
Abstract
In this work, 1-phenyl-β-carboline-3-carboxamide-1,2,3-triazole-N-phenylacetamide skeleton as a novel scaffold was designed based on hybridization of moieties that were found in the potent α-glucosidase inhibitors. Fourteen derivatives 14a-n of this scaffold were synthesized by the efficient chemical reactions. In vitro anti-α-glucosidase assay demonstrated that all the new fourteen derivatives with IC50 values ranging from 64.0 to 661.4 µM were more potent than positive control acarbose with IC50 value of 750.0 and in vitro kinetic study revealed that the most potent compound among them, compound 14b, was an uncompetitive α-glucosidase inhibitor. Moreover, determination of the circular dichroism (CD) spectra demonstrated that compound 14b altered the secondary structure of α-glucosidase. Prediction of the pharmacokinetics and toxicity of the most potent compound 14b showed that our new compound had good toxicity profile as an oral drug candidate. Based on these findings, compound 14b can be considered as a promising candidate for the development of a new α-glucosidase inhibitor.
Similar content being viewed by others
Introduction
Diabetes mellitus (DM) is the most common metabolic disorder that has two main types: type 1 and type 21. In the all types of this disease, the patient faces increased blood glucose level. Type 2 of DM (90% occurrence) is the most common type of it and controlled by the oral blood glucose-lowering medications2,3,4. Preventing glucose absorption by inhibition of carbohydrate degradation is an essential approach in the treatment of Type 2 DM and obesity5,6. The most important digestive enzyme for the hydrolysis of carbohydrates and converting them into glucose is α-glucosidase7. Function of this enzyme increased postprandial hyperglycemia (PPH) due to the release of glucose into the bloodstream8. Therefore, inhibition of α-glucosidase as an important digestive enzyme, can delay the absorption of glucose into the bloodstream and decreased PPH9. Several α-glucosidase inhibitors such as acarbose, voglibose, and miglitol are available in the pharmaceutical market for diabetic patients10. However, due to the gastrointestinal side effects of these drugs and the inadequacy of the oral medications for the treatment of diabetes, the search for new drugs in this field continues11,12,13,14,15,16.
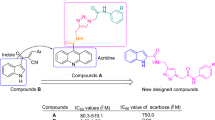
Recently, many research groups have been introduced potent α-glucosidase inhibitors by molecular hybridization theory17. Based on this theory, active pharmacophores of the synthetic and natural derivatives were selected and were attached together by simple chemical reactions18. β-Carboline is a nitrogen containing tricyclic structure that found in many natural and synthetic bioactive compounds with various activities such as antiplasmodial, antioxidant, antimicrobial, antitumor, cytotoxic, antimutagenic, hallucinogenic, and antigenotoxic properties19. Moreover, β-carboline ring in the many compounds with anti-α-glucosidase activity was found20,21,22,23. For example, β-carboline-3-amide derivative A and 1-phenyl-β-carboline-3-amide derivatives B and C exhibited significant inhibition effects against α-glucosidase (Fig. 1)21,22,23. Therefore, we selected 1-phenyl-β-carboline-3-amide moiety as an active pharmacophore against α-glucosidase. On the other hand, carboxamide-1,2,3-triazole-N-phenylacetamide moiety with simple derivation capability is an attractive structure for design of the novel α-glucosidase inhibitors (compounds D and E, Fig. 1)24,25. Therefore, we with consideration of 1-phenyl-β-carboline-3-amide structure and carboxamide-1,2,3-triazole-N-phenylacetamide moiety designed a new scaffold with 1-phenyl-β-carboline-3- carboxamide-1,2,3-triazole-N-phenylacetamide skeleton (Fig. 1). Fourteen derivatives 14a-n were synthesized of this skeleton and after the characterization, were evaluated their inhibitory activates against yeast form of α-glucosidase. Furthermore, in vitro and in silico evaluations such as kinetic analysis, circular dichroism (CD), and prediction of ADMET were also performed on the most potent compounds.

Design strategy for 1-phenyl-β-carboline-3- carboxamide-1,2,3-triazole-N-phenylacetamide derivatives as new α-glucosidase inhibitors.
Results and discussion
Chemistry
In the present work, we synthesized new 1-phenyl-β-carboline-3-carboxamide-1,2,3-triazole-N-phenylacetamide derivatives 14a-n by described procedure in Scheme 126,27,28. As can be seen in Scheme 1, L-tryptophan (1) converted to L-tryptophan methyl ester (2) in the presence of thionyl chloride and NaHCO3. After that, L-tryptophan methyl ester (2) reacted with various benzaldehydes 3 to give 2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indole derivatives 4. The latter derivatives were first aromatized by KMnO4 in anhydrous THF to give β-carboline derivatives 5 and then were converted to acid derivatives 6 by NaOH in methanol. After that, compounds 6 reacted with propargylamine (7) to give propargel derivatives 8. The latter derivatives 8, sodium ascorbate, and CuSO4 were added to the in situ prepared azide derivatives 13 and the reaction was continued at RT for 20–27 h to give the target compounds 14a-n.

(a) SOCl2, MeOH, NaHCO3, Reflux, 8 h; (b) TFA, CH2Cl2, RT, 24 h; (c) KMnO4,THF, RT, 12 h; (d) NaOH, MeOH, Reflux,12 h; (e) TBTU, NEt3, DMF, RT, 4 h; (f) DMF, RT, 30 min; (g) Et₃N, H2O/t-BuOH, RT, 1 h; (h) CuSO4.5H2O, Sodium Ascorbate, RT, 20–27 h.
In vitro anti-α-glucosidase assay and structure–activity relationships (SARs)
The in vitro results of the α-glucosidase inhibitory assay of β-carboline-3-carboxamide-1,2,3-triazole-N-phenylacetamide derivatives 14a-n were shown in Table 1. All the target compounds were more potent than the used positive control (acarbose). Among these compounds, compound 14b showed a significant anti-α-glucosidase activity (IC50 value = 64.0 µM) in comparison to acarbose (IC50 value = 750.0 µM).
