
Abstract
Ovarian cancer is the deadliest gynecological malignancy, owing to its late-stage diagnosis and high rates of recurrence and resistance following standard-of-care treatment, highlighting the need for novel treatment approaches. Through an unbiased drug screen, we identified the kinase inhibitor, lestaurtinib, as a potent antineoplastic agent for chemotherapy- and PARP-inhibitor (PARPi)-sensitive and -resistant ovarian cancer cells and patient derived xenografts (PDXs). RNA-sequencing revealed that lestaurtinib potently suppressed JAK/STAT signaling and lestaurtinib efficacy was shown to be directly related to JAK/STAT pathway activity in cell lines and PDX models. Most ovarian cancer cells exhibited constitutive JAK/STAT pathway activation and genetic loss of STAT1 and STAT3 resulted in growth inhibition. Lestaurtinib also displayed synergy when combined with cisplatin and olaparib, including in a model of PARPi resistance. In contrast, the most well-known JAK/STAT inhibitor, ruxolitinib, lacked antineoplastic activity against all ovarian cancer cell lines and PDX models tested. This divergent behavior was reflected in the ability of lestaurtinib to block both Y701/705 and S727 phosphorylation of STAT1 and STAT3, whereas ruxolitinib failed to block S727. Consistent with these findings, lestaurtinib additionally inhibited the serine/threonine kinases, JNK and ERK, leading to more complete suppression of STAT phosphorylation. Concordantly, combinatorial treatment with ruxolitinib and a JNK or ERK inhibitor resulted in synergistic antineoplastic effects at dose levels where the single agents were ineffective. Taken together, these findings indicate that lestaurtinib, and other treatments that converge on JAK/STAT signaling, are worthy of further pre-clinical and clinical exploration for the treatment of highly aggressive and advanced forms of ovarian cancer.
Similar content being viewed by others

Introduction
Epithelial ovarian cancer is the 6th leading cause of cancer related deaths in women and the most fatal of all female reproductive cancers1,2. Patient outcomes following the diagnosis of ovarian cancer are dismal with 5-year overall survival of less than 30%2. These poor survival rates reflect an insufficiency of routine screening methods, lack of early detection, and de novo or rapidly acquired resistance to current standard-of-care regimens3. Problematically, over 75% of patients are diagnosed with stage III or IV disease, where there is already metastatic spread throughout the peritoneum with the involvement of other organ sites3. Current treatment guidelines for patients with newly diagnosed advanced ovarian cancer include debulking surgery with adjuvant or neoadjuvant therapy using carboplatin and paclitaxel4,5. More recently, a few targeted therapies have been FDA approved, including multiple PARP inhibitors (PARPi)6, the anti-VEGF inhibitor, bevacizumab, for use in combination with chemotherapy or a PARPi7, and mirvetuximab soravtansine, an antibody-drug conjugate targeting the folate receptor alpha (FRα)8. Despite these advances, the majority of patients experience only modest increases in progression-free survival (PFS) with PARPi or VEGFi therapies6, and mirvetuximab soravtansine is currently only approved for patients who have received 1–3 lines of prior systemic therapy8. Importantly, none of these additional lines of treatment have led to meaningful increases in overall survival.
To combat these challenges, an increasing number of studies have aimed to define mechanisms of ovarian cancer resistance, particularly to platinum-based and PARPi therapies, and to characterize the molecular alterations developed during treatment that may be therapeutically actionable. Thus far, identified mechanisms of resistance include restoration of homologous recombination (HR) activity, increased expression of drug efflux pumps, mitigation of replication stress and alterations in growth factor signaling pathways, among others9. Despite these efforts and the many resulting early phase clinical trials, the 5- and 10-year survival rates of ovarian cancer patients has only improved by 6.1% and 2.5% respectively since the 1980’s10. These realities highlight the need to better understand the basis for systemic treatment failure and to devise alternative, more effective, treatment strategies.
In this study, we employed an unbiased drug screen to identify small molecules that potently inhibit and/or kill both treatment-sensitive and treatment-resistant ovarian cancer cell line models. We identified the kinase inhibitor lestaurtinib as a drug of interest and, through RNA-sequencing (RNAseq) analysis, demonstrated that this agent primarily inhibits the JAK/STAT signaling pathway in ovarian cancer cells. Further analysis comparing lestaurtinib to the FDA approved JAK inhibitor, ruxolitinib, found that lestaurtinib inhibited STAT1 and STAT3 serine as well as tyrosine phosphorylation. Finally, lestaurtinib showed monotherapy activity in many ovarian cancer cell line and PDX models and enhanced the antineoplastic effects of cisplatin and olaparib in both treatment naïve and treatment refractory models.
Results
Drug screen identifies lestaurtinib as an inhibitor of therapy-sensitive and -resistant ovarian cancer cells
To identify novel inhibitors of ovarian cancer cell viability, we performed a drug screen using 1 µM concentrations of a panel of small molecules targeting epigenetic regulators. To enhance generalizability of the results, we employed representative cell lines of the two most common ovarian cancer subtypes11, high-grade serous (OVSAHO) and endometrioid (MDAH), and their isogenic cisplatin-resistant clones12. Approximately 15% of the compounds were found to inhibit cell viability in at least 1 of the 4 cell lines (Fig. 1a) and hits were prioritized as indicated in the schema (Fig. 1b). Four compounds were identified that met these criteria: (S)-HDAC-42, SB939, CPI-203, and lestaurtinib. (S)-HDAC-42 and SB939 are both HDAC inhibitors and are part of a drug class that is widely studied in ovarian cancer13. CPI-203 is an epigenetic reader domain inhibitor that binds to the bromodomains of BRD4 and has previously been studied in the context of ovarian cancer14. In contrast, lestaurtinib (CEP-701, Teva), a broad spectrum tyrosine kinase inhibitor known to target FLT3, JAK2, and PRK115, has never been studied in the context of ovarian cancer, thus we elected to further evaluate this compound. To validate the antineoplastic activity of lestaurtinib, we conducted dose response cell viability assays in an expanded panel of ovarian cancer cell lines, including MDAH, MDAH cisplatin-resistant (Cis Res), OVSAHO, OVSAHO Cis Res, SKOV3 (serous cystadenocarcinoma), OVCAR7 (high grade serous), JHOC5 (clear cell carcinoma), PEO1 (high grade serous), two isogenic therapy resistant PEO1-related clones (PEO4; cisplatin-resistant and ABTR#2; PARPi-resistant), COV362 (high grade serous), and its isogenic PARPi-resistant clone (COV362-OlapR). Across this panel, the lestaurtinib IC50 concentrations ranged from 10-410 nM (Fig. 1c), which is far below the known Cmax and Ctrough values for lestaurtinib (7-12 µM and 1-12 µM, respectively) in patients receiving a standard dose of 80 mg twice daily16,17.

a Viability of indicated cell lines following 48 h of treatment with 1 µM of the 145 compounds contained in the Cayman Chemical epigenetic drug library (#11076) relative to DMSO vehicle. N = 2. b Drug screen hit prioritization schema. Figure created with BioRender.com. c Dose response curves (two-fold dilution starting at 2.5 µM) and IC50 concentrations for lestaurtinib in a panel of therapy-sensitive and -resistant ovarian cancer cell lines following 5–10 days of treatment. N = 8. Graphs represent mean ± standard error. Abbreviations: Cis Res cisplatin-resistant, OlapR olaparib-resistant.
Lestaurtinib induces cell cycle arrest, apoptosis, and inhibits colony formation
To determine whether lestaurtinib is cytostatic or cytotoxic, we conducted cell cycle profiling and assessed levels of apoptosis. OVSAHO, MDAH, PEO1, and ABTR#2 cell cycle reporter lines were treated with escalating doses of lestaurtinib. Following 24 h of treatment, lestaurtinib induced a G1 cell cycle arrest with a corresponding G2/M-phase diminution in all 4 cell lines (Fig. 2a, Supplementary Fig. 1a). Longer treatment (2–5 days) with lestaurtinib induced substantial levels of apoptosis as measured by Caspase-3/7 and Annexin V reporter assays at these same concentrations (Fig. 2b, Supplementary Fig. 1b, c). Finally, lestaurtinib concentrations of ≥250 nM markedly inhibited colony formation in all cell models (Fig. 2c, Supplementary Fig. 1d). These results indicate that lestaurtinib rapidly induces cell cycle arrest followed by progressive induction of apoptosis, resulting in significant suppression of cell viability at clinically achievable concentrations.

a Indicated cell lines expressing a fluorescent cell cycle reporter were treated with multiple concentrations of lestaurtinib for 24 h and cell cycle phase distribution was monitored. The percentage of cells in each phase of the cell cycle was quantified via the IncuCyte S3 system based on the presence of nuclear red (G1), nuclear green (G2/M) and nuclear yellow (S). Representative images of the ABTR#2 cell line are shown. N = 5–8. Graphs represent mean ± standard error. ANOVA p-values compared to DMSO for each cell cycle stage: *<0.0332. b Nuc Red expressing cells were incubated with Caspase 3/7-GFP or Annexin V-GFP reporters and subsequently treated with lestaurtinib for 2 days. Percent apoptotic cells were defined as # green positive cells relative to # of red positive cells. Representative images of the ABTR#2 cell line are shown. N = 3–5. c Colony formation was assessed in the indicated cell lines following 3 weeks of treatment with multiple concentrations of lestaurtinib and the number of colonies were quantified by crystal violet staining. N = 3. Representative images of the ABTR#2 cell line are shown. Abbreviations: Nuc nuclear. Graphs represent mean ± standard error. ANOVA p-values: *<0.0332,**<0.0021,***<0.0002,****<0.0001.
