysis, and handling of missing values, used in regression, classification, and ranking tasks. Dr. Chen from Washington University introduces XGBoost, an ensemble learning model based on Boosting. It constructs multiple CART trees based on splitting nodes, reduces variance, and optimizes CPU multi-threads for improved accuracy. It has applications in artificial intelligence, data analysis, statistics, and mining21.
Support vector machine
Support Vector Machine (SVM) is a popular Supervised Learning algorithm used for classification and regression problems in Machine Learning. It aims to create the best decision boundary, a hyperplane, for separating n-dimensional space into classes. Vapnik and colleagues developed SVMs in the 1990s and presented their findings in 199522.
Support vector regression
Support vector regression (SVR) is a continuous regression method that extends linear SVMs, primarily used for time series prediction, by finding a hyperplane with the maximum margin between data points, unlike linear regression which requires specifying relationships between variables.
Differential gene expression
DGE analysis is a crucial bioinformatics technique for understanding biological processes by comparing gene expression levels between conditions or treatments. Differential expression analysis involves analyzing normalized read count data to identify quantitative changes in expression levels between experimental groups through statistical analysis. Differential expression testing aims to identify genes expressed at different levels between conditions, providing biological insights into the processes impacted by the conditions of interest23.
Feature importance
“Feature importance” is a method that assigns a score to each input feature in a model, with higher scores indicating a greater influence of the characteristic on the model used to forecast a variable24.
Understanding the importance of a feature can improve a model’s clarity and effectiveness by providing insights into the relationships between input and output variables. This can help remove unnecessary or redundant features and identify important qualities that may have gone unnoticed, opening up new research possibilities. Methods like Gradient Boosting and XGBoost feature importance can be used to determine feature importance, offering diverse perspectives on feature significance and identifying the most influential ones in a model’s predictions25.
Molecular pathway enrichment and gene ontology analysis
A framework and a collection of ideas for characterizing the roles of gene products from all species are provided by the Gene Ontology (GO)26. The Gene Ontology (GO) knowledge base is a significant resource for understanding gene functions, enabling computational analysis of large-scale molecular biology and genetics studies in biomedical research, making it both machine- and human-readable.
A significant introspective effort to categorize biological viewpoints, such as biological mechanisms or chromosomal regions linked to a range of related disorders, is known as a functional enrichment evaluation. A comprehensive web-based tool for gene set enrichment, EnrichR was used to study pathway enrichment and gene ontology, which includes biological processes, cellular components, and molecular functions. several KEGG27, Reactome28, WikiPathways29, Elsevier, and BioCarta30 were utilized to characterise the major pathways, with an acceptable adjusted P-value of less than 0.05.
Protein–protein interactions (PPIs) analysis
Protein–protein interactions (PPIs) are physical connections formed through biochemical or electrostatic processes between proteins, which are crucial for various biological processes like cell-to-cell contacts, cell cycle progression, signal transmission, and metabolic pathways.Online software called the Search Tool for the Retrieval of Interacting Genes (STRING)31 is widely used to evaluate PPI data. The dataset was utilized to evaluate DEG relationships, and a PPI network was created using Cytoscape, revealing significantly correlated proteins based on topological characteristics32.
Hub gene identification
Hub genes are those in the gene network that collaborate with a large number of other genes and are frequently essential for biological functions and gene regulation. Fur- thermore, hub genes were shown to be the most strongly linked to illness33.
A Cytoscape plugin called CytoHubba (Version 0.1)34 was utilized to determine the network hub genes. Another plugin for Cytoscape that may be used to generate and display functionally grouped networks of biological concepts and pathways is called ClueGO35. A useful addition to ClueGO, the CluePedia Cytoscape plugin searches for additional markers that may be connected to routes.
Assessing transcription factors (TF) and miRNAs network
Proteins known as transcription factors control how genes are copied into RNA, which is the first step in the process of creating proteins. Transcription factors, are proteins that bind to DNA, and activate or deactivate genes, with enhancers and silencers binding sites in specific parts of the body, enabling cells to perform logic operations36.
We utilized the Network Analyst tool to explore the JASPAR37 and ChEA38 databases and identify topographically plausible transcription factors likely to link to our DEGs. JASPAR is a publicly available database containing carefully selected, non-redundant transcription factor (TF) binding profiles for TFs in many species spanning six taxonomic groupings. These profiles are kept as TF flexible models (TFFMs) and position frequency matrices (PFMs).
A gene-set enrichment analysis technique called ChIP-X39. Enrichment Analysis is designed to determine whether query gene sets are enriched with genes that may be transcription factor targets. In ChEA, sets of probable target genes are selected from published ChIP-chip, ChIP-seq, ChIP-PET, and DamID investigations and labeled with transcription factors using a gene-set library (Fig. 2).

Supervised learning model to diagnosis tuberculosis.
MTIs, or miRNA-target interactions, are stored in the miRTarBase database. By using reporter assays, western blots, or microarray studies with miRNA overexpression or knockdown, the gathered MTIs are experimentally confirmed.