Structure-activity relationships (SARs)
Based on the obtained enzymatic inhibition results, SAR study was performed on various derivatives of the designed scaffold. Structurally, our new synthesized compounds were divided into three groups based on the substitution on the pendant phenyl ring of β-carboline moiety: group (1) un-substituted phenyl derivatives 14a-e, group (2) 4-methoxyphenyl derivatives 14f-i, and group (3) 4-chlorophenyl derivatives 14j-n. In each group, the substitutions on the phenyl ring of N-phenylacetamide moiety were changed. SARs of these groups were schematically in Figs. 2, 3 and 4.

SAR diagram of compounds 14a-e in anti-α-glucosidase assay.

SAR diagram of compounds 14j-n in anti-α-glucosidase assay.
As can be seen in Table 1, the most potent compound in the present work was 2-methylphenylacetamide derivative 14b. This compound was belonged to group 1. SAR diagram in this group was showed in Fig. 2. As can be seen in this figure and Table 1, removing the 2-methyl substituent of compound 14b negligibly decreased anti-α-glucosidase activity as observed in compound 14a while introduction of a nitro group on 4-position of phenyl ring of N-phenylacetamide moiety of compound 14b, as in the case of compound 14c, decreased anti-α-glucosidase activity to 10-folds. On the other hand, 3-chloro derivative 14e and 2-nitro-4-methoxy derivative 14e of this group were moderate α-glucosidase inhibitors in comparison to compound 14b.
In the group 2, with 4-methoxy substituent on pendant phenyl ring of β-carboline moiety, only 3-chlorophenylacetamide derivative 14i is a significant inhibitor in comparison to acarbose and the rest compounds 14f-h considered as the weak α-glucosidase inhibitors (Table 1; Fig. 3).

SAR diagram of compounds 14f-i in anti-α-glucosidase assay.
From the screening data of compound 14j-n (group 3), it was revealed that phenylacetamide derivative 14j and 2-fluorophenylacetamide derivative 14 m were the most potent compounds (Fig. 4). These compounds were around 5-folds more active than acarbose against α-glucosidase. In group 3, 3-chlorophenylacetamide derivative 14n and 2-methyl-4-nitrophenylacetamide derivative 14 L were good α-glucosidase inhibitors in comparison to acarbose. Our data also showed that removing 4-nitro substituent of compound 14 L, as in the case of compound 14k, inhibitory activity diminished to 3-folds.
SAR diagrams of corresponding analogs in each series of the synthesized compounds were showed in Fig. 5. As can be seen this figure, in addition to substituent on phenyl ring of N-phenylacetamide moiety, anti-anti-α-glucosidase activity of the new synthesized compounds dramatically depended on type of substituent on pendant phenyl ring of β-carboline moiety.

SAR diagrams of corresponding analogs in each series of the synthesized compounds in anti-α-glucosidase assay.
Comparison of the new compounds 14 with used template compounds
As can be seen in Fig. 1, we used of compounds A-E as templates for designing of new compounds 1421,22,23,24,25. An overview of the effects of the template compounds A-E and new compounds 14 shows that the most potent compound among the new compounds 14, compound 14b, was more potent than compound A and the most potent compound of series D. On the other hand, the most potent compounds in the series B, C, and D were more potent than compound 14b. It is worthy to note that several inactive compounds were observed among the reported derivatives of templates B and E while all the newly synthesized derivatives of series 14 were potent. In general, it can be mentioned that only thiosemicarbazide-based β-carboline derivatives C can inhibit α-glucosidase better than our new compounds.
The comparison of IC50 values of the new derivatives 14 with their corresponding analogs of the template compounds D and E revealed that new compounds 14 were more potent than their corresponding analogs of the reported compounds D and E (Fig. 6)24,25.

Comparison of IC50 values of new derivatives 14 against α-glucosidase with their corresponding analogs of template derivatives D and E.
Kinetic study
Kinetics of compound 14b as the most potent new α-glucosidase inhibitor with 1-phenyl-β-carboline-3-carboxamide-1,2,3-triazole-N-phenylacetamide skeleton was evaluated. Our result was shown in Fig. 7. As can be seen in this figure, increase in the concentration of compound 14b as inhibitor led to decrease in Km and Vmax. This finding demonstrated that compound 14b was an uncompetitive α-glucosidase inhibitor.

The Lineweaver–Burk plots in the absence and presence of different concentrations of compound 14b.
CD examination
The secondary structure of α-glucosidase was examined at three molar ratios of compound 14b (α-glucosidase to compound 14b: 1 to 0, 1 to 1, 1 to 2) using CD measurements between 190 and 260 nm (Fig. 8). The negative peaks at roughly 205–225 nm in the CD spectra of the target enzyme alone suggested a typical signal of an α-helix-containing protein. The CD spectra of α-glucosidase revealed that 205–225 nm peaks were decreased when the compound 14b was added, especially in the ratio of 1 to 2 of α-glucosidase to compound 14b (Table 2). This finding revealed that the interaction of the compound 14b with the α-glucosidase led to alterations in the secondary structure of α-glucosidase’s conformation.

CD Spectra of interaction of α-glucosidase with compound 14b. Molar ratios of α-glucosidase to compound 14b: 1:0 (Black line), 1:1 (Orange line), 1:2 (Blue line).
In Silico studies on druglikeness, pharmacokinetics, and toxicity
Druglikeness study, prediction of pharmacokinetics (ADME), and toxicity (T) profile of acarbose and the most potent compounds 14b and 14a were performed by an online server and the obtained results were listed in Table 329. As can be seen in Table 3, acarbose did not follow of Lipinski ‘Rule of five’ while the selected new compounds 14b and 14a followed of Lipinski Rule. In term of pharmacokinetics, compounds 14b, 14a, and acarbose had poor permeability to Caco-2 cells, and favorable permeability to blood brain barrier (BBB) and skin. Moreover, acarbose did not have human intestinal absorption (HIA) while compounds 14b and 14a had high HIA. In term of toxicity profile, our new compounds 14b and 14a demonstrated excellent in silico properties in comparison to acarbose. In this regard, acarbose was mutagen and had carcinogen effect on mouse while compound 14b and 14a were non-mutagen and did not have carcinogenicity on rat and mouse.
