
Abstract
Zebrafish have emerged as an alternative vertebrate model for both basic and applied science. While two zebrafish lines, AB and TU, have been extensively used, recent studies suggest that complex behaviors and susceptibility to adult phenotypes vary among lines. Given the increasing demand across diverse research fields, establishing a phylogenetically distinct wild-type zebrafish line without deleterious genetic variants would greatly benefit the research community. In this study, we documented the establishment of the RIKEN Wild-type (RW) line and conducted comparative genome analyses to investigate the genetic characteristics of various wild-type zebrafish lines such as AB, TU, TL, WIK, SAT, NHGRI-1, PET, *AB, IND, M-AB and IM with a particular focus on the genetic characterization of the RW line. We identified numerous genetic variants in each line that may affect coding proteins, some of which are unique to each line, conferring specific genetic traits. Notably, the RW line was found to carry such genetic variants in 13 genes. Furthermore, our phylogenetic analysis revealed that the RW line is genetically distinct from other commonly used lines. Collectively, the RW line is a robust zebrafish line with excellent breeding characteristics, making it valuable for studies exploring genetic diversity and line-specific traits within the species.
Similar content being viewed by others

Introduction
Zebrafish (Danio rerio) have unique advantages such as high fecundity and optical transparency during early development and thus has served as a model organism in genetics and developmental biology. Over several decades, numerous zebrafish lines have been established. The most commonly used zebrafish line is AB, which originated from two lines, A and B, both purchased from a pet shop in Albany, Oregon in the 1970s1. The AB has been established by eliminating early deleterious genetic variants in the large population through mass mating and eventually maintained by a Round Robin mating to preserve genetic variability by in vitro fertilization2. The second major line is Tübingen (TU), originating from a pet shop in Germany3. This line has been maintained by mass mating after removing lethal mutations2 and was used for the zebrafish genome project4. The Tüpfel long fin (TL) line carries homozygous mutations in leot1 and lofdt2, which respectively affect gap junction function and potassium channel expression, resulting in a spotting pigment pattern and elongated fins5,6. The Sanger AB Tübingen (SAT) and NHGRI-1 lines have been generated by crossing of AB and TU fish and subsequent mass mating7,8. Wild-caught lines such as Wild India Kolkata (WIK), Cooch Behar (CB), Nadia (NA), India (IND) and Darjeeling (DAR) have also been reported9,10,11. Isogenic lines such as C29, C32 and SJD have been generated by heat shock or pressure techniques12,13,1. Recently, two inbred lines, M-AB and IM, have been created by sib-pair mating over 20 generations from *AB and IND, respectively14,15. The *AB was generated from the AB line by selecting good parthenogenesis-derived females16. Pet shop-derived wild-type zebrafish such as Ekkwill (EK) and PET have also been used to compare characteristics of zebrafish lines17,18. Collecting various lines in addition to the major lines is useful for the study of genetic mapping and genetic variability.
In this study, we describe the characterization of a zebrafish line, Riken Wild-type (RW), which has been established in Japan, and has been used for various genetic researches19,20,21,22,23. Our whole genome sequence (WGS) and phylogenetic analysis of AB, TU, TL, WIK, SAT, NHGRI-1, PET and RW along with our previous sequencing data of *AB, IND, M-AB and IM revealed that RW forms an independent group apart from the other lines and carries unique genetic characteristics. We also identified genetic variants that affect coding proteins in RW and the other lines.
Results
Establishment and maintenance of the RW line
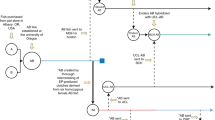
Since 1991, the RW line has been maintained through mass mating in Japan, after being originally sourced from a pet shop in Michigan, USA. For each mating, two female and two male adult zebrafish were placed in a mating tank to produce embryos (Fig. 1A). The embryos were carefully examined for spontaneous developmental defects or mortality by 5 days post-fertilization (dpf). Healthy clutches in which nearly all embryos exhibited normal development up to 5 dpf were selected for rearing to adulthood. After several months, healthy adult fish were used for reproduction (Fig. 1B). Through this rigorous selection process, we successfully eliminated undesirable genetic variants responsible for developmental malformations, fragility, and impaired reproduction. This breeding protocol has been carried out approximately every month since the breeding of the fish colony started in RIKEN in 1997. As a result, clutches with unhealthy embryos are now rarely observed. Originally referred to as the Michigan line, this line was officially renamed RW.

