
Abstract
Research in Inflammatory Bowel Disease (IBD) assessing the genetic structure and its association with IBD phenotypes is needed, especially in IBD-underrepresented populations such as the South American IBD population. Aim. We examine the correlation between Amerindian ancestry and IBD phenotypes within a South American cohort and investigate the association between previously identified IBD risk variants and phenotypes. We assessed the ancestral structure (IBD = 291, Controls = 51) to examine the association between Amerindian ancestry (AMR) and IBD variables. Additionally, we analyzed the influence of known IBD genetic risk factors on disease outcomes. We used Chi-square and Fisher’s tests to analyze the relationship between phenotypes and ancestry proportions, calculating odds ratios (OR) and confidence intervals (CI). Logistic regression examined genetic variants associations with IBD outcomes, and classification models for predicting prolonged remission were developed using decision tree and random forest techniques. The median distribution of global ancestry was 58% European, 39% Amerindian, and 3% African. There were no significant differences in IBD risk based on ancestry proportion between cases and controls. In Ulcerative colitis (UC), patients with a high Amerindian Ancestry Proportion (HAAP) were significantly linked to increased chances of resective surgery (OR = 4.27, CI = 1.41–12.94, p = 0.01), pouch formation (OR = 7.47, CI = 1.86–30.1, p = 0.003), and IBD reactivation during COVID-19 infection (OR = 5.16, CI = 1.61–6.53, p = 0.005). Whereas, in the Crohn’s Disease (CD) group, the median Amerindian ancestry proportion was lower in the group with perianal disease (33.5% versus 39.5%, P value = 0.03). CD patients with High Amerindian Ancestry proportion had lower risk for surgery (OR = 0.17, CI = 0.03–0.83, P value = 0.02). Our study highlights the impact of Amerindian ancestry on IBD phenotypes, suggesting a role for genetic and ancestral factors in disease phenotype. Further investigation is needed to unravel the underlying mechanisms driving these associations.
Similar content being viewed by others


Introduction
Inflammatory Bowel Disease (IBD) includes Crohn’s disease (CD) and ulcerative colitis (UC). The etiology of IBD is multifactorial, involving a complex interplay of genetic predispositions, environmental influences, and dysregulation of gut microbiota, leading to an aberrant immune response1. While the precise cause of IBD remains elusive, the clinical progression of the disease is notably heterogeneous among patients, which makes it an unpredictable disease2.
More than 240 genetic variants related to IBD have been identified3,4,5. Unlike classical Mendelian disorders, IBD is a genetically complex disease, and traditional genetic analytics are not enough to shape the disease´s complexity6. However, genetic studies have helped to answer in some cases which individuals have more IBD risk and which IBD patients will suffer a disabling course of the disease7. Host genetic factors are known to influence susceptibility to IBD. Furthermore, ancestry influences the risk to develop the disease and disease presentation8. An important limitation of the first genetic studies in IBD is that they were primarily conducted on people of European Ancestry9. Recent genetic studies have incorporated diverse populations, revealing that IBD prediction is enhanced when utilizing data from multiple ancestry groups compared to single population data. This approach has significant implications in identifying population-specific variants, which could facilitate the development of targeted treatments10. South American populations, such as the Chilean population, are underrepresented in Genomic Wide Association Studies (GWAs). Our study aimed to explore, in a South American sample, the relationship between ancestry proportion and IBD clinical phenotypes. Additionally, we assessed the impact of previously identified IBD risk variants from IBD GWAs on the disease clinical outcomes. We used traditional statistical analysis and machine learning tools to develop predictive models to accomplish this objective.
Methods
Patient recruitment
We conducted an observational and prospective study at Hospital San Borja Arriarán (HSBA), a tertiary referral center for IBD in Santiago, Chile. The study included patients from similar socioeconomic backgrounds, classified as working class (D) and lower middle class (C3) according to the scale of the Association of Market Researchers and Public Opinion, Chile11. Patients were enrolled if they had an IBD diagnosis supported by clinical, endoscopic, histologic, and imaging data according to clinical guidelines12,13,14 and International Disease Classification criteria. Patients were invited to participate during scheduled colonoscopies as requested by their doctors. A comprehensive database of relevant clinical data was compiled for all participants at the time of recruitment. We collected clinical data common data for UC and CD including sex, age, age at diagnosis, age less than 40 years at diagnoses, alcohol consumption/smoking habits, IBD family history, extraintestinal manifestations (EIMs). Main IBD phenotypes were classified according to the Montreal classification15. Moreover, history of infection for cytomegalovirus, Clostridioides difficile, Coronavirus 19 infection (COVID-19), laboratory parameters, IBD resective surgery, pouch, current use of steroids, immunomodulators, biological therapies, naïve anti-TNF, history of discontinuation or failure to anti-TNF, primary no response to anti-TNF, loss of response to anti-TNF, history of immunogenicity to biological therapy, clinical activity (Harvey Bradshaw index for CD and Total Mayo score for UC), endoscopy activity (Simple Endoscopic Activity Score in Crohn’s Disease (SES-CD) for CD and Mayo Score for UC) was registered. Clinical remission was defined for CD as a Harvey Bradshaw less than 5 and for UC as a total Mayo score less than 316. Endoscopy remission for CD as a SES-CD less than 3 and for UC as an Endoscopy Mayo score 016. Histologic remission defined by absence of erosion, ulceration, and epithelial damage and absence of neutrophils17,18. The authors defined prolonged Clinical and endoscopic remission as clinical and endoscopic remission over the last five years and frequent relapse by more than one flare per year over the previous five years. Furthermore, a control group comprised patients who underwent a colonoscopy indicated by their doctor. These patients did not have any conditions such as IBD, immune disorders, or cancer, and obtained normal findings on the exam. Ethics approval was obtained from the Institutional Review Boards of the Servicio de Salud Metropolitano Central/HSBA (IRB:43/2022) and the Pontificia Universidad Católica de Chile (IRB:220228001). All individuals provided written informed consent. All methods were performed in accordance with the relevant guidelines and regulations.