Lestaurtinib inhibits JAK/STAT signaling
To elucidate the genome-wide effects of lestaurtinib on ovarian cancer cells, we conducted bulk RNAseq in MDAH and OVSAHO cells treated with 500 nM lestaurtinib for 24 h. In the MDAH cells, there were 371 genes downregulated and 614 genes upregulated (Fig. 3a), while there were 473 genes downregulated and 466 genes upregulated in the OVSAHO cells (Fig. 3b). Interrogation of these differentially regulated genes in each cell line via Ingenuity Pathway Analysis (IPA) revealed significant alterations to many inflammatory pathways (IL-17 signaling, rheumatoid arthritis, neuroinflammation, acute phase response, S100 family signaling, cytokine production and signaling, TNFR1/2 signaling, inflammatory airway diseases, and iNOS signaling), which are all comprised of JAK/STAT signaling components (Fig. 3c, d). Cell-cell communication pathways (tumor microenvironment, EMT by growth factors pathway, and neuregulin signaling), pro-cancer/proliferation pathways (cholesterol biosynthesis, ceramide signaling, p53 signaling, glioma signaling), and HIF1α signaling were also impacted (Fig. 3c, d). Upstream regulator analysis indicated that inhibition of STAT-mediated transcription likely contributed to the differential gene expression profiles following lestaurtinib treatment (Fig. 3e, f). In addition, we conducted Gene Set Enrichment Analysis (GSEA) on the MDAH and OVSAHO datasets and found that many JAK/STAT signaling hallmark and KEGG pathways, as well as hallmark interferon gamma and inflammatory response pathways, were enriched in vehicle treated samples and depleted in lestaurtinib treated samples (Supplementary Fig. 2).

Volcano plots depicting differentially expressed genes in (a) MDAH and (b) OVSAHO cells, where blue indicates downregulated and red indicates upregulated genes ( | fold change | ≥ 1.5, p < 0.05 and FDR < 0.1). The number of of significantly regulated genes are indicated. Bubble plots depicting significantly regulated pathways following lestaurtinib treatment of (c) MDAH and (d) OVSAHO cells as identified by Ingenuity Pathway Analysis. Bubble blots depicting predicted upstream regulators that likely contribute to lestaurtinib mediated differential gene expression in (e) MDAH and (f) OVSAHO cells. g PCA plot generated from RNAseq results of MDAH and OVSAHO cells treated with vehicle and lestaurtinib. h Venn diagram depicting the overlap of lestaurtinib regulated genes in MDAH and OVSAHO cells. i Line graph showing concordance/discordance of the 161 commonly regulated by lestauritnib in both MDAH and OVSAHO cell lines. Abbreviations: Lest lestaurtinib, Veh vehicle. Pathways and regulators with p < 0.05 and |z-score | > 2 are shown.
We next compared the MDAH and OVSAHO cell line responses to lestaurtinib with each other and found that they vastly differed, a result that primarily reflects the basal expression differences observed between these two cell lines which is likely driven by the ovarian cancer subtype of origin (endometrioid vs high grade serous, respectively) (Fig. 3g). Specifically, a total of 824 and 778 uniquely regulated genes following lestaurtinib treatment were identified in the MDAH and OVSAHO cells, respectively, in addition to 161 commonly regulated genes (Fig. 3h). Of the 161 commonly regulated genes, 61.5% were directionally concordant (Fig. 3i). Despite these differences in cell line dependent gene expression changes, the overall impact of lestaurtinib on JAK/STAT signaling and inflammatory pathways was conserved between the two cell lines.
Activity of lestaurtinib in ovarian cancer PDX models ex vivo
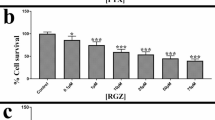
To further extend the potential clinical relevance of these observations in ovarian cancer, the inhibitory effects of lestaurtinib were assessed in a panel of ovarian cancer PDXs grown ex vivo as spheroid cultures (Fig. 4a). All the PDX models were derived from high grade serous ovarian cancer tumors and their characteristics can be found in Supplementary Data 1. Of the 8 models tested, 4 were responsive to lestaurtinib while another 4 were not (Fig. 4b). Four of the PDX models represent PARPi-sensitive and -resistant pairs (Supplementary Data 1). PH039S and PH077S were derived from treatment naïve patients who responded to cisplatin treatment and have previously been confirmed to be sensitive to paclitaxel/carboplatin treatment both with and without the PARPi, niraparib18, as well as to carboplatin and niraparib as single agents19. PH039R and PH077R represent derivatives of these two models that were developed to be PARPi-resistant through 4 serial cycles of niraparib treatment in mice19. Of note, the PARPi-resistant PH039R derivative was exquisitely sensitive to lestaurtinib whereas the treatment naïve parental model was not (Fig. 4b). In contrast, neither of the PH077 models responded to lestaurtinib (Fig. 4b).

a Depiction of ovarian cancer PDX processing for rapid evaluation of drug efficacy in ex vivo cultures. Created with BioRender.com. b Viability of PDX models following 3 days of lestaurtinib treatment as assessed via 3D CellTiter-Glo. N = 3–6. Volcano plots depicting differentially expressed genes in (c) PH039R vs PH039S, d PH077R vs PH077S, and e lestaurtinib sensitive (PH039R, PH730, PH580) vs lestaurtinib resistant (PH039S, PH077S, PH077R, PH354) PDXs as determined by RNAseq, where blue indicates significantly downregulated genes and red indicates significantly upregulated genes ( | fold change | ≥ 1.5, p < 0.05 and FDR < 0.1). The number of significantly regulated genes are shown. f–k Ingenuity Pathway Analysis and upstream regulator assessment of differentially expressed genes between the indicated PDX models. Pathways and up-stream regulators with p < 0.05 and |z-score | > 2 were considered as significant. Abbreviations: Lest lestaurtinib, Sen sensitive, Res resistant. Graphs depict mean ± standard error. ANOVA p-values: *<0.0332,**<0.0021,***<0.0002,****<0.0001.
Given these differences, we compared RNAseq data generated from the two PDX pairs (PH039 and PH077) that were derived from untreated mice, as well as the sequencing data derived from untreated PDX specimens of the 3 lestaurtinib-sensitive vs the 4 lestaurtinib-resistant models to assess inherent gene expression differences that may contribute to lestaurtinib sensitivity (Fig. 4c–e). Differentially expressed genes in untreated PH039R vs PH039S were subjected to IPA which revealed that the top three upregulated pathways in PH039R were interferon signaling, hypercytokinemia in influenza, and ISGylation signaling, all of which comprise JAK/STAT signaling pathway components (Fig. 4f). Additionally, STAT1 was identified as a top transcriptional regulator likely driving the more highly expressed genes in PH039R (Fig. 4g). These results suggest that upregulation of JAK/STAT signaling and inflammatory pathways is predictive of lestaurtinib sensitivity. In contrast, we compared the matched pair, PH077R vs PH077S, neither of which were responsive to lestaurtinib, and found that none of the pathways or upstream effectors identified involved JAK/STAT signaling (Fig. 4h, i); further supporting the notion that active JAK/STAT signaling predicts lestaurtinib sensitivity. When pooling sequencing data from the lestaurtinib-sensitive vs -resistant PDXs, we discovered substantial upregulation of JAK/STAT related pathways in models that responded to lestaurtinib vs those that did not, including interferon induction, IRF activation, IFNα/β signaling, and cytokine storm signaling, with IRF1, 5, and 7 as identified upstream regulators (Fig. 4j, k). These findings, in addition to those reported in Fig. 3, strongly suggest that the antineoplastic activity of lestaurtinib may only be observed in cells/tumors with high JAK/STAT signaling and inflammatory pathway activity.
JAK/STAT pathway activation in ovarian cancer cell lines
Considering the observation that lestaurtinib suppresses genes involved in JAK/STAT signaling and elicits potent antineoplastic activity in PDX models with highly active JAK/STAT signaling, we characterized key components of this pathway in two immortalized fallopian tube epithelial cell lines (FT194 and FT282) and across a panel of ovarian cancer cell lines for which isogenic treatment-resistant derivatives were available. At the RNA level, JAK1 was the most abundant of the kinases, while STAT1 and STAT3 were the predominant STATs detected (Fig. 5a). At the protein level, JAK1 and JAK2 were robustly expressed, while JAK3 was only detected in one of the ovarian cancer cell lines (Fig. 5b, Supplementary Fig. 3a). JAK2 exhibited similarly high expression in the two immortalized fallopian tube epithelial cell lines, while JAK1 was much less abundant relative to cancer cells (Fig. 5b, Supplementary Fig. 3a). Total and phosphorylated forms of STAT1, STAT3 and STAT5 protein were highly variable, but were universally detected, apart from STAT5 in OVSAHO and the PEO resistant models (Fig. 5b, Supplementary Fig. 3a).

