
Abstract
Liquid biopsy is a promising non-invasive technology that is capable of diagnosing cancer. However, current ctDNA-based approaches detect only a minority of early-stage disease. We set out to improve the sensitivity of liquid biopsy by harnessing tumor recognition by T cells through the sequencing of the circulating T-cell receptor repertoire. We studied a cohort of 463 patients with lung cancer (86% stage I) and 587 subjects without cancer using gDNA extracted from blood buffy coats. We performed TCR β chain sequencing to yield a median of 113,571 TCR clonotypes per sample and built a TCR sequence similarity graph to cluster clonotypes into TCR repertoire functional units (RFUs). The TCR frequencies of RFUs were tested for association with cancer status and RFUs with a statistically significant association were combined into a cancer score using a support vector machine model. The model was evaluated by 10-fold cross-validation and compared with a ctDNA panel of 237 mutation hotspots in 154 lung cancer driver genes and 17 cancer related protein biomarkers in 85 subjects. We identified 327 cancer-associated TCR RFUs with a false discovery rate (FDR) ≤ 0.1, including 157 enriched in cancer samples and 170 enriched in controls. Levels of 247/327 (76%) RFUs were correlated with the presence of an HLA allele at FDR ≤ 0.1 and tumor-infiltrating lymphocyte TCRs from multiple RFUs bound HLA presented tumor antigen peptides, suggesting antigen recognition as a driver of the cancer-RFU associations found. The RFU cancer score detected nearly 50% of stage I lung cancers at a specificity of 80% and boosted the sensitivity by up to 20 percentage points when added to ctDNA and circulating proteins in a multi-analyte cancer screening test. Overall, we show that circulating TCR repertoire functional unit analysis can complement established analytes to improve liquid biopsy sensitivity for early-stage cancer.
Similar content being viewed by others

Introduction
Despite recent advances in cancer therapies, more than 600,000 cancer-related deaths are expected annually in the United States and stark differences in outcomes persist between individuals diagnosed with early vs. late-stage disease1. Large studies have established the benefit of cancer screening across a range of indications and modalities. These include the landmark national lung screening trial (NLST) that demonstrated the benefit of using low-dose CT (LDCT) to detect early-stage lung cancer2,3, and the DeeP-C study that established the benefit of multitarget stool DNA testing for colorectal-cancer screening4. Unfortunately, many individuals do not undergo recommended testing, and cancer types responsible for 70% of all cancer-related deaths are still unaddressed by current screening tools1,5. Furthermore, current inclusion criteria for screening for lung cancer, based on age and tobacco exposure, are flawed and fail to include many individuals at risk while exposing others at low risk to complications from invasive testing or anxiety derived from false positive findings. Better characterization of screening findings to discriminate between true positives and false positives is also an unmet clinical need. Thus, further technological improvements in early detection are needed.
Recent studies have focused on complementary approaches to established cancer screening techniques. In particular, the feasibility of cancer detection in blood samples is being explored given the value of circulating tumor DNA (ctDNA) in identifying actionable mutations6,7,8 and of protein biomarkers in monitoring cancer progression and treatment response9. Because blood is an excellent source of diagnostic information due to its ease of sampling and rich analyte content, blood-based cancer screening creates opportunities for both improving single-cancer detection and more comprehensive multi-cancer early detection (MCED) tests. Several analytes found in blood plasma are being explored as stand-alone assays or as complements to imaging10, including methylated ctDNA11,12, mutated ctDNA13, protein biomarkers13, and ctDNA fragmentomics14.
The performance of early cancer detection using a blood plasma analyte is determined by the analyte’s concentration and the sensitivity/specificity of the test. In early-stage cancer, relatively lower tumor burden and invasiveness leads to low plasma analyte tumor fraction and, consequently, limited sensitivity15,16. For example, while blood plasma biomarkers can be used to detect up to 90% of late-stage lung cancers17, the sensitivity for detecting early-stage lung cancer at high specificity has been reported to be only ~20% with methods based on ctDNA17,18. If blood-based liquid biopsy is to achieve its potential as a cancer screening method, additional analytes and techniques are needed to increase the sensitivity for early-stage disease.
The immune response to tumors is an essential element of cancer biology19 and a potential source of biomarkers in early-stage disease20. T cells recognizing the same antigen have been shown to harbor recurrent TCRs or TCR motifs21,22, and this phenomenon has been used successfully to develop diagnostic tests for infectious diseases such as COVID-19 and CMV23,24. Further studies have shown that TCRs with putative shared antigen specificity based on TCR sequence similarity can be grouped together to increase the statistical power for detecting disease associations, including those in the cancer domain25,26,27. The goal of our study was to develop TCR repertoire sequencing as a novel component of blood-based cancer screening tests.
Lung cancer was selected as the initial indication for developing a TCR repertoire sequencing method for blood-based cancer screening, given the known clinical benefit of its early detection, limitations of current blood-based methods, and the presence of an adaptive immune response28,29. We collected blood samples from a large cohort of patients with lung cancer and additional blood samples from a similarly sized cohort of individuals who had undergone low-dose CT screening or bronchial biopsy and were found not to have lung cancer or were not otherwise known to have the disease.
We developed an NGS assay to sequence the TCR β chain from blood buffy coats and implemented computational algorithms to organize a dataset of tens of millions of TCRs into TCR repertoire functional units (“RFUs”) based on TCR sequence similarity30. Grouping TCRs into RFUs that likely recognize the same or related antigens30 allowed a cross-sample comparison of immune responses, enabling a case-control association study of RFU TCR counts with cancer status. We used cancer-associated RFUs to train a machine learning model for lung cancer prediction and demonstrated that this TCR-based predictor is complementary to established biomarkers.
We detail how this was achieved in the sections below by (1) describing the lung cancer case and control sample cohort collected in the study; (2) defining the concept of circulating TCR repertoire functional units and summarizing the size and distribution of these units across all the samples in the cohort; (3) identifying which TCR RFUs are associated with the presence of lung cancer through a cancer case / non-cancer control statistical association study; and (4) combining cancer-associated RFUs into a unified prediction model of lung cancer status. We then estimate the additional predictive value of this TCR RFU lung cancer status prediction model when considered in the context of current circulating tumor DNA (ctDNA) and protein biomarker cancer prediction approaches. Finally, we explore the biological basis of this TCR cancer prediction signal by analyzing a separate set of lung cancer tumor-infiltrating lymphocytes (TIL). Overall, we show that incorporating TCR repertoire sequencing in a liquid-biopsy early detection assay is a promising avenue for improving the sensitivity for early-stage disease.
Results
Dataset for the discovery of lung cancer-associated TCR RFUs
To identify the RFUs associated with cancer status (Fig. 1), we assembled a cohort of blood samples from 463 patients diagnosed with lung cancer (Supplementary Data 1). The cohort was enriched for subjects with stage I disease (Fig. 2a) and spanned all the major lung cancer subtypes (Fig. 2b). Blood samples from 587 subjects without lung cancer were collected as control samples, with the majority of individuals meeting the current inclusion criteria for lung cancer screening (Fig. 2c–d). We used a custom TCR sequencing assay (Methods) to sequence 128,902,511 productive TCR clonotypes. The median productive TCR clonotypes per sample was 113,571, which was comparable between cancer patients and non-cancer controls (Fig. 2f, g). After filtering for the most abundant CDR3 lengths of 10–16 residues and removing any clonotypes with unique molecular identifier (UMI) read count below each sample’s median UMI read count × 0.75 to preferentially remove naïve T cell clonotypes, 69,027,705 total clonotypes remained and were used for further analysis.

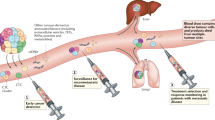
(1) TCR β chain of circulating T cells from blood buffy coats is deeply sequenced using an NGS-based assay. (2) Filtered TCRs are clustered into repertoire functional units (RFU) using sequence similarity as the distance metric. (3) A generalized linear regression model is applied to each RFU to discover RFUs that are individually associated with cancer status after accounting for demographic and technical covariates. (4) Significantly cancer-associated RFUs are used jointly to train a machine learning model to predict cancer status. Figure created in part using BioRender.com.