Schematic illustration of the maintenance protocol for the RW line. (A) During each breeding cycle, two pairs of adult zebrafish were housed together in a mating tank to produce embryos. The embryos were screened for developmental abnormalities or mortality up to 5 dpf. Only healthy clutches in which the majority of embryos exhibited normal development by 5 dpf were selected for rearing to adulthood. (B) After reaching maturity, healthy adult fish were used for subsequent breeding. This selection process effectively eliminated harmful genetic variants associated with developmental defects, fragility, and reduced fertility. Breeding has been conducted approximately once a month to sustain the RW line.
Phylogenetic analysis of the zebrafish wild-type lines
To investigate the phylogenetic relationship between the RW line and other wild-type lines, we conducted WGS on three individual fish from each of the following zebrafish lines: RW, AB, TU, TL, WIK, SAT, NHGRI-1 and PET. Additionally, genomic data for inbred lines (three individuals each from M-AB and IM) and their parental lines (three individuals each from *AB and IND), previously obtained in our recent study14, were retrieved from the NCBI Sequence Read Archive (SRA). Using the zebrafish genome assembly of the TU line (TU_GRCz11) as reference data, we searched for single nucleotide polymorphisms (SNPs) in a total of 36 fish (Table 1), identifying 39,421,676 SNP positions, which account for 2.93% of the entire zebrafish genome. As expected, the number of SNPs identified in the TU line was lower than that in the other lines, because TU_GRCz11 was used as the reference genome. The RW line exhibited a higher number of SNPs compared to AB, SAT, and NHGRI-1 line, suggesting that the genetic distance of RW from TU is greater than that of AB, SAT, and NHGRI-1 from TU.
We conducted a phylogenetic analysis of 12 zebrafish lines using Danio aesculapii and Danio nigrofasciatus as closely related outgroups. Our SNP-based phylogenetic analysis confirmed that AB, TU, SAT and NHGRI-1 clustered into a subgroup along with *AB, M-AB, IND and IM (Fig. 2; Supplementary Figure S1). In contrast, RW formed a distinct and independent subgroup. Similarly, TL and PET grouped into a separate cluster. Furthermore, the WIK line was distinctly separated from the other zebrafish lines.

Phylogenetic tree of zebrafish wild-type lines. Phylogenetic relationships among 12 zebrafish lines were analyzed using Danio aesculapii and Danio nigrofasciatus as closely related outgroups. The phylogenetic tree was constructed with 10,000 bootstrap replicates, and the coefficients indicate bootstrap values at the tree nodes.
Genomic characteristics of the zebrafish wild-type lines
We next assessed the heterozygosity of wild-type zebrafish lines. Heterozygosity was defined by the ratio of heterozygous variants to the total genome size (1.345 GB). The heterozygosity of RW (0.348 ± 0.007%) was comparable to those of the AB (0.337 ± 0.010) and WIK (0.345 ± 0.010), higher than those of TU (0.180 ± 0.011%), SAT (0.204 ± 0.0115) and NHGRI-1 (0.285 ± 0.006%) and lower than that of PET (0.408 ± 0.077%) (Fig. 3A). These findings suggest that the genetic variation within the RW line is maintained at levels similar to those observed in AB and WIK lines. In contrast, the heterozygosity of inbred lines (M-AB, 0.011 ± 0.002%; IM, 0.008 ± 0.001%) was lower than that of their parental lines (*AB line, 0.086 ± 0.004%; IND, 0.197 ± 0.0016%) as previously reported14. We also estimated nucleotide diversity (p), confirming that the M-AB and IM lines exhibited extremely low genetic diversity (Fig. 3B).

Genetic heterozygosity of zebrafish lines. (A) Heterozygosity was calculated as the ratio of heterozygous variants to the total nucleotide count in the whole genome. Error bars represent the mean ± standard deviation. Each point in the bar graph corresponds to a biological replicate (n = 3). (B) Nucleotide diversity (p) for each line was depicted using violin plots and box plots.
Genetic variants that may affect gene products in wild-type zebrafish lines
To characterize the genetic features of wild-type zebrafish lines, we focused on SNPs that exhibit nucleotide differences from the reference TU_GRCz11 genome. Some of these SNPs (differences from the database) were found to be homozygous in an individual. We hereafter refer to such SNPs as “homozygous SNPs”, which may potentially cause gene disruption in an individual. These homozygous SNPs were identified in protein-coding regions, 2-bp splice junctions in introns, 5’-untranslated regions (UTRs), 3’-UTRs, other intronic regions or intergenic regions, based on Ensembl annotations (Table 2). The location of homozygous SNPs across these genomic region was comparable among the wild-type lines.