a Relative mRNA expression levels of primary JAK/STAT family members in normal fallopian tube epithelial cells (FT194, FT282) and ovarian cancer cell lines under normal growth conditions. b Western blots depicting the total protein and phosphorylation levels of primary JAK/STAT signaling components in indicated cell lines under normal growth conditions. Actin and vinculin served as loading controls. c Western blots depicting the levels of STAT1 and STAT3 phosphorylation in response to 10 ng/mL IFNγ and increasing concentrations of lestaurtinib for 30 min. Actin served as the loading control. d Western blots depicting total STAT1 and STAT3 protein following 72 h treatment with scrambled control, STAT1 or STAT3 siRNA. Actin served as the loading control. e Relative cell proliferation rates in response to siRNA-mediated STAT1 or STAT3 knockdown 5 days after transfection. N = 5–10. f Representative brightfield images depicting cell density 5 days post-transfection. g Western blots depicting total STAT1 and STAT3 protein levels in MDAH and OVSAHO parental, STAT1 (S1) and STAT3 (S3) CRISPR knockout (KO) cells. Actin served as the loading control. h Relative proliferation rates of control, STAT1 and STAT3 KO cells and i representative brightfield images depicting cell density 5 days after plating. N = 8–10. j STAT1 and STAT3 essentiality in a panel of 59 ovarian cancer cell lines assessed via RNAi or CRISPR KO screens from the DepMap database. Gene Effect <0 indicates dependency, <-1 indicates essentiality. Abbreviations: Par parental, Cis Res cisplatin resistant, OlapR olaparib-resistant, Con control, WT wildtype, KO knockout, S1 STAT1, S3 STAT3. Graphs depict mean ± standard error. ANOVA p-values: *<0.0332,**<0.0021,***<0.0002,****<0.0001.
In further studies, we assessed the effects of IFNγ and lestaurtinib on STAT1 and STAT3 activation in ovarian cancer cells. IFNγ robustly induced pY701-STAT1, pS727-STAT1, and pY705-STAT3 protein levels (Fig. 5c, Supplementary Fig. 3b). Little to no impact on pS727-STAT3 by IFNγ was observed, but this residue was found to be constitutively activated across all models (Fig. 5c, Supplementary Fig. 3b). Lestaurtinib treatment inhibited IFNγ-mediated phosphorylation of STAT1 and STAT3 in a dose dependent manner and largely suppressed pS727 phosphorylation below basal levels for both STAT1 and STAT3 (Fig. 5c, Supplementary Fig. 3b). These results indicated that JAK1 and JAK2 are the primary upstream effectors of the JAK/STAT pathway in ovarian cancer cells and that lestaurtinib effectively inhibits phosphorylation of two key residues on STAT1 and STAT3 that are required for their activity.
Contribution of STAT1 and STAT3 to ovarian cancer cell viability and proliferation
To directly assess the roles of STAT1 and STAT3 in ovarian cancer cell viability and proliferation, siRNAs were used to selectively knockdown STAT1 and STAT3 in MDAH and OVSAHO cells (Fig. 5d, Supplementary Fig. 3c). Following STAT1 or STAT3 knockdown, MDAH and OVSAHO cells exhibited significant growth inhibition (Fig. 5e, f). Given that siRNA-mediated knockdown of STAT1 and STAT3 were incomplete, we used CRISPR-Cas9 to interrupt the genes encoding these transcription factors. Successful knockouts (KO) were identified by western blotting and confirmed by DNA Sanger sequencing (Fig. 5g, Supplementary Fig. 3d, e). Both STAT1-KO and STAT3-KO isogenic cell lines grew significantly slower than their respective parental controls (Fig. 5h, i). These findings are in agreement with DepMap Portal20 data of 59 ovarian cancer cell lines indicating that knockdown or deletion of STAT1 and STAT3 are largely growth inhibitory (gene effect scores = –0.5 to 0), but not lethal (gene effect scores ≤ –1, Fig. 5j). These genetic loss of function studies further implicate STAT1 and STAT3 as drivers of ovarian cancer cell proliferation.
Lestaurtinib synergizes with standard-of-care agents
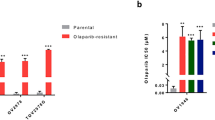
Because complete ablation of STAT1 or STAT3 was not sufficient to kill ovarian cancer cells, we next sought to determine whether inhibition of JAK/STAT signaling would enhance the cytotoxicity of standard-of-care therapies. We designed synergy studies based on the individual drug IC50s (Fig. 1, Supplementary Fig. 4), altered the ratios between the two inhibitors, and used both the Loewe synergy score (synergy >10)21 and the Chou-Talalay combination index (CI, synergy <1 and highly synergistic <0.3)22 to assess synergistic effects. Lestaurtinib was found to synergize with cisplatin in all cell lines tested via the Loewe synergy score, though PEO1 cells (inherently platinum sensitive due to a BRCA2 mutation) exhibited a lower degree of synergy (Fig. 6a). Using the CI method, synergy was found for specific dose ratios for each cell line that were within clinically achievable dose ranges for both drugs, with highly synergistic combinations identified in certain dose ratios for MDAH and ABTR#2 cells (Fig. 6b, Supplementary Data 2). Lestaurtinib also synergized with the PARPi, olaparib, across all cell lines according to the Loewe synergy score (Fig. 6c). The CI method indicated that there were multiple dose ratios of lestaurtinib and olaparib that were strongly synergistic in MDAH, OVSAHO, and PEO1 cells with most synergistic combinations falling within clinically achievable concentrations (Fig. 6d, Supplementary Data 2). Importantly, lestaurtinib also synergized with olaparib in the ABTR#2 cells, a PARPi-resistant cell line, indicating its potential to resensitize cells to PARPi therapy (Fig. 6d, Supplementary Data 2). These data support the potential utility of lestaurtinib, not only as a monotherapy or salvage strategy, but also as a component of combination therapies, even in the setting of advanced and resistant forms of disease.

Indicated cells were treated with cisplatin (25,000 nM or 10,000 nM, threefold dilution) or olaparib (25000 nM, threefold dilution), alone and in combination with lestaurtinib (5000 nM or 1000 nM, threefold dilution), for 5–7 days. Relative cell viability was measured with crystal violet staining. a, c Synergy plots depicting Loewe scores generated using SynergyFinder3.0. b, d Combination index (CI) scores were calculated using the Chou-Talalay method and Compusyn software. CI-ED50 = combination index at 50% fraction effected, CI-ED95 = combination index at 95% fraction effected, where the fraction effected represents precent cell death. Antagonism = >1 (black), additive = 1 (gray), synergy = < 1 (blue), and highly synergistic = < 0.3 (red). Pink highlighting = synergism identified using clinically achievable concentrations of both drugs.
Comparison of lestaurtinib and ruxolitinib in ovarian cancer cell lines
At present, the most well characterized JAK/STAT inhibitor is ruxolitinib23. To determine whether ruxolitinib would induce similar effects to that of lestaurtinib, we performed cell proliferation assays and derived IC50 values in our panel of ovarian cancer cell lines. Ruxolitinib IC50 values were near or above 20 µM (Fig. 7a), a concentration far above the plasma Cmax of 1 µM and Ctrough of 100 nM24 achieved in humans receiving the standard dose of 25 mg twice daily23. Similarly, ruxolitinib did not significantly inhibit any of the 8 PDX models treated ex vivo (Fig. 7b), including those models with high levels of JAK/STAT pathway activity (Fig. 4). Ruxolitinib did not induce cell cycle arrest after 24 h of treatment nor did it induce apoptosis after 5 days of treatment (Supplementary Fig. 5a, b). However, ruxolitinib did modestly inhibit colony formation following 3 weeks of treatment (Supplementary Fig. 5c).

a Dose response curves (twofold dilution starting at 200 µM) and IC50 concentrations for ruxolitinib in a panel of therapy-sensitive and -resistant ovarian cancer cell lines following 5–10 days of treatment. N = 8. b Viability of PDX models following 3 days of ruxolitinib treatment ex vivo as assessed via 3D CellTiter-Glo. N = 3–6. c Expression of indicated STAT1 and STAT3 regulated genes following 24 h of treatment with DMSO, 250 nM lestaurtinib or 250 nM ruxolitinib. d Western blots depicting the levels of STAT1 and STAT3 phosphorylation following treatment with DMSO or indicated concentrations of lestaurtinib and ruxolitinib for 30 min. Actin served as the loading control. Abbreviations: Cis Res cisplatin resistant, OlapR olaparib-resistant, Lest lestaurtinib, Ruxo ruxolitinib. Graphs depict mean ± standard error. ANOVA p-values: *<0.0332,**<0.0021,***<0.0002,****<0.0001.
To examine the basis for the disparate effects of ruxolitinib and lestaurtinib, we assessed the ability of these two drugs to alter the expression of lestaurtinib regulated genes identified from the RNAseq studies (Fig. 3a, b) that are components of the JAK/STAT and inflammatory pathways shown to be altered by lestaurtinib treatment (Fig. 3c, d). Following 24 h of lestaurtinib treatment, CCN2 (4/4 cell lines), KLF2 (3/4 cell lines), and SAA1 (4/4 cell lines) gene expression was increased as expected, while 24 h of ruxolitinib treatment did not induce these genes with the exception of CCN2 and SAA1 in ABTR#2 cells (Fig. 7c). For downregulated genes, lestaurtinib treatment suppressed N-CAD (3/4), FN1 (1/4), and p21 (3/4) while ruxolitinib elicited no inhibitory effects, and instead actually increased expression of these genes in the PEO1 and ABTR#2 cells (Fig. 7c). Further, we compared the ability of lestaurtinib and ruxolitinib to inhibit phosphorylation of the Y701/705 and S727 residues of STAT1 and STAT3. Similar to Fig. 5c, lestaurtinib suppressed phosphorylation of these residues in a dose dependent manner. In contrast, ruxolitinib potently inhibited Y701/705 phosphorylation, but had little to no effect on S727 (Fig. 7d, Supplementary Fig. 6a). These results unveil a substantial difference between lestaurtinib and ruxolitinib regarding their effects on the phosphorylation profiles, and thus the activities, of STAT1 and STAT3 in ovarian cancer cells.