Bioavailability radars of compounds 14b, 14a, and acarbose were obtained by SwissADME30. In these bioavailability radars, the suitable physicochemical space for oral bioavailability was showed with pink color. As can be seen in Fig. 9, compound 14b more than two other studied compounds was in the pink area and probably has better oral bioavailability in comparison to compound 14a and acarbose.

Bioavailability radars of the compounds 14b, 14a, and acarbose.
Conclusion
In summary, we designed and synthesized a novel series of 1-phenyl-β-carboline-3-carboxamide-1,2,3-triazole-N-phenylacetamide derivatives 14a-n as potent agents against α-glucosidase. In vitro assay of these compounds showed that all the new compounds 14a-n were potent than positive control and compound 14b with IC50 value of 64.0 µM exhibited highest inhibitory activity. Kinetic analysis suggested that compound 14b is an uncompetitive inhibitor against α-glucosidase. CD spectra result indicated that compound 14b had the ability to alter conformational changes of α-glucosidase. ADMET prediction of compound 14b demonstrated that this compound can be an oral drug and had better toxicity profile in comparison to positive control acarbose. In total, compound 14b was significantly more potent than acarbose in in vitro assay and had better pharmacokinetics and toxicity profile in comparison of acarbose. Although this study requires further studies such as in vivo study to further confirm the effectiveness of compound 14b.
Experimental
Synthetic of Methyl tryptophanate (2)
To a stirring solution of tryptophan (1, 10 mmol) in methanol (40 mL), thionyl chloride (11 mmol) was added dropwise at 0 °C. After 30 min, it mixture was refluxed for 8 h. Following completion of the reaction, excess methanol was removed under vacuum conditions. The resulted intermediate was extracted with dichlorometan (3 × 40 mL) and was washed with saturated NaHCO3 solution. Then, dichlorometan layer was dried over anhydrous Na2SO4 and this solvent was evaporated under vacuum to obtain methyl tryptophanate (2)26.
General synthetic procedure for Methyl 1-phenyl-2,3,4,9-tetrahydro-1 H-pyrido[3,4-b]indole-3-carboxylate derivatives 4
Compound 2 (5 mmol) and various aldehydes 3 (5 mmol) were dissolved in 60 mL of dichlorometan and chilled to 0 °C in an ice bath and TFA (5%, 12.5 mmol) was added dropwise. After that, the obtained mixture was stirred at room temperature (RT) for 24 h. After completion (checked by TLC), the reaction mixture was made alkaline with dilute NaHCO3 solution and extracted with dichlorometan (3 × 50 mL). The dichlorometan layer was washed successively with water and brine, followed by drying over Na2SO4, filtration, and solvent evaporation under reduced pressure to give products 427.
General synthetic procedure for Methyl 1-phenyl-9 H-pyrido[3,4-b]indole-3-carboxylate derivatives 5
A suspension of compounds 4 (5 mmol) and KMnO4 (12.5 mmol) in anhydrous THF (50 mL) was stirred for 12 h at RT. Then, KMnO4 was removed by buchner filtration and THF was evaporated under reduced pressure to give derivatives 6 that were used without further purification for the following step28.
General synthetic procedure for 1-phenyl-9 H-pyrido[3,4-b]indole-3-carboxylic acid derivatives 6
A mixture containing compounds 6 (5 mmol) and aqueous NaOH (1 M, 50 mL) in methanol (10 mL) was stirred. After 30 min, this mixture was heated at reflux condition for 12 h. Then, the mixture reaction was cooled to RT, the pH of it was adjusted to 7 by HCl solution (1 M), and formed precipitates were filtered to give compounds 628.
General synthetic procedure for 1-phenyl-N-(prop-2-yn-1-yl)-9 H-pyrido[3,4-b]indole-3-carboxamide derivatives 8
A mixture of 2 compounds 6 (5 mmol), propargylamine (7), TBTU, and NEt3 in DMF (50 mL) was stirred at RT for 4 h. Then, water was added to the reaction mixture and obtained participates were filtered off and recrystallized in ethanol to obtain pure 1-phenyl-N-(prop-2-yn-1-yl)-9H-pyrido[3,4-b]indole-3-carboxamide derivatives 8.
In situ preparation of azide derivatives 13
Azide derivatives 13 were prepared according to our previously reported works25.
General synthetic procedures for 1-phenyl-β-carboline-3-carboxamide-1,2,3-triazole-N-phenylacetamide derivatives 14a-n
In the final step, compounds 8 (1mmol), sodium ascorbate (15 mol %, 0.13 g), and CuSO4 (7 mol%) were added to a stirred mixture of in situ prepared azide derivatives 13 in H2O/t-BuOH (50:50, 10 mL) at RT. After 20–27 h, the reaction mixture was poured into crushed ice and precipitated products 14a-n were filtered off, washed with cold water, and purified by recrystallization in ethanol1H NMR and13C NMR spectra of the new compounds 14a-n were showed in support information (Fig. 1-14 s).
