
Abstract
Lymphatic vessels are essential for preventing the accumulation of harmful components within peripheral tissues, including the artery wall. Various endogenous mechanisms maintain adequate lymphatic function throughout life, with platelets being essential for preserving lymphatic vessel integrity. However, since lymph lacks platelets, their impact on the lymphatic system has long been viewed as restricted to areas where lymphatics intersect with blood vessels. Nevertheless, platelets can also exert long range effects through the release of extracellular vesicles (EVs) upon activation. We observed that platelet EVs (PEVs) are present in lymph, a compartment to which they could transfer regulatory effects of platelets. Here, we report that PEVs in lymph exhibit a distinct signature enabling them to interact with lymphatic endothelial cells (LECs). In vitro experiments show that the internalization of PEVs by LECs maintains their functional integrity. Treatment with PEVs improves lymphatic contraction capacity in atherosclerosis-prone mice. We suggest that boosting lymphatic pumping with exogenous PEVs offers a novel therapeutic approach for chronic inflammatory diseases characterized by defective lymphatics.
Similar content being viewed by others

Introduction
The lymphatic system is a body-wide network programed to maintain fluid balance, immune cell trafficking, lipid absorption in the intestine, and pathogen clearance from peripheral tissues. These functions are also essential for maintaining the integrity of large blood vessels1,2. In the aorta, lymphatics are located throughout the adventitial layer of the artery wall3. More than four decades ago, it has been suggested that stasis of the interstitial fluid in the arterial wall could be due to dysfunctional lymphatic vessels4. In the past few years, the emergence of genetic and surgical interventions in animal models along with imaging tools have greatly contributed to our knowledge of the interplay between lymphatic function and vascular health and disease5,6,7,8,9,10,11,12,13,14. This nexus is of considerable interest in the context of prevalent, inflammatory, degenerative disease states within the vascular wall, such as atherosclerosis, which represents a major conceptual and clinical challenge and a source of considerable morbidity and mortality. Among several manifestations of this state is the atherosclerosis-driven coronary artery disease (CAD), raising questions as to the possible involvement of the lymphatic microcirculation.
Adventitial lymphatics have been found to be involved in the mobilization of cholesterol out of the artery wall before it reaches the bloodstream during the reverse cholesterol transport (RCT) process5. Milasan et al. reported that a defect in the propelling capacity of the collecting lymphatic vessels occurs prior to the onset of atherogenesis in low-density lipoprotein receptor (Ldlr)-/- mice15. Changes in membrane lipids of lymphatic endothelial cells (LECs), but not hypercholesterolemia per se, could explain, at least in part, the loss of integrity of lymphatic vessels in atherogenesis14. Our studies aiming to pre-empt cardiovascular diseases suggested that boosting lymphatic transport early in the atherosclerotic process limits plaque progression while also abrogating immune cell accumulation in peripheral tissues13. Considering these data, a promising approach would be to specifically target the contractile capacity of collecting lymphatic vessels to prevent lymphatic function loss in chronic inflammatory diseases.
In the past few years, lymphatic pumping regulators have been proven to augment lymphatic contractility and function ex vivo16,17. Yet, a significant obstacle for employing these novel compounds in vivo is the absence of suitable drug delivery mechanisms, which could enhance lymphatic absorption and accessibility while reducing systemic drug exposure and subsequent side effects. Recently, the injection of Bay K8644 (BayK), a small-molecule agonist of L-type calcium channels, as a nanoparticle (NP) formulation, has been reported to improve lymphatic vessel pumping function in a mouse tail model of lymphedema18. Nevertheless, due to the widespread presence of L-type calcium channels on nontarget cells19 and the complexity of using NP formulations in humans, certain active compounds, such as Bay K8644, raised concerns as to off-target side effects.
Here, we show that taking advantage of the endogenous mechanisms of nanocarrier formation by platelets may both have a role in LEC regulation, and serve as a prototypic delivery system for activities directed at the lymphatic vasculature within the large blood vessel wall. Platelets have emerged as central regulators in assuring lymphatic maintenance20. Both at the embryonic stage and throughout life, platelets sustain blood-lymphatic separation at the lympho-venous junctions21. They do so via the interaction of their C-type lectin-like, type II (CLEC-2) receptor with podoplanin on LECs. It has been assumed that since platelets are absent from the lymphatic circulation, their effects are restricted to the lympho-vascular boundaries22 .