Basis for lestaurtinib-mediated inhibition of STAT1 and STAT3 S727 phosphorylation
Given the ability of lestaurtinib to inhibit S727 phosphorylation of STAT1 and STAT3, we sought to identify the mechanistic basis for this inhibition. Kinases found to bind lestaurtinib with a KD of ≤500 nM (82 total) were curated from a Kinobead screen25. We then interrogated the PhosphoSitePlus database to identify kinases predicted or known to phosphorylate the S727 residue on STAT1 and STAT326 and pulled the top 30 kinases from each of these lists. When comparing the overlap of these lists (Supplementary Data 3), we found three kinases (MAPK8, MAPK9, and MAPK10 encoding JNK1-3) that were identified in all 3 datasets (Fig. 8a). Ten additional kinases, predominated by the ERK family (MAPK1, MAPK3, MAPK7 and MAPK12), were commonly predicted to phosphorylate S727, but have not yet been shown to bind lestaurtinib (Fig. 8a). Based on these findings, we assessed the effects of lestaurtinib on JNK and ERK via western blotting in a panel of ovarian cancer cells. Lestaurtinib not only inhibited S727 phosphorylation of STAT1 and STAT3 as previously shown (Fig. 5c), but also suppressed phosphorylation of ERK1/2 and JNK1/2 in a dose-dependent manner (Fig. 8b, Supplementary Fig. 6b). Given these findings, we next assessed the effects of selective ERK (LY3214996) and JNK (CC-90001) inhibitors on ovarian cancer cell line growth and STAT1 and STAT3 S727 phosphorylation. The IC50 values for LY3214996 and CC-90001 ranged from 2-6 µM and 2-20 µM, respectively (Supplementary Fig. 6c). As shown in Fig. 8c, d and Supplementary Fig. 6d, e, both inhibitors significantly reduced STAT3 S727 phosphorylation in a dose dependent manner, with some inhibitory effects on STAT1 in these ovarian cancer cell lines. Of note, variable effects were observed in the levels of phosphorylation of ERK and JNK following LY3214996 and CC-90001 treatment respectively. This is well documented in the literature where both increased and decreased ERK and JNK phosphorylation have been reported following treatment with these drugs27,28,29. In the ovarian cancer models employed here, neither inhibitor had any impact on the phosphorylation of ERK or JNK (Fig. 8c, d, Supplementary Fig. 6d, e). To confirm their on-target activity, we also assessed the ability of lestaurtinib, LY3214996, and CC-90001 to inhibit phosphorylation of cJUN, a canonical substrate of both ERK and JNK. These three drugs robustly suppressed phosphorylation of cJUN in a dose dependent manner across multiple ovarian cancer cell line models (Fig. 8e, Supplementary Fig. 6f). Given the selective inhibition of LY3214996 and CC-90001 on S727 phosphorylation, we determined whether their combination with ruxolitinib, a potent inhibitor of Y701/705 phosphorylation, would recapitulate the effects of lestaurtinib. Interestingly, multiple dose ratios of ruxolitinib in combination with LY3214996 or CC90001 were found to be synergistic in all cell lines tested (Fig. 9a–d, Supplementary Data 2). Although most synergistic combinations did not fall within clinically achievable dose ranges of these drugs (Supplementary Data 2), the results support the necessity of blocking both STAT phosphorylation residues for optimal cell growth inhibition, an effect achieved by lestaurtinib monotherapy, but not by other monotherapies examined here.

a Venn diagram of kinases known to be inhibited by lestaurtinib and predicted to phosphorylate S727 of STAT1 and/or STAT3. Western blots depicting total and phosphorylated levels of STAT1, STAT3, ERK and JNK following 30 min of (b) lestaurtinib, c LY3214996 (ERKi) or d CC-90001 (JNKi) treatment. e Western blots depicting total and phosphorylated levels of cJUN following 30 mins of lestaurtinib, LY3214996 (ERKi), or CC-90001 (JNKi) treatment. Vinculin served as the loading control.

Indicated cells were treated with LY3214996 (70 µM, threefold dilution) or CC-90001 (70 µM, threefold dilution), alone and in combination with ruxolitinib (25 µM, threefold dilution) for 5–7 days. Relative cell viability was measured with crystal violet staining. a, c Synergy plots depicting Loewe scores were generated using SynergyFinder3.0. b, d Combination index (CI) scores were calculated using the Chou-Talalay method and Compusyn software. CI-ED50 = combination index at 50% fraction effected, CI-ED95 = combination index at 95% fraction effected, where the fraction effected represents precent cell death. Antagonism = >1 (black), additive = 1 (gray), synergy = <1 (blue), and highly synergistic = <0.3 (red). Pink highlighting = synergism identified using clinically achievable concentrations of both drugs.
Discussion
In the present study, we identified lestaurtinib as a potent inhibitor of ovarian cancer cells with low nM IC50 values that are substantially less than clinically achievable plasma concentrations. We demonstrate that lestaurtinib uniquely and robustly inhibits JAK/STAT signaling through blockade of both Y701/705 and S727 phosphorylation of STAT1 and STAT3. Lestaurtinib treatment of PDX models ex vivo confirmed antineoplastic activity in 4 of 8 models tested. Intriguingly, high JAK/STAT pathway activity was predictive of lestaurtinib efficacy across these models. Moreover, lestaurtinib synergized with both cisplatin and olaparib, even in PARPi-resistant cells. Conversely, the most well-known JAK/STAT inhibitor, ruxolitinib, failed to suppress S727 phosphorylation of STAT1 and STAT3 and displayed no efficacy as a monotherapy in any cell line or PDX model tested. Intriguingly, when ruxolitinib was combined with either a JNK or ERK inhibitor, which were shown to inhibit S727 phosphorylation, synergistic antineoplastic activity was observed. Collectively, these observations have defined novel mechanisms of action by which lestaurtinib functions and highlight the potential of blocking multiple kinases upstream of STAT1 and STAT3 for the treatment of ovarian cancer.
The STAT family of transcription factors consists of seven members that share similar domain structures, including the transactivating domain (TAD) where Y701/705 and S727 are located. The TAD domain is essential for STAT transcriptional activity as it provides binding sites for essential co-regulators and stabilization at target gene promoters30. Phosphorylation of the tyrosine residue of both STAT1 and STAT3 is primarily mediated by JAKs, which are complexed with various cytokine receptors in response to cytokine mediated activation31. However, these tyrosine residues can also be directly phosphorylated by other receptor tyrosine kinases such as EGFR, PDGFR, and CSF1R, among others32. Upon phosphorylation of Y701/705, STAT proteins homo- or hetero-dimerize and translocate into the nucleus where they regulate gene expression32. The consequences of S727 phosphorylation are less well documented but have been reported to play a role in recruitment of transcriptional machinery33,34. Mutation of S727 to alanine, which prevents phosphorylation, has no impact on Y701/705 phosphorylation or STAT DNA binding in vitro or in vivo, but is necessary for maximal transcriptional output35. Further, blockade of STAT3 S727 phosphorylation has been shown to prevent DNA association of STAT3 homodimers, but not STAT1/STAT3 heterodimers or STAT1 homodimers36. Interestingly, high levels of phospho-S727 STAT3 have been reported in endometrial cancer cells, and treatment of these cells with a STAT3 inhibitor significantly decreased S727 phosphorylation resulting in decreased cell viability and induction of cell cycle arrest37. These observations provide a potential explanation for the importance of inhibiting S727 phosphorylation in the present study.
JAK/STAT signaling is reported to be oncogenic in many forms of cancer, including ovarian cancer38,39. A recent study utilizing single cell RNA-sequencing of ovarian tumors and PDX models reported that JAK1, STAT1 and STAT3 were expressed in 80-90% of all tumor cells and were among the most highly abundant transcripts within those cells40. Further, JAK/STAT activating cytokines were among the most highly expressed genes in cells of the tumor microenvironment40 and have previously been linked to chemotherapy resistance and decreased PFS41,42. In ovarian tumors, discrepant roles of STAT1 have been reported. For example, STAT1 was found to be overexpressed in ovarian tumors compared to normal ovarian tissues and this overexpression was associated with better overall survival43,44,45. Additionally, elevated expression of STAT1 has been shown to suppress ovarian cancer metastasis, inhibit angiogenesis, and enhance the anti-tumor immune response46,47,48. Conversely, STAT1 upregulates iNOS in ovarian cancer tissues, which promotes ovarian cancer cell proliferation and suppresses apoptosis49. ITGB1, a known inducer of STAT1 activity via activation of FAK, is also upregulated in ovarian tumors and drives proliferation, inhibition of apoptosis, and increased migration and invasion43,50. Finally, STAT1 has been shown to contribute to chemotherapy resistance, as its elevated expression is associated with cisplatin resistance in multiple studies51,52. Taken together, STAT1 is capable of eliciting both anti-tumor and pro-tumor effects in ovarian cancer, and these discrepant roles are likely dependent on the cell line model, tumor stage, and treatment status. STAT3 is widely accepted to be an oncogene in many cancers, including ovarian cancer53. Specifically, STAT3 is reported to be more highly expressed, with increased Y705 phosphorylation, in ovarian tumors compared to normal ovarian tissues and its expression rises with increasing tumor grade54,55. STAT3 has been shown to induce proliferation and survival of ovarian cancer cells56, enhance angiogenesis57,58,59, induce metastasis60,61,62,63, maintain cancer stem cell populations64,65,66, and drive drug resistance, including chemotherapy-67,68,69 and PARPi-resistance70. These findings, like those presented here, strongly support oncogenic roles for STAT3 in ovarian cancer progression and treatment failure.