Analysis of potential drugs
The term “Potential Drug-Drug Interaction” (or "potential DDI”) or Potential Drugs describes the potential for one drug to affect another’s effects when taken concurrently. A medication has to interact with a matching protein or enzyme on the receptor to affect it. The interaction between proteins and bioactive molecules is crucial for many pharmacological or prophylactic approaches.
Drug-protein interaction refers to the interaction between a protein molecule and a drug molecule, which significantly impacts the activity of medications.
DrugBank Online is a crucial tool in the drug industry, enabling significant advancements in the data-driven medicine sector due to its comprehensive referencing and detailed data descriptions.
The program Analyst was used to predict drug-protein correlations.
Result
Evolutionary metrics and results of statistical models
Here, we attempt to interpret the numerous signs that might be employed to determine the effectiveness of the recommended approach. More accuracy is required when assessing models employing RNa-sequence data (Fig. 3). Even though the efficacy of a model is frequently evaluated using accuracy, Therefore, in addition to accuracy, the evaluation metrics RocAuc, Precision, Recall, Specificity, F1-Score, TPR, and FPR are utilized to obtain an additional thorough comprehension of a prototype’s effectiveness. Each of these metrics was used in the research we conducted to evaluate the suggested model’s reliability. The matrix of uncertainty compiles the several metrics that are employed to assess how effective a classification model is. The following four components are required for this confusion matrix: The acronyms TP, FP, TN, and FN represent the terms “True Positive,” “True Negative,” and “False Negative,” in that order (FN). Four things may happen. True positives are tallied when an incident is identified as positive and is deemed positive; false negatives are counted when an event is labeled as negative. A true negative is counted if the instance is classed as negative; a false positive is tallied if it is labeled as positive. The most frequent results are appropriate labeling (TP) and identification (TN), as opposed to incorrect labeling (FP and FN). The percentage of accurate forecasts to all predictions is known as accuracy. True positives (TP) and true negatives (TN) make up correct forecasts. The totality of the positive (P) and negative (N) instances make up each forecast. N is made up of false negatives (FN) and TN, whereas P is made up of TP and false positives (FP).

Evaluation among the ML model.
When evaluating a classification model’s capacity to forecast tuberculosis, precision, and recall are essential metrics to consider. Precision is the percentage of correctly estimated victims of tuberculosis to all expected patients40. So the precision metric quantifies the proportion of accurate forecasts generated by the model and the amount of accurate positive predictions, or true positives, divided by the total number of positive predictions (including true and false positives) that the model correctly anticipated is the precision.
Conversely, recall, which is often referred to as responsiveness, is calculated by dividing the sum of true positive labels by the sum of all real positive labels41. It compares the percentage of correctly estimated TB patients with the total number of TB patients. The formulas used for these metrics are:
The average of the harmonics of a classification model’s recall and precision is known as the F1 score, or F-measure42. The F1 measure accurately reflects a models depend- ability since both metrics have an equivalent role in the outcome. The equation used for the F1 score is:
The classification assessment statistic for a model, called specificity, measures the pro-portion of true negatives that the framework correctly detects. This suggests that a further percentage of real negative data was misinterpreted as positive; One may call these “false positives.” The model’s elevated specificity means that most of the negative findings are being correctly classified by the model. In contrast, a low specificity indicates that many negative results are being incorrectly labeled as positive. Since the expense of false negatives is substantial like when it comes to medical treatment, high specificity is desired43. Specificity can be computed using the formula below:
In the context of a matrix of confusion, sensitivity or recall are other names for the True Positive Rate (TPR). True Negative Rate (TNR) is also often utilized, much like specificity. A low FPR is crucial to prevent needless additional testing and possible patient damage, whereas a high TPR is crucial to guarantee that every single cancer case is identified during medical treatment (Fig. 4). Maintaining the efficacy and security of tests used for diagnosis and screening for medical conditions requires striking a balance between TPR and FPR. It is computed as follows:

Supervised learning to diagnosis tuberculosis.
We employed five untrained models—XG Boost, Logistic Regression, Random Forest Classifier, AdaBoost, and Support Vector Machine—to predict tuberculosis from RNA-Sequence count data. From Table 2 the results of the confusion matrices for Precision 0.95, Recall 0.964, RocAuc 0.985, Specificity 0.962, F1-Score 0.957, TPR 0.964, and FPR 0.038 demonstrate that the XG Boost model worked effectively, with the greatest prediction accuracy at 0.963% and lowest Log Loss at 0.139%. Furthermore, with a prediction accuracy of 0.866%, the AdaBoost plus Support Vector Machine model demonstrated the second-highest accuracy. Their respective Log Losses are 0.666% and 0.661%, making them the highest. AdaBoost Precision 0.845, F1-Score 0.844, TPR 0.840, FPR 0.114, Recall 0.840, Specificity 0.886, and RocAuc 0.060 which is the lowest for this specific dataset are shown in the sections that follow. AdaBoost’s ROC AUC value, which measures a model’s ability to distinguish between positive and negative classes, is the lowest. Moreover, for Support Vector Machine F1-Score 0.838, TPR 0.804, FPR 0.087, Precision 0.874, Recall 0.804, RocAuc 0.115, Specificity 0.913. On the other hand, with an accuracy of 0.739%, Logistic Regression is the least accurate model overall. In addition, the Random Forest Classifier’s success rate in third place was 0.772%.