Cohort distribution of cancer stage (a), cancer histology (b), smoking status (c), age (d), gender and race (e), TCR repertoire depth as measured by clonotype count (f) and TCR repertoire depth as measured by TCR UMI count (g).
TCR repertoire functional unit definition
A dataset of approximately 70 million TCR clonotypes is computationally prohibitive to standard clustering algorithms such as hierarchical clustering, owing to their iterative approach and reliance on a distance matrix. To group our TCRs into RFUs, we first created an approximate nearest neighbor graph on the TCRs26 using a CDR3 sequence dissimilarity metric27,31, with each graph node corresponding to a distinct deduplicated TCR V gene and a CDR3 amino acid sequence. We applied a non-parametric (no prior assumption of cluster count or shape) \(O(n\log n\,)\) time complexity TCR clustering algorithm to this graph to assign nodes to RFUs or as non-clustered singletons32. An RFU is defined as a cluster with at least two nodes or a singleton node with at least two instances of the TCR sequence in the dataset.
We generated several candidate RFU sets by varying the maximum TCR dissimilarity cutoff (parameter dc), which controlled the clustering sparsity. Five of the dc cutoffs were 0.5, 5, 11, 12, and 22; these values correspond to sequence similarities ranging from full CDR3 amino acid identity (0.5) to one mismatch or indel + an additional conservative mismatch allowed (22). Three dc cutoffs, 1.1, 1.2, and 2.2, were additionally considered after dividing the sequence distance by the number of considered residues in the CDR3 alignment.
The clustering analysis generated between ~74 K and 7 M RFUs depending on the dc setting (Fig. 3a, Supplementary Table 1). The RFUs followed a power law distribution in size (Fig. 3b, c), with a small number of large RFUs and many small RFUs.

(a) The tallies of TCR clonotypes clustered into an RFU (green) or remaining as an unclustered clonotype (brown) for each dc setting. (b) The number of RFUs generated by using each dc cutoff. (c) The number of RFUs (Y-axis) that are composed of a given range of TCR clonotypes (X axis) for the clustering results of each clustering setting (panels).
Lung cancer-associated RFU discovery
With the defined RFUs, we next turned to cancer case/non-cancer control association testing to identify the cancer-associated RFUs. Owing to the large number of RFUs and their predominantly small size, there is a significant multiple testing burden imposed by small RFUs, for which we have minimal statistical power to detect cancer associations at our sample size. To address this for the case-control analysis, we restricted the set of candidate RFUs to the most common RFUs with TCR clonotypes observed in at least 15 individuals and with multiple (≥8) distinct clonotypes present in at least three individuals, regardless of cancer status. This resulted in 6375 RFUs being tested for cancer association (Supplementary Table 1). While RFUs obtained with different dc settings partially overlap, clustering TCRs across a range of dc cutoffs allows us to find the optimal balance between the population prevalence and the degree of putative shared antigen specificity of each RFU for cancer association testing.
We observed that the per-subject distribution of RFU TCR counts (number of TCR clonotypes from an RFU present in an individual) can be modeled analogously to gene expression levels measured using RNA-seq, with the level of an RFU computed as the sum of its constituent TCR clonotype counts. Therefore, we used the well-established gamma-Poisson generalized linear model33 to test for RFU association with cancer status. This model accounts for variable depth of sequencing and RFU count overdispersion and allows us to incorporate demographic and technical covariates such as age, gender, race, and TCR repertoire depth into the analysis.
We identified a total of 327 RFUs associated with cancer status at false discovery rate (FDR) ≤ 0.1 across the eight dc cutoffs, including 157 that were enriched in cancer samples with fold change between 1.03 and 2.26, and 170 that were enriched in non-cancer controls with a fold change between 1.05 and 17.2 (Fig. 4a, Supplementary Data 2). Of the 327 cancer-associated RFUs, TCR clonotype counts for 157 RFUs were also correlated with subject age, while 136 were correlated with race, and 88 to gender at FDR ≤ 0.1 (Supplementary Data 3). Of the 327 RFUs, 124 had a repeated TCR centroid across different dc cutoffs, indicating that they were overlapping RFUs.

(a) Volcano plot of RFU-cancer associations. The boxplots on either side of the volcano plot show covariate-adjusted RFU TCR counts of a positively and a negatively cancer-associated RFU, as indicated by the dashed arrows. (b) ROC curves for train and test folds from 10-fold cross-validation, broken down by cancer stage. (c) Stage I cancer sensitivity based on cross-validated test fold cancer scores as shown in (b). (d) Comparison of cross-validated cancer prediction scores by cancer stage or benign lung nodule status.
HLA type correlation with cancer-associated RFUs
To explore the biological basis of TCR RFU associations with cancer status, we evaluated the correlation between RFU TCR counts and per-subject HLA types. We reasoned that if TCR RFU cancer associations are driven by antigen recognition, then RFU TCR counts should be correlated with the presence of particular HLA alleles, and the identity of correlated alleles may shed light on the biology at play. Subject HLA types were imputed from TCR sequence data using HLAGuessr34 (Methods) and resulted in average of 9.7 called alleles (3.8 Class I, 5.9 Class II) per subject, with the expected distribution of common and rarer alleles (Supplementary Fig. 1a, b). No HLA allele was significantly enriched in either cancer cases or non-cancer controls (Fisher’s exact test, FDR ≤ 0.1).
Of the 327 cancer-associated RFUs, TCR counts of 163 (50%) were found to be significantly correlated with the presence of one or more HLA alleles in cancer subjects, and 212 (65%) were correlated in the non-cancer controls (t-test, FDR ≤ 0.1). A total of 247/327 (76%) RFUs correlated with at least one HLA allele when all subjects were considered together. Interestingly, most significant associations (92% in cancer patients and 81% in non-cancer controls) were with HLA class II alleles (Supplementary Fig. 1c, d, Supplementary Data 4), highlighting a potentially key role of CD4 T cell responses in the tumor microenvironment in the TCR RFU cancer associations we found35,36.
Cancer prediction from RFUs
We next investigated whether lung cancer-associated RFUs could be used as biomarkers for the early detection of lung cancer. To this end, we implemented a machine learning (ML) model to predict cancer status from cancer-associated RFUs and evaluated it using 10-fold cross-validation (CV); the overall dataset was thus split into 10 sets of 90% train/10% test sample subset pairs randomly. Importantly, for this analysis, both clustering and RFU cancer case-control association testing were independently repeated within each train fold, thus avoiding train-test leakage introduced by using cancer RFUs defined using the whole dataset. Additionally, to minimize RFU-level bias arising from demographic and technical covariate imbalances between the cancer and control cohorts, we used each RFU’s TCR counts corrected for the fitted effect of the demographic and technical covariates as the ML features (Supplementary Fig. 2, Supplementary Data 3). The covariate-adjusted RFU features were used to train a bagging classifier of support vector machine (SVM) classifiers with a linear kernel (Methods). Performance is reported as an average across the 10 test sample subsets. We observed an average cross-validation train fold performance receiver-operator curve (ROC) area under the curve (AUC) of 0.81 and test ROC AUC of 0.72 (stage 0-I: 0.71, and stage II-IV: 0.74) (Fig. 4b).
Importantly, model predictions did not appear to be driven by batch effects related to the source of the samples (Supplementary Fig. 3d, Supplementary Data 1) or technical factors leading to variable TCR repertoire depth (Supplementary Fig. 3b, c). Likewise, model scores were not driven by various demographic covariates (Supplementary Fig. 3e–g) and were uniformly higher in cancer patients than in age-matched non-cancer controls (Supplementary Fig. 3a). Model scores were also higher in cancer patients than in non-cancer controls known to have heart disease and/or chronic obstructive pulmonary disease (COPD) (Supplementary Fig. 3h), and were uniform across various lung cancer subtypes (Supplementary Fig. 3i). Notably, 48% of stage I subjects (test samples of each cross-validation fold) could be detected by the model at a specificity of 80% (Fig. 4c), and the model could differentiate between lung cancer and benign nodules (Fig. 4d), highlighting the promise of this early detection approach. TCR cancer prediction score did not significantly differ between cancer stages (p = 0.42) (Supplementary Fig. 4).