Lestaurtinib has previously been studied in clinical trials for acute myeloid leukemia16 (NCT00469859 and NCT00030186), myelofibrosis71,72, multiple myeloma (NCT00242827), leukemia73, polycythemia vera and essential thrombocytosis74, psoriasis (NCT00236119), neuroblastoma75, and prostate cancer (NCT00081601). Lestaurtinib was shown to be overall well-tolerated, with gastrointestinal issues being the most common serious adverse event. Significant clinical benefit was achieved for some patients in both the myelofibrosis trials71,72. Interestingly, lestaurtinib was found to decrease levels of phospho-STAT3 in myelofibrosis patients who responded to lestaurtinib therapy71. Disappointingly, lestaurtinib failed to show clinical benefit for most multiple myeloma (NCT00242827) and leukemia16,73 (NCT00469859 and NCT00030186) patients. With the exception of one trial in prostate cancer (NCT00081601), which has not yet reported results, lestaurtinib has not been studied in solid tumors and is no longer undergoing clinical development. It is therefore unlikely that lestaurtinib will ever be employed in the ovarian cancer setting. Nevertheless, the data presented here utilizing lestaurtinib as a tool compound lend further support to exploring the utility of other compounds, or combination therapies, that converge on the JAK/STAT pathway for the treatment of ovarian cancer, particularly those agents that are capable of blocking multiple phospho-residues on STAT1 and STAT3.
Ruxolitinib, which is FDA approved for the treatment of myelofibrosis, polycythemia and graft-versus-host disease23, is also the only JAK/STAT inhibitor that has been evaluated in a clinical trial for ovarian cancer (NCT02713386). This recently completed phase I/II study identified the maximum tolerated dose of ruxolitinib in the presence of standard-of-care paclitaxel and carboplatin, and subsequently evaluated the efficacy of this combination on PFS relative to paclitaxel/carboplatin alone76. Ruxolitinib was found to be well tolerated with acceptable toxicity and offered a modest improvement in PFS of 3 months (14.6 months vs 11.6 months) with a hazard ratio of 0.702 (p = 0.059) without any improvement in overall survival (HR = 0.785; p = 0.24) in the neoadjuvant setting for patients with newly diagnosed stage III-IV ovarian/fallopian tube/primary peritoneal carcinoma76. In the present report, we demonstrate that ruxolitinib does not inhibit the viability or proliferative capacity of ovarian cancer cells or PDX models, even at concentrations much higher than can be clinically achieved. Nevertheless, the recent ruxolitinib trial has demonstrated the feasibility of implementing early-phase randomized studies for front line treatment of ovarian cancer and suggests that similar trials with more potent JAK/STAT inhibitors should be considered.
It is important to note that multiple classes of drugs have been designed to disrupt JAK/STAT signaling by targeting different nodes of this complex pathway. These include antibodies to block specific cytokines or their cognate receptors, such as interleukins and TNF, which are used to treat rheumatoid arthritis, inflammatory bowel disease and psoriasis77; small molecules similar to ruxolitinib, aimed at broadly or selectively inhibiting specific JAK proteins78; peptidomimetics targeting the SH2 domains of specific STATs designed to prevent dimerization and/or nuclear translocation77; computationally designed non-peptide based small molecules that can selectively bind to STAT proteins and disrupt their transcriptional output77; and nucleotide-based molecules, such as decoy oligonucleotides, antisense oligonucleotides, and siRNAs that disrupt STAT protein-DNA interactions or cause STAT protein downregulation77. The present study not only adds lestaurtinib, an agent with a well understood safety profile, to the list of JAK/STAT pathway inhibitors, but also demonstrates the ability of lestaurtinib to inhibit S727 as well as Y701/705 phosphorylation, which appears to contribute to its potent antineoplastic activity in ovarian cancer models.
While the present study has demonstrated important roles of STAT1 and STAT3 in ovarian cancer progression, and has found that blockade of both Y701/705 and S727 is necessary to achieve potent antineoplastic effects, additional studies are needed to evaluate the exact mechanism by which these transcription factors drive cancer progression. First, it is not yet known how differential phosphorylation of various STAT1 and STAT3 sites contributes to their oncogenic activity and future studies of phospho-mutant and phospho-mimetic versions of STAT1 and STAT3 are needed to address this question. Second, tumor associated macrophages and fibroblasts in the ovarian tumor microenvironment not only produce JAK/STAT activating cytokines, but also exhibit high expression of JAK1, STAT1, and STAT340. Additional studies are needed to delineate the effects of lestaurtinib and other JAK/STAT inhibitors on STAT phosphorylation in these cell types, the resulting consequences on the tumor microenvironment, and ultimately the combined impact on tumor biology and clinically relevant outcomes. Such understanding will allow for the rational design and development of highly specific molecules that precisely disrupt STAT-mediated oncogenic cascades, while potentially sparing essential functions in anti-tumor immune cells and normal tissues. Finally, our RNAseq data revealed that lestaurtinib also impacts additional pathways, which remain to be further explored regarding their contributions to lestaurtinib’s antineoplastic activity.
In summary, we have shown that JAK1, STAT1, and STAT3 are among the most abundantly expressed components of the JAK/STAT pathway in ovarian cancer cells; that constitutive activation of STAT1 and STAT3 frequently occur in ovarian cancer cell lines and PDX models; that lestaurtinib is a potent inhibitor of JAK/STAT signaling with the ability to block multiple phosphorylation events that are essential for STAT transcriptional activity; and that blockade of these phosphorylation events leads to cell cycle arrest followed by high levels of apoptosis that result in lestaurtinib-mediated killing of ovarian cancer cells, especially in settings where JAK/STAT signaling is elevated or constitutively active. These observations provide support for lestaurtinib, and other inhibitors that converge on this pathway, to undergo further preclinical testing and future clinical study for the treatment of highly aggressive and refractory forms of ovarian cancer.
Methods
Drugs and recombinant proteins
The epigenetics screening library (#11076) and lestaurtinib (#12904) were purchased from Cayman Chemicals. Ruxolitinib (#S1378), olaparib (#S1060), LY3214996 (#S8534), SN-38 (#S4908), and rucaparib (#S4948) were purchased from SellekChem. CC-90001 (#HY-138304) was purchased from MedChemExpress. Cisplatin was purchased from Fresenius Kabi (#100351). Recombinant proteins, IFNγ (#285-IF) and IL6 (#7270-IL), were purchased from R&D Systems.
Cell culture
MDAH-2774 (MDAH) and OVSAHO cells, and their respective platinum-resistant clones12, were generously provided by Dr. Gustavo Baldassarre (National Cancer Institute, Italy). PEO1 and PEO4 cells were derived from the same patient during early and later points of her care79, and were generously provided by Dr. Scott Kaufmann (Mayo Clinic, Rochester, MN). The ABTR#2 cell line was derived from PEO1 cells by PARPi selection and were characterized as previously described80. COV362 olaparib-resistant cells (COV362-OlapR) were generated by continuous selection in olaparib concentrations ranging from 0.5 µM to 10 µM, characterized as illustrated in Supplementary Fig. 7, and were generously provided by Dr. Scott Kaufmann (Mayo Clinic, Rochester, MN). JHOC5 and OVCAR7 cells were generously provided by Drs. Evette Radisky (Mayo Clinic, Jacksonville, FL) and Viji Shridhar (Mayo Clinic, Rochester, MN), respectively. SKOV3 (#HTB-77), FT194 (#CRL-3445), and FT282 (#CRL-3449) cell lines were purchased from ATCC. MDAH, OVSAHO, COV362, SKOV3, OVCAR7, and JHOC5 cells were maintained in phenol red-free RPMI medium (#11835050, Gibco) containing 10% (v/v) FBS (#900-308, Gemini Bioproducts) and 1% (v/v) antibiotic/antimycotic (#15240-062, Gibco). COV362-OlapR cells were maintained as the parental line with the addition of 5 µM olaparib. PEO1, PEO4, and ABTR#2 cells were maintained in phenol red-free DMEM/F12 medium (#41121800, Jango Bio) containing 10% (v/v) FBS, 1% (v/v) non-essential amino acids (#M7145, Corning), 10 µg/ml insulin (#IO516, Sigma-Aldrich), and 1% (v/v) Pen/Strep (#15070063, Invitrogen). FT194 and FT282 were maintained in phenol red-free DMEM/F12 medium containing 10% (v/v) FBS and 1% (v/v) Pen/Strep. All cells were cultured in a humidified 37 °C incubator with 5% CO2. Cells were authenticated by IDEXX BioAnalytics (last tested March 2023) and tested for mycoplasma infection every 6 months using a Mycoplasma Detection Kit (#131001, SouthernBiotech).