Comparative transcription sequencing utilizing the significance of features
To discover extremely expressed genes, Differential Gene Expressions (DEGs) have been performed in this study by using two feature-importance methodologies utilizing algorithms that use machine learning using count data of RNA-Sequence of TB with patients and non-TB. The maximum effectiveness was attained by training five controlled techniques: XG Boost, Logistic Regression, Random Forest Classifier, AdaBoost, and Support Vector Machine. XGBoosting, on the other hand, worked effectively, showing excellent prediction accuracy levels at 0.963% whereas the rest of the algorithms were below 0.9, which is why we selected it. The top 100 frequently occurring genes were then selected from the XGBoost algorithm to minimize uncertainty. Furthermore, the Extended data contain our expected expressed genes in the supplementary file Table S1. Typically, P-value, Adjusted P-value, and Log-FC are used to identify important genes; however, we focused on picking out features to identify them in a new way, resulting in an effective result.
Assessment of gene ontology and pathway enrichment analysis
Considering Gene Ontology (GO) provides a comprehensive description of protein functions, it is considered one of the essential components of physiological description. GO refers to a controlled and structured phrase set of words called GO terms44. The study usually yields an ordered set of GO terms having P-values corresponding to every phrase45. Pathway analysis is an effective method for identifying genes, proteins, and metabolites that function differentially and are generated by present high volumes screening. It is also useful in studying physiology46. Pathway analysis is a technique used in genome-wide association research or genomics tools for the preliminary identification and understanding of a diseased or physiological state47. Ontology and pathways designed to carry out a comprehensive physiological simulation method are essential components of physiological treatments. We used an expression set enhancement strategy to identify networks using the machine learning program EnrichR. Five pathway resources were used to perform tests using DEGs of TB. Figure 5 displays the 20 major parameters of the signaling pathways. The following Table 3 lists the top 10 functions related to cellular components, biological processes, and the top 4 for molecular processes. Both the GO and the Pathways are filtered by the adj. P-value, which is often less than 0.05. The results are then arranged in ascending order.

An overview of the network abundance for tuberculosis DEGs. Y axis signaling pathways and X-axis denoted as negative log10 P value. Asthma has highest negative log10 P value.
Protein–protein interactions (PPIs) analysis and hub genes identification
The capacity of compounds to operate as drugs and the target protein’s activity are largely determined by protein-protein interactions. Most proteins and genes recognize the activities of the ensuing phenotype as a collection of interconnections. Cell-to-cell contacts, regulation of metabolism, evolutionary supervision, and other functions in biology are all managed by protein-protein interactions or PPIs48. The PPI network was analyzed using STRING, and compliance networks and recurring connections among DEGs were predicted using a Cytoscape visualization. By using topological measures, such as a degree greater than 15°, PPI analysis was used to designate highly communicative proteins. The PPI network (Fig. 6) has 40 nodes and 76 edges connecting them, which are the most notable DEGs. Hub genes exhibit the top 10% interconnectedness and a significant correlation with potential units. Because of these interactions, hub genes usually have a major function in biological systems. We utilized the Cytohubba plugin in Cytoscape to identify the top 20 DEGs or hub genes. Notably, Fig. 7 depicts the hub genes identified using the MCC approach: CREBBP, SPR, H2AX, CD84, LILRB2, UTY, TFEB, LILRB1, FOXI2, and HVCN1, while the Bottleneck approach identified LILRB2, CD84, LILRB4, HLA-DPB1, HLA-DQB1, HLA-DRB1, LILRB1, C1QB, CD160, and CREBBP as hub genes.

PPI network is made up of DEGs for tuberculosis. Differentially ex- pressed protein genes are represented by the circular nodes in the picture, and the interaction between the nodes is shown by the edges. The PPI is made up of 40 nodes and 76 edges. STRING was used to build the PPI network, and Cytoscape was used to view it.

Identification of hub genes within the cluster using cytohubba: application of MCC (Maximal Clique Centrality) and bottleneck algorithms and network comparison. The linkages between the top 10 hub genes from each method and additional genes (yellow) are indicated by dark green high- lights. While the (A) BottleNeck has 30 nodes and 65 edges (B) MCC network has 22 nodes and 55 edges.