The greatest imbalances between our cases and controls involved age and TCR repertoire depth (Fig. 2). To confirm that the ML model scores were not driven by these covariates, we fitted a linear regression model using cancer status, age, TCR repertoire clonotype counts, and UMI read counts as predictors for the RFU TCR score as the response. The resulting p values for the cancer status, age, TCR clonotype count, and TCR UMI count were \(9.5\times {10}^{-22}\), \(9.1\times {10}^{-4}\), \(5.5\times {10}^{-4}\) and \(2.3\times {10}^{-3}\), indicating that the final cancer prediction score was predominantly driven by a sample’s cancer status as opposed to its age or TCR repertoire depth.
Uncovering the cancer signal requires TCR grouping by sequence similarity
To estimate the benefit of the TCR clustering (RFU formation) step for cancer prediction, we applied the same cross-validation procedure to features derived from individual unclustered TCRs. Using filtered TCRs as the starting point, we fitted the GLM model to the TCR counts of each unique TCR sequence based on the V gene, J gene, and CDR3 amino acid sequence. Significantly cancer-associated unique TCR sequences (FDR ≤ 0.1) were used as features in the SVM model. Across CV folds, there were, on average, only 5.6 significantly cancer-associated TCRs (range: 3–8), which were all used for the prediction model. The mean CV AUC for this TCR model was only 0.59. The significantly lower feature count and CV AUC of TCRs vs. RFUs are consistent with the hypothesis that TCR-cancer associations are too weak to be discovered individually, and that a stronger signal can be achieved by combining TCRs with similar sequences26.
Lung cancer-associated plasma protein biomarkers
We next sought to assess the potential contribution of this TCR-based signature to the early detection of cancer in the context of established tumor analytes. We first reviewed the literature on protein biomarkers with known or suggested roles in lung cancer detection in plasma. Two large, well-designed case-control studies have recently evaluated multiplexed protein biomarker panels for their association with either imminent lung cancer diagnosis or pulmonary nodule malignancy37,38. A total of 54 distinct protein markers were reported to be potentially associated with either diagnosed lung cancer or malignant nodules in the two studies.
To compare the predictive performance of the published protein biomarkers with our TCR signature, we generated circulating protein level data from 235 study subjects, including 109 cancer patients and 126 non-cancer controls. The demographic and tumor property distributions of these subjects closely matched the overall distribution (Supplementary Figure 5, and Figure 2). We used the Olink Oncology and Inflammation Explore® panels to assay protein markers associated with these biological pathways. Of the 54 published cancer-associated protein markers, 26 were covered by these panels and could be used to replicate the reported results (Supplementary Table 2).
Of these 26 proteins, 18 were significantly associated with cancer status in our cohort at an FDR < 0.05. Notably, 17 of the 18 proteins were positively associated with cancer status in both the published reports and our dataset. One additional protein (ALPP) was associated with cancer status in our cohort (FDR < 0.05) but in the opposite direction (positive in the published study38 but negative in our data). The eight remaining proteins tested were not significantly associated with cancer status in our dataset. The 17 successfully replicated protein biomarkers were used in the subsequent analyses.
Lung cancer prediction using protein biomarkers
We trained a support vector machine classifier (linear basis function kernel, regularization parameter C = 0.01) for lung cancer prediction using the 17 validated protein biomarkers. Forward feature selection with 5-fold internal CV resulted in models with an average of 4.4 features selected, achieving an overall cross-validated ROC AUC of 0.70 (Fig. 5a). In line with expectations, this protein-based model showed much stronger performance for the prediction of late-stage cancer (stage IV CV AUC = 0.89) than for early-stage disease (stage I CV AUC = 0.66).

(a) Cross-validated ROC curve of SVM model score using the protein expression features only. (b) Circulating DNA mutation counts in the assayed subjects by cancer stage. (c) Cross-validated ROC curve of prediction score from logistic regression with forward feature selection using the mutation features only. Cancer detection sensitivity using different subset of analytes for stage I and stage II-IV lung cancer is shown in (d) and (e), respectively.
Cancer detection with recurrent ctDNA lung cancer driver mutations
Next, we implemented a broadly used ctDNA assessment approach to further evaluate the TCR RFU-based cancer prediction in the context of established plasma-based early detection methods. We generated ctDNA mutation data for 100 subjects comprising 61 patients with cancer and 39 non-cancer controls (Supplementary Table 3). Targeted sequencing was performed on 237 mutation hotspots in 154 lung cancer driver genes (Supplementary Data 5)13,39 using commercially available Illumina® sequencing library construction and hybridization target capture reagents (Methods). The matching genomic DNA (gDNA) from each subject was sequenced alongside the ctDNA samples to identify and exclude ctDNA mutations derived from clonal hematopoiesis of indeterminate potential (CHIP). The average unique molecule coverage on the targeted mutation sites was >1,500x and >875x for ctDNA and gDNA samples, respectively.
After NGS variant calling and filtering (Methods) and excluding mutations found in matching gDNA samples, 28 mutations were called across the 100 subjects (Fig. 5b, Supplementary Data 6). A logistic regression (LR) model with forward feature selection using mutation count and average mutation allele frequency as features was trained using 5-fold CV to classify the subjects as positive or negative for ctDNA. As expected, more mutations were identified in individuals with stage III-IV disease, allowing the majority of these patients to be detected. In contrast, the sensitivity was lower for early-stage disease, in line with prior literature13,16 (Fig. 5c).
The only genes with driver mutations observed in multiple patients were TP53 (15/100) and KRAS (3/100). The TCR RFU-based cancer prediction scores were not associated with either TP53 (p = 0.65) or KRAS mutation status (p = 0.40) (Supplementary Fig. 6).
Lung cancer prediction with a multi-analyte liquid biopsy incorporating TCR RFUs
Having trained individual cancer prediction models for TCR RFUs, protein biomarkers, and ctDNA mutations, we sought to measure the added contribution of the TCR component to detecting early-stage cancer. Of the 1050, 235, and 100 subjects with TCR, protein, and mutation dat,a respectively, 85 were processed for all three analytes (Supplementary Table 4). For each analyte class, we recorded whether each sample’s cross-validation score passed the threshold determined by a given target specificity level when the sample was in the held-out set during the cross-validation. This provided an unbiased, cross-validated sensitivity estimate for each individual analyte and allowed us to compare which cases were called positive by various subsets of analytes (Supplementary Table 5).
We observed a substantial gain in sensitivity for stage I cancer when TCR RFU biomarkers were added to ctDNA mutations and proteins, with an up to ~20%-point increase observed at the target specificity levels typical for single cancer type screening tests (Fig. 5d). In contrast, TCR RFUs achieved limited improvement in the detection of stage II-IV cancers (Fig. 5e), which could be explained by the high level of performance achieved by current plasma analytes in advanced disease.
Analysis of RFU TCRs in tumor infiltrating lymphocytes
Lastly, we sought to further characterize the biological basis for the association of circulating TCR RFUs with cancer status by studying tumor infiltrating lymphocytes (TIL) in a separate set of 20 lung cancer patients who underwent surgical resection (Supplementary Data 7). Surgical tumor specimens were processed into single cell suspensions, and TIL were isolated by fluorescence-activated cell sorting (FACS) using standard cell surface marker stains and 19 antigen-specific dextramers (Methods). The dextramers targeted common cancer antigens, including the MAGE genes, viral antigens, and nonsense peptide (i.e., negative) HLA binding controls, and had unique sequence feature barcodes to allow multiplexing. Dextramers were matched to each subject according to HLA type (Supplementary Data 8). Isolated TIL were subjected to single-cell gene expression and TCR sequencing using the 10x Genomics platform, resulting in an average of 2111 T cells analyzed per patient (Supplementary Data 9). An average of 1218 genes were observed to be expressed in the analyzed T cells, with an average of 0.98 TCR β chains and 0.87 TCR alpha chains observed per cell. Sequencing of feature barcodes corresponding to antigen-specific dextramers resulted in an average of 37 feature barcodes per cell.