IncuCyte compatible cell line generation
OVSAHO, MDAH, PEO1, and ABTR#2 cells were infected at an MOI of 3−5 with the IncuCyte Nuclight Red (#4476 Sartorius) or IncuCyte Cell Cycle Green/Red Lentivirus Reagent (#4779, Sartorius), and pooled stably expressing cell lines were selected with and maintained in indicated medium further supplemented with 500 µg/L puromycin (#A11138, Gibco).
Drug library screen
Indicated cell line models were plated at 2000 cells per well in 96 well plates (#3595, Corning). Twenty-four hours later, the 145 compounds contained within the Cayman epigenetics screening library, including DMSO as a vehicle control, were added at a 1 µM final concentration in duplicate. Following 48 h of treatment, cell viability was determined using CellTiter-Glo (#G9243, Promega) following the manufacturer’s protocol and assayed using a GloMax luminometer (Promega). Luminescence values for each compound, in duplicate, were averaged and normalized to the DMSO control wells to generate drug response values.
Proliferation assays
Cells were plated at a density of 1000–2000 cell per well in 96 well plates in 100 μL of cell type specific medium and allowed to adhere overnight. For basal growth monitoring, indicated cells were placed in the IncuCyte S3 instrument (Sartorius) and allowed to proliferate for 5−7 days at which point growth was compared by confluence and/or red nuclei count at the same time point across each comparison. Dose response curves were performed for indicated drugs using 9 rounds of twofold serial dilutions starting at 2.5 µM for lestaurtinib, 200 µM for ruxolitinib, 50 µM for olaparib, LY3214996, and CC-90001, or the dose range of 50, 25, 10, 7.5, 5, 2.5, 1, 0.5 µM for cisplatin. Cells were maintained until vehicle treated wells neared confluence (between 5 and 10 days) at which point cells were fixed with 25% (v/v) glutaraldehyde (# G6257, Sigma-Aldrich) for 10 min, washed four times with water, stained with crystal violet (#C6158, Sigma-Aldrich), and washed again. Crystal violet was solubilized with 100 µL of a solution containing a 1:1 ratio of 100 nM sodium citrate (#W302600, Sigma-Aldrich) and 100% ethanol and quantified using a plate reader at 550 nm excitation. IC50 values were calculated using a sigmoidal dose-response curve (log(inhibitor) vs normalized response-Variable slope) in GraphPad Prism.
Cell cycle profiling
Indicated cell lines expressing the cell cycle reporter were seeded in 96-well plates at a density of 2000–5000 cells per well and treated 24 h later with DMSO or the indicated drugs. Immediately following treatment, cells were imaged every 2−4 h in an IncuCyte S3 instrument (Sartorius) and the percent of cells in each cell cycle phase was calculated according to the manufacturer’s protocol.
Apoptosis assays
Cells expressing Nuclight Red were seeded in 96-well plates at a density of 1000−2000 cells per well and treated 24 h later with DMSO or the indicated drugs plus a 1:1000 dilution of Annexin V-Green (#4642, Sartorius) or a 1:2000 dilution of Caspase3/7-Green (#4440, Sartorius). Cells were imaged every 6 h for 2−5 days in an IncuCyte S3 instrument and the ratio of apoptotic cells (green) to total cells (red) was determined.
Colony formation assays
Cells were plated in triplicate at a density of 200 cells per well in 6-well plates and treated with DMSO or the indicated drugs for approximately 3 weeks. Colonies were stained with crystal violet as described above and the number of colonies was quantified.
RNA-sequencing and analysis
Following treatment of cells with DMSO or lestaurtinib (500 nM) for 24 h, total RNA was isolated using the Qiagen miRNeasy mini kit (#217004, Qiagen) following the manufacturer’s instructions. Library preparation and sequencing was performed by the Mayo Clinic Genome Analysis Core using an Illumina HiSeq 4000. Reads were aligned using STAR (version 2.7.3a), and featureCounts (version 2.8.2) was used to assign mapped reads. Any gene with an RPKM < 1 in all samples was considered to be lowly expressed and removed from further analysis. Sample collection, RNA sequencing, and data processing of treatment naïve PDX models was previously reported19,81. Comparison tests were performed using edgeR82 and significance was measured by |fold change | ≥ 1.5, P < 0.05 and FDR < 0.1. Ingenuity Pathway Analysis (IPA) software (Ingenuity Systems Inc.) was used to identify significantly altered biological pathways and upstream regulators from the differentially expressed genes (P < 0.05 and |z-score | > 2). Association of gene expression patterns from the RNAseq datasets with publicly available databases was performed using Gene Set Enrichment Analysis (GSEA)83,84.
PDX ex vivo culture
Experiments were approved by the Institutional Animal Care and Use Committee (IACUC) at the Mayo Clinic. PDXs were developed at Mayo Clinic and maintained in female SCID mice as previously described85. The PDX models minimal information standard (PDX-MI) is detailed in Supplementary Data 1. Freshly harvested PDXs were dissociated using a tumor dissociation kit (#130-095-929, Miltenyi Biotec), and tumor cells, including tumor associated mouse cells, were plated in nano-culture plates (#NG-PLH9010, OrganoGenix) at a density of 40,000 cells per well and allowed to form spheroids for 3 days86. Spheroids were treated as indicated for an additional 3 days and viability was measured using 3D CellTiter-Glo (#G9682, Promega) and a GloMax luminometer (Promega).
Real-time qPCR
Cells were seeded at 50% confluence and treated as indicated, followed by total RNA isolation using TRIzol™ Reagent (#15596018, Qiagen). cDNA was generated from 1 µg of total RNA via an iScript™ cDNA Synthesis Kit (#1708891, Bio Rad), and subsequently used for real-time quantitative PCR (RT-qPCR) using PerfeCTa™ SYBR Green Fast Mix™ (#95071-012, Quanta Biosciences) and a Bio-Rad CFX Real-Time PCR detection system. Primers were purchased from Integrated DNA Technologies (IDT) and their sequences are listed in Supplementary Data 4.
Western blots
Cells were seeded at approximately 70% confluence and treated as indicated for 30 mins. Cell lysates were generated using NETN buffer (150 mM NaCl, 1 mM EDTA, 20 mM pH 8.0 Tris, 0.5% NP-40) containing 1X cOmplete™ Protease Inhibitor Cocktail without EDTA (PI) (#11836170001, Roche) and 1X PhosSTOP™ phosphatase inhibitor (#049068370001, Roche). Twenty µg of protein was loaded into each well of SDS-containing Criterion XT gels and western blotting was performed as previously described87. Antibody information is reported in Supplementary Data 5. All original western blots can be found in the supplementary data. Western blots were quantified using ImageJ software and total protein levels were normalized to the indicated loading control. When appropriate, phosphorylated forms of indicated proteins were then normalized to their respective total protein levels after adjusting for any loading discrepancies. All quantification data were derived from the images included in each figure and are representative of at least 3 biological replicates.
siRNA knockdown
siRNAs designed to specifically target human STAT1 or STAT3 were purchased from Dharmacon (#L-003543-00-0010, # L-003544-00-0010). Cells were transfected with 5 nM ON-TARGETplus SMARTpool siRNAs using DharmaFECT 1 reagent (#T-2005-01, Dharmacon) according to the manufacturer’s protocol. Non-Targeting siRNA Pool 1 (#D-001206-13, Dharmacon) was used as the negative control.
CRISPR knockout cell line generation
The STAT1 and STAT3 CRISPR guide RNAs (crRNAs, Supplementary Data 6) were designed using Synthego and the UCSC genome browser, where they were predicted as high-confidence guide RNAs88. Guide RNAs, along with Alt-R CRISPR-Cas9-tracrRNA 5’ATTO550 (tracrRNA, #1075927) and Alt-R Cas9 protein (#1081058), were purchased from IDT. Cells were plated in 10 cm dishes at approximately 50% confluence. Twenty-four hours later, equimolar amounts of guide RNA in 1 mL nuclease free water and tracrRNA in 1 mL duplex buffer were heated to 95 °C for 5 min, allowed to cool to 21 °C, and placed on ice for 30 min to form the sgRNA complex. The Lipofectamine™ CRISPRMAX™ Cas9 Transfection kit (#CMAXX00-001, Thermo-Fisher Scientific) was used to transfect Cas9 protein and the sgRNA complex into MDAH and OVSAHO cells following the manufacture’s protocol. The top 10% ATTO550 positive cells were collected by flow sorting and seeded into 10 cm dishes at low density. Following 1−2 weeks of growth, individual colonies were picked, expanded, and screened by western blotting. Clones with no detectable STAT1 or STAT3 protein expression were further screened at the genomic level via Sanger sequencing.
Drug synergy
Cells were plated in 96 well plates at a density of 1000−2000 cells per well in triplicate and allowed to adhere overnight. Cells were then treated with monotherapy or combination therapy as indicated using threefold serial dilutions, with the concentration ranges determined by individual drug IC50s with the highest dose killing close to 100% of cells and the lowest dose killing close to 0% of cells. Following 5−10 days of treatment, cell viability was measured using CellTiter-Glo. Synergy was assessed using SynergyFinder21 and the Compusyn software22.