Our present study documents that platelets can exert their influence throughout the lymphatic system, through the release and circulation of molecularly rich extracellular vesicles (EVs). Moreover, we suggest that platelet EVs (PEVs) may regulate extravascular tissues to which platelets have no direct access. We reasoned that since plasma contains PEVs and is continuously filtered through the extracellular space by a semipermeable layer of blood endothelial cells (BECs), PEVs may reach lymphatic capillaries along with other components of the interstitial fluid and modulate lymphatic function per se23,24.
In this study, we sought explore this hypothesis to determine whether the effect of PEVs in the interior of the lymphatic vessels could mirror the beneficial effects of CLEC-2-positive platelets occurring at the exterior of the lymphatic network. We show that CD41+ PEVs positive for CLEC-2 could be detected in lymph, albeit at levels lower than those in blood. This suggests that CD41+ EVs present in lymph can bind to podoplanin expressed on LECs and potentially exert regulatory influences. The unique composition of lymph PEVs confers upon them the ability to stimulate LECs and preserve of the integrity of the lymphatic vasculature. We document that PEVs may maintain the lymphatic function in large blood vessels of atherosclerosis-prone mice, a context associated with lymphatic dysfunction at a very young age15,25. We propose that boosting lymphatic pumping with the use of exogenous PEVs may offer a novel therapeutic approach for chronic inflammatory diseases characterized by defective lymphatics, such as atherosclerosis.
Results
Lymph is rich in platelet- and leukocyte-derived EVs but contains less CD41+ CLEC-2+ EVs than plasma
Platelets are prerequisite players in the preservation of lymphatic vessel function. We have previously demonstrated that PEVs are present in mouse lymph26,27, but whether they have any role in the maintenance of lymphatic function remains largely unstudied. To test this possibility, we first assessed the proportion of platelet EVs in mouse plasma and lymph by flow cytometry. EVs expressing CD41, a marker of platelets and megakaryocytes, constituted the majority among major histocompatibility complex class I (MHCI)-positive EVs (i.e. EVs derived from all nucleated cells and platelets) in lymph and plasma (Fig. 1a). Whereas the absolute concentration of CD41+ EVs was greater in lymph than in plasma (Fig. 1b), a specific subset of EVs double-positive for CD41 and CLEC-2 (CD41+CLEC-2+ EVs) was reduced in lymph compared with plasma (Fig. 1c). Thus, in plasma as many as 84% of the CD41+ EVs expressed CLEC-2, whereas only 7% of circulating PEVs were CLEC-2+ in lymph. Interestingly, EVs derived from the various leukocyte subsets investigated were more concentrated in lymph than in plasma (Fig. 1d–f) while the concentration of red blood cell (RBC) EVs (TER-119+) was not significantly different between plasma and lymph (Fig. 1g). These results support the notion that blood cells communicate with the lymphatic system by transmitting their EVs to lymph. However, in the case of platelets, this passage is somewhat selective.

Plasma and lymph CFSE-positive EVs were identified by flow cytometry. a The proportion of EVs positive for CD41 amongst total MHCI+ EVs was identified in plasma and lymph. Different EV subsets were characterized using antibodies against a combination of anti-MHCI and (b) anti-CD41, (c) anti-CD41 and anti-CLEC-2, (d) anti-CD45 and anti-CD11b, (e) anti-CD45 and anti-CD3e and (f) anti-CD45 and anti-LY6G. g EVs derived from red blood cells were also identified using an anti-TER-119 antibody. n = 6 ± SEM. MHCI: major histocompatibility complex class I; EVs: extracellular vesicles.