N-((1-(2-oxo-2-(phenylamino)ethyl)-1 H-1,2,3-triazol-4-yl)methyl)-1-phenyl-9 H-pyrido[3,4-b]indole-3-carboxamide (14a)
White powder; Yield: 72%, m.p. > 200 °C1H NMR (301 MHz, DMSO-d6) δ 11.93 (s, 1 H), 10.57 (s, 1 H), 9.33 (s, 1 H), 8.94 (s, 1 H), 8.44 (d, J = 7.7 Hz, 1 H), 8.23 (d, J = 7.2 Hz, 2 H), 8.13 (s, 1 H), 7.79–7.55 (m, 8 H), 7.33 (t, J = 8.1 Hz, 1 H), 7.18 (t, J = 8.6 Hz, 2 H), 5.37 (s, 2 H), 4.77 (d, J = 6.2 Hz, 2 H)13C NMR (76 MHz, DMSO-d6) δ 165.46, 164.74, 160.31, 157.14, 142.10, 141.20, 140.11, 137.98, 135.33, 134.78, 130.41, 129.45, 129.27, 129.10, 128.81, 122.47, 121.73, 121.57, 121.47, 120.69, 115.82, 113.65, 113.20, 52.62, 35.28. Anal. Calcd for C29H23N7O2: C, 69.45; H, 4.62; N, 19.55; Found: C, 69.32; H, 4.54; N, 19.62.
N-((1-(2-oxo-2-(o-tolylamino)ethyl)-1 H-1,2,3-triazol-4-yl)methyl)-1-phenyl-9 H-pyrido[3,4-b]indole-3-carboxamide (14b)
White powder; Yield: 75%, m.p. > 200 °C1H NMR (301 MHz, DMSO-d6) δ 11.94 (s, 1 H), 9.83 (s, 1 H), 9.34 (t, J = 6.1 Hz, 1 H), 8.95 (s, 1 H), 8.44 (d, J = 7.9 Hz, 1 H), 8.24 (d, J = 7.5 Hz, 2 H), 8.14 (s, 1 H), 7.76 (d, J = 8.2 Hz, 1 H), 7.71–7.55 (m, 4 H), 7.48 (d, J = 7.7 Hz, 1 H), 7.34 (t, J = 7.5 Hz, 1 H), 7.21 (t, J = 8.0 Hz, 1 H), 7.11 (d, J = 7.3 Hz, 1 H), 5.43 (s, 2 H), 4.79 (s, 2 H), 2.25 (s, 3 H)13C NMR (76 MHz, DMSO-d6) δ 165.49, 165.00, 142.10, 141.21, 140.10, 136.01, 134.79, 132.05, 130.91, 130.42, 129.47, 129.28, 129.12, 126.54, 126.01, 125.22, 121.73, 120.72, 113.66, 113.21, 52.44, 35.30, 18.28. Anal. Calcd for C30H25N7O2: C, 69.89; H, 4.89; N, 19.02; Found: C, 69.71; H, 4.95; N, 19.15.
N-((1-(2-((2-methyl-4-nitrophenyl)amino)-2-oxoethyl)-1 H-1,2,3-triazol-4-yl)methyl)-1-phenyl-9 H-pyrido[3,4-b]indole-3-carboxamide (14c)
White powder; Yield: 73%, m.p. > 200 °C1H NMR (301 MHz, DMSO-d6) δ 11.88 (s, 1 H), 10.00 (s, 1 H), 9.28 (s, 1 H), 8.88 (s, 1 H), 8.41 (d, J = 6.4 Hz, 1 H), 8.19 (d, J = 7.2 Hz, 2 H), 8.11 (s, 2 H), 8.03 (d, J = 8.1 Hz, 1 H), 7.93 (d, J = 7.8 Hz, 1 H), 7.69–7.57 (m, 14 H), 7.32 (d, J = 7.4 Hz, 1 H), 5.48 (s, 2 H), 4.72 (s, 2 H), 2.37 (s, 3 H)13C NMR (76 MHz, DMSO) δ 165.36, 164.95, 143.40, 142.14, 141.61, 140.68, 139.62, 137.48, 134.28, 131.21, 129.91, 128.99, 128.79, 128.64, 128.60, 125.50, 124.10, 123.15, 122.01, 121.80, 121.23, 120.23, 113.15, 112.72, 52.15, 34.82, 17.84. Anal. Calcd for C30H24N8O4: C, 64.28; H, 4.32; N, 19.99; Found: C, 64.35; H, 4.44; N, 19.81.
N-((1-(2-((3-chlorophenyl)amino)-2-oxoethyl)-1 H-1,2,3-triazol-4-yl)methyl)-1-phenyl-9 H-pyrido[3,4-b]indole-3-carboxamide (14d)
White powder; Yield: 78%, m.p. > 200 °C1H NMR (301 MHz, DMSO-d6) δ 11.89 (s, 1 H), 10.04 (s, 1 H), 9.27 (s, 1 H), 8.89 (s, 1 H), 8.42 (d, J = 7.1 Hz, 1 H), 8.18 (s, 1 H), 8.07 (s, 1 H), 7.71 (d, J = 8.6 Hz, 2 H), 7.68–7.56 (m, 3 H), 7.48 (s, 1 H), 7.31 (s, 2 H), 7.19 (s, 1 H), 5.43 (s, 2 H), 4.70 (s, 2 H)13C NMR (76 MHz, DMSO) δ 164.96, 164.89, 141.62, 140.72, 139.63, 137.49, 134.29, 134.20, 129.93, 129.62, 129.00, 128.81, 128.67, 127.56, 126.69, 126.24, 125.84, 124.72, 122.05, 121.24, 120.25, 113.17, 112.73, 51.93, 34.71. Anal. Calcd for C29H22ClN7O2: C, 64.99; H, 4.14; N, 18.29; Found: C, 64.86; H, 4.21; N, 18.37.