T cell subtypes of cancer-associated RFUs
T cell subtypes were evaluated from the gene expression profiles using CellTypist40,41 (Methods), which revealed that the majority of assayed T cells were cytotoxic, while regulatory T cells and helper T cells were the second and third most abundant groups (Supplementary Fig. 7). Rarer T cell subtypes, such as mucosal-associated invariant T (MAIT) cells, appear in a subset of patients, highlighting the inter-subject heterogeneity in the anti-tumor immune response (Supplementary Fig. 8).
We sought to assess whether TCRs identified as members of cancer-associated RFUs in our case-control cohort were preferentially carried by any specific T cell subtype. To do this, we intersected 17,551 distinct TCR β chain clonotypes carried by T cells in the 20 TIL surgical patients with TCRs found in the 327 cancer-associated RFUs. Because the 20 patients subjected to TIL single cell sequencing were distinct from the 1,050 subjects in the blood buffy coat RFU discovery, this analysis only revealed patterns apparent for public TCRs that are shared across unrelated subjects. A total of 1772 such public TCR clonotypes were found to overlap between single cell TIL and buffy coat case-control datasets.
Using these shared cancer-associated TCRs, we evaluated whether clonotypes carried by each cell type were more likely to be found in the cancer-associated set. Although no single cell-type enrichment reached statistical significance after correction for multiple testing, MAIT cells tended to be enriched in cancer-associated RFU TCRs (p < 0.05; Supplementary Fig. 9). The clonotypes associated with MAIT cells were also preferentially associated with TRBV6-2 (p = 6.8e−04), TRBV6-4 (p = 5.1e−15), and TRBV20-1 (p = 1.9e−05).
Antigen recognition of TCR clonotype in cancer-associated RFUs
We then considered which of the studied antigens might be recognized by the clonotypes identified as members of cancer-associated RFUs. To this end, we tested the association between dextramer-specific feature barcodes and clonotypes in each subject separately. Clonotypes that bound to negative control dextramers were excluded. Using Fisher’s exact test, we found 24 significant associations involving eight subjects with FDR ≤ 0.1, including both cancer and viral antigens (Supplementary Data 10).
Of the 15 clonotypes found to be associated with cancer antigen-specific dextramers, four clonotypes in three subjects were found to be members of a cancer-associated RFU. All four were associated with dextramer binding cancer MAGE antigens and were found in T cells of multiple subtypes (Supplementary Data 11). One clonotype in subject NSC028 demonstrated cross-reactivity and bound dextramers to both MAGEA1 and CMV epitopes.
Discussion
In this study, we aimed to address the current limitations of blood-based cancer screening tests for early-stage lung cancer. Given that early-stage tumors shed little ctDNA into the blood, we leveraged the anti-tumor T cell response, which was assessed by sequencing the circulating T-cell receptor repertoire and analyzing cancer-associated TCR repertoire functional units (RFUs). Cancer TCR RFUs combined with a machine learning model detected up to half of stage I lung cancer cases, and this signal proved complementary and additive to established tumor-derived analytes, such as circulating proteins or tumor DNA.
There is a strong rationale for the inclusion of TCR repertoire sequencing in blood-based cancer screening tests. Tumor immune surveillance by T cells is a key mechanism of cancer control and offers an orthogonal principle of detection, assessing host response rather than a tumor-shed analyte such as ctDNA. T cell proliferation may even act as an amplifier of subtle signals from small tumors, in a way that a rapidly cleared analyte such as ctDNA cannot. Tumor antigen recognition is encoded by T-cell receptor sequences, which can easily be assayed in circulation by NGS using buffy coat genomic DNA. T-cell receptor sequencing also integrates seamlessly into existing liquid biopsy workflows using the remaining buffy coat fraction from the same blood draw currently used to obtain plasma for ctDNA.
To put our work in context, we note that the refinement of high-throughput TCR-sequencing technologies and the deeper knowledge of the biological relevance of T-cells in the development of disease states has led to an increasing interest in TCR profiling as a diagnostic, monitoring, or treatment adjunct tool in multiple clinical settings. TCR sequencing allows for the analysis of immune response, providing information on the composition of TCR repertoires and facilitating the discovery of TCR-antigen interactions. Multiple diseases may benefit from the application of TCR sequencing to the clinical setting, from infectious diseases to cancer42. For instance, as of May 2025, over 200 trials using TCR sequencing/repertoire profiling were included in clinicaltrials.gov, with over 150 focused on cancer. In the context of oncology, TCR sequencing helps to understand intratumor heterogeneity and thus to learn about cancer immunity and predicting therapeutic responses to immunotherapy43. TCR sequencing is also useful to follow tumor evolution through immune monitoring and to refine current T cell–based therapies, improving their specificity or helping in the monitoring and tracking of T-cell clonotypes after administration.
Unlike other biomarkers, sensitivity for early-stage lung cancer using TCR RFUs remained nearly the same as for late-stage disease, which is in line with a recent study on ovarian cancer early detection30. A possible explanation is immunoediting44, where during early tumor progression the immune system enters the ‘elimination’ phase by production of anti-tumor T cells, which leads to an amplified signal in the peripheral blood repertoire. Indeed, the T cell response against malignant transformation can occur as early as the pre-cancer stage45. Hence, it is not surprising to see a similar level of cancer-associated RFU signal in patients with stage I lung cancer as in patients with more advanced disease.
We examined the biological basis of these new TCR RFU biomarkers by evaluating their association with each subject’s HLA type and their potential for tumor antigen recognition in a separate TIL dataset. We found that TCR RFUs were likely dominated by CD4 T cell/class II HLA targeted responses, suggesting that the immune response in the tumor microenvironment plays a pivotal role. Furthermore, several relevant class I/CD8 T cell responses and even possible MAIT responses were suggested by TIL analysis, highlighting additional possible contributions to the overall signature found. MAIT cells localize to mucosal tissues such as the lung epithelium, function through antigen recognition that does not rely on classical major histocompatibility complex, and have innate-like properties that can respond quickly to infections and stress46,47. Our observations suggest that this unconventional subtype may also contribute to a circulating T cell signature of cancer. Although the target antigens of our cancer-associated RFUs are currently unknown, collectively, these observations are consistent with low-level immune responses mounted by highly prevalent, broadly cross-reactive T cells against self-antigens present in the tumor microenvironment or overexpressed in cancer cells.
Although intriguing, our study had several limitations that should be considered in its interpretation. Blood for our cancer cases was generally obtained close to or after the time of diagnosis (though before treatment), which may have overestimated the robustness of the biomarker in the lung cancer screening setting. In addition, a minority of our controls were not confirmed by CT scan to be free of lung cancer, which may have reduced the observed TCR RFU signal if some of these cases had undetected, asymptomatic disease. The case-control analysis relied on TCR β chain sequencing only, which likely reduced the antigen specificity inherent in the RFUs due to missing TCR α chain pairing information. Notably, although our cross-validation approach was comprehensive and comprised the entire RFU discovery process, additional unseen samples for held-out RFU validation were not available. Finally, the TIL analysis was limited because it used a small number of unrelated subjects to assess only a small portion of the possible relevant antigenic space.
Notwithstanding these limitations, we demonstrated that it is possible to detect the presence of lung cancer in blood by analyzing the circulating TCR repertoire using the abundantly available and currently unused buffy coat blood fraction. Indeed, most liquid biopsy trials archive unused buffy coat as a matter of course, creating an opportunity for supplementary analyses of previous plasma-based studies in the near term. When combined with established analytes, TCR repertoire analysis has the potential to enable cancer detection at earlier stages and prevent cancer death.