Discovering the miRNAs and transcription factors (TF) that bind to their neighboring DEGs
We employed a system that uses a network approach to analyze the controlling TFs and miRNAs to identify significant expression alterations and discover additional signaling molecules associated with the hub protein. Proteins known as transcription factors are substances that control transcription as well as gene activity in all living organisms49. MiRNAs, which are minuscule RNA molecules, are involved in the modulation of post-transcriptional expression. Figure 8 depicts the interaction between DEGs and TFs, while Fig. 9 shows the relationship between DEGs and miRNAs. TFs of genes with differential expression have major regulators that were STAT3, GATA2, KLF4, MYC, FLI1, TP53, REST, HNF4A, FOXP1, POU5F1, SPI1, NANOG, SOX2, PPARG, CREM, GATA2, NFKB1, E2F1, JUN, USF2, PPARG, HOXA5, FOXC1, and YY1 (Table 4). has-mir-383-3p, has-mir-520h, has-mir-520g-3p, has-mir-7977, has-mir-218-5p, has-mir-6499- 3p, has-mir-30a-5p, has-let-7b-5p, has-mir-26b-5p, has-mir-27a-3p, has-mir-129-2-3p, has-mir-34a-5p, has-mir-1-3p, has-mir-16-5p, has-mir-124-3p, and has-let-7b-5p were defined to create a succinct summary of the DEGs acting as post-transcriptional regulators. The transcriptional and post-transcriptional regulatory elements of the genes asso ciated with TB that are differently regulated are compiled in Tables 4 and 5 respectively.

The Network Analyst’s framework for integrated regulated collaboration among DEGs and TFs, using (a) ChEA and (b) Jasper database. (a) The network contains 47 nodes and 196 edges, where (b) has 31 and 95, nodes and edges respectively. Transcription factors are represented by square nodes, while genes that are connected to transcription factors are represented by circular nodes.

The interconnectedness of regulated relationships between miRNAs and DEGs. Here, the circular gene representations link to the miRNAs, which are represented by the square node. Network (a) contains 22 nodes and 29 edges while (b) has 23 nodes and 54 edges both are constructed using miRTarBase and TarBase databases respectively.
Potential medication
To comprehend the molecular components associated with the transmission of signals, a protein-drug interaction study must be carried out50. Using NetworkAnalyst approaches based on drug-protein interactions from the DrugBank library, we identified 22 prospective therapy medications for frequently occurring DEGs as promising medicinal options in TB. Figure 10 displays 22 widely used medicinal substances, Bevacizumab, Daclizumab, Palivizumab, Natalizumab, Efalizumab, Alefacept, Alemtuzumab, Tositumomab, Ibritumomab tiuxetan, Muromonab, Basiliximab, Rituximab, Trastuzumab, Gem- tuzumab ozogamicin, Abciximab, Adalimumab, Etanercept, Cetuximab, N-Acetyle Sero- tonin, Biopterin, and 2’-Monophosphoadenosine 5’-Diphosphoribose which had been identified in the DEGs of TB Protein Drug Associations.

This figure depicted 22 potential medications for tuberculosis treatment identified through the protein-drug interaction approach. Among them, 18 drugs target the C1QB gene, while the others interact with the SPR gene. In the diagram, medications are represented by rectangular nodes, and their corresponding gene targets are depicted as spherical symbols.
Discussion
Nowadays, machine learning is becoming increasingly important in the bioinformatics industry as artificial intelligence advances quickly, allowing for in-depth data analysis. While most RNA-Sequence data may be processed using machine learning approaches, tuberculosis dataset has been examined in this study since it is a infectious disease with potentially fatal consequences. In the investigation, we introduced a count- oriented classification workflow for RNA sequence information analysis that uses well- established machine learning techniques to effectively detect tuberculosis. Additionally, the methods employed here allow us to deal with substantial transcriptome data and make trustworthy inferences about TB proteins. Notably, we made an effort to create a novel approach by using the machine learning technique’s key characteristic technique to the TB data in order to identify genes with enhanced expression of RNA-Sequence data. Also, we employed a variety of bioinformatics tools in our investigation, which enables us to fully comprehend tuberculosis and pinpoint its related biomarkers. We used multiple trained machine learning methods, such as Random Forest, AdaBoost, Gradient Boosting, Logistic Regression, Decision Tree, and XGBoost, in our detailed examination of tuberculosis (TB). Figure 3 showed their accuracy, losses, and performance indicators in graphic style. The Table 2 provides more information on each of these indicators. we investigated Pathway enrichment assessments in Fig. 5, Gene Ontology analysis in Table 3, Protein-Protein Interaction examinations in Fig. 6, and investigated Hub-Protein interactions in Fig. 7, Transcriptional Factor interactions in Fig. 8, miRNA interactions in Fig. 9, and drug-protein interactions in Fig. 10.
There are various reasons why our top model, XGBoost, performed better on our dataset than the others. With integrated L1 and L2 regularization and the capacity to represent intricate non-linear relationships, it excelled in managing high-dimensional data. Its efficiency is further increased by special features including tree pruning, effective column block computation, and internal handling of missing values. The model’s edge was probably influenced by its flexibility in adjusting hyperparameters, resistance to outliers, and capacity to record features engagement. Furthermore, some datasets’ intrinsic characteristics may be more compatible with gradient-boosted trees, indicating that the underneath patterns in our data were especially suitable to XGBoost. Below is the mathematical formula for XGBoost model’s objective and procedure: The model’s prediction for the ith instance at the tth iteration of a dataset comprising n samples and m characteristics is represented by the symbol yˆit). In every iteration of XGBoost, the following objective function needs to be optimized:
When the regularization term, Ω, is defined as follows and l is the loss function with differentiable convexity:
In this case, wj is the score given to the jth leaf, and T shows the number of leaves in the tree. The tree’s ultimate configuration is determined by minimizing:
where g, and h denote the corresponding sum of the cases that reach the child node on the left, G, and H stand for the total of the loss function’s first- and second-order gradients for the instances in the current node.