N-((1-(2-((4-methoxy-2-nitrophenyl)amino)-2-oxoethyl)-1 H-1,2,3-triazol-4-yl)methyl)-1-phenyl-9 H-pyrido[3,4-b]indole-3-carboxamide (14e)
White powder; Yield: 71%, m.p. > 200 °C1H NMR (301 MHz, DMSO-d6) δ 11.90 (s, 1 H), 10.54 (s, 1 H), 9.28 (s, 1 H), 8.91 (s, 1 H), 8.45 (d, J = 7.9 Hz, 1 H), 8.21 (d, J = 7.4 Hz, 2 H), 8.06 (s, 1 H), 7.94–7.47 (m, 7 H), 7.34 (t, J = 8.2 Hz, 2 H), 5.39 (s, 2 H), 5.00–4.58 (m, 3 H), 3.84 (s, 3 H);13C NMR (76 MHz, DMSO-d6) δ 165.42, 157.03, 144.00, 142.08, 141.17, 140.09, 137.95, 134.75, 130.39, 129.45, 129.26, 128.05, 125.14, 123.55, 122.50, 121.71, 120.75, 113.63, 113.18, 109.73, 56.50, 52.25, 35.23. Anal. Calcd for C30H24N8O5: C, 62.50; H, 4.20; N, 19.43; Found: C, 62.61; H, 4.11; N, 19.32.
1-(4-methoxyphenyl)-N-((1-(2-oxo-2-(phenylamino)ethyl)-1 H-1,2,3-triazol-4-yl)methyl)-9 H-pyrido[3,4-b]indole-3-carboxamide (14f)
White powder; Yield: 75%, m.p. > 200 °C1H NMR (301 MHz, DMSO-d6) δ 11.82 (s, 1 H), 10.43 (s, 1 H), 9.22 (t, J = 5.9 Hz, 1 H), 8.80 (s, 1 H), 8.403 (d, J = 7.8 Hz, 1 H), 8.14 (d, J = 8.6 Hz, 2 H), 8.03 (s, 1 H), 7.69 (d, J = 8.2 Hz, 1 H), 7.57 (dd, J = 13.3, 7.6 Hz, 3 H), 7.30 (t, J = 7.7 Hz, 2 H), 7.19 (d, J = 8.7 Hz, 2 H), 7.06 (t, J = 7.4 Hz, 1 H), 5.29 (s, 2 H), 4.66 (s, 2 H), 3.88 (s, 3 H)13C NMR (76 MHz, DMSO) δ 164.27, 159.96, 155.90, 141.52, 140.30, 139.48, 138.43, 130.15, 129.36, 128.90, 128.51, 123.73, 121.27, 120.22, 119.17, 114.17, 113.18, 112.56, 55.39, 50.43, 34.74. Anal. Calcd for C30H25N7O3: C, 67.79; H, 4.74; N, 18.44; Found: C, 67.69; H, 4.64; N, 18.28.
1-(4-methoxyphenyl)-N-((1-(2-oxo-2-(o-tolylamino)ethyl)-1 H-1,2,3-triazol-4-yl)methyl)-9 H-pyrido[3,4-b]indole-3-carboxamide (14 g)
White powder; Yield: 77%, m.p. > 200 °C1H NMR (301 MHz, DMSO-d6) δ 11.86 (s, 1 H), 9.78 (s, 1 H), 9.26 (t, J = 6.1 Hz, 1 H), 8.84 (s, 1 H), 8.42 (d, J = 7.8 Hz, 1 H), 8.18 (d, J = 8.2 Hz, 2 H), 8.08 (s, 1 H), 7.72 (d, J = 8.1 Hz, 1 H), 7.61 (t, J = 7.6 Hz, 1 H), 7.44 (d, J = 7.6 Hz, 1 H), 7.33 (t, J = 7.4 Hz, 1 H), 7.30–7.15 (m, 3 H), 7.11 (d, J = 7.2 Hz, 1 H), 5.38 (s, 2 H), 4.71 (d, J = 6.0 Hz, 2 H), 2.23 (s, 3 H)13C NMR (76 MHz, DMSO-d6) δ 165.44, 164.94, 160.43, 142.00, 141.11, 139.95, 135.99, 134.48, 132.03, 130.89, 130.62, 130.40, 130.16, 128.98, 126.53, 126.00, 125.20, 122.43, 121.75, 120.64, 114.64, 113.17, 113.05, 55.84, 52.36, 35.22, 18.26. Anal. Calcd for C31H27N7O3: C, 68.24; H, 4.99; N, 17.97; Found: C, 68.35; H, 4.86; N, 17.81.
N-((1-(2-((2-fluorophenyl)amino)-2-oxoethyl)-1 H-1,2,3-triazol-4-yl)methyl)-1-(4-methoxyphenyl)-9 H-pyrido[3,4-b]indole-3-carboxamide (14 h)
White powder; Yield: 72%, m.p. > 200 °C1H NMR (301 MHz, DMSO-d6) δ 11.83 (s, 1 H), 10.28 (s, 1 H), 9.23 (s, 1 H), 8.81 (s, 1 H), 8.40 (d, J = 7.7 Hz, 1 H), 8.15 (d, J = 8.1 Hz, 2 H), 8.04 (s, 1 H), 7.87 (d, J = 8.7 Hz, 1 H), 7.69 (d, J = 8.0 Hz, 1 H), 7.59 (t, J = 7.6 Hz, 1 H), 7.29 (d, J = 7.4 Hz, 2 H), 7.18 (d, J = 10.8 Hz, 3 H), 5.39 (s, 2 H), 4.67 (s, 2 H), 3.88 (s, 3 H)13C NMR (76 MHz, DMSO) δ 164.95, 159.97, 155.04, 150.52, 141.53, 140.64, 139.48, 134.01, 130.16, 129.94, 129.69, 125.71, 125.42, 124.49, 123.75, 121.98, 121.28, 120.16, 115.73, 115.42, 114.18, 112.72, 112.57, 55.38, 51.93, 34.73. Anal. Calcd for C30H24FN7O3: C, 65.57; H, 4.40; N, 17.84; Found: C, 65.64; H, 4.33.