Genotyping
Five mL of blood was retrieved from each participant and stored in ethylenediaminetetraacetic acid disodium salt (EDTA) tubes. Then DNA was extracted with Invisorb Blood Universal (Invitek) # ref 1,031,150,200 purification kit, according to the manufacturer’s suggestions. Samples were stored at -80º C until genotyped at Erasmus MC-Netherlands, and 725.497 single nucleotide polymorphisms (SNPs) were investigated using Illumina’s Infinium Global Screening Array.
Genotyping QC
Genotype quality control (QC) was performed using R Studio version 4.2.2 with the plinkQC library. The perMarkerQC function was utilized to assess missingness rates across samples, deviation from Hardy-Weinberg Equilibrium (HWE), and minor allele frequencies (MAF) by applying a threshold of 0.01 (Supplementary Fig. 1). Additionally, the perIndividualQC function was employed to evaluate the total heterozygosity rates, missingness, concordance of assigned sex with SNP sex, relatedness to other study individuals, and genetic ancestry of the samples in the PLINK dataset. Supplementary data shows the QC for individuals and markers (Supplementary Figs. 1 and 2).
Estimation of genetic ancestry
We performed a global ancestry analysis using the admixture19. We employed a reference panel of populations obtained from the 1000 Genome Project and HapMap for our analyses. This included the Native American population (AMR = 43 unrelated individuals), the European population (CEU = 56 unrelated individuals), the African population (YRI = 55 unrelated individuals), and our dataset of 342 individuals from the Chilean population. Of Note, the 43 native American samples exhibited 99% or higher Native American ancestry. This cohort was assembled from a collective of populations, including ten individuals from the Nahua, six from the Maya, two from the Quechua, and twenty-five from the Aymara. Additionally, reference populations from 1000 Genomes Project (ASW: African Ancestry in Southwest USA, TSI: Toscani in Italy, IBS: Iberian Population in Spain, MXL: Mexican Ancestry in Los Angeles, USA, PUR: Puerto Ricans in Puerto Rico, CLM: Colombians from Medellin, Colombia, PEL: Peruvians from Lima, Peru) were included for population structure and admixture analysis comparison.
PLINK20 was used to manipulate the VCF and bed file formats from HAPMAP obtaining a total of 41,193 SNPs with genotypes for all 496 individuals in the study. To reduce the impact of linkage disequilibrium on our ancestry estimation, we pruned and filtered the SNPs using the Plink options (--indep-pairwise 50 10 0.1 –-geno 0.01), resulting in a refined set of 23,716 SNPs suitable for ADMIXTURE analysis. We then leveraged the ADMIXTURE cross-validation option (--cv) to ascertain the optimal number of ancestral populations, or clusters, for a supervised analysis (Supplementary Fig. 3). ADMIXTURE analysis was performed for two to six possible ancestral groups (K = 2 … K = 6), aiming to pinpoint the number of ancestral populations corresponding to the lowest CV error, as detailed in Supplementary Fig. 3. Our iterative approach, which involved testing various K values, determined that a K value of 3 yielded the lowest average CV error. This indicates that three ancestral populations most accurately represent the genetic foundation of the Chilean individuals in this study. For comparison, we conducted an additional ADMIXTURE analysis incorporating 1000 Genomes (1000G) data. Using PLINK, we merged genotype data from 962 individuals across 232,297 SNPs, including reference populations ASW (55), TSI (107), IBS (107), MXL (64), PUR (104), CLM (97), PEL (86), and CHI (342). After variant pruning and minor allele frequency (MAF) filtering (MAF > 0.05), a total of 96,786 markers were retained for ADMIXTURE analysis (Fig. 1).

Population Structure and Genetic Ancestry Composition of Chilean Individuals. (A) Principal Component Analysis (PCA): PCA plot of SNP data showing clustering by superpopulation (AFR: African populations, AMR: American populations, EAS: East Asian populations, EUR: European populations, SAS: South Asian populations). PC1 (47%) and PC2 (26%) capture most of the genetic variation, with Chilean individuals clustering with the Admixed American populations, reflecting their admixed ancestry. (B) Global ancestry proportions of Chilean individuals inferred using ADMIXTURE (K = 3). Three primary ancestry components are identified: European (EUR, green), Amerindian (AMR, orange), and African (AFR, red). Reference populations from 1000 Genomes Project (ASW, TSI, IBS, MXL, PUR, CLM, PEL) were included for comparison. (C) Ancestry Proportions: Boxplots displaying the proportion of European, Amerindian, and African ancestry in the Chilean cohort. The results highlight the predominance of European (x = 58%) and Amerindian(x = 39%) ancestry, with a smaller African (x = 3%) component.
Furthermore, a genetic Principal Component Analysis (PCA) was conducted using PLINK and the 1000Genome data. The PCA was performed using a total of 107,891 SNPs from 2915 unrelated individuals, and 20 components (Fig. 1). Finally, the admixture and PCA results were visualized using the libraries ggplot and tidyverse from R studio version 4.2.2.
Statistical methods
We analyzed association between the phenotype and ancestry proportions using a Chi-square test and Fisher test. Additionally, we calculated the odds ratio using the Wald method. For these analyses, we utilized the epitools, readxl, and rapportools libraries from R version 4.2.2. Next, we explored the association between Amerindian ancestry proportion (AMR) and categorical (demographical and clinical) variables. We also examined the relationship between AMR and numerical variables. Statistical analyses were performed using Python libraries such as pandas, seaborn, matplotlib, and scipy.stats. To compare the median of the quantitative variables between the two categorical groups, we employed the Mann-Whitney U test, a non-parametric test. Regarding the categorical variables, we utilized the Chi-square test and Fisher’s test to assess significant differences between the groups. Odds ratios (OR) and confidence intervals (CI) were calculated to further evaluate the associations. We considered a p-value < 0.05 as indicative of significance. From published IBD GWAs studies,1,2,21 we investigated 226 SNPs related to IBD among 291 IBD Chilean genotypes obtained from the bim, fam, and ped files from the Illumina array after performing the GWAS quality control. Using R studio version 4.2.3 and the libraries genio, plinkFile, readr, and tidyverse, we filtered the226 mentioned variants (Supplementary Table 1). A total of171 variants were found in our Chilean cohort. This information was integrated to build a database merging the clinical data with the genotypes. We aimed to explore the potential association between SNP genotypes related to IBD and High Amerindian Ancestry Proportion (HAAP), defined as greater than 43%, representing the third quartile of the AMR population in our sample. A contingency table was constructed, and a Chi-square test was conducted using Python programming and libraries such as pandas, seaborn, and matplotlib.pyplot to determine the statistical significance of the association. The significance threshold was set at 0.05. Subsequently, we conducted logistic regression analyses, incorporating covariates such as age, sex, smoking, and current anti-TNF therapy to account for potential confounders. These analyses utilized the readxl library for data import, dplyr for data manipulation, and stats for statistical modeling in R. The same analysis was performed to explore the association between prolonged clinical and endoscopy remission and SNPs related to IBD.