Homozygous SNPs in protein-coding regions can be classified into nonsense, missense and synonymous variants. Among these, nonsense variants result in the truncation of protein synthesis. Disruption of the initiation or termination codon significantly impacts protein function. Similarly, homozygous SNPs in 2-bp splice junctions cause mis-splicing, which leads to a frameshift in translation. Consequently, homozygous SNPs in these categories have the potential to disrupt gene function. We focused on genes harboring such homozygous SNPs that were consistently present in all three individuals of each line (Supplementary Figures S2, S3). Although a considerable number of potentially disrupted genes were identified in each line, the same gene disruptions were detected across multiple lines (Fig. 4). In such cases, gene annotations in Ensembl often comprise intron-containing transcripts, suggesting that some predicted protein-coding sequences may be invalid due to the annotation of minor or inappropriate transcripts24,14. Therefore, we focused on potential gene disruptions uniquely identified in each line as distinct genetic characteristics. The AB and TU lines carried mutations in or124-4 and bcan, respectively (Supplementary Tables S1, S2; Supplementary Figures S4, S5). The TL line uniquely harbored nine gene disruptions including the leot1 mutation, which alters the skin pattern from stripes to spots as a specific trait of the TL line6 (Supplementary Table S3; Supplementary Figures S6-S14). Similarly, unique gene disruptions were identified in the WIK, SAT, NHGRI-1, PET, *AB, IND, M-AB and IM lines (Supplementary Tables S4-S12; Supplementary Figures S15-S52).The RW line carried 13 disrupted genes, specifically sult3st1, ighv 1–2, nexmifa, nitr3r.1 L, nitr7a, mvb12bb, steap3, sctr, stk39, golga4, urb1, invs, cyp2 × 6 (Table 3; Supplementary Figures S53-S65). Furthermore, we identified all missense variants in protein-coding regions and assessed their potential to disrupt protein function based on PROVEAN scores for each line (Supplementary Tables S1-S12).

Number of disrupted genes. Homozygous SNPs that introduce nonsense codons or disrupt start codons, stop codons, or 2-bp splice junctions within protein-coding genes were classified as gene-disrupting genetic variants. The white bars represent the total number of genes disrupted in each line, while the colored bars represent the number of genes uniquely disrupted in each line.
Discussion
This study described the establishment of the wild-type zebrafish RW line and outlined genetic characteristics of the RW and other zebrafish lines. Given its robustness in reproduction and development, the RW line is well-suited for genetic and behavioral studies in zebrafish19,20,21,22,23.
Phylogenetic analysis and genetic divergence among zebrafish lines
Our WGS and SNP-based analysis has created a comprehensive phylogenetic tree of zebrafish wild-type lines that aligns with previous phylogenetic studies25,26,14,11. Specifically, earlier analyses suggested that the AB and TU lines are genetically close compared to wild-caught lines11. Our phylogenetic study confirmed that AB and TU lines are genetically close to SAT and NIGRI-1 lines, which derive from hybrids of AB and TU lines. Furthermore, the subgroup of *AB and *AB-derived M-AB lines as well as that of wild-caught IND and IND-derived IM lines were genetically closer to AB and TU lines rather than RW lines. This suggests that the RW line forms a unique subgroup distinct from AB and TU lines. Additionally, our analysis confirms that the WIK line, originating from wild-caught fish in Kolkata, is highly divergent from the other lines, thereby validating its usefulness in the screening of genetic variation.
Genetic characteristics of wild-type lines
We have identified unique gene variants affecting coding proteins in each line. In the TL genome, we successfully detected the leot1 mutation, a well-known nonsense variant (p.Arg68Ter) of the gja5b gene that encodes for connexin 41.8 protein6. This mutation alters the pigment pattern of the skin from stripes to spots, which is a TL-specific trait in appearance.