N-((1-(2-((3-chlorophenyl)amino)-2-oxoethyl)-1 H-1,2,3-triazol-4-yl)methyl)-1-(4-methoxyphenyl)-9 H-pyrido[3,4-b]indole-3-carboxamide (14i)
White powder; Yield: 74%, m.p. > 200 °C1H NMR (301 MHz, DMSO-d6) δ 11.85 (s, 1 H), 10.05 (s, 1 H), 9.27 (s, 1 H), 8.83 (s, 1 H), 8.40 (s, 1 H), 8.15 (s, 2 H), 8.08 (s, 1 H), 7.73 (s, 2 H), 7.50 (s, 2 H), 7.31 (s, 2 H), 7.20 (s, 3 H), 5.43 (s, 2 H), 4.70 (s, 2 H), 3.88 (s, 3 H)13C NMR (76 MHz, DMSO) δ 164.99, 159.99, 141.56, 140.67, 139.47, 134.19, 134.06, 130.18, 129.95, 129.63, 128.54, 127.58, 126.72, 126.26, 125.86, 121.99, 121.30, 120.20, 114.20, 112.73, 112.61, 55.39, 51.97, 34.76. Anal. Calcd for C30H24ClN7O3: C, 63.66; H, 4.27; N, 17.32; Found: C, 63.72; H, 4.16; N, 17.25.
1-(4-chlorophenyl)-N-((1-(2-oxo-2-(phenylamino)ethyl)-1 H-1,2,3-triazol-4-yl)methyl)-9 H-pyrido[3,4-b]indole-3-carboxamide (14j)
White powder; Yield: 73%, m.p. > 200 °C1H NMR (301 MHz, DMSO-d6) δ 11.94 (s, 1 H), 10.47 (s, 1 H), 9.31 (s, 1 H), 8.92 (s, 1 H), 8.45 (d, J = 7.9 Hz, 1 H), 8.26 (d, J = 8.0 Hz, 2 H), 8.09 (s, 1 H), 7.92–7.66 (m, 3 H), 7.71–7.53 (m, 3 H), 7.33 (t, J = 7.6 Hz, 3 H), 7.09 (t, J = 7.4 Hz, 1 H), 5.34 (s, 2 H), 5.09–4.71 (m, 2 H)13C NMR (76 MHz, DMSO-d6) δ 142.10, 140.17, 139.82, 138.91, 136.71, 134.71, 134.23, 131.09, 130.63, 129.37, 129.22, 124.21, 122.56, 121.68, 120.80, 119.68, 113.92, 113.13, 52.66, 35.26. Anal. Calcd for C29H22ClN7O2: C, 64.99; H, 4.14; N, 18.29; Found: C, 64.78; H, 4.02; N, 18.17.
1-(4-chlorophenyl)-N-((1-(2-oxo-2-(o-tolylamino)ethyl)-1 H-1,2,3-triazol-4-yl)methyl)-9 H-pyrido[3,4-b]indole-3-carboxamide (14k)
White powder; Yield: 69%, m.p. > 200 °C1H NMR (301 MHz, DMSO-d6) δ 11.96 (s, 1 H), 9.81 (s, 1 H), 9.33 (s, 1 H), 8.93 (s, 1 H), 8.44 (d, J = 7.6 Hz, 1 H), 8.25 (d, J = 7.7 Hz, 2 H), 8.17–8.06 (m, 1 H), 7.92–7.56 (m, 4 H), 7.46 (d, J = 7.6 Hz, 1 H), 7.34 (t, J = 7.3 Hz, 1 H), 7.29–7.04 (m, 3 H), 5.41 (s, 2 H), 4.75 (s, 2 H), 2.24 (s, 3 H)13C NMR (76 MHz, DMSO-d6) δ 165.37, 164.97, 142.11, 140.14, 139.84, 136.70, 136.00, 134.72, 134.25, 132.04, 131.08, 130.90, 130.63, 129.23, 126.53, 126.01, 125.21, 122.53, 121.68, 120.81, 113.93, 113.15, 52.42, 18.27. Anal. Calcd for C30H24ClN7O2: C, 65.51; H, 4.40; N, 17.83; Found: C, 65.43; H, 4.28; N, 17.63.
1-(4-chlorophenyl)-N-((1-(2-((2-methyl-4-nitrophenyl)amino)-2-oxoethyl)-1 H-1,2,3-triazol-4-yl)methyl)-9 H-pyrido[3,4-b]indole-3-carboxamide (14 L)
White powder; Yield: 74%, m.p. > 200 °C1H NMR (301 MHz, DMSO-d6) δ 11.91 (s, 1 H), 10.00 (s, 1 H), 9.29 (s, 1 H), 8.88 (s, 1 H), 8.41 (d, J = 6.9 Hz, 1 H), 8.26–8.18 (m, 2 H), 8.12 (s, 1 H), 8.03 (d, J = 8.7 Hz, 1 H), 7.92 (d, J = 8.6 Hz, 1 H), 7.74–7.65 (m, 2 H), 7.63–7.57 (m, 1 H), 7.31 (t, J = 7.3 Hz, 1 H), 5.47 (s, 2 H), 4.70 (s, 2 H), 2.37 (s, 3 H)13C NMR (76 MHz, DMSO) δ 165.33, 164.82, 143.41, 142.11, 141.60, 139.64, 139.30, 136.19, 134.20, 133.75, 131.24, 130.60, 130.13, 128.73, 125.50, 123.18, 122.07, 121.80, 121.18, 120.32, 113.44, 112.65, 52.13, 34.73, 17.83. Anal. Calcd for C30H23ClN8O4: C, 60.56; H, 3.90; N, 18.83; Found: C, 60.65; H, 3.81; N, 18.76.