Furthermore, leveraging our previous study, where we developed a regression model for various binary clinical outcomes22, our current research focuses on constructing a classification model specifically for prolonged clinical/endoscopic remission. The aim was to examine the relevance of various features in predicting this outcome. These features encompassed clinical outcomes, laboratory parameters, ancestry proportions, and SNPs. To achieve this, tree decision and random forest techniques were employed to understand better the genetic and clinical factors associated with prolonged clinical/endoscopic remission. In our model-building process, we utilized Python and various libraries. Pandas aided in data manipulation and analysis, numpy facilitated mathematical operations, and matplotlib.pyplot and seaborn were used for data visualization. Data preprocessing involved scaling with StandardScaler and handling missing values using SimpleImputer. The data was split into training and testing sets using the train_test_split function from sklearn.model_selection.
We experimented with algorithms for classification models, including Logistic Regression, Decision Tree Classifier, and Random Forest Classifier from sklearn.linear_model, sklearn.tree and sklearn.ensemble Python libraries. Model performance evaluation employed metrics such as confusion matrix, classification report, precision-recall curve, and recall score from sklearn.metrics. Data preprocessing techniques like MinMaxScaler, Label Encoder, and One Hot Encoder from sklearn.preprocessing were applied as needed. To optimize the models, we utilized GridSearchCV from sklearn.model_selection for hyperparameter tuning, enabling fine-tuning of the models to improve performance and accuracy.
Results
We genotyped 384 IBD patients and controls at Erasmus MC-Netherlands using Illumina’s Infinium Global Screening Array, resulting in the genotyping of 725,497 SNPs. However, after applying standard filters and quality control, 357,392 variants and 342 individuals (291 cases and 51 controls) remained for further analysis. Most of the SNPs discarded (n = 232,141) were not polymorphic in the Chilean individuals (MAF < 0.01, Supplementary Fig. 1–2). The genetic structure of the Chilean population, as revealed by PCA and Admixture analysis, demonstrates the admixed nature of the Chilean Population (Fig. 1). The PCA shows Chilean individuals are positioned with the Admixed American (AMR) populations, confirming their mixed genetic background. This positioning aligns with historical admixture events involving European colonizers, indigenous Native American groups, and African descendants. Figure 1B, the admixture analysis, further supports this by identifying three primary ancestral clusters (African, European, and Native American) within the Chilean population. The analysis reveals the most predominant ancestries: Native American and European ancestry, with a more minor but significant African contribution. Figure 1C quantifies these proportions, confirming that most Chilean individuals have a substantial Native American ancestry component, a notable European contribution, and a minor African component. Together, these results highlight the diverse genetic heritage of the Chilean population, shaped by historical migration, colonization, and the transatlantic slave trade, and distinguish Chileans from other reference populations such as African Americans (ASW), Puerto Ricans (PUR), and Peruvians (PEL). The ancestry structure composition is illustrated in Fig. 1, revealing the presence of three primary ancestry groups. The green color represents European ancestry, the orange color represents Amerindian ancestry, and the red color represents African ancestry. Figure 1B presents the results of the ADMIXTURE analysis, showing the cross-validation error for ancestral population clusters (K = 2–6) within the Chilean cohort. The optimal number of ancestral populations is identified at K = 3, utilizing CEU (European), AMR (Amerindian), and YRI (African) as reference panels. The PCA was conducted to examine the ancestry relationships (Fig. 1C). The distribution of ancestry proportions in the studied population is provided in Table 1. This cohort’s median ancestry distribution comprised 58% European, 39% Amerindian, and 3% African ancestry. Notably, the third quartile of Amerindian ancestry proportion was calculated as 42.9%, prompting us to classify a high Amerindian ancestry proportion as 43% for subsequent analysis. The analysis of IBD risk did not reveal any significant differences based on ancestry proportion when comparing cases and controls (Supplementary Tables 2 and 3).
A total of 291 patients with IBD were included in the study, with 216 (74%) diagnosed with UC and 75 (26%) with CD. The clinical characteristics of the investigated IBD patients are summarized in Table 2. The median age of the patients was 50 years (range: 15–81), and the median duration of disease was nine years (range: 0–49 years). Extra-intestinal manifestations were reported by 36% of the IBD patients, and more than 50% had a history of hospitalization. Most patients were non-smokers. Surgical resection was reported by 16% of the patients, and 10% had a history of Clostridioides difficile infections. At the time of the study, 15% of the patients were using steroids, 15% were on anti-TNF therapy, 81% were anti-TNF naive, and 31% were using thiopurines. According to the Montreal Classification, 55% of UC cases had extensive colitis, 26% had left-sided colitis, 18% had proctitis, and information on disease extent was unavailable for 1% of cases. In the CD group, only 8% were diagnosed before 17. The most common disease extension in CD was colonic (L2) involvement, observed in 51% of cases, followed by ileocolonic (L3) involvement in 33% of cases. Upper digestive tract involvement (L4) was present in only 9% of CD cases, and 43% had perianal involvement. The most frequently observed CD phenotype was inflammatory (B1, 41%), followed by penetrating (B3, 33%) and structuring (B2, 25%).