Statistical analyses
All experiments were conducted in at least 3 biological replicates with 3−8 technical replicates per assay. Representative data sets are shown. Student’s t test and two-way ANOVAs were used to determine significant differences between treatments and p-values < 0.05 were considered statistically significant after adjusting for multiple comparisons (Tukey adjustment) when necessary. Graphical representations and statistical analyses were performed using GraphPad Prism.
Data availability
All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. The data generated in this study are publicly available in Gene Expression Omnibus (GEO) at GSE291796. Sequencing for PDX models PH39S and PH39R can be found in GEO at GSE165052, and for models PH77S and PH77R at https://github.com/Liuy12/PDX_paper. Sequencing for PH730, PH580, and PH354 are available upon request from JW. All other relevant data are included in the manuscript or available from the corresponding author upon request.
Code availability
No custom codes were used to analyze the data presented in this manuscript and all codes are publicly available.
References
Siegel, R. L., Giaquinto, A. N. & Jemal, A. Cancer statistics, 2024. CA Cancer J. Clin. 74, 12–49 (2024).
Wei, W. et al. Clinical outcome and prognostic factors of patients with early-stage epithelial ovarian cancer. Oncotarget 8, 23862–23870 (2017).
Doubeni, C. A., Doubeni, A. R. & Myers, A. E. Diagnosis and management of ovarian cancer. Am. Fam. Physician 93, 937–944 (2016).
Morgan, R. J. Jr. et al. Ovarian cancer, version 1.2016, NCCN clinical practice guidelines in oncology. J. Natl. Compr. Canc Netw. 14, 1134–1163 (2016).
Colombo, N. et al. ESMO-ESGO consensus conference recommendations on ovarian cancer: pathology and molecular biology, early and advanced stages, borderline tumours and recurrent disease. Dagger Ann. Oncol. 30, 672–705 (2019).
Garrido, M. P. et al. Current treatments and new possible complementary therapies for epithelial ovarian cancer. Biomedicines 10 https://doi.org/10.3390/biomedicines10010077 (2021).
Tewari, K. S. et al. Final overall survival of a randomized trial of bevacizumab for primary treatment of ovarian cancer. J. Clin. Oncol. 37, 2317–2328 (2019).
Matulonis, U. A. et al. Efficacy and safety of mirvetuximab soravtansine in patients with platinum-resistant ovarian cancer with high folate receptor alpha expression: Results from the SORAYA study. J. Clin. Oncol. 41, 2436–2445 (2023).
Liu, Z., Jing, C. & Kong, F. From clinical management to personalized medicine: novel therapeutic approaches for ovarian clear cell cancer. J. Ovarian Res. 17, 39 (2024).
Wu, J. et al. Improved survival in ovarian cancer, with widening survival gaps of races and socioeconomic status: a period analysis, 1983–2012. J. Cancer 9, 3548–3556 (2018).
Domcke, S., Sinha, R., Levine, D. A., Sander, C. & Schultz, N. Evaluating cell lines as tumour models by comparison of genomic profiles. Nat. Commun. 4, 2126 (2013).
Sonego, M. et al. Common biological phenotypes characterize the acquisition of platinum-resistance in epithelial ovarian cancer cells. Sci. Rep. 7, 7104 (2017).
Guo, F. & Wang, H. Potential of histone deacetylase inhibitors for the therapy of ovarian cancer. Front Oncol. 12, 1057186 (2022).
Wu, Y. et al. Targeting CCR2(+) macrophages with BET inhibitor overcomes adaptive resistance to anti-VEGF therapy in ovarian cancer. J. Cancer Res Clin. Oncol. 148, 803–821 (2022).
Shabbir, M. & Stuart, R. Lestaurtinib, a multitargeted tyrosine kinase inhibitor: from bench to bedside. Expert Opin. Investig. Drugs 19, 427–436 (2010).
Levis, M. et al. Results from a randomized trial of salvage chemotherapy followed by lestaurtinib for patients with FLT3 mutant AML in first relapse. Blood 117, 3294–3301 (2011).
Smith, B. D. et al. Single-agent CEP-701, a novel FLT3 inhibitor, shows biologic and clinical activity in patients with relapsed or refractory acute myeloid leukemia. Blood 103, 3669–3676 (2004).
AlHilli, M. M. et al. In vivo anti-tumor activity of the PARP inhibitor niraparib in homologous recombination deficient and proficient ovarian carcinoma. Gynecol. Oncol. 143, 379–388 (2016).
Hurley, R. M. et al. Characterization of a RAD51C-silenced high-grade serous ovarian cancer model during development of PARP inhibitor resistance. NAR Cancer 3, zcab028 (2021).
DepMap, B. DepMap 23Q4. Figshare+ (2023).
Ianevski, A., Giri, A. K. & Aittokallio, T. SynergyFinder 3.0: an interactive analysis and consensus interpretation of multi-drug synergies across multiple samples. Nucleic Acids Res. 50, W739–W743 (2022).
Chou, T. C. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 70, 440–446 (2010).
Shawky, A. M., Almalki, F. A., Abdalla, A. N., Abdelazeem, A. H. & Gouda, A. M. A Comprehensive Overview of Globally Approved JAK Inhibitors. Pharmaceutics 14 https://doi.org/10.3390/pharmaceutics14051001 (2022).
Corporation, I.JAKAFI, ruxolitinib phosphate, Clinical Pharmacology and biopharmaceutics Review. Center fro Drug Evaluation and Research REFERENCE ID: 3040809 (2011).
Klaeger, S. et al. The target landscape of clinical kinase drugs. Science 358 https://doi.org/10.1126/science.aan4368 (2017).
Hornbeck, P. V. et al. PhosphoSitePlus, 2014: mutations, PTMs and recalibrations. Nucleic Acids Res. 43, D512–D520 (2015).
Ma, Y. et al. LY3214996 relieves acquired resistance to sorafenib in hepatocellular carcinoma cells. Int J. Med Sci. 18, 1456–1464 (2021).
Shi, M. et al. Caveolae-associated protein 3 promotes adipogenic differentiation of porcine preadipocytes by promoting extracellular signal-regulated kinase phosphorylation. Anim. Sci. J. 94, e13822 (2023).
Lombard, C. K., Davis, A. L., Inukai, T. & Maly, D. J. Allosteric modulation of JNK docking site interactions with ATP-competitive inhibitors. Biochemistry 57, 5897–5909 (2018).
Frietze, S. & Farnham, P. J. Transcription factor effector domains. Subcell. Biochem 52, 261–277 (2011).
Wang, Y. et al. Deciphering JAK/STAT signaling pathway: A multifaceted approach to tumorigenesis, progression and therapeutic interventions. Int. Immunopharmacol. 131, 111846 (2024).
Akira, S. Functional roles of STAT family proteins: Lessons from knockout mice. Stem Cells 17, 138–146 (1999).
Loh, C. Y. et al. Signal transducer and activator of transcription (STATs) proteins in cancer and inflammation: Functions and therapeutic implication. Front Oncol. 9, 48 (2019).
Nitulescu, I. I. et al. Mediator kinase phosphorylation of STAT1 S727 promotes growth of neoplasms with JAK-STAT activation. EBioMedicine 26, 112–125 (2017).
Wen, Z., Zhong, Z. & Darnell, J. E. Jr Maximal activation of transcription by Stat1 and Stat3 requires both tyrosine and serine phosphorylation. Cell 82, 241–250 (1995).
Zhang, X., Blenis, J., Li, H. C., Schindler, C. & Chen-Kiang, S. Requirement of serine phosphorylation for formation of STAT-promoter complexes. Science 267, 1990–1994 (1995).
Tierney, B. J. et al. Aberrantly activated pSTAT3-Ser727 in human endometrial cancer is suppressed by HO-3867, a novel STAT3 inhibitor. Gynecol. Oncol. 135, 133–141 (2014).
Yu, H. & Jove, R. The STATs of cancer–new molecular targets come of age. Nat. Rev. Cancer 4, 97–105 (2004).
Kong, H. et al. Prognostic significance of STAT3/phosphorylated-STAT3 in tumor: a meta-analysis of literatures. Int J. Clin. Exp. Med. 8, 8525–8539 (2015).
Izar, B. et al. A single-cell landscape of high-grade serous ovarian cancer. Nat. Med. 26, 1271–1279 (2020).
Isobe, A. et al. Interleukin 6 receptor is an independent prognostic factor and a potential therapeutic target of ovarian cancer. PLoS One 10, e0118080 (2015).
Wang, L. et al. CAFs enhance paclitaxel resistance by inducing EMT through the IL6/JAK2/STAT3 pathway. Oncol. Rep. 39, 2081–2090 (2018).
Tian, X. et al. Physical interaction of STAT1 isoforms with TGF-beta receptors leads to functional crosstalk between two signaling pathways in epithelial ovarian cancer. J. Exp. Clin. Cancer Res. 37, 103 (2018).
Liu, F. et al. Expression of STAT1 is positively correlated with PD-L1 in human ovarian cancer. Cancer Biol. Ther. 21, 963–971 (2020).
Josahkian, J. A. et al. Increased STAT1 expression in high grade serous ovarian cancer is associated with a better outcome. Int. J. Gynecol. Cancer 28, 459–465 (2018).
Groom, J. R. & Luster, A. D. CXCR3 in T cell function. Exp. Cell Res. 317, 620–631 (2011).
Bronger, H. et al. CXCL9 and CXCL10 predict survival and are regulated by cyclooxygenase inhibition in advanced serous ovarian cancer. Br. J. Cancer 115, 553–563 (2016).