In the AB line, we identified two nonsense variants (p.Glu95Ter and p.Leu259Ter) in the or124-4 gene encoding a putative odorant receptor protein, but neither the expression nor function of this odorant receptor has been investigated. A previous study reported that the AB line carries a 1-bp deletion in the coding sequence of the fga gene, which encodes the fibrinogen alpha-chain27,28. This frame-shift mutation results in delayed platelet adhesion after laser-induced venous injury, a specific trait of the AB line. Although our analysis missed this mutation because we focused on SNPs rather than insertions/deletions, manual analysis confirmed the presence of this 1-bp deletion in the fga gene in the AB, SAT and NIGRI-1 lines but not in the other lines (Supplementary Figure S66).
We identified numerous genetic variants that affect protein-coding genes in all lines. However, some genes disrupted by these variants might be pseudogenes or misannotated in the Ensembl database.
Genomic characteristics of the RW line
In the RW line, we identified 13 genetic variants that potentially disrupt genes. Among these, three genes have been characterized in zebrafish. The neurite extension and migration factor a (nexmifa) gene encodes for a cell adhesion molecule and its deficiency causes morphological defects of motor axons at 48 and 72 hpf in zebrafish29. Although the initiation codon of the nexmifa gene was disrupted in the RW line (Supplementary Figure S55), zebrafish embryos of the RW line do not show motor neuron deficits30. Another in-frame methionine codon located downstream of the genetic variant in the nexmifa gene may compensate for gene product translation in this case. The urb1 gene encodes an essential ribosome biogenesis protein, and zebrafish embryos homozygous for a missense mutation (p.Phe80Leu) exhibited impaired proliferation of digestive organs at 48 hpf31. A variant of the splice donor site of exon 1 in the urb1 gene was identified in the RW line (Supplementary Figure S63), whereas defects in the digestive organs have not been reported in embryos from the RW line. The inversin (invs) gene encodes a protein containing ankyrin domains and IQ calmodulin-binding domains. Antisense morpholino-mediated knockdown of invs resulted in renal cysts in zebrafish32. In the RW line, a nonsense mutation was identified near the termination codon of the invs gene, resulting in the deletion of four amino acids at the C-terminus of the protein (Supplementary Figure S64). This truncation may not affect the function of the invs gene product. Indeed, embryos of the RW line do not exhibit renal defects, which are observed in invs morphants. We also identified potentially gene-disrupting variants in the sult3st1, ighv1-2, nitr3r.1 L, nitr7a, mvb12bb, steap3, sctr, stk39, golga4 and cyp2 × 6 genes (Supplementary Figure S53, S54, S56-S62, S65), but the loss of any of these genes has not been previously reported. It should also be noted that gene annotations in zebrafish Ensembl database often include intron-containing transcripts due to annotation errors24. In such cases, SNPs that have no impact on gene products may appear in our findings.
Usefulness of the RW line
In this study, we described the establishment and maintenance of the RW line. Since unwanted genetic variants affecting development and reproduction were removed during the early establishment process, RW fish can be easily maintained in a healthy condition throughout all life stages from embryos to adults. In addition, RW embryos are suitable for microinjection techniques, including antisense knockdown, Tol2 transgenesis and CRISPR/Cas9-mediated genome editing19,20,21,22,23. Taken together, the zebrafish RW line serves as a valuable resource for analyzing genetically susceptible phenotypes such as behaviors, microbiomes and drug susceptibility. The RW line is available from the National BioResource Project (NBRP), an international resource center based in Japan33.
Methods
Ethics declarations
This study was approved by the Animal Care and Ethics Committee of RIKEN Center for Brain Science and Aoyama Gakuin University and carried out according to the Animal Research Reporting of In VIVO Experiments (ARRIVE) guidelines and relevant regulations.
Animals
Zebrafish AB, TU, TL, WIK, SAT, NHGRI-1 lines were purchased from ZIRC. Inbred M-AB and IM lines and their parental *AB and IND lines were described as previously14. The PET was the name of breeding stock of zebrafish, which can be purchased from a local pet store in Japan (Charm, Oizumi, Gunma). The RW line originated from a pet shop in Michigan, USA and has been maintained by mass mating in the fish facilities of National Institute for Basic Biology (1991–1993), Keio University, School of Medicine (1993–1997), and RIKEN Center for Brain Science (1997-present) under the standard laboratory condition. The details of removing unwanted genetic variants are illustrated in Fig. 1 and described in the Results section.