Methods
Blood sample collection and processing
Generally, blood samples for the study were collected from subjects using two 10 mL Streck Cell-Free DNA Blood Collection Tubes (Streck-BCT). Buffy coat and plasma samples were prepared with a single spin fractionation protocol. Whole blood within 48 h of collection was centrifuged at 1600 g for 10 min at room temperature. The plasma was then slowly removed so as not to disturb the buffy coat layer below and aliquoted into cryovials. The remaining buffy coat was then removed and stored in cryovials. (An exception was the University of Navarra cancer patient cohort and a subset of the U.S. CRO B cancer cohort, which were collected in EDTA tubes and processed into fractions the same day, usually within 5 h). All Buffy coat and plasma aliquots were stored at −80 °C until needed for the genomic, proteomic, or cell-free DNA assays.
Participants signed written informed consent, and study-related procedures were conducted in accordance with the Declaration of Helsinki and applicable national, state, and local regulations. Sample collection at each site was approved by a local institutional review board (IRB) or independent ethics committee (IEC). Samples and data from patients included in the study provided by the Biobank of the University of Navarra were processed following standard operating procedures under approval by the University of Navarra Research Ethics Committee (reference #2023.159). Additional approvals were given by Dana-Farber Cancer Institute IRB #98-063 and Rush University Medical Center IRB #22012505.
Extraction of cfDNA and gDNA
Nucleic acids were extracted from the frozen archived patient plasma and buffy coat using Promega Maxwell® instruments and associated kits. Extraction of genomic DNA from buffy coat was performed using the Maxwell Blood Kit (ASB1400) and protocol. Because of the high mass input requirements of TCR repertoire analysis, two 300 µL aliquots of buffy coat were used per patient, and the resulting gDNA combined at the end of extraction. Briefly, each aliquot was first combined with a cell lysis buffer and Proteinase K and incubated at 56 °C for 15 min. These digested samples were then loaded into wells of the Maxwell Blood Kit cartridges and run on the Maxwell RSC Instrument with the associated extraction program. The final gDNA was quantified using the Thermofisher Qubit Instrument.
Extraction of cell-free DNA from plasma was performed using the Maxwell cfDNA LV Plasma Kit (AS1840) and protocol. The thawed single-spun plasma was prepared for loading on the kit by first performing a second high-speed centrifugation, to remove cell debris, at 20,000 x g for 20 min at room temperature. This double-spun plasma was removed and added to a 50 mL conical tube without disturbing the cell pellet. An equal volume of Promega bead binding buffer, relative to input plasma volume, was added to each sample along with Promega binding beads. This plasma and bead slurry was then incubated and shaken on the Promega HSM Instrument for 90 min. This mixture was put on a magnet to capture the binding beads. The plasma and binding buffer were discarded, and the unpurified, concentrated, cell-free DNA was eluted from the pelleted beads. This final elution was added to the wells of the Maxwell Kit cartridge and run on the Maxwell RSC Instrument with the associated extraction program. The final cfDNA was quantified using the Thermofisher Qubit Instrument and fragment lengths profiled using the Agilent Tapestation. Samples were quality controlled for library construction by requiring both a total cfDNA yield greater than 5 ng and an absence of gDNA contamination using Agilent cfDNA Screen Tapes (Agilent #5067-5630).
Protein analysis with Olink platform
One of the single-spun plasma aliquots described above was provided to Olink® as an input for the Proximity Extension Assay (PEA). Plasma samples were stored before plating at −80oC. Each sample was plated using 100 µL of plasma and shipped to Olink on dry ice. The PEA Assay was conducted to determine expression levels of proteins in cancer and inflammation-related pathways, including the 17 proteins used above48.
Multiplex PCR Assay (mRFU) for characterizing the rearranged TCR β chain receptor sequences
Extracted genomic DNA from the buffy coat was used as input to the assay. The TCR β chain was sequenced by targeting 58 TCRB V gene segments and 13 TCRB J gene segments for PCR-based enrichment. Candidate gene-specific primers were generated in silico using the GRCh38 reference genome and Primer3 software. The gene-specific sequences were used to generate mRFU assay primers by adding Illumina-compatible sequences to the 5’ end of the sequence, as shown in Supplementary Fig. 8. The V primers also contained a unique molecular identifier (UMI) sequence made up of 12 random nucleotides for error correction and quantitation. Two primer pools were created by equimolar mixing of the V primers (Pool 1) and the J primers (Pool 2).
The mRFU Assay was made up of 3 reactions designed to enrich for the target TCR rearrangements and to add Illumina sequencing adaptors. The first reaction was a single primer extension using DNA polymerase and primer Pool 1 as shown in Supplementary Fig. 9. After a 1.0x Ampure clean up, the extension product was taken into a low-cycle PCR reaction to amplify the genomic regions with fully rearranged V-J sequences. This PCR used the 20 unique bases of the Illumina Read1 Primer Sequence as a forward primer and the multiplexed J gene-specific primer Pool 2 as a reverse primer. This product was cleaned up using a dual-sided Ampure clean-up to remove the long genomic sequences and to remove the primer and short off-target products. Lastly, an 18-cycle PCR (PCR2) was run to attach full-length Illumina P5/P7 sequences with sample barcodes. These libraries were then sequenced on an Illumina® Novaseq targeting a depth of ~50 M reads per sample.
RFU TCR clonotype count model
A negative binomial generalized linear model (NB-GLM) was fitted for each RFU using DESeq249. DESeq2 size factors (the NB-GLM offset term) were calculated using function “pooledSizeFactors()” in R package Scuttle50. The response variable per sample was computed as the total number of TCR clonotypes assigned to the RFU. The linear predictors of the model are as follows (Supplementary Data 12).
-
Cancer status
-
Age
-
Gender
-
Race (Caucasian, Black, Hispanic, and Unknown/Other)
-
TCR amplification V-J primer lot
-
Third-order polynomial terms for median UMI count across clonotypes of each sample
-
Third-order polynomial terms for the total UMI count of each sample
RFU cancer associations p-values were calculated using the likelihood ratio test. Only RFUs with a depth-normalized RFU count of ≥8 in at least 3 subjects were considered for multiple testing correction using Benjamini-Hochberg FDR.
Clustering/RFU formation
We implemented a fast non-parametric clustering algorithm, CFSFDP, to cluster TCRs32. The original algorithm requires a dissimilarity matrix between all data points, which is computationally prohibitive for a dataset of tens of millions of TCRs. We thus implemented the following improvements to the original CFSFDP algorithm:
-
Instead of computing and storing a pairwise dissimilarity matrix exhaustively, we build an approximate nearest neighbor (ANN) index26 using PynnDescent (https://github.com/lmcinnes/pynndescent). We used a previously developed TCR dissimilarity metric27,31 and used the ANN index to find the nearest k neighbors of a given TCR in the index in a computationally efficient fashion. Since this dissimilarity metric is applicable to comparing TCRs of different V genes and CDR3 lengths, we are able to cluster the entire TCDR dataset into a single RFU clustering.
-
In the original CFSFDP algorithm, the density of each data point is calculated by exhaustively enumerating the number of neighbors within a dissimilarity cutoff of dc, which is an O(n2) operation for n data points. Instead, we use the ANN index to search for all the neighbors of each TCR within dc. Since an ANN index returns neighbors in the order of similarity, this involves only searching for the neighbors of each TCR up to a dissimilarity ≤ dc. Similarly, we query the ANN index to search for the nearest TCR of higher density for each TCR instead of performing an exhaustive pairwise search.
We skip the computationally expensive step of computing the “halo” regions of each cluster. Instead, we accept the joining of two TCRs if their similarity ≤ dc.