In bioinformatics, gene ontology (GO) and enrichment analysis are widely utilized statistical methods that help researchers uncover the biological significance of large gene sets. GO suggests that biological processes can involve molecular interactions. The molecular aspect focuses on molecular processes, while the cellular component refers to the location where a gene exerts its function. Meanwhile, pathway analysis offers a distinct approach by exploring the connections between physiologically or molecularly complex diseases. This is the most efficient way to make an organism react to changes occurring inside of it. From Table 3 the leading 10 genes of biological process, cellular component, and molecular function are mentioned in turn according to their P values. The most significant genes are HLA-DRB3; and HLA-DRB1 for these above-mentioned three processes. HLA-DRB1 alleles could represent an increased risk of developing active TB51. Other important DEGs are TEPSIN, TMED3, HVCN1, and ATP2B2 for these three categories. Figure 5 showed the pathway enrichment summary of tuberculosis DEGs. According to the figure, the top pathways are asthma, allograft rejection, Graft-versus-host disease, and Type I diabetes mellitus.
In our study, the protein-protein interaction (PPI) network was analyzed using Cytoscape and STRING. Our method combined the robust display and analysis capabilities of Cytoscape with the huge PPI database of STRING. With the use of Cytoscape’s sophisticated network analysis tools and STRING’s carefully selected data, this integration enables a thorough investigation of the interaction network, resulting in a more in-depth and understandable knowledge of protein interactions. This, in our opinion, makes our method a useful addition to already-existing computational techniques, especially in situations where computational resources or data availability may be restricted. In contrast to certain machine learning techniques that could need a lot of training data and feature building, our methodology uses Cytoscape’s network measurements and STRING’s preset interaction scores directly to identify key proteins. In conclusion, our suggested workflow presents a well-balanced strategy for combining the interactive features of Cytoscape with reliable data from STRING, making it easy to use and understand for PPI network analysis.
Hub proteins of high or low expression levels in TB patients have been found. TB quickly triggers the transcription factor cAMP Response Element Binding Protein (CREBP), which controls a variety of cellular reactions in macrophages52. Experiments on the M. tuberculosis challenge utilizing CD84-deficient C57BL/6 mice indicate that CD84 expression probably contributes to immunosuppression of T and B cells during M. tuberculosis pathogenesis and also inhibits B cell activation53. Myeloid-derived suppressor cells (MDSCs) are immunosuppressive cells that are reprogrammed to become pro-inflammatory by LILRB2 antagonists, which kills Mtb54. Compared to healthy controls, patients with active tuberculosis had reduced CD160 expression in their B cells and monocytes55.
The TFs we found are connected to TB. Table 5 shows the overview of TF biomolecules of differentially expressed genes. NFKB1, one of the most influential hub genes was determined as a potential therapeutic target in tuberculosis56. GATA2 is downregulated by sustained stimulation with M. tuberculosis antigen57. After M. tuberculosis infection, TLR4 which is YY1-activated may exacerbate mycobacterial damage in human microbes58.
Tuberculosis is also associated with our discovered miRNAs. Serum exosomes may be a useful marker for exosomal miR-7977, which is crucial for lung cancer diagnosis59. In both MTB-infected and uninfected THP-1 cells, forced overexpression of miR- 30a had a restraining action on TLR/MyD88 activation and cytokine production60. mir- 26b-5p might function as biomarker for IOTB(Intraocular Tuberculosis)61.
We want to delineate the particular benefits of utilizing machine learning strategies like AdaBoost, XGBoost, and Logistic Regression classifiers in comparison to more conventional approaches like R-based statistical methodologies and other well-established bioinformatics tools. Multiple weak learners are used by machine learning models, especially ensemble techniques like AdaBoost and XGBoost, to create a strong prediction model. When compared to single statistical methods, this approach frequently yields better generalization and higher accuracy. ML classifiers were able to identify complicated, non-linear correlations in the data, which improved the accuracy of identifying genes that were differentially expressed in differential gene analysis. Gene expression profiles, genetic variants, and route information are just a few of the data types that our machine-learning algorithms effectively manage and integrate. Even if they are strong, traditional approaches could have trouble integrating diverse data sources. With their capacity to process and learn from intricate, multi-dimensional datasets, machine learning techniques offer a more comprehensive perspective and reveal interactions that traditional tools would overlook. Because machine learning algorithms are flexible, they can find new patterns and connections in the data that might be missed by conventional tools. They can find new patterns and connections in the data. For instance, when we used XGBoost to identify hub genes, we were able to identify possible important regulators that were previously missed by traditional centrality metrics. Large-scale genomic investigations can benefit from our ML algorithms’ better computing efficiency and quicker processing times as compared to other conventional methods.