WGS library preparation and sequencing
WGS library preparation was conducted according to previous study14. Briefly, fish were anesthetized in 0.01% ethyl 4-aminobenzoate (Sigma-Aldrich, St. Louis, MO, USA) and their fins clipped, homogenized, and the genomic DNA was extracted using a phenol/chloroform method. The genomic DNA was converted into a sequencing library through fragmentation and ligation processes using the 5X WGS Fragmentation mix & Ligase Mix (QIAGEN, Hilden, Germany), followed by purification using AMpure XP beads (Beckman Coulter, Brea, CA, USA), adapter annealing, as describe previously. The adapter for the ligation step was prepared by annealing of C*A*C*TCTTTCCCTACACGACGCTCTTCCGA*T*C*T and /5Phos/G*A*T*CGGAAGAGCACACGTCTGAACTCCAGT*C*A*C (* and /5Phos/ signify a phosphorylated bond and a phosphorylation, respectively). PCR amplification with indexing involves adding unique index sequences to each sample during PCR using Fw_i5 primer (AATGATACGGCGACCACCGAGATCTACACXXXXXXXXACACTCTTTCCCTACACGACGC) and Rev_i7 primer (CAAGCAGAAGACGGCATACGAGATXXXXXXXXGTGACTGGAGTTCAGACGTGT). XXXXXXXX shown in the above primers are index sequences for multiplex sequencing (Supplementary Table S13). Then, 150 bp paired-end sequencing with DNBSEQ-T7 (MGI Tech, Shenzhen, China) was conducted by BGI Genomics (Shenzhen, China). Only TU line, sequencing of 150 bp paired-end reads was performed using HiSeq X Ten (Illumina, San Diego, CA, USA) by Macrogen, Inc. (Seoul, Korea).
Data analysis
The genome sequence of Danio rerio (Danio_rerio.GRCz11.dna.primary_assembly.fa.gz) was available at https://ftp.ensembl.org/pub/release-109/fasta/danio_rerio/dna34,4. The reference sequence file for mapping was created for these genomic data, excluding extra data other than chromosomes (Chr 1 ~ 25) and the mitochondrial genome sequence (Danio_rerio.GRCz11.dna.primary_assembly.fa.gz). After trimming reads and removing adapter sequences using fastp version 0.20.135,36 with the default parameters except for “--detect_adapter_for_pe: Specify that the sample is paired-end”, the data were mapped to the assembly genome (Danio_rerio.GRCz11.dna.primary_assembly-only-chr.fa) with BWA-MEM version 0.7.17-r118837. The sequence coverage was confirmed to be around 20 for each line. Potential PCR duplicates were marked using the Mark Duplicates tool in Genome Analysis Toolkit GATK version 4.4.0.038. SNPs were detected by the Haplotype Caller tool in GATK using the default parameters except for “-mbq 20: Minimum base quality needed to consider a base for calling” and the Select Variants tool in GATK. VCF-merge in the VCFtools version 0.1.16 package was used to merge all vcf files. Base quality recalibration was performed using the Variant Filtration tool in GATK, and raw SNPs were filtered with the following parameters: QD < 2.0: Variant confidence normalized by unfiltered depth of variant samples; FS > 60.0: Strand bias estimated using Fisher’s Exact Test; MQ < 60: Root Mean Square of the mapping quality of reads across all samples; MQRankSum < -12.5: Rank Sum Test for mapping qualities of REF versus ALT reads; ReadPosRankSum < -8.0: Root Mean Square of the mapping quality of reads across all samples; and DP < 10: Depth of informative coverage for each sample. To assess how amino acids are affected by SNPs, genetic variants at the amino acid level were annotated using SnpEff version 4.3t and GRCz11 10939. The gene annotations were classified by SnpSift version 4.3t40.
Phylogenetic analysis
To reveal the phylogenetic relationships of diverse zebrafish lines, we constructed a phylogenetic tree using The merged vcf file was converted to a PHYLIP file using vcf2phylip version 2.041. A Python 3 script ascbias.py available at https://github.com/btmartin721/raxml_ascbias was used to remove inverted sites from the PHYLIP file. The edited PHYLIP files were evaluated with the bioconda package ModelTest-NG version 0.1.742,43 to select the best-fit model of evolution for DNA alignments. ML phylogenetic tree construction was carried out with IQ-tree version 2.0.744 using the GTR + G4 + ASC model and 10,000 bootstrap replicates. iTOL version 645 was used for final editing (https://github.com/Hirata-lab-2023/RW_line/phy.sh). The sequencing data for Danio aesculapii (ERR3332304) and Danio nigrofasciatus (ERR034323) were obtained from the NCBI SRA.