Several findings were observed when examining the association between AMR and clinical variables in the UC group. Firstly, the median AMR was higher in patients diagnosed before the age of 40 compared to those diagnosed later (39.9% versus 37.4%, P value = 0.01). Conversely, it was lower in the patients who achieved maintained clinical and endoscopic remission in the last five years (35% versus 39%, P value = 0.02). Interestingly, a higher median AMR was associated with IBD reactivation during a COVID-19 infection (43% versus 39%, P value = 0.006). See Table 3 Among the studied variables was a family history of IBD; however, we did not find any association with the median AMR (40% vs. 39%, P value = 0.39). In addition to these associations, we further explored the impact of a HAAP (High Amerindian Ancestry Proportion ≧ of 43%) on clinical outcomes within the UC group. This HAAP was significantly associated with resective surgery (57, OR = 4.27, CI = 1,41-12.94, p-value = 0.01), pouch (70%, OR = 1.86–3.01, p-value = 0.003), clinical and endoscopic remission over one year (19%, OR = 0.46,CI = 0.24-0,87), and IBD reactivation during a COVID-19 infection (62%, OR = 5.16, CI = 1.61–6.53) as shown in Table 4. Interestingly, 70% of UC patients who required pouch formation had a HAAP. Moreover, among the UC patients who maintained clinical and endoscopic remission over one year, 81% had a lower Amerindian ancestry proportion, while only 19% had HAAP. Additionally, 62% of UC patients who experienced a COVID-19 infection had a HAAP.
In the CD group, we observed that the median AMR was lower in the group with perianal disease than the group without perianal disease (33.5% versus 39.5%, P value = 0.03). Additionally, only 6% of the CD patients who underwent resective surgery had HAAP (OR = 0.17, CI = 0.03–0.83, P value = 0.02). These findings suggest that there may be a potential association between Amerindian ancestry and a lower likelihood of developing perianal disease and requiring resective surgery in the CD group (Table 5). These results provide valuable insights into the potential role of Amerindian ancestry in influencing the phenotype of CD within this specific cohort.
In our analysis, we integrated data from both UC and CD patients to explore the potential association between Amerindian ancestry and standard clinical variables in IBD. We observed that in IBD patients diagnosed before the age of 40, the median AMR was higher compared to those diagnosed later (40% versus 38%, P value = 0.03). Similarly, we found a similar trend in the group of IBD patients who experienced one or more outcomes associated with severe disease, such as surgery, failure to anti-TNF treatment, pouch, or flares in the last five years. In this group, the median AMR was 39.4% compared to 34.9% in the reference group (P value = 0.0007). Conversely, a lower median Amerindian ancestry proportion was observed in the group of IBD patients currently on biological therapy (36.6% versus 39.3%, P value = 0.03) and those who achieved sustained clinical and endoscopic remission in the last five years (36.2% versus 39.6%, P value = 0.0006). Additionally, in the IBD group, we found a significant association between Amerindian ancestry proportion and a history of gastrointestinal infection, previous Clostridioides difficile infection, as well as prolonged clinical and endoscopic remission (over five years). See Tables 6 and 7. Figure 2, summarize these results.

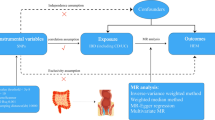
Impact of Amerindian Ancestry on Clinical Variables in IBD Subgroups. (A) Median Amerindian ancestry proportion across various clinical features in IBD (Median group yes versus Median group no). A higher median Amerindian ancestry proportion was associated with early-onset IBD/UC, a severe disease course (IBD), and UC flare during COVID-19 infection. Conversely, a lower median Amerindian ancestry proportion is linked to prolonged clinical and endoscopic remission in UC and IBD, current use of biological therapy in IBD, and perianal disease in CD (B) High Amerindian Ancestry Proportion Impact on Clinical Outcomes. We defined HAAP as an Amerindian ancestry proportion equal to or greater than 43%. In the UC group, a high proportion of patients with HAAP had a history of pouch formation, surgical resection, and IBD flare during a COVID-19 infection. Conversely, most patients who achieved clinical and endoscopic remission over a year (UC), underwent resective surgery (CD), had a previous history of gastrointestinal infection (IBD), experienced past infection by Clostridioides (IBD), or had prolonged clinical and endoscopic remission (IBD) did not have HAAP. IBD: Inflammatory Bowel Disease, UC: Ulcerative Colitis, CD: Crohn’s Disease, HAAP: High Amerindian Ancestry Proportion, COVID-19=Coronavirus 19 infection.
We also investigated the potential association between HAAP and genotypes of SNPs previously associated with IBD. The SNPs significantly associated with this outcome are shown in SupplementaryTable 5. Furthermore, we performed a gene set enrichment analysis using gProfiler, using the genes to which these SNPs were mapped. Our analysis revealed a significant enrichment of cellular response to interleukin-6 (GO:0071354, p-value adjusted = 0.05) and histone H3Y41 kinase activity (GO:00035401, p-value adjusted = 0.03) within the gene/protein set. These findings might suggest a potential involvement of IL-6 cytokine according to Amerindian ancestry. Further studies will be interesting in exploring how histone modifications influence gene expression patterns in IBD within populations, such as in Latin American countries, where epigenetic changes may account for the rising incidence of IBD23 (Table 8).
We found a significant association between previously identified SNPs linked to IBD and prolonged clinical and endoscopy remission, as shown in Table 9 and Supplementary Table 4. Additionally, a gene set enrichment analysis (Table 10) revealed that the genes associated with these SNPs were connected to specific enzyme activities, including L-cystine L-cysteine-lyase (deaminating) (GO:0044540, adjusted P value = 0.04), homocysteine desulfhydrase activity (GO:0047982, adjusted P value = 0.04), cystathionine gamma-lyase activity (GO:0004123, adjusted P value = 0.04), selenocystathionine gamma-lyase activity (GO:0098606, adjusted p-value = 0.04), and L-cysteine desulfhydrase activity (GO:0080146, adjusted P value = 0.04). The connection between these enzymes, IBD prolonged clinical and endoscopy remission, and microbiota interaction presents an intriguing avenue for future research24,25,26.