1-(4-chlorophenyl)-N-((1-(2-((2-fluorophenyl)amino)-2-oxoethyl)-1 H-1,2,3-triazol-4-yl)methyl)-9 H-pyrido[3,4-b]indole-3-carboxamide (14 m)
White powder; Yield: 78%, m.p. > 200 °C1H NMR (301 MHz, DMSO-d6) δ 11.94 (s, 1 H), 10.30 (s, 1 H), 9.30 (t, J = 6.0 Hz, 1 H), 8.90 (s, 1 H), 8.45 (d, J = 7.8 Hz, 1 H), 8.25 (d, J = 8.0 Hz, 2 H), 8.06 (s, 1 H), 7.93 (d, J = 8.0 Hz, 1 H), 7.71 (d, J = 8.0 Hz, 3 H), 7.64 (d, J = 7.3 Hz, 1 H), 7.32 (dt, J = 14.7, 7.6 Hz, 2 H), 7.17 (dd, J = 6.7, 3.5 Hz, 2 H), 5.42 (s, 2 H), 4.71 (d, J = 6.0 Hz, 2 H)13C NMR (76 MHz, DMSO-d6) δ 165.39, 165.31, 155.53, 142.08, 140.14, 139.81, 136.69, 134.69, 134.23, 131.09, 130.62, 129.22, 129.04, 126.05, 124.99, 124.21, 122.57, 121.66, 120.81, 116.19, 115.94, 113.91, 113.12, 52.40, 35.23. Anal. Calcd for C29H21ClFN7O2: C, 62.88; H, 3.82; N, 17.70; Found: C, 62.75; H, 3.71; N, 17.59.
1-(4-chlorophenyl)-N-((1-(2-((3-chlorophenyl)amino)-2-oxoethyl)-1 H-1,2,3-triazol-4-yl)methyl)-9 H-pyrido[3,4-b]indole-3-carboxamide (14n)
White powder; Yield: 70%, m.p. > 200 °C1H NMR (301 MHz, DMSO-d6) δ 11.95 (s, 1 H), 10.07 (s, 1 H), 9.32 (t, J = 6.0 Hz, 1 H), 8.92 (s, 1 H), 8.44 (d, J = 7.8 Hz, 1 H), 8.25 (d, J = 8.0 Hz, 2 H), 8.10 (s, 1 H), 7.95–7.59 (m, 5 H), 7.52 (d, J = 7.8 Hz, 1 H), 7.34 (t, J = 7.7 Hz, 2 H), 7.23 (d, J = 7.7 Hz, 1 H), 5.46 (s, 2 H), 4.73 (d, J = 5.9 Hz, 2 H)13C NMR (76 MHz, DMSO-d6) δ 165.47, 165.35, 142.10, 140.15, 139.82, 136.70, 134.71, 134.65, 134.24, 131.09, 130.63, 130.08, 129.22, 128.01, 127.14, 126.69, 126.30, 122.56, 121.67, 120.81, 113.93, 113.14, 52.40, 35.26. Anal. Calcd for C29H21Cl2N7O2: C, 61.06; H, 3.71; N, 17.19; Found: C, 61.18; H, 3.86; N, 17.05.
In vitro α-glucosidase Inhibition assay and kinetics
The in vitro α-glucosidase inhibitory activity of β-carboline-3-carboxamide-1,2,3-triazole-N-phenylacetamide derivatives 14a-n and kinetics of the most active compound 14b was determined according to the previously reported methods25.
Measurement of CD spectra
For the measurement of CD spectra, alteration in secondary structure of α-glucosidase in the absence and presence of the most potent compound 14b were examined at 25 °C in a J-810 spectropolarimeter (JASCO, Tokyo). For this propose, a mixture of Milli-Q water and α-glucosidase was incubated at 37 °C for 1 h in three ratio of α-glucosidase to the selected compound 14b (1:0, 1:1, 1:2). Data were collected with the following specifications: anellipticity sensitivity of 100 mdeg, a band width of 1.0 nm, a range of 190–260 nm, a speed of 45 nm/min, and a data pitch of 1.0 nm31.
Druglikeness study and prediction of pharmacokinetics and toxicity
In silico druglikeness study and ADME/T prediction were obtained from the preADMET online server (https://preadmet.bmdrc.org/).
Data availability
All data generated or analyzed during this study are included in this published article and its supplementary information files.
Change history
30 June 2025
A Correction to this paper has been published: https://doi.org/10.1038/s41598-025-08983-3
References
Mukhtar, Y., Galalain, A. & Yunusa, U. A modern overview on diabetes mellitus: a chronic endocrine disorder. Eur. J. Biol. Biotechnol. 5, 1–4 (2020).
Whalen, K., Miller, S. & Onge, E. S. The role of sodium-glucose co-transporter 2 inhibitors in the treatment of type 2 diabetes. Clin. Ther. 37, 1150–1166 (2015).
Corder, A. M. & Henry, R. J. Carbohydrate-degrading enzymes in germinating wheat. Cereal Chem. 66, 435–439 (1989).
Eldor, R., DeFronzo, R. A. & Abdul-Ghani, M. Vivo actions of peroxisome proliferator–activated receptors: glycemic control, insulin sensitivity, and insulin secretion. Diabetes Care. 36, 162–174 (2013).
Braunschweig, C. L. et al. Nutritional status and risk factors for chronic disease in urban-dwelling adults with down syndrome. Am. J. Ment Defic. 109, 186–193 (2004).
VSS, P. et al. Nutritional components relevant to type-2-diabetes: dietary sources, metabolic functions and glycaemic effects. J. Adv. Med. Dent. Sci. Res. 52–75 (2018).
Chiba, S. Molecular mechanism in α-glucosidase and glucoamylase. Biosci. Biotechnol. Biochem. 61, 1233–1239 (1997).
Kim, M. J. et al. Comparative study of the Inhibition of α-glucosidase, α-amylase, and cyclomaltodextrin glucanosyltransferase by acarbose, isoacarbose, and acarviosine–glucose. Arch. Biochem. Biophys. 371, 277–283 (1999).
Im, K. H., Choi, J., Baek, S. A. & Lee, T. S. Hyperlipidemic inhibitory effects of Phellinus Pini in rats fed with a high fat and cholesterol diet. Mycobiology 46, 159–167 (2018).