SNP analysis
Heterozygous and homozygous SNP analysis was performed with a homemade script (https://github.com/Hirata-lab-2023/RW_strain/anlysis.R) using the core tools of R 4.2.3 and the R package – ggplot2 version 3.4.246, openxlsx version 4.2.5.2, patchwork version 1.1.2, ggsignif version 0.6.4, dplyr version 1.1.2, ggbreak version 0.1.147, stringr version 1.5.0, UpSetR version 1.4.048, reshape2 version 1.4.446, and sets version 1.0–2449. The numbers of heterozygous SNPs were counted in each individual, and the percentages per total genome nucleotides were calculated. Nucleotide diversity (π) was calculated using VCFtools version 0.1.16 with the --window-pi and --window-pi-step options. A sliding window approach was applied with a window size of 10,000 base and a step size of 2,000 base across the genome to estimate local nucleotide diversity levels. To evaluate the functional effects of nonsynonymous mutations, we constructed a computational pipeline, Z-VCFAA, which converts VCF files into corresponding protein FASTA sequences. This pipeline enables downstream functional analysis of amino acid substitutions and is publicly available for reproducibility and accessibility (https://github.com/Hirata-lab-2023/Z-VCFAA-). Based on the generated FASTA sequences, we performed functional impact prediction of missense variants using PROVEAN version 1.1.550. All figures were edited using Adobe Illustrator version 26.4.1.
Statistical analysis
Data are presented as the mean ± standard deviation of at least three independent experiments. The results of the statistical test are indicated as * (P ≤ 0.05), ** (P ≤ 0.01), *** (P ≤ 0.001) or **** (P ≤ 0.0001). P ≤ 0.05 was considered statistically significant. Graphical presentations were made with the R package ggplot246.
Data availability
Our sequencing data are deposited in the NCBI Sequence Read Archive as follows (Bio Project: PRJNA1110785): AB_1: SRR29004176; AB_2: SRR25514324; AB_3: SRR29004174 ; TU_1: SRR30229261; TU_2: SRR29006348; TU_3: SRR29006347 TL_1: SRR29004373; TL_2: SRR29004372, TL_3: SRR29004371; WIK_1: SRR29004179; WIK_2: SRR29004178; WIK_3: SRR29004177; SAT_1: SRR29004367; SAT_2: SRR29004366; SAT_3: SRR29004365; NHGRI-1_1: SRR29004370; NHGRI-1_2: SRR29004369; NHGRI-1_3; SRR29004368; PET_1; SRR29004364; PET_2: SRR29004363; PET_3: SRR29004362; RW_1: SRR29014430; RW_2: SRR29014429; RW_3: SRR29014428. Whole-genome sequences of M-AB (M-AB_1: SRR25514300; M-AB_2: SRR25514299; M-AB_3: SRR25514298), *AB (*AB_1: SRR25514325; *AB_2: SRR25514324; *AB_3: SRR25514323), IM (IM_1: SRR25514304; IM_2: SRR25514303; IM_3: SRR25514302) and IND (IND_1: SRR25514329; IND_2: SRR25514328; IND_3: SRR25514327.) were obtained from the NCBI Sequence Read Archive.
References
Streisinger, G., Walker, C., Dower, N., Knauber, D. & Singer, F. Production of clones of homozygous diploid zebra fish (Brachydanio rerio). Nature 291 (5813), 293–296 (1981).
Trevarrow, B. & Robison, B. Genetic backgrounds, standard lines, and husbandry of zebrafish. Zebrafish:2nd Edition Genetics Genomics and Informatics 77, 599–616. (2004).
Mullins, M. C., Hammerschmidt, M., Haffter, P. & Nusslein-Volhard, C. Large-scale mutagenesis in the zebrafish: In search of genes controlling development in a vertebrate. Curr. Biol. 4 (3), 189–202 (1994).
Howe, K. et al. The zebrafish reference genome sequence and its relationship to the human genome. Nature 496 (7446), 498–503 (2013).
Stewart, S. et al. J. A. and ‘longfin causes cis-ectopic expression of the kcnh2a ether-a-go-go K + channel to autonomously prolong fin outgrowth. Development 148 (11). (2021).
Watanabe, M. et al. Spot pattern of Leopard Danio is caused by mutation in the zebrafish connexin 41.8 gene. EMBO Rep. 7 (9), 893–897 (2006).