Au, K. K. et al. STAT1-associated intratumoural T(H)1 immunity predicts chemotherapy resistance in high-grade serous ovarian cancer. J. Pathol. Clin. Res. 2, 259–270 (2016).
Burke, A. J., Garrido, P., Johnson, C., Sullivan, F. J. & Glynn, S. A. Inflammation and nitrosative stress effects in ovarian and prostate pathology and carcinogenesis. Antioxid. Redox Signal 26, 1078–1090 (2017).
Yang, Z. et al. Activation of integrin beta1 mediates the increased malignant potential of ovarian cancer cells exerted by inflammatory cytokines. Anticancer Agents Med. Chem. 14, 955–962 (2014).
Roberts, D. et al. Identification of genes associated with platinum drug sensitivity and resistance in human ovarian cancer cells. Br. J. Cancer 92, 1149–1158 (2005).
Stronach, E. A. et al. HDAC4-regulated STAT1 activation mediates platinum resistance in ovarian cancer. Cancer Res. 71, 4412–4422 (2011).
Standing, D. et al. The role of STATs in ovarian cancer: exploring their potential for therapy. Cancers (Basel) 15 https://doi.org/10.3390/cancers15092485 (2023).
Shang, A. Q. et al. Relationship between HER2 and JAK/STAT-SOCS3 signaling pathway and clinicopathological features and prognosis of ovarian cancer. Cancer Biol. Ther. 18, 314–322 (2017).
Meinhold-Heerlein, I. et al. Molecular and prognostic distinction between serous ovarian carcinomas of varying grade and malignant potential. Oncogene 24, 1053–1065 (2005).
Huang, M., Page, C., Reynolds, R. K. & Lin, J. Constitutive activation of stat 3 oncogene product in human ovarian carcinoma cells. Gynecol. Oncol. 79, 67–73 (2000).
Chen, H., Ye, D., Xie, X., Chen, B. & Lu, W. VEGF, VEGFRs expressions and activated STATs in ovarian epithelial carcinoma. Gynecol. Oncol. 94, 630–635 (2004).
Anglesio, M. S. et al. IL6-STAT3-HIF signaling and therapeutic response to the angiogenesis inhibitor sunitinib in ovarian clear cell cancer. Clin. Cancer Res. 17, 2538–2548 (2011).
Wei, L. H. et al. Interleukin-6 promotes cervical tumor growth by VEGF-dependent angiogenesis via a STAT3 pathway. Oncogene 22, 1517–1527 (2003).
Colomiere, M. et al. Cross talk of signals between EGFR and IL-6R through JAK2/STAT3 mediate epithelial-mesenchymal transition in ovarian carcinomas. Br. J. Cancer 100, 134–144 (2009).
Yue, P. et al. Hyperactive EGF receptor, Jaks and Stat3 signaling promote enhanced colony-forming ability, motility and migration of cisplatin-resistant ovarian cancer cells. Oncogene 31, 2309–2322 (2012).
Chong, K. Y. et al. Inhibition of Heat Shock Protein 90 suppresses TWIST1 Transcription. Mol. Pharm. 96, 168–179 (2019).
Saini, U. et al. Elevated STAT3 expression in ovarian cancer ascites promotes invasion and metastasis: A potential therapeutic target. Oncogene 36, 168–181 (2017).
Chen, M. W. et al. The STAT3-miRNA-92-Wnt signaling pathway regulates spheroid formation and malignant progression in ovarian cancer. Cancer Res. 77, 1955–1967 (2017).
Burgos-Ojeda, D. et al. CD24+ ovarian cancer cells are enriched for cancer-initiating cells and dependent on JAK2 signaling for growth and metastasis. Mol. Cancer Ther. 14, 1717–1727 (2015).
Ning, Y. et al. Co-culture of ovarian cancer stem-like cells with macrophages induced SKOV3 cells stemness via IL-8/STAT3 signaling. Biomed. Pharmacother. 103, 262–271 (2018).
Sheng, W. J., Jiang, H., Wu, D. L. & Zheng, J. H. Early responses of the STAT3 pathway to platinum drugs are associated with cisplatin resistance in epithelial ovarian cancer. Braz. J. Med Biol. Res. 46, 650–658 (2013).
Duan, Z. et al. Signal transducers and activators of transcription 3 pathway activation in drug-resistant ovarian cancer. Clin. Cancer Res 12, 5055–5063 (2006).
Abubaker, K. et al. Inhibition of the JAK2/STAT3 pathway in ovarian cancer results in the loss of cancer stem cell-like characteristics and a reduced tumor burden. BMC Cancer 14, 317 (2014).
Martincuks, A. et al. PARP inhibition activates STAT3 in both tumor and immune cells underlying therapy resistance and immunosuppression in ovarian cancer. Front Oncol. 11, 724104 (2021).
Santos, F. P. et al. Phase 2 study of CEP-701, an orally available JAK2 inhibitor, in patients with primary or post-polycythemia vera/essential thrombocythemia myelofibrosis. Blood 115, 1131–1136 (2010).
Hexner, E. O. et al. Phase I dose escalation study of lestaurtinib in patients with myelofibrosis. Leuk. Lymphoma 56, 2543–2551 (2015).
Brown, P. A. et al. FLT3 inhibitor lestaurtinib plus chemotherapy for newly diagnosed KMT2A-rearranged infant acute lymphoblastic leukemia: Children’s Oncology Group trial AALL0631. Leukemia 35, 1279–1290 (2021).
Hexner, E. et al. Open-label study of oral CEP-701 (lestaurtinib) in patients with polycythaemia vera or essential thrombocythaemia with JAK2-V617F mutation. Br. J. Haematol. 164, 83–93 (2014).
Minturn, J. E. et al. Phase I trial of lestaurtinib for children with refractory neuroblastoma: a new approaches to neuroblastoma therapy consortium study. Cancer Chemother. Pharm. 68, 1057–1065 (2011).
Landen, C. N. et al. Phase I and Randomized Phase II Study of Ruxolitinib With Frontline Neoadjuvant Therapy in Advanced Ovarian Cancer: An NRG Oncology Group Study. J. Clin. Oncol. 42, 2537–2545 (2024).
Hu, X., Li, J., Fu, M., Zhao, X. & Wang, W. The JAK/STAT signaling pathway: From bench to clinic. Signal Transduct. Target Ther. 6, 402 (2021).
Coricello, A., Mesiti, F., Lupia, A., Maruca, A. & Alcaro, S. Inside perspective of the synthetic and computational toolbox of JAK inhibitors: Recent updates. Molecules 25 https://doi.org/10.3390/molecules25153321 (2020).
Langdon, S. P. et al. Characterization and properties of nine human ovarian adenocarcinoma cell lines. Cancer Res. 48, 6166–6172 (1988).
Ray, U. et al. PG545 sensitizes ovarian cancer cells to PARP inhibitors through modulation of RAD51-DEK interaction. Oncogene 42, 2725–2736 (2023).
Liu, Y. et al. Gene expression differences between matched pairs of ovarian cancer patient tumors and patient-derived xenografts. Sci. Rep. 9, 6314 (2019).
Robinson, M. D., McCarthy, D. J. & Smyth, G. K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2010).
Mootha, V. K. et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet 34, 267–273 (2003).
Subramanian, A. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 102, 15545–15550 (2005).
Weroha, S. J. et al. Tumorgrafts as in vivo surrogates for women with ovarian cancer. Clin. Cancer Res. 20, 1288–1297 (2014).
Shuford, S. et al. Prospective validation of an ex vivo, patient-derived 3D spheroid model for response predictions in newly diagnosed ovarian cancer. Sci. Rep. 9, 11153 (2019).
Aspros, K. G. M. et al. Estrogen receptor beta repurposes EZH2 to suppress oncogenic NFkappaB/p65 signaling in triple negative breast cancer. NPJ Breast Cancer 8, 20 (2022).
Kent, W. J. et al. The human genome browser at UCSC. Genome Res. 12, 996–1006 (2002).
Acknowledgements
Research reported in this publication was supported by the National Cancer Institute of the National Institutes of Health through the Mayo Clinic Ovarian Cancer SPORE Grant P50CA136393; the Department of Defense Ovarian Cancer Research Program OC210132: W81XWH-22-1-0634; the Minnesota Ovarian Cancer Alliance; the Mayo Clinic Graduate School of Biomedical Sciences; and the Mayo Clinic Foundation. The funders played no role in study design, data collection, analysis and interpretation of data, or the writing of this manuscript.
Author information
Authors and Affiliations
Contributions
Experimental design, E.P.B.R., J.R.H.; experimental execution: E.P.B.R., M.J.E., X.H., A.B., N.A.P., A.J.J., Y.O., A.B.D., S.S., J.R.H.; experimental resources, G.B., S.H.K., S.J.W.; initial manuscript writing, E.P.B.R., J.R.H.; manuscript review and editing, E.P.B.R., M.J.E., X.H., A.B., N.A.P., A.J.J., Y.O., A.D.B., S.S., G.B., S.H.K., S.J.W., J.R.H.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Rodman, E.P.B., Emch, M.J., Hou, X. et al. Lestaurtinib’s antineoplastic activity converges on JAK/STAT signaling to inhibit treatment naïve and therapy resistant forms ovarian cancer. npj Precis. Onc. 9, 236 (2025). https://doi.org/10.1038/s41698-025-00947-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41698-025-00947-0