Prior to clustering, TCR clonotypes were deduplicated to distinct V gene, J gene, and CDR3 amino acid sequences, given that TCRs with an identical V gene, J gene, and CDR3 amino acid sequence have a TCR dissimilarity of 0 and are trivially clustered together regardless of dc. The three N-terminal and two C-terminal residues in the CDR3 were excluded from the dissimilarity calculation in line with past studies25,26, and the V gene sequence of each TCR was combined with the CDR3 sequence dissimilarity into an overall TCR-TCR dissimilarity score27.
The approximate runtime of TCR deduplication and indexing using PyNNDescent on our dataset ( ~ 70 M input TCR clonotypes) is 4–5 h with 24 provided Intel Xeon CPU cores and 140 GB of peak memory usage. The approximate runtime of the density calculation and TCR clustering (using the TCR index) is 1 h with 24 provided Intel Xeon CPU cores and 100 GB peak memory usage.
TCR to RFU assignment in cross-validation
TCRs from samples in the CV test splits were matched to the most similar TCR in the repertoires of the corresponding CV train split using the approximate nearest neighbor index of the train samples’ TCRs. If the dissimilarity between a query TCR and its nearest train TCR ≤ dc, the query TCR was assigned to the RFU of the train TCR. If the train TCR was a singleton TCR, then the query TCR was left unassigned.
HLA genotype inference
HLA inference was carried out using HLAGuesser (https://github.com/statbiophys/HLAGuessr) using default parameters. HLAGuesser probability of 0.45 was used as the threshold to define an HLA allele as present. We analyzed HLA-A, B, C, DRB1, DPB1, DQA1, and DQB1 gene alleles that had a frequency ≥ 0.05 in an in-house dataset of ~100 subjects for which we had performed sequencing-based HLA genotyping and could verify inference accuracy. This yielded a set of 70 called HLA alleles for correlation analysis against RFUs counts (see below). Average AUC between HLAGuessr and HLA sequencing was 0.94.
HLA gene tested | # alleles tested |
|---|---|
A | 10 |
B | 12 |
C | 10 |
DPB1 | 8 |
DQA1 | 10 |
DQB1 | 10 |
DRB1 | 10 |
Feature selection and machine learning modeling
RFUs with a multiple testing corrected false discovery rate ≤ 0.1 were used as input features for ML. To derive input feature values for ML modeling, the fitted GLM coefficients were used to derive the likelihood for each sample under the assumption that the sample is a cancer (\({L}_{\text{cancer}}=L(y| \text{cancer},\text{other covariates})\)) or a non-cancer sample (\({L}_{\text{non}-\text{cancer}}=L(y| \text{not cancer},\text{other covariates})\)). The covariate-adjusted ML input feature is then defined as \(\log {L}_{\text{cancer}}-\log {L}_{\text{non}-\text{cancer}}\). The features were centered to 0 and scaled to 1 in train samples before being passed to ML modeling.
We employed a bagging classifier of 100 support vector machine classifiers with a linear kernel. Model training and evaluation was performed using Python package scikit-learn with the following parameters: C = 0.001, max_features = 0.5.
For the ML model using single-TCR derived features, the same approach was followed for deriving ML feature values. The same bagged SVM model was used, except max_features was set to 1.0.
Circulating tumor DNA mutation analysis
Library construction and hybridization-based target capture using Integrated DNA Technologies (IDT) xGen™ reagents was used to prepare the DNA samples for next-generation sequencing on the Illumina® platform. The circulating tumor DNA assay required preparation of two libraries for each subject: a cell free (cfDNA) library from the plasma and a genomic DNA (gDNA) library from 300 ng of the buffy coat. While the extracted cfDNA was naturally present as the needed short fragments, gDNA required an upfront shearing on the Covaris® ML230 Platform to enter library construction. After gDNA shearing, both gDNA and cfDNA libraries went through the same IDT xGen ccfDNA and FFPE Kit library preparation protocol. Briefly, the DNA ends were repaired to generate blunt ends, followed by a single-stranded ligation of a sequencing adapter to the 3’ end of the insert. A second ligation step followed, using an adaptor UMI fragment that acts as a primer to gap fill the 5’ side of the insert. Adapter-ligated DNA library underwent PCR amplification to enrich fragments containing adapters on both ends and to incorporate sample-specific indices that allow for multiplexing during sequencing. The final libraries were quantified using the Thermofisher Qubit Instrument and quality controlled for amplicon size using the Agilent Tapestation Instrument.
Following library preparation, hybridization was performed using the xGen™ NGS Hybridization Capture Kit and Biotinylated xGen™ Lockdown Probes custom-designed to target the mutated genes of interest. Briefly, the cfDNA and gDNA sequencing libraries were heat denatured and allowed to reanneal in the presence of a high concentration of biotinylated target probes. After an overnight annealing, streptavidin-coated magnetic beads were added to the hybridization mixture. The streptavidin-biotin bonding allowed enrichment of target sequences using a magnet to pellet the paramagnetic streptavidin-coated beads. The pelleted beads were washed to remove non-specifically bound DNA fragments and any remaining excess probes, and the bound library was put through a final round of PCR amplification to generate sufficient material for sequencing. The final hybrid captured libraries were quantified using the Thermofisher Qubit Instrument and quality controlled for amplicon size using the Agilent Tapestation Instrument.
The final target-captured library was sequenced using Illumina® Novaseq platform, targeting 400 M read pairs for cfDNA and 200 M read pairs for gDNA.
Bioinformatics methods for ctDNA mutation analysis
Sequencing data were demultiplexed using Illumina BaseSpace, and read pairs were aligned to the human reference genome GRCh38 using BWA MEM (v0.7.17). Downstream Unique Molecular Identifier (UMI) processing and read collapsing to duplex consensus sequences were generated with fgbio (v2.0.2). Consensus sequences were then re-aligned to the human genome using BWA MEM (v0.7.17) and quality filtering with fgbio (v2.0.2) performed, requiring a minimum of 2 reads for a consensus sequence. Variant calling was performed using VarDict (v1.8.3). VarDict VCF (Variant Call Format) output files were annotated using snpEff (v5.1).
Both cfDNA and gDNA samples were processed separately with the analysis pipeline above, generating a VCF file for each sample. Raw mutation calls were first filtered by retaining position overlap with regions from 154 cancer driver genes and the entire TP53 gene. For gDNA variant calls, we required the “FILTER” field to be “PASS”. For the cfDNA variant calls we applied the following filters: Allele Frequency (AF) must be 0.1% < AF < 40.0%, Total Depth (DP) > 100, FILTER field must be “PASS”, mutation type must be “coding” or “splice site” only, and mutation must not be present in the matched gDNA sample. We did not require a minimum variant depth for gDNA mutations.
TCR calling from raw sequencing data
-
1.
Sequencing data from each sample was combined into a single pair of FASTQ files.
-
2.
Raw reads were aligned to the GRCh38 reference genome using BWA MEM (https://github.com/lh3/bwa). Only reads with mapping quality ≥ 10 were kept.
-
3.
Raw reads with the same UMI were collapsed into UMI families using Fgbio, generating unmapped “UMI read pairs” (http://fulcrumgenomics.github.io/fgbio/). Only UMI reads constituted from at least two raw reads were kept.
-
4.
Only UMI read pairs with BQ ≥ 45 for all the consensus bases were kept.
-
5.
Forward and reverse mates of the UMI read pairs were collapsed into a single consensus molecule using AdapterRemoval v251.
-
6.
The UMI consensus read reads were aligned to the GRCh38 reference genome. Split read alignment information was recorded by the soft clipping and hard clipping of each read’s alignment positions. Only consensus reads mapping to both a V and a J were kept. The highest-scoring V and J alignments were used as the V-J genes of each consensus read.
-
7.
The nucleotides spanning the CDR3 region of each consensus read were inferred from the split read alignments (soft and hard clipping information) using the conserved 5’-cysteine and 3’-phenylalanine as the boundary positions. This region was translated to the CDR3 amino acid sequence of the TCR. TCRs with an in-frame CDR3 (no frame shifts between the 5’-C and 3’-F) and without stop codons in the CDR3 were used as productive TCRs in the cancer prediction analyses.