LaFave, M. C., Varshney, G. K., Vemulapalli, M., Mullikin, J. C. & Burgess, S. M. A defined zebrafish line for high-throughput genetics and genomics: NHGRI-1. Genetics 198 (1), 167–170 (2014).
Nasiadka, A. & Clark, M. D. Zebrafish breeding in the laboratory environment. ILAR J. 53 (2), 161–168 (2012).
Anderson, J. L. et al. Multiple sex-associated regions and a putative sex chromosome in zebrafish revealed by RAD mapping and population genomics. PLoS ONE. 7 (7), e40701 (2012).
Rauch, G. J., Granato, M. & Haffter, P. A polymorphic zebrafish line for genetic mapping using SSLPs on high-percentage agarose gels. Tech. Tips Online. 2 (1), 148–150 (1997).
Wilson, C. A. et al. rd, ‘Wild sex in zebrafish: Loss of the natural sex determinant in domesticated strains. Genetics 198 (3): 1291 – 308. (2014).
Chakrabarti, S., Streisinger, G., Singer, F. & Walker, C. Frequency of gamma-Ray induced specific locus and recessive lethal mutations in mature germ cells of the zebrafish, BRACHYDANIO RERIO. Genetics 103 (1), 109–123 (1983).
Johnson, S. L., Africa, D., Horne, S. & Postlethwait, J. H. Half-tetrad analysis in zebrafish: Mapping the Ros mutation and the centromere of linkage group I. Genetics 139 (4), 1727–1735 (1995).
Sadamitsu, K. et al. Establishment of a zebrafish inbred strain, M-AB, capable of regular breeding and genetic manipulation. Sci. Rep. 14 (1), 7455 (2024).
Shinya, M. & Sakai, N. Generation of highly homogeneous strains of zebrafish through full sib-pair mating. G3 (Bethesda). 1 (5), 377–386 (2011).
Walker, C. Chapter 3 haploid screens and Gamma-Ray mutagenesis. Methods Cell. Biol. 60, 43–70 (1998).
Stachel, S. E., Grunwald, D. J. & Myers, P. Z. Lithium perturbation and goosecoid expression identify a dorsal specification pathway in the pregastrula zebrafish. Development 117 (4), 1261–1274 (1993).
Wakamatsu, Y., Ogino, K. & Hirata, H. Swimming capability of zebrafish is governed by water temperature, caudal fin length and genetic background. Sci. Rep. 9 (1), 16307 (2019).
Higashijima, S., Hotta, Y. & Okamoto, H. Visualization of cranial motor neurons in live Transgenic zebrafish expressing green fluorescent protein under the control of the islet-1 promoter/enhancer. J. Neurosci. 20 (1), 206–218 (2000).
Masai, I. et al. N-cadherin mediates retinal lamination, maintenance of forebrain compartments and patterning of retinal neurites. Development 130 (11), 2479–2494 (2003).
Ohata, S. et al. Dual roles of Notch in regulation of apically restricted mitosis and apicobasal Polarity of neuroepithelial cells. Neuron 69 (2), 215–230 (2011).
Wada, H. et al. Dual roles of zygotic and maternal Scribble1 in neural migration and convergent extension movements in zebrafish embryos. Development 132 (10), 2273–2285 (2005).
Wada, H., Tanaka, H., Nakayama, S., Iwasaki, M. & Okamoto, H. Frizzled3a and Celsr2 function in the neuroepithelium to regulate migration of facial motor neurons in the developing zebrafish hindbrain. Development 133 (23), 4749-59. (2006).
Sadamitsu, K. et al. Characterization of zebrafish GABA(A) receptor subunits. Sci. Rep. 11 (1), 6242 (2021).
Guryev, V. et al. Genetic variation in the zebrafish. Genome Res. 16 (4), 491–497 (2006).
McCluskey, B. M. & Postlethwait, J. H. Phylogeny of zebrafish, a model species, within Danio, a model genus. Mol. Biol. Evol. 32 (3), 635–652 (2015).
Fish, R. J., Di Sanza, C. & Neerman-Arbez, M. Targeted mutation of zebrafish Fga models human congenital afibrinogenemia. Blood 123 (14), 2278–2281 (2014).
Freire, C. et al. A genetic modifier of venous thrombosis in zebrafish reveals a functional role for fibrinogen aalphae in early hemostasis. Blood Adv. 4 (21), 5480–5491 (2020).