For TB patients, our identified potential therapeutic compounds may be beneficial. Treatment with bevacizumab reduces hypoxia in TB granulomas, enhances small molecule delivery, and encourages vascular normalization62. There is some ambiguity on how daclizumab, palivizumab, natalizumab, efalizumab, tositumomab, alefacept, muromonab, and others react with TB. Thus, preclinical and clinical trials together with additional research are needed.
Conclusion
Our study endeavors to employ a machine learning method to detect important genes using the feature significance tools and to confront the overarching global challenge of tuberculosis and advance the well-being and livelihoods of individuals. Here, we explored both machine learning techniques and conventional bioinformatics methods.
We now have a more comprehensive understanding of the molecular pathways behind tuberculosis sickness because of the integration of machine learning algorithms, statistical analysis, and bioinformatics tools. This approach would allow us to more effectively identify expressed genes, moving beyond reliance only on conventional bioinformatics methods with a specific focus on detecting DEGs. In addition, when dealing with a massive amount of features, employing a machine learning approach proves to be effective as well as more reliable in predicting DEGs. We’ve analyzed vast transcriptomic data successfully using multiple ML models. XGBoost model demonstrating strong performance achieved 96.3% accuracy. This shows that ML can enhance TB diagnosis, enabling tailored treatments and revolutionizing clinical practice.
In our TB biomarker study, we analyzed various molecular components, such as 20 pathways, 24 gene ontologies, 23 transcription factors, 20 hub genes, 16 miRNAs, and 22 possible drugs. Based on our verification, we validated our study. The three main pathways are hematopoietic cell lineage, Th1 and Th2 cell development, and antigen processing, as well as presentation Fig. 5. The reported Hub proteins such as CD84, LILRB2, LILRB4, and CD160 might provide encouraging pathways for advancing disease study and therapy in Table 6. Similarly, transcriptional factors like FOXC1, GATA2, YY1, SPI1, MYC, and SOX2 in Fig. 8, coupled with miRNAs like has-mir-7977, hsa-let-7b-5p, hsa-mir-26b-5p, hsa-mir-16-5p, hsa-mir-124-3p, and hsa-mir-1-3p in Fig. 9, explore the regulatory dynamics at the core of TB. Moreover, our study pinpointed prospective drug candidates such as biopetrin, oxaloacetate ion, bevacizumab, and other compounds, showing potential for therapeutic application in Fig. 10. Although more preclinical and clinical research is required to confirm their safety and efficacy profiles, these discoveries present opportunities to transform TB therapy approaches and enhance patient outcomes. This research marks a notable step forward in utilizing machine learning for detecting genes that are expressed from RNA sequence count data. By combining essential bioinformatics techniques and verifying results opposing previous studies on TB, we provide a strong method for comprehending and tackling TB on a molecular level.
Limitations of the studying
A limitation of this research is the requirement for improved precision for the models. Although our present methodology has yielded some insightful findings, the incorporation of machine learning models in further research endeavors may considerably augment the precision and accuracy of our results. Furthermore, we still need to use the evidence of research currently available for validating a subset of biomarkers. Thoroughly validating those biomarkers is crucial, as it may provide researchers with a better understanding of expressed genes, eventually allowing more accurate analysis and enhanced TB management tactics
Data availability
All data generated or analysed during this study are included in this published article.
Code availability
Code available on git-hub, can be accessed by this link: https://github.com/saddam017/ML-for-differential-expressed-genes.
References
Karim, M. R. et al. Explainable ai for bioinformatics: Methods, tools and applications. Brief. Bioinform. 24(5), bbad236 (2023).
Han, H. & Liu, X. The challenges of explainable AI in biomedical data science. BMC Bioinform. 22(Suppl 12), 443 (2022).
Kaisar, S. & Chowdhury, A. Integrating oversampling and ensemble-based machine learning techniques for an imbalanced dataset in dyslexia screening tests. ICT Express 8(4), 563–568 (2022).
Sprang, M., Andrade-Navarro, M. A. & Fontaine, J.-F. Batch effect detection and correction in RNA-seq data using machine-learning-based automated assessment of quality. BMC Bioinform. 23(Suppl 6), 279 (2022).
Bagcchi, S. WHO’s global tuberculosis report 2022. The Lancet Microbe 4(1), e20 (2023).
Pai, M., Dewan, P. K. & Swaminathan, S. Transforming tuberculosis diagnosis. Nat. Microbiol. 8(5), 756–759 (2023).
Coombes, C. E. et al. Unsupervised machine learning and prognostic factors of survival in chronic lymphocytic leukemia. J. Am. Med. Inform. Assoc. 27(7), 1019–1027 (2020).
Hyland, S. L. et al. Early prediction of circulatory failure in the intensive care unit using machine learning. Nat. Med. 26(3), 364–373 (2020).
Kouchaki, S. et al. Application of machine learning techniques to tuberculosis drug resistance analysis. Bioinformatics 35(13), 2276–2282 (2019).
Singh, M. et al. Evolution of machine learning in tuberculosis diagnosis: A review of deep learning-based medical applications. Electronics 11(17), 2634 (2022).