As mentioned, we have developed a classification model to evaluate the prolonged clinical and endoscopy remission. This classification model provides an opportunity to explore the feasibility of utilizing this model in identifying individuals with a less aggressive disease course and a more favorable prognosis, evaluating the importance features (clinical variables, laboratory parameters, ancestries proportion, and SNPs) for this outcome. The development of such a model holds great potential in evaluates the influence of both clinical and genetic factors on disease progression. Our study’s two most effective models were the Tree Decision (TD) and Random Forest (RF) models.
The TD model exhibited exceptional performance on the training data, achieving 100% accuracy, precision, recall, and F1 score (weighted average). The model demonstrated a precision of 97%, recall of 97%, and an F1 score of 97% (weighted average) on the testing data. See Supplementary Figure S4. Upon analyzing the variable importance in the TD model, we identified that the most significant factor was the history of outcomes related to a severe course (such as surgery, failure of anti-TNF treatment, pouch, or flares within the last five years), accounting for 80% of the model’s importance. Other influential factors included female sex (6%) and creatinine levels (5%). Please refer to Fig. 3 for further details.
Similarly, the RF model also exhibited strong performance on the training data, achieving 100% accuracy, precision, recall, and F1 score (weighted average). On the testing data, the model achieved a precision of 100%, recall of 73%, and an F1 score of 84% (weighted average). Supplementary Figure S4. Consistent with the TD model, the most important variable for this classifier was the history of outcomes related to a severe course, accounting for 75% of its importance. Other significant factors included clinical and endoscopy remission in the last year (5%), creatinine levels (3%), hemoglobin levels (2%), age of diagnosis (1%), and loss of response to anti-TNF treatment (1%). Please refer to Fig. 4 for further details.

Top ten features identified in the Decision Tree model for predicting Prolonged Clinical and Endoscopic Remission. In this classifier model for predicting prolonged clinical and endoscopic remission, the most important features were associated with a severe phenotype, including a history of surgical failure, use of anti-TNFa medication, and relapse within the past years. Other significant predictors included sex, creatinine levels, and the genetic variant rs921720.

Top ten features identified in the Random Forest model for predicting Prolonged Clinical and Endoscopy Remission. The Random Forest classifier for Prolonged Clinical and Endoscopic Remission identified several key clinical features. These included characteristics associated with a severe phenotype, such as a history of surgical failure, use of anti-TNF medication, and relapse within the past years. Additionally, features such as clinical and endoscopic remission over a year, creatinine levels, hemoglobin levels, age at diagnosis, loss of response to anti-TNF medication, Glutamato Piruvate Transaminase (GPT) levels, white cell count, the genetic variant rs7236492, and clinical remission were also found to be significant predictors.
Discussion
While inflammatory bowel disease (IBD) was initially believed to affect individuals of European ancestry primarily, there has been a significant shift in the epidemiological landscape, with an increasing prevalence observed among individuals in Latin America as well as the Latino population in the United States. In Latin America, IBD is currently in an accelerating stage, marked by rising incidence and prevalence rates27. Meanwhile, the reported prevalence of IBD among Latin communities in the United States is approximately 383 per 100,000 person-year28,29.
Latin American populations differ from Caucasian populations as they are the result of genetic admixture among ancestral populations from Europe, Native Americans, and Africa30. Each Latin American population presents a unique pattern of these three ancestral groups, contributing to their distinct genetic makeup. Mixing genetic backgrounds from multiple continents has led to a rich diversity within Latin American populations. This diversity is reflected in the wide range of genetic variations and phenotypic characteristics observed across Latin American countries and regions31. Therefore, assessing how variations in ancestry may impact the phenotype of IBD across populations can reveal differences that could facilitate the implementation of personalized medicine approaches. In our cohort, the predominant subtype of IBD was UC, accounting for 74% of cases, which is similar to previously reported rates in Latin America32. The average age of onset was 36 years, and approximately 36% of patients reported extraintestinal manifestations, like in previous studies in Latin America28,32,33. When looking at the extension of the disease, it was found that pancolitis was the most common in UC patients (55%), which aligns with findings from other Latin American studies32. Nevertheless, there is variation in the prevalence of UC extension across different regions in Latin America. In Puerto Rico, distal proctitis (Montreal classification E1) was found to be as high as 55.3%34. Meanwhile, in Peru, the extent of left-sided colitis (Montreal classification E2) varied between 11.1% and 62.9% in different studies35,36. As for extensive colitis (Montreal classification E3), one Brazilian study reported a prevalence of 12%.37 However, in Argentina, the prevalence of extensive colitis was reported to be as high as 77%38. In CD patients, colonic extension was the most prevalent disease localization (51%). In comparison, only 16% showed isolated ileal involvement. This differs from other IBD studies where Latin-American CD patients mainly developed ileocolonic disease28,32. Another difference observed was the rate of upper gastrointestinal involvement, which was found in 9% of the population, twice the rate reported in other Latin American IBD studies32. Previous studies have shown that African American or Black, Hispanic, and Asian patients with CD tend to have a more extensive distribution of intestinal inflammation compared to White-non-Hispanic patients. Specifically, higher proportions of White-non-Hispanic patients were found to have isolated ileal disease when directly compared to African American, Hispanic, or Asian patients with CD in studies that examined disease location among different ancestries39. Furthermore, the perianal CD was present in 43% of Crohn’s patients, higher than the 16.7% reported in other Latin American studies32. Interestingly, in Latin America, the perianal compromise varies from 12% in Brazil to 53% in Peru40,41. Despite these differences, the inflammatory behavior in CD was the most prevalent, which is consistent with observations in other Latin American IBD populations38,32. Overall, our findings demonstrate both similarities and differences in the characteristics of IBD in our cohort compared to previous studies conducted in Latin America.