Specific lipids travel through lymph as cargo of extracellular vesicles
With these insights in the composition of EVs in plasma and lymph, we set out to explore the role of platelet EVs, which is the most abundant subset of MHCI+ EVs, in lymph. We recently reported that lymph PEVs cannot be distinguished from blood PEVs by their gene-regulatory microRNA profiles26. However, our knowledge on other molecular cargo contained in various PEV subsets is limited, especially with regards to the lipid composition. Recently, PEVs have been identified as important lipid carriers in severe allergic reaction, a condition in which the resolution of inflammation is dysregulated28. Therefore, we sought to identify lipids that could travel preferentially in PEVs to either blood or lymph and may reach their recipient cells, including the respective endothelia. We first sorted MHCI+ EVs from mouse plasma and lymph with a small-particle option cell sorter and proceeded to characterize their lipid composition. Given the unique lipid repertoires associated with different EV subpopulations, we employed untargeted lipidomics to compare the lipid content of total lymph, total plasma, lymph EVs and plasma EVs (Supplementary data 1). Matched mouse lymph and plasma samples and the corresponding isolated EVs were analyzed by LC-QTOF-MS, revealing notable differences. Principal component analysis (PCA) of both fluids (Fig. 2a) and EVs (Fig. 2b) highlighted the distinctive distribution of lipid subsets in plasma and lymph.

Matched mice lymph and plasma, and EVs isolated from those two respective biological fluids, were analyzed by LC-QTOF-MS. Principal component analysis (PCA) plots for (a) plasma in blue and lymph in red and for (b) plasma EVs in blue and lymph EVs in red (PC 1, PC 2 and PC 3 refer to Principal Components 1, 2 and 3 on the Y, X and Z axes, respectively). c Volcano plot of untargeted lipidomics in lymph and plasma. The x axis represents the fold changes (in log2) of MS signal intensities values in lymph vs. plasma and the Y-axis represents the p-values (in –log10). Selection criteria were established with corrected p < 0.05 (horizontal blue line) and FC > 10 or < 0.1 (vertical blue lines), and with less stringent criteria of selection using FC > 1.3 or < 0.77 (vertical red lines). d Volcano plot of untargeted lipidomics in lymph EVs and plasma EVs. Selection criteria were established with corrected p < 0.05 (horizontal red line) and FC > 1.2 or < 0.83 (vertical red lines). Features that were discriminating lymph and plasma (or lymph EVs and plasma EVs) were annotated and validated using MS/MS analysis (color symbols). n = 3 (each n is a pool of 2–4 mice). PC: Principal Component; Car: acylcarnitines; CE: cholesteryl esters; FFA: free fatty acid; HexCer: hexosyl Ceramides; LPC: lysoglycerophosphatidylcholine; LPE: lysoglycerophosphatidylethanolamine; LPCO-: ether-linked lysoglycerophosphatidylcholine; PC: diacylglycerophosphatidylcholine; PE: diacylglycerophosphatidylethanolamine; PCO-: ether diacylglycerophosphatidylcholine; DG: diacylglycerols; SM: sphyngomyelins; TG: triacylglycerols.
In fluids, among the 1200 features obtained following MS data processing (p-value < 0.05 and fold change (FC) >10 or <0.1), we identified 55 features that significantly discriminated lymph from plasma (Fig. 2c). Using MS/MS analysis, 29 unique lipids were annotated and validated, as indicated by color symbols in the volcano plot. Using less stringent criteria of selection (FC > 1.3 or < 0.77; vertical red lines), we annotated 51 additional lipid entities by manual alignment with a previously acquired human database for which the lipid entities have already been validated using MSMS analysis. Lysophosphatidylcholines (LPC) and triglycerides (TG) were respectively down- and upregulated in lymph compared with plasma (Table 1). In EVs, amongst 1867 features obtained following MS data processing (p < 0.05 and FC > 1.2 or < 0.83), we selected 82 features that were discriminating lymph EVs from plasma EVs (Fig. 2d). We then identified eight unique lipids that were either down- or upregulated in lymph EVs compared to plasma EVs. Whereas LPC was also downregulated in lymph EVs, one phosphatidylcholine (PC(36:4)) was upregulated in lymph EVs compared with plasma EVs (p ≤ 0.02) (Table 2). Phosphatidylcholines are lipids bearing anti-oxidative properties and account for ~40% of total phospholipids in all mammalian cells29,30. Thus, lymph EVs harbor unique lipid signatures that distinguish them from their plasma counterparts which may influence their function within the microenvironments they reach.