Barrett, T. et al. NCBI GEO: Archive for functional genomics data sets—10 years on. Nucleic Acids Res. 39(suppl_1), D1005–D1010 (2010).
Scriba, T. J. et al. Sequential inflammatory processes define human progression from M. tuberculosis infection to tuberculosis disease. PLoS Pathog. 13(11), e1006687 (2017).
Mahi, N. A. et al. GREIN: An interactive web platform for re-analyzing GEO RNA-seq data. Sci. Rep. 9(1), 7580 (2019).
Nguyen, T. et al. RNA-Seq count data modelling by grey relational analysis and nonparametric Gaussian process. PLoS ONE 11(10), e0164766 (2016).
McKinney, W. pandas: A foundational Python library for data analysis and statistics. Python High Perform. Sci. Comput. 14(9), 1–9 (2011).
Breiman, L. Random forests. Mach. Learn. 45, 5–32 (2001).
Jackins, V. et al. AI-based smart prediction of clinical disease using random forest classifier and Naive Bayes. J. Supercomput. 77(5), 5198–5219 (2021).
Ying, C. et al. Advance and prospects of AdaBoost algorithm. Acta Automatica Sinica 39(6), 745–758 (2013).
Sevinç, E. An empowered AdaBoost algorithm implementation: A COVID-19 dataset study. Comput. Ind. Eng. 165, 107912 (2022).
Tolles, J. & Meurer, W. J. Logistic regression: Relating patient characteristics to outcomes. JAMA 316(5), 533–534 (2016).
Xu, Z. & Z. Wang. A risk prediction model for type 2 diabetes based on weighted feature selection of random forest and xgboost ensemble classifier. in 2019 eleventh international conference on advanced computational intelligence (ICACI). 2019. IEEE.
Vapnik, V. Support-vector networks. Mach. Learn. 20, 273–297 (1995).
Adaikalakoteswari, A. et al. Differential gene expression of NADPH oxidase (p22phox) and hemoxygenase-1 in patients with Type 2 diabetes and microangiopathy. Diabet. Med. 23(6), 666–674 (2006).
Rajbahadur, G. K. et al. The impact of feature importance methods on the interpretation of defect classifiers. IEEE Trans. Software Eng. 48(7), 2245–2261 (2021).
Mehta, S. et al. A galaxy of informatics resources for MS-based proteomics. Expert Rev. Proteomics 20(11), 251–266 (2023).
Liu, Z. J. Bioinformatics in aquaculture: Principles and methods (John Wiley & Sons, Hoboken, 2017).
Kanehisa, M. The KEGG database. in ‘In silico’simulation of biological processes: Novartis Foundation Symposium 247. 2002. Wiley Online Library.
Jassal, B. et al. The reactome pathway knowledgebase. Nucleic Acids Res. 48(D1), D498–D503 (2020).
Kelder, T. et al. WikiPathways: Building research communities on biological pathways. Nucleic Acids Res. 40(D1), D1301–D1307 (2012).
Nishimura, D. BioCarta. Biotech Softw. Internet Rep.: Comput. Softw. J. Sci. 2(3), 117–120 (2001).
Szklarczyk, D. et al. STRING v11: Protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 47(D1), D607–D613 (2019).
Rao, V. S. et al. Protein-protein interaction detection: Methods and analysis. Int. J. Proteomics 2014(1), 147648 (2014).
Liu, Y. et al. Identification of hub genes and key pathways associated with bipolar disorder based on weighted gene co-expression network analysis. Front. Physiol. 10, 1081 (2019).
Chin, C.-H. et al. cytoHubba: Identifying hub objects and sub-networks from complex interactome. BMC Syst. Biol. 8, 1–7 (2014).
Bindea, G. et al. ClueGO: A Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 25(8), 1091–1093 (2009).
An, X. et al. Transcriptome analysis and transcription factors responsive to drought stress in Hibiscus cannabinus. PeerJ 8, e8470 (2020).
Khan, A. et al. JASPAR 2018: Update of the open-access database of transcription factor binding profiles and its web framework. Nucleic Acids Res. 46(D1), D260–D266 (2018).
Lachmann, A. et al. ChEA: Transcription factor regulation inferred from integrating genome-wide ChIP-X experiments. Bioinformatics 26(19), 2438–2444 (2010).
Uijtendaal, E. V. et al. Analysis of potential drug-drug interactions in medical intensive care unit patients. Pharmacother.: J. Hum. Pharmacol. Drug Ther. 34(3), 213–219 (2014).
Fawcett, T. Roc analysis in pattern recognition. Pattern Recogn. Lett. 8, 861–874 (2005).
Sofaer, H. R., Hoeting, J. A. & Jarnevich, C. S. The area under the precision-recall curve as a performance metric for rare binary events. Methods Ecol. Evol. 10(4), 565–577 (2019).
Lever, J., Krzywinski, M. & Altman, N. Points of significance: Model selection and overfitting. Nat. Methods 13(9), 703–705 (2016).