Zheng, Y. Q. et al. Nexmifa regulates axon morphogenesis in motor neurons in zebrafish. Front. Mol. Neurosci. 15, 848257 (2022).
Uemura, O. et al. Comparative functional genomics revealed conservation and diversification of three enhancers of the isl1 gene for motor and sensory neuron-specific expression. Dev. Biol. 278 (2), 587–606 (2005).
He, J. et al. Ribosome biogenesis protein Urb1 acts downstream of mTOR complex 1 to modulate digestive organ development in zebrafish. J. Genet. Genomics. 44 (12), 567–576 (2017).
Simons, M. et al. Inversin, the gene product mutated in nephronophthisis type II, functions as a molecular switch between Wnt signaling pathways. Nat. Genet. 37 (5), 537–543 (2005).
Okamoto, H. & Ishioka, A. Zebrafish research in Japan and the National bioresource project. Exp. Anim. 59 (1), 9–12 (2010).
Broughton, R. E., Milam, J. E. & Roe, B. A. The complete sequence of the zebrafish (Danio rerio) mitochondrial genome and evolutionary patterns in vertebrate mitochondrial DNA. Genome Res. 11 (11), 1958–1967 (2001).
Chen, S. Ultrafast one-pass FASTQ data preprocessing, quality control, and deduplication using Fastp. Imeta 2 (2), e107 (2023).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. Fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34 (17), i884–i890 (2018).
Li, H. ‘Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM (2013). arXiv:1303.3997
McKenna, A. et al. The genome analysis toolkit: A mapreduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20 (9), 1297–1303 (2010).
Cingolani, P. et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly. (Austin). 6 (2), 80–92 (2012b).
Cingolani, P. et al. Using Drosophila melanogaster as a model for genotoxic chemical mutational studies with a new program, SnpSift. Front. Genet. 3, 35 (2012a).
Ortiz, E. M. ‘vcf2phylip v2.0: convert a VCF matrix into several matrix formats for phylogenetic analysis. zenodo. (2019).
Darriba, D. et al. ModelTest-NG: A new and scalable tool for the selection of DNA and protein evolutionary models. Mol. Biol. Evol. 37 (1), 291–294 (2020).
Flouri, T. et al. The phylogenetic likelihood library. Syst. Biol. 64 (2), 356–362 (2015).
Nguyen, L. T., Schmidt, H. A., von Haeseler, A. & Minh, B. Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32 (1), 268–274 (2015).
Letunic, I. & Bork, P. Interactive tree of life (iTOL) v6: Recent updates to the phylogenetic tree display and annotation tool. Nucleic Acids Res. 52 (W1), W78–W82 (2024).
Wickham, H. ‘Reshaping Data with the reshape Package. Journal of Statistical Software 21 (12). (2007).
Xu, S. B. et al. Use ggbreak to effectively utilize plotting space to deal with large datasets and outliers. Front. Genet. 12. (2021).
Conway, J. R., Lex, A. & Gehlenborg, N. UpSetR: An R package for the visualization of intersecting sets and their properties. Bioinformatics 33 (18), 2938–2940 (2017).
Meyer, D. & Hornik, K. Generalized and customizable sets in R. J. Stat. Softw. 31 (2), 1–27 (2009).
Choi, Y., Sims, G. E., Murphy, S., Miller, J. R. & Chan, A. P. Predicting the functional effect of amino acid substitutions and indels. PLoS One. 7 (10), e46688 (2012).
Acknowledgements
We thank members of the Okamoto laboratory and Hirata laboratory for fish care. We also thank Drs. Yo Yamasaki and Shigehiro Kuraku (National Institute of Genetics) for critical discussions on phylogenetic analysis. This work was supported by the Japan Agency for Medical Research and Development (AMED) (23mk0101223h0602), and the Long-Range Research Initiatives of the Japan Chemical Industry Association to HH and National Bioresource Project of Japan-Zebrafish to HO.
Author information
Authors and Affiliations
Contributions
K.S., M.K., H.O. and H.H. designed research. K.S., M.K., S.W., A.I., S.N. and R.N. performed experiments and analyzed data. K.S. and H.H. prepared Figures and Tables. K.S., H.O. and H.H. wrote the manuscript. All authors reviewed and approved the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Sadamitsu, K., Kashima, M., Wada, S. et al. Establishment and genetic characterization of zebrafish RW line. Sci Rep 15, 14512 (2025). https://doi.org/10.1038/s41598-025-98674-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-98674-w