On average, Chileans are 42% Amerindian and 53% European (disaggregated into 25% Mapuche and 18% Aymara)42. The ancestry distribution in our IBD Chilean cohort was 58% European, 39% Amerindian, and 3% African. In our previous work, we discovered a significant association between a high Mapuche ancestry proportion and the risk of IBD43. However, we did not observe risk differences according to Amerindian ancestry proportion in this cohort. In this study, we utilized a native American ancestry proportion derived from a reference panel that included a broader Latin population rather than specifically focusing on the Mapuche population, which could explain these differences. The proportion of native American ancestry in Chile represents a combination of various native American groups, including the Mapuche and Aymara populations44. Therefore, the observed differences in this study may be attributed to including multiple Native American groups in the analysis rather than solely focusing on the Mapuche population. Considering this issue, we estimated the ancestry proportions for Mapuches and Aymara by utilizing the K = 4 clustering results, which included European, Aymara, Mapuche, and African groups, as opposed to the K = 3 clustering that only included European, Amerindian, and African groups. However, no significant differences in IBD risk were observed (Supplementary Table 3). Another potential explanation could be attributed to the utilization of a larger and different control group in our previous study (3,147 individuals of Chilean descent from a gallbladder cancer study)45. Hence, further investigation with a larger sample size is warranted to definitively explore the potential influence of ancestry on IBD risk. We made some notable observations when exploring the relationship between ancestry and clinical outcomes in UC. Firstly, we found a higher median Amerindian ancestry in the group of patients diagnosed before age 40, suggesting a potential association between ancestry and early-onset UC. On the other hand, patients who achieved prolonged clinical and endoscopic remission had a lower median Amerindian ancestry, indicating a possible negative correlation between Amerindian ancestry and UC sustained remission.
Furthermore, interesting findings emerged among UC patients who underwent pouch surgery. Approximately 70% of these patients had HAAP. Similarly, 57% of UC patients who required surgery exhibited HAAP. These findings may suggest an association between a high Amerindian ancestry and a more severe phenotype in UC. Conversely, a lower median proportion of Amerindian ancestry was observed in CD patients with perianal disease. Furthermore, among CD patients who required surgery, a significant majority (94%) had a lower Amerindian ancestry. These contrasting observations suggest that the influence of Amerindian ancestry on disease severity and surgical outcomes may differ between UC and CD patients. While a higher Amerindian ancestry appears to be associated with a more severe phenotype in UC, a lower Amerindian ancestry may be linked to perianal disease and the need for surgery in CD patients. The observed differences in the association between Amerindian ancestry and disease characteristics in UC and CD patients could be attributed to various factors, including genetic, environmental, and immunological influences. Maybe certain genetic variants or alleles associated with Amerindian ancestry contribute to an increased risk or severity of UC or are protective for CD in these patients. Additionally, environmental factors prevalent in populations with higher Amerindian ancestry may play a role in exacerbating disease severity. It is important to note that these associations between ancestry and disease characteristics are complex and multifactorial. Genetic and environmental factors interact in intricate ways, and additional research is needed to understand further the underlying mechanisms driving these differences. When analyzing the IBD group, it is important to consider the divergent effects of ancestry on UC and CD. Interestingly, like the observations in UC, we found a higher median Amerindian ancestry in the subgroup of patients diagnosed younger than 40. In contrast, a lower median Amerindian ancestry was associated with prolonged clinical and endoscopy remission. However, it is worth noting that these results should be interpreted in the context of the sample size discrepancy between UC and Crohn’s disease, with the UC cohort being almost 3 times larger. Apart from the genetic variability linked to the general risk of developing IBD, there has been significant attention given to exploring the relationship between genetic variants and specific subtypes or characteristics of IBD, such as prolonged clinical and endoscopy remission. Tables 9 and 10. Importantly, our analyses indicated no significant differences in the use of anti-TNF therapy between high and low Amerindian ancestry groups, suggesting that treatment disparities are unlikely to influence the observed outcomes. These patient groups belong to the same socioeconomic stratum and had equal access to biological therapies.
Several SNPs exhibit significant associations with prolonged clinical and endoscopy remission, as shown in Table 9. For rs6871626, the AA genotype shows a stronger association with remission, with a multivariate OR of 5.86, CI of 1.80-19.08, and p-value of 0.002. The AG and AA genotypes of rs7134599 are associated with remission, with multivariate ORs of 2.43 (CI 1.06–5.82, p = 0.04) and 3.88 (CI 1.09–12.44, p = 0.02), respectively. rs11150589’s TT genotype shows a multivariate OR of 4.02 (CI 1.40-11.23, p = 0.007). Finally, rs2651244’s AA genotype strongly associates with remission, with a multivariate OR of 5.12, CI 1.55-16.00, and p-value of 0.005. These findings highlight significant genetic influences on IBD remission outcomes.
The gene set enrichment Analysis derived from SNPs associated with IBD and HAAP revealed a significant association with IL-6, a key inflammatory mediator in IBD. It influences the differentiation of T helper 17 (Th17) cells and supports Th1 cell survival, contributing to inflammation in CD and ulcerative UC46.
A key point to highlight is that our analysis did not specifically target SNPs directly associated with Amerindian ancestry; instead, we aimed to explore which IBD genetic variants are linked to HPAA. Our investigation seeks to identify any genetic variants that may also have a connection to ancestry, thereby indirectly enhancing our understanding of how ancestry influences IBD phenotypes. Interestingly, we made an intriguing observation during the development of classifiers for predicting clinical and endoscopy remission over the past five years. At least one outcome associated with a severe disease course emerged as the primary distinguishing feature. These outcomes encompassed surgery, failure of anti-TNF treatment, pouch, or flares within the last five years. None of the SNPs exhibited an importance level exceeding 5% in these models. Our findings suggest that clinical features play a more significant role in predicting these outcomes within our population. These results motivate us to expand our sample size and plan for future whole-genome sequencing to identify new genetic variants that may be relevant to our population.
Due to our relatively small size of the dataset we focused on established genetic associations to address a specific query regarding the potential risk associated with previously identified variants in the phenotype of these IBD individuals.
Strengths and limitations
Our study gives valuable insights into the clinical and genetic dimensions of IBD within a South American cohort, emphasizing the role of ancestry in disease phenotype. A main strength lies in the integration of clinical characteristics with comprehensive genotyping data of SNPs previously associated with IBD risk in other ancestral cohorts. This allows for a more detailed analysis of the genetic influences on IBD among a historically underrepresented population. The study helps to explore how Amerindian ancestry influences IBD phenotype, contributing to personalized medicine.