-
8.
Each unique V gene, J gene, and CDR3 nucleotide sequence combination was considered a distinct TCR clonotype. The number of UMI-collapsed consensus reads of each TCR clonotype was recorded as its “UMI count”.
Single-cell TCR sequencing analysis
Subjects for the TIL studies were prospectively consented lung cancer patients undergoing clinically indicated resection of their cancers at Brigham and Women’s Hospital, without history of neoadjuvant treatment for lung cancer or history of other cancers. Patients with Stage I, II, and resectable stage III NSCLC were included, taking consecutively consented cases of all types to reflect population prevalence, with consideration given to the appropriateness of available tissue for the planned single-cell analyses. After allocating portions of the resected specimen to clinical needs, excess fresh remaining tissue was provided for research in tubes containing DMEM over ice.
Fresh tumor specimens were minced in a 10 cm plate with media (DMEM + 10% FBS), penicillin-streptomycin (Fisher Scientific), 100 U/mL collagenase type IV (Life Technologies), and 2.5 mg/mL DNAse I (Sigma Aldrich), then incubated for 45 min at 37 C. Single-cell suspensions were isolated by straining through a 40 µm filter. Red blood cells were lysed using RBC Lysis Buffer (BioLegend). Cells were incubated with Zombie Green Fixable Viability Kit (BioLegend), blocked with Human TruStain FcX (BioLegend), and stained with human anti-CD3-PECy7 (clone UCHT1, BioLegend), anti-CD8-PerCPCy5.5 (clone RPA-T8, Fisher), and patient HLA-specific PE-conjugated dextramers (Immudex, Supplementary Data 8). Viable T cells were isolated via the FACS Melody instrument (BD Biosciences) according to the gating schema (Supplementary Fig. 12).
Once isolated, the target cell population was input to a Chromium X via manufacturer’s protocol described in Chromium Next GEM Single Cell 5’ Reagent Kits v2 (Dual Index) with Feature Barcode technology for Cell Surface Protein & Immune Receptor Mapping with Feature Barcoding technology for Cell Surface Protein (CG000330 Rev G). Briefly, Gel Bead-in-Emulsion reverse transcription (GEM-RT) reaction, clean-up, and PCR amplification steps were performed to generate sequencing libraries. A portion of the cleaned cDNA was used to construct cell surface protein libraries (Chromium 5’ Feature Barcode Kit). TCR V(D)J targeted enrichment library preparation (Chromium Single Cell V(D)J Enrichment Kit, Human T cell) was then performed. Libraries were uniquely indexed for multiplexed sequencing and sequenced on a NovaSeq 6000 using 150 bp paired-end reads targeting 50 M reads total (Supplementary Fig. 11).
Cell Ranger v7.1 (https://www.10xgenomics.com/support/software/cell-ranger/latest) multi was run to generate FASTQ files from gene expression, VDJ, and feature barcode libraries. Human reference refdata-gex-GRCh38-2020-A and refdata-cellranger-vdj-GRCh38-alts-ensembl-7.1.0 were provided as references for alignment. Dextramer feature barcode sequences were also provided and used as input. Cell Ranger was run as a single command, providing a configuration file for each of the inputs to the multi-tool.
CellTypist v1.6.3 (https://github.com/Teichlab/celltypist) was used to annotate each cell with cell type. The sample_filtered_feature_bc_matrix.h5 output from Cell Ranger was used as the input to CellTypist. Cell type predictions were generated using the Immune_All_High.pkl model.
Data availability
Source data for the study is either included in the paper supplement or deposited at https://figshare.com/. This includes TCR sequences for the N = 1050 subjects in the case/control cohort (https://doi.org/10.25452/figshare.plus.28063118), Olink® protein measurement results for N = 235 subjects (https://doi.org/10.25452/figshare.plus.28067195), the somatic mutation calls for N = 100 subjects (Supplementary Data 6), and the single cell data for the N = 20 TIL cohort. (https://doi.org/10.25452/figshare.plus.28067336).
Code availability
Data was analyzed as described in the Methods section using R (v4), Python (v3.11), and Linux shell-based tools. The end-to-end Snakemake workflow used to discover cancer-associated RFUs and generate machine learning features for train and test data is available at https://github.com/serumdetect/cdp-analysis-public. The workflow depends on our package for clustering TCRs using an approximate nearest neighbor TCR index, which is available at https://github.com/serumdetect/tcrnn-public. The package depends on a custom branch of PyNNDescent where an overflow bug was fixed (https://github.com/serumdetect/pynndescent/tree/768d050fa54eb66311bafacf02071faf84a53d74). Machine learning model training and testing were performed on the machine learning features using the Python package scikit-learn v1.6.1.
References
American Cancer Society. Cancer Facts & Figures 2022. Atlanta: American Cancer Society; 2022.
National Lung Screening Trial Research, T. et al. Reduced lung-cancer mortality with low-dose computed tomographic screening. N. Engl. J. Med. 365, 395–409 (2011).
National Lung Screening Trial Research, T. et al. Results of initial low-dose computed tomographic screening for lung cancer. N. Engl. J. Med. 368, 1980–1991 (2013).
Imperiale, T. F. et al. Multitarget stool DNA testing for colorectal-cancer screening. N. Engl. J. Med. 370, 1287–1297 (2014).
Grail, L. Multi-Cancer Early Detection (MCED) Tests: A New Approach in the War on Cancer. (https://grail.com/wp-content/uploads/2022/09/GRAIL-MCED-Fact-Sheet.pdf, 2022).
U.S. Food and Drug Administration, Summary of Safety and Effectiveness Data for Premarket Approval (PMA) P190032 FoundationOne Liquid CDx (2020)
U.S. Food and Drug Administration, Summary of Safety and Effectiveness Data for Premarket Approval (PMA) P200010 Guardant360 CDx (2020)
Rolfo, C. et al. Liquid biopsy for advanced NSCLC: A Consensus Statement From the International Association for the Study of Lung Cancer. J. Thorac. Oncol. 16, 1647–1662 (2021).
Diamandis, E. P., Bast, R. C. Jr., Gold, P., Chu, T. M. & Magnani, J. L. Reflection on the discovery of carcinoembryonic antigen, prostate-specific antigen, and cancer antigens CA125 and CA19-9. Clin. Chem. 59, 22–31 (2013).
Mathios, D. et al. Detection and characterization of lung cancer using cell-free DNA fragmentomes. Nat. Commun. 12, 5060 (2021).
Liu, M. C. et al. Sensitive and specific multi-cancer detection and localization using methylation signatures in cell-free DNA. Ann. Oncol. 31, 745–759 (2020).
Shen, S. Y. et al. Sensitive tumour detection and classification using plasma cell-free DNA methylomes. Nature 563, 579–583 (2018).
Cohen, J. D. et al. Detection and localization of surgically resectable cancers with a multi-analyte blood test. Science 359, 926–930 (2018).
Cristiano, S. et al. Genome-wide cell-free DNA fragmentation in patients with cancer. Nature 570, 385–389 (2019).
Bettegowda, C. et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci. Transl. Med. 6, 224ra224 (2014).
Jamshidi, A. et al. Evaluation of cell-free DNA approaches for multi-cancer early detection. Cancer Cell 40, 1537–1549 e1512 (2022).
Klein, E. A. et al. Clinical validation of a targeted methylation-based multi-cancer early detection test using an independent validation set. Ann. Oncol. 32, 1167–1177 (2021).
Lennon, A. M. et al. Feasibility of blood testing combined with PET-CT to screen for cancer and guide intervention. Science 369, eabb9601 (2020).
Hanahan, D. & Weinberg, R. A. Hallmarks of cancer: the next generation. Cell 144, 646–674 (2011).