Aoki, K., Muraoka, T., Ito, Y., Togashi, Y. & Terauchi, Y. Comparison of adverse Gastrointestinal effects of acarbose and Miglitol in healthy men: a crossover study. Intern. Med. 49, 1085–1087 (2010).
Nadeem, M. S., Hayat, S., Zeyadi, M. A., Kazmi, I. & Ullah, H. Design, synthesis, biological and computational analysis of isatin-based bis-thiourea analogues as anti-diabetic and anti-nematode agents. J. Mol. Struct. 1323, 140698 (2025).
Iqbal, T. et al. Insights into the role of potent thiadiazole based schiff base derivatives in targeting type-II diabetes: a combine in-vitro and in-silico approaches. J. Mol. Struct. 1321, 140000 (2025).
Taha, M. et al. Synthesis, biological evaluation and molecular docking study of indazole based schiff base analogues as new anti-diabetic inhibitors. J. Mol. Struct. 1300, 137189 (2024).
Aleid, G. et al. Design, synthesis, biological evaluation and molecular Docking study of thiadiazole-isatin hybrid analogues as potential anti-diabetic and anti-bacterial agents. Results Chem. 11, 101805 (2024).
Ullah, H. et al. Synthesis, in vitro α-amylase activity and molecular Docking study of Benzoxazole derivatives. Chem. Data Collect. 51, 101133 (2024).
Rocha, S. et al. A study towards drug discovery for the management of type 2 diabetes mellitus through Inhibition of the carbohydrate-hydrolyzing enzymes α-amylase and α-glucosidase by chalcone derivatives. Food Funct. 10, 5510–5520 (2019).
Mushtaq, A., Azam, U., Mehreen, S. & Naseer, M. M. Synthetic α-glucosidase inhibitors as promising anti-diabetic agents: recent developments and future challenges. Eur. J. Med. Chem. 249, 115119 (2023).
Viegas-Junior, C., Danuello, A., da Silva Bolzani, V., Barreiro, E. J. & Fraga, C. A. M. Molecular hybridization: a useful tool in the design of new drug prototypes. Curr. Med. Chem. 14, 1829–1852 (2007).
Samala, A., Murthy, M. S. & Gottumukkala, K. M. A comprehensive review on recent advances in synthesis and pharmacotherapeutic potential of betacarbolines. Res. J. Pharm. Technol. 11, 3547–3560 (2018).
Iqbal, S. et al. New carbazole linked 1, 2, 3-triazoles as highly potent non-sugar α-glucosidase inhibitors. Bioorg. Chem. 74, 72–81 (2017).
Ravinder, K. et al. Isolation and synthesis of a novel β-Carboline guanidine derivative Tiruchanduramine from the Indian Ascidian Synoicum macroglossum. Tetrahedron Lett. 46, 5475–5478 (2005).
Lin, J. et al. New β-carboline derivatives as potential α-glucosidase inhibitor: synthesis and biological activity evaluation. J. Mol. Struct. 1283, 135279 (2023).
Liang, B., Xiao, D., Wang, S. H. & Xu, X. Novel thiosemicarbazide-based β-carboline derivatives as α-glucosidase inhibitors: synthesis and biological evaluation. Eur. J. Med. Chem. 275, 116595 (2024).
Sepehri, N. et al. New acridine-9-carboxamide linked to 1, 2, 3-triazole-N-phenylacetamide derivatives as potent α-glucosidase inhibitors: design, synthesis, in vitro, and in Silico biological evaluations. Med. Chem. Res. 29, 1836–1845 (2020).
Yousefnejad, F. et al. Design, synthesis, in vitro, and in Silico evaluations of benzo [d] imidazole-amide-1, 2, 3-triazole-N-arylacetamide hybrids as new antidiabetic agents targeting α-glucosidase. Sci. Rep. 13, 12397 (2023).
Kovvuri, J. et al. Design, synthesis and biological evaluation of new β-carboline-bisindole compounds as DNA binding, photocleavage agents and topoisomerase I inhibitors. Eur. J. Med. Chem. 143, 1563–1577 (2018).
Zhao, Y. et al. Design, synthesis and evaluation of novel bivalent β-carboline derivatives as multifunctional agents for the treatment of Alzheimer’s disease. Bioorg. Med. Chem. Lett. 26, 3812–3824 (2018).
Tokala, R. et al. Development of β-carboline-benzothiazole hybrids via Carboxamide formation as cytotoxic agents: DNA intercalative topoisomerase IIα Inhibition and apoptosis induction. Bioorg. Chem. 106, 104481 (2021).
Pradeepkiran, J. A., Sainath, S. B. & Shrikanya, K. V. L. In silico validation and ADMET analysis for the best lead molecules. In Brucella Melitensis 133–176 (Academic Press, 2021).
Daina, A., Michielin, O. & Zoete, V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 7, 42717 (2017).
Miles, A. J., Janes, R. W. & Wallace, B. A. Tools and methods for circular dichroism spectroscopy of proteins: a tutorial review. Chem. Soc. Rev. 50, 8400–8413 (2021).
Funding
This work was financially supported by the National Institute for Medical Research Development (NIMAD) (the grant number: 4021352).
Author information
Authors and Affiliations
Contributions
E.S., M.H.S., N.D., A.Z., A.T-R., and M.N contributed in the synthesis and characterization of compounds. S.M., M.A.F., and B.L. performed in vitro biological assay. M.H. and M.M-K performed in silico assays. M.M. and M.A. conceived the idea and designed the experiments. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original online version of this Article was revised: In the original version of this Article, Mohammad Mahdavi was omitted as a corresponding author. Correspondence and requests for materials should also be addressed to momahdavi@tums.ac.ir.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Safaie, E., Sayahi, M.H., Dastyafteh, N. et al. 1-Phenyl-β-carboline-3-carboxamide-1,2,3-triazole-N-phenylacetamide hybrids as new α-glucosidase inhibitors. Sci Rep 15, 17418 (2025). https://doi.org/10.1038/s41598-025-99807-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-99807-x