A study limitation is a relatively small sample size, particularly in the CD group, which may constrain the generalizability of the findings. Besides, the focus on a Chilean cohort restricts the ability to extrapolate results to other Latin American populations with different genetic admixtures.
The study’s cross-sectional nature also limits causal interpretations of the observed associations. Further extensive longitudinal multi-ethnic cohort studies that include a larger Latin population with a yet characterized population would be beneficial in deciphering the complicated interactions between IBD phenotypes and ancestry and their relationship with the immune response. These enhancements will aid in confirming genetic associations and exploring the influence of environmental and lifestyle factors.
Conclusion
Our findings demonstrate differences in IBD phenotypes based on Amerindian ancestry proportion, suggesting that genetic or ancestral factors may contribute to the disease’s phenotype and severity. Additionally, the results indicate a difference in the direction and effect of the influence of Amerindian ancestry on UC and CD patients. Further research is necessary to gain a deeper understanding of the underlying mechanisms that drive these associations.
Data availability
Data availability statement: This study employed the Genotyping by Sequencing Array (GSA) service utilizing the Illumina GSA array platform at Erasmus MC to ensure accurate and comprehensive analysis. The data generated has been deposited in the European Variation Archive (EVA), and the accession number is PRJEB87649.
References
Park, S. C. & Jeen, Y. T. Genetic studies of inflammatory bowel disease-focusing on Asian patients. Cells 8 https://doi.org/10.3390/cells8050404 (2019).
Zhao, M. et al. Predictors of response and disease course in patients with inflammatory bowel disease treated with biological therapy - The Danish IBD biobank project: protocol for a multicentre prospective cohort study. BMJ Open. ;10. (2020).
Jostins, L. et al. Host–microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 491, 119–124 (2012).
Liu, J. Z. et al. Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat. Genet. 47, 979–986 (2015).
De Lange, K. M. et al. Genome-wide association study implicates immune activation of multiple integrin genes in inflammatory bowel disease. Nat. Genet. 49, 256–261 (2017).
Ye, B. D. & McGovern, D. P. B. Genetic variation in IBD: progress, clues to pathogenesis and possible clinical utility. Exp. Rev. Clin. Immunol. 12, 1091–1107. https://doi.org/10.1080/1744666X.2016.1184972 (2016).
Alvarez-Lobos, M. et al. Crohn’s disease patients carrying Nod2/CARD15 gene variants have an increased and early need for first surgery due to stricturing disease and higher rate of surgical recurrence. Ann. Surg. 242, 693–700 (2005).
Walker, D. G. et al. Ethnicity differences in genetic susceptibility to ulcerative colitis: A comparison of Indian Asians and white Northern Europeans. Inflamm. Bowel Dis. 19, 2888–2894 (2013).
Ananthakrishnan, A. N. IBD risk prediction using multi-ethnic polygenic risk scores. Nat. Rev. Gastroenterol. Hepatol. 18, 217–218 (2021).
Gettler, K. et al. Common and rare variant prediction and penetrance of IBD in a large, multi-ethnic, health system-based biobank cohort. Gastroenterology 160, 1546–1557 (2021).
AIM. AIM (Asociación de Investigadores de Mercado y opinión Publica) Grupos Socioeconómicos de Chile. (2024). https://aimchile.cl/gse-chile/. Accessed April 3.
Maaser, C. et al. ECCO-ESGAR guideline for diagnostic assessment in IBD part 1: initial diagnosis, monitoring of known IBD, detection of complications. J. Crohns Colitis. 13, 144–164 (2019).
Rubin, D. T., Ananthakrishnan, A. N., Siegel, C. A., Sauer, B. G. & Long, M. D. ACG clinical guideline: ulcerative colitis in adults. Am. J. Gastroenterol. 114, 384–413 (2019).
Lichtenstein, G. R. et al. ACG clinical guideline: management of Crohn’s disease in adults. Am. J. Gastroenterol. 113, 481–517. https://doi.org/10.1038/ajg.2018.27 (2018).
Satsangi, J. The Montreal classification of inflammatory bowel disease: controversies, consensus, and implications. Gut 55, 749–753 (2006).
Turner, D. et al. STRIDE-II: an update on the selecting therapeutic targets in inflammatory bowel disease (STRIDE) initiative of the international organization for the study of IBD (IOIBD): determining therapeutic goals for treat-to-target strategies in IBD. Gastroenterology 160, 1570–1583 (2021).
Magro, F. et al. ECCO position paper: harmonization of the approach to ulcerative colitis histopathology. J. Crohns Colitis. 14, 1503–1511 (2020).
Magro, F. et al. ECCO position on harmonisation of Crohn’s disease mucosal histopathology. J. Crohns Colitis. 16, 876–883 (2022).
Alexander, D. H., Novembre, J. & Lange, K. Fast model-based Estimation of ancestry in unrelated individuals. Genome Res. 19, 1655–1664 (2009).
Purcell, S. et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575 (2007).
de Lange, K. M. et al. Genome-wide association study implicates immune activation of multiple integrin genes in inflammatory bowel disease. Nat. Genet. 49 (2), 256–261 (2017).
Pérez, T. et al. P904 genetic variants in IBD Chilean patients are related to clinical outcomes. J. Crohns Colitis. 17, i1017–i1018 (2023).
Liang, B., Wang, Y., Xu, J., Shao, Y. & Xing, D. Unlocking the potential of targeting histone-modifying enzymes for treating IBD and CRC. Clin. Epigenetics. 15, 146 (2023).
Fukamachi, H., Nakano, Y., Yoshimura, M. & Koga, T. Cloning and characterization of the l-cysteine desulfhydrase gene of Fusobacterium nucleatum. FEMS Microbiol. Lett. 215, 75–80 (2002).
Engevik, M., Danhof, H., Britton, R. & Versalovic, J. 20 Elucidating the role of Fusobacterium nucleatum in intestinal inflammation. Inflamm. Bowel Dis. 26, S29–S29 (2020).