Sullivan, F. M. et al. Earlier diagnosis of lung cancer in a randomised trial of an autoantibody blood test followed by imaging. European Respiratory Journal 57, 2000670 (2021).
Glanville, J. et al. Identifying specificity groups in the T cell receptor repertoire. Nature 547, 94–98 (2017).
Dash, P. et al. Quantifiable predictive features define epitope-specific T cell receptor repertoires. Nature 547, 89–93 (2017).
Emerson, R. O. et al. Immunosequencing identifies signatures of cytomegalovirus exposure history and HLA-mediated effects on the T cell repertoire. Nat. Genet. 49, 659–665 (2017).
Nolan, S. et al. A large-scale database of T-cell receptor beta sequences and binding associations from natural and synthetic exposure to SARS-CoV-2. Front. Immunol. 16, 1488851 (2025).
Beshnova, D. et al. De novo prediction of cancer-associated T cell receptors for noninvasive cancer detection. Sci Transl Med. 12, eaaz3738 (2020).
Zhang, H., Zhan, X. & Li, B. GIANA allows computationally-efficient TCR clustering and multi-disease repertoire classification by isometric transformation. Nat. Commun. 12, 4699 (2021).
Mayer-Blackwell, K. et al. TCR meta-clonotypes for biomarker discovery with tcrdist3 enabled identification of public, HLA-restricted clusters of SARS-CoV-2 TCRs. eLife 10, e68605 (2021).
Maleki Vareki, S. High and low mutational burden tumors versus immunologically hot and cold tumors and response to immune checkpoint inhibitors. J. Immunother. Cancer 6, 157 (2018).
Gajewski, T. F. et al. Cancer immunotherapy targets based on understanding the T cell-inflamed versus non-T cell-inflamed tumor microenvironment. Adv. Exp. Med. Biol. 1036, 19–31 (2017).
Yu, X. et al. Quantifiable TCR repertoire changes in prediagnostic blood specimens among patients with high-grade ovarian cancer. Cell Rep. Med. 5, 101612 (2024).
Zhang, H. et al. Investigation of antigen-specific T-cell receptor clusters in human cancers. Clin. Cancer Res 26, 1359–1371 (2020).
Rodriguez, A. & Laio, A. Machine learning. Clustering by fast search and find of density peaks. Science 344, 1492–1496 (2014).
Ahlmann-Eltze, C. & Huber, W. glmGamPoi: fitting Gamma-Poisson generalized linear models on single cell count data. Bioinformatics 36, 5701–5702 (2021).
Ruiz Ortega, M. et al. Learning predictive signatures of HLA type from T-cell repertoires. PLoS Comput Biol. 21, e1012724 (2025).
Tay, R. E., Richardson, E. K. & Toh, H. C. Revisiting the role of CD4(+) T cells in cancer immunotherapy-new insights into old paradigms. Cancer Gene Ther. 28, 5–17 (2021).
Veatch, J. R. et al. Endogenous CD4(+) T cells recognize neoantigens in lung cancer patients, including recurrent oncogenic KRAS and ERBB2 (Her2) driver mutations. Cancer Immunol. Res. 7, 910–922 (2019).
Khodayari Moez, E. et al. Circulating proteome for pulmonary nodule malignancy. J. Natl. Cancer Inst. 115, 1060–1070 (2023).
Lung Cancer Cohort, C. The blood proteome of imminent lung cancer diagnosis. Nat. Commun. 14, 3042 (2023).
Consortium, A.P.G. AACR Project GENIE: Powering precision medicine through an International Consortium. Cancer Discov. 7, 818–831 (2017).
Dominguez Conde, C. et al. Cross-tissue immune cell analysis reveals tissue-specific features in humans. Science 376, eabl5197 (2022).
Xu, C. et al. Automatic cell-type harmonization and integration across Human Cell Atlas datasets. Cell 186, 5876–5891 e5820 (2023).
Mazzotti, L. et al. T-cell receptor repertoire sequencing and its applications: Focus on infectious diseases and cancer. Int J Mol Sci. 23, 8590 (2022).
Frank, M. L. et al. T-cell receptor Repertoire sequencing in the era of cancer immunotherapy. Clin. Cancer Res 29, 994–1008 (2023).
Dunn, G. P., Old, L. J. & Schreiber, R. D. The three Es of cancer immunoediting. Annu. Rev. Immunol. 22, 329–360 (2004).
Yu, X. et al. Dissection of transcriptome dysregulation and immune characterization in women with germline BRCA1 mutation at single-cell resolution. BMC Med. 20, 283 (2022).
Godfrey, D. I., Koay, H. F., McCluskey, J. & Gherardin, N. A. The biology and functional importance of MAIT cells. Nat. Immunol. 20, 1110–1128 (2019).
Yigit, M., Basoglu, O. F. & Unutmaz, D. Mucosal-associated invariant T cells in cancer: dual roles, complex interactions and therapeutic potential. Front. Immunol. 15, 1369236 (2024).
Wik, L. et al. Proximity extension assay in combination with next-generation sequencing for high-throughput proteome-wide analysis. Mol. Cell Proteom. 20, 100168 (2021).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
McCarthy, D. J., Campbell, K. R., Lun, A. T. & Wills, Q. F. Scater: pre-processing, quality control, normalization and visualization of single-cell RNA-seq data in R. Bioinformatics 33, 1179–1186 (2017).
Schubert, M., Lindgreen, S. & Orlando, L. AdapterRemoval v2: rapid adapter trimming, identification, and read merging. BMC Res. Notes 9, 88 (2016).
Acknowledgements
Overall, study funding was provided by Serum Detect, Inc. Single-cell sequencing work was supported by the Robert A. and Renée E. Belfer Foundation and the Expect Miracles Foundation. L. M. Montuenga was also supported by CIBERONC (CB16/12/00443), the Spanish Ministry of Science and Innovation, and Fondo de Investigación Sanitaria Fondo Europeo de Desarrollo Regional (PI22/00451). We acknowledge the patients for their participation and the Biobank of the University of Navarra for its collaboration. All rights, title, and interest in the protein biomarker data are owned by Delfi Diagnostics, Inc.
Author information
Authors and Affiliations
Contributions
Study leadership: R.Y., L.M., L.S., J.A.B., R.B., P.L., M.N., Y.L. Assay development and sample processing: M.N., K.F., E.H., D.C., A.L., M.B., S.K., I.G. Sample and data collection: M.A.F., Md.M.O., A.P., I.L.V., H.M., SLK, A.C., M.R., C.S. Data analysis: Y.L., D.S., J.B. Manuscript preparation: R.Y., L.M., L.S., Y.L.
Corresponding authors
Ethics declarations
Competing interests
The authors declare the following competing financial or non-financial interests: Y. Li, Serum Detect, Inc., employment, stock, patent. M. Nahas, Serum Detect, Inc. employment, stock, patent. D. Stephens, Serum Detect, Inc., employment, stock, patent. K. Froburg, Serum Detect, Inc. employment, stock. E. Hintz, Serum Detect, Inc. employment, stock. D. Champagne, Serum Detect, Inc., employment, stock. A. Lochab, Serum Detect, Inc. employment, stock. M. Brown, Serum Detect, Inc. employment, stock. J. Braun, Serum Detect, Inc. employment, stock. C. W. Seder, Serum Detect, Inc. grant/contract. J. A. Borgia, Serum Detect, Inc. grant/contract. Abbott grant/contract. Rational Vaccines Diagnostics, independent contractor. Luminex Corporation independent contractor. L. Seijo, Serum Detect, Inc., independent contractor, grant/contract. L. M. Montuenga, Serum Detect, Inc., independent contractor, grant/contract. R. Yelensky, Serum Detect, Inc. employment, fiduciary officer, stock, patent.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Li, Y., Nahas, M., Stephens, D. et al. Circulating T-cell receptor repertoire for cancer early detection. npj Precis. Onc. 9, 245 (2025). https://doi.org/10.1038/s41698-025-01036-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41698-025-01036-y