Lange, R. T. & Lippa, S. M. Sensitivity and specificity should never be interpreted in isolation without consideration of other clinical utility metrics. Clin. Neuropsychol. 31(6–7), 1015–1028 (2017).
Subramanian, A. et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. 102(43), 15545–15550 (2005).
Yon Rhee, S. et al. Use and misuse of the gene ontology annotations. Nat. Rev. Genet. 9(7), 509–515 (2008).
Papin, J. A. et al. Comparison of network-based pathway analysis methods. Trends Biotechnol. 22(8), 400–405 (2004).
García-Campos, M. A., Espinal-Enríquez, J. & Hernández-Lemus, E. Pathway analysis: State of the art. Front. Physiol. 6, 383 (2015).
Braun, P. & Gingras, A. C. History of protein–protein interactions: From egg-white to complex networks. Proteomics 12(10), 1478–1498 (2012).
Cheng, C. et al. Understanding transcriptional regulation by integrative analysis of transcription factor binding data. Genome Res. 22(9), 1658–1667 (2012).
Lahti, J. L. et al. Bioinformatics and variability in drug response: A protein structural perspective. J. R. Soc. Interface 9(72), 1409–1437 (2012).
Odera, S. et al. Association between human leukocyte antigen class II (HLA-DRB and-DQB) alleles and outcome of exposure to Mycobacterium tuberculosis: A cross-sectional study in Nairobi, Kenya. Pan African Med. J. https://doi.org/10.11604/pamj.2022.41.149.30056 (2022).
Leopold Wager, C. M. et al. Activation of transcription factor CREB in human macrophages by Mycobacterium tuberculosis promotes bacterial survival, reduces NF-kB nuclear transit and limits phagolysosome fusion by reduced necroptotic signaling. PLoS Pathog. 19(3), e1011297 (2023).
Zheng, N. et al. CD84 is a suppressor of T and B Cell activation during mycobacterium tuberculosis pathogenesis. Microbiol. Spectr. 10(1), e01557-e1621 (2022).
Singh, V. K. et al. Antibody-mediated LILRB2-receptor antagonism induces human myeloid-derived suppressor cells to kill mycobacterium tuberculosis. Front. Immunol. 13, 865503 (2022).
Yang, B. et al. Expression of CD160 in natural killer (NK) cells from patients with active tuberculosis and its relationship with cell functions. Xi bao yu fen zi Mian yi xue za zhi= Chinese J. Cell. Mol. Immunol. 38(10), 918–924 (2022).
Alam, A. et al. Human gene expression profiling identifies key therapeutic targets in tuberculosis infection: A systematic network meta-analysis. Infect. Genet. Evol. 87, 104649 (2021).
Li, F. et al. Persistent stimulation with Mycobacterium tuberculosis antigen impairs the proliferation and transcriptional program of hematopoietic cells in bone marrow. Mol. Immunol. 112, 115–122 (2019).
Yang, X. et al. YY1 Contributes to the inflammatory responses of mycobacterium tuberculosis-infected macrophages through transcription activation-mediated upregulation TLR4. Mol. Biotechnol. 67(2), 778–789 (2024).
Biadglegne, F. et al. Composition and clinical significance of exosomes in tuberculosis: A systematic literature review. J. Clin. Med. 10(1), 145 (2021).
Wu, Y., Sun, Q. & Dai, L. Immune regulation of miR-30 on the Mycobacterium tuberculosis-induced TLR/MyD88 signaling pathway in THP-1 cells. Exp. Ther. Med. 14(4), 3299–3303 (2017).
Chadalawada, S. et al. Dysregulated expression of microRNAs in aqueous humor from intraocular tuberculosis patients. Mol. Biol. Rep. 49(1), 97–107 (2022).
Datta, M. et al. Anti-vascular endothelial growth factor treatment normalizes tuberculosis granuloma vasculature and improves small molecule delivery. Proc. Natl. Acad. Sci. 112(6), 1827–1832 (2015).
Acknowledgements
The authors would like to extend their sincere appreciation to the Ongoing Research Funding Program (ORF-2025-189), King Saud University, Riyadh, Saudi Arabia.
Funding
This work is financially supported by the Ongoing Research Funding Program (ORF-2025-189), King Saud University, Riyadh, Saudi Arabia.
Author information
Authors and Affiliations
Contributions
Conceptualization, original draft writing, reviewing, and editing: Md. Saddam Hossain, Md. Parvez Khandocar, Farzana Akter Riti. Formal analysis, investigations, funding acquisition, reviewing, and editing: Md. Yeakub Ali, Prithbey Raj Dey, S M Jahurul Haque, Khalid S. Almaary. Resources, data validation, data curation, and supervision: Amira Metouekel, Atrsaw Asrat Mengistie, Mohammed Bourhia, Farid Khallouki.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
Not applicable.
Ethical consideration
Not applicable.
Consent for publication
Not applicable.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Hossain, M.S., Khandocar, M.P., Riti, F.A. et al. A comprehensive machine learning for high throughput Tuberculosis sequence analysis, functional annotation, and visualization. Sci Rep 15, 25866 (2025). https://doi.org/10.1038/s41598-025-98654-0
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-98654-0