Su, W. et al. Fusobacterium nucleatum promotes the development of ulcerative colitis by inducing the autophagic cell death of intestinal epithelial. Front. Cell. Infect. Microbiol. ;10. (2020).
Windsor, J. W. & Kaplan, G. G. Evolving epidemiology of IBD. Curr. Gastroenterol. Rep. 21, 40 (2019).
Kotze, P. G. et al. Progression of inflammatory bowel diseases throughout Latin America and the Caribbean: a systematic review. Clin. Gastroenterol. Hepatol. 18, 304–312 (2020).
Damas, O. M. & Maldonado-Contreras, A. Breaking barriers in dietary research: strategies to diversify recruitment in clinical studies and develop culturally tailored diets for Hispanic communities living with inflammatory bowel disease. Gastroenterology 165, 324–328 (2023).
Norris, E. T. et al. Genetic ancestry, admixture and health determinants in Latin America. BMC Genom. 19, 861 (2018).
Avalos, D. J. et al. Latin Americans and US Hispanics show differences in IBD phenotype: a systematic review with meta-analysis. J. Investig Med. 70, 919–933 (2022).
Juliao-Baños, F. et al. Trends in the epidemiology of inflammatory bowel disease in Colombia by demographics and region using a nationally representative claims database and characterization of inflammatory bowel disease phenotype in a case series of Colombian patients. Medicine 100, e24729 (2021).
Pérez-Jeldres, T. et al. Ethnicity influences phenotype and clinical outcomes: comparing a South American with a North American inflammatory bowel disease cohort. Medicine 101, e30216 (2022).
Moreno, J. M., Rubio, C. E. & Torres, E. A. Inflammatory disease of the Gastrointestinal tract at the university hospital, medical center, Puerto Rico. 1980-87. Bol. Asoc. Med. P R. 81, 214–218 (1989).
Calderón, A. V., Velarde, O. F., Yoshidaira, M. Y. & Barahona, E. R. [Clinical and epidemiological profile of ulcerative colitis in a hospital in Lima]. Rev. Gastroenterol. Peru. 24, 135–142 (2004).
Illescas, L., García, L., Faggioni, F. & Velasco, L. [Ulcerative colitis: a 52 years retrospective study]. Rev. Gastroenterol. Peru. 19, 116–123 (1999).
Delmondes, L. M., Nunes, M. O., Azevedo, A. R., de Santana Oliveira, M. M. & Coelho, L. E. Da Rocha Torres-Neto J. Clinical and sociodemographic aspects of inflammatory bowel disease patients. Gastroenterol. Res. 8, 207–215 (2015).
Arcucci, M. S. et al. Pediatric inflammatory bowel disease: a multicenter study of changing trends in Argentina over the past 30 years. Pediatr. Gastroenterol. Hepatol. Nutr. 25, 218 (2022).
Barnes, E. L., Loftus, E. V. & Kappelman, M. D. Effects of race and ethnicity on diagnosis and management of inflammatory bowel diseases. Gastroenterology 160, 677–689 (2021).
Bendaño, T. & Frisancho, O. [Clinical and evolutive profile of Crohn’s disease in hospital rebagliati (Lima-Peru)]. Rev. Gastroenterol. Peru. 30, 17–24 (2010).
de Souza, M. M., Belasco, A. G. S. & de Aguilar-Nascimento, J. E. Perfil epidemiológico Dos Pacientes portadores de Doença inflamatória intestinal do Estado de Mato Grosso. Revista Brasileira De Coloproctologia. 28, 324–328 (2008).
Verdugo, R. A. et al. Development of a small panel of SNPs to infer ancestry in Chileans that distinguishes Aymara and Mapuche components. Biol. Res. 53, 15 (2020).
Pérez-Jeldres, T. et al. Amerindian ancestry proportion as a risk factor for inflammatory bowel diseases: results from a Latin American Andean cohort. Front. Med. (Lausanne) ;10. (2023).
Klimentidis, Y. C., Miller, G. F. & Shriver, M. D. Genetic admixture, self-reported ethnicity, self-estimated admixture, and skin pigmentation among Hispanics and native Americans. Am. J. Phys. Anthropol. 138, 375–383 (2009).
Barahona Ponce, C. et al. Gallstones, body mass index, C-reactive protein, and gallbladder cancer: Mendelian randomization analysis of Chilean and European genotype data. Hepatology 73, 1783–1796 (2021).
Shahini, A. & Shahini, A. Role of interleukin-6-mediated inflammation in the pathogenesis of inflammatory bowel disease: focus on the available therapeutic approaches and gut Microbiome. J. Cell. Commun. Signal. 17, 55–74 (2023).
Acknowledgements
This study would not have been possible without the exceptional support of the patients, nurses, and technicians from the Endoscopy Unit of the Hospital San Borja Arriarán, Santiago, Chile.
Funding
Tamara Pérez-Jeldres was supported by ANID, Chile. Project Fondecyt Initiation [Grant Number 11220147]. Danilo Alvares was supported by the UKRI Medical Research Council [Grant number MC_UU_00002/5]. Manuel Alvarez-Lobos was supported by ANID, Chile. Project Fondecyt Regular [Grant 1211344]. Alex Di Genova was supported by ANID, Chile. Project Fondecyt Regular [Grant 1221029 ] and SA77210017.
Author information
Authors and Affiliations
Contributions
TP-J, ML-B were the guarantor of the article.NA, LA, VS, EA, ADV, MG collected the data.TP-J, A-DG, DA, LK, CM Analyzed the data.TP-J, A-DG, ML-B Prepared the first draft of the article.TP-J, ML-B, DA, MA, LA, RS, GA, NA, RE, CH, RC, MG, VS, ADLV, EA, CP, CS, JFM, DGA, LK, CM reviewed the manuscript for important intellectual content.Finalized the manuscript TP-J, ML-B, A-DG.All the authors approved the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Pérez-Jeldres, T., Bustamante, M.L., Alvares, D. et al. Impact of Amerindian ancestry on clinical outcomes in Crohn’s disease and ulcerative colitis in a Latino population. Sci Rep 15, 15331 (2025). https://doi.org/10.1038/s41598-025-99543-2
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-99543-2