PEVs preserve the integrity of lymphatic endothelial cells by decreasing ROS production and necrosis, and by hampering their EV release
In order to glean new insights into the function of lymph PEVs, we employed a model of cultured LECs. LEC dysfunction is thought to play a role in vascular diseases observed in atherosclerosis-prone mice, such as Ldlr-/- strain in which lymphatic dysfunction occurs at a very young age15 and LECs are implicated in this early defect14. We reasoned that if PEVs play a beneficial role in the maintenance of lymphatic vessels through a direct effect on LECs, such effect could be observed in PEV/LEC co-culture system. Therefore, one million human PEVs per mL were incubated with human LECs for up to 24 h and the integrity of the latter cells was evaluated.
PEVs produced from human platelet-rich plasma (PRP) were isolated and confirmed to be 90% positive for CD62P, a platelet activation marker, as assessed by flow cytometry (Supplementary Fig. 1a). As recommended by the MISEV2023 guidelines and to further confirm the purity of the produced PEVs, morphological assessment, size distribution and protein expression were determined by transmission electron microscopy (TEM), nanoparticles tracking analysis (NTA) and immunoblotting, respectively31. Images obtained by TEM indicated that small particles could be visualized, with sizes mostly below 200 nm in diameter, in samples that were cleared of cells and debris (Supplementary Fig. 1b). The size distribution of the PEVs ranged between 25 and 725 nm, with a peak at 150 nm (Supplementary Fig. 1c). To compare the effect of PEVs to those of other EVs derived from blood cells, we also produced and characterized EVs derived from red blood cells (rbEVs) of the same donors. Similar to PEVs, the morphological analysis and size distribution of the rbEVs yielded size range of 25–750 nm, but with a peak at 75 nm (Supplementary Fig. 1d, e). Both PEVs and rbEVs expressed the anchored-cytoplasmic membrane bilayer flotillin but not calnexin, an endoplasmic reticulum luminal protein expressed in whole cells and used to test for organelle integrity, showing that both EV isolates were free of cells and debris (Supplementary Fig. 1f). These respective EV preparations were added to cultures of LECs.
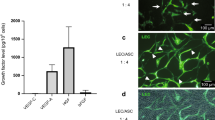
After 2 h of incubation, few PEVs positive for CellVue were detected within LECs, delimited with the plasma membrane marker wheat germ agglutinin (WGA) (Fig. 3a). The internalization increased >4-fold after a 24 h incubation (65 and 276 µm3 of PEVs per cell, after 2 h and 24 h, respectively, Fig. 3a, b). As lymph rbEVs are more concentrated in atherosclerotic-prone animal than in healthy wild-type (wt) mice27, we also compared the effect of PEVs to that of rbEVs. Like rbEVs (Fig. 3c), PEVs generated reactive oxygen species (ROS) when incubated with a fluorescent probe, chloromethyl-dichlorodihydrofluorescein diacetate, acetyl ester (CM-H2DCFDA), as detected by flow cytometry (Fig. 3d). Superoxide dismutase (SOD) efficiently abrogated ROS production in PEVs, but rather modestly decreased it in rbEVs. Similar to their effect on blood endothelial cells32, rbEVs also increased ROS production by LECs (Fig. 3e). To the contrary, PEVs decreased ROS production by LECs (Fig. 3f). PEVs have been implicated in different stages of atherogenesis, including endothelial dysfunction, mainly through triggering of endothelial cell death33. At variance with these reports, our data reveal that PEVs decreased cell death, as displayed by the decreased proportion of PEVs-treated LECs that was positive for propidium iodide (PI) but negative for AnnexinV compared with control-treated LECs (Fig. 3g). EVs derived from red blood cells did not affect the cell necrosis ratio (Fig. 3h).

PEVs stained with CellVue (red) were incubated on LEC in vitro for (a) 2 h and (b) 24 h and incubated with WGA (green) and Hoechst (blue). Z-stacks were acquired using a LSM 710 Confocal Microscope (Zeiss) equipped with a 63/1.4 oil DIC objective (white scale bar, 20 µm). (c) rbEVs, (d) PEVs and LECs treated with (e) rbEVs or (f) PEVs for 24 h were incubated with CM-H2DCFDA and ROS production was measured using flow cytometry. Superoxide dismutase (100 U/mL) was used as a negative control. The percentage of AnnexinV-Propidium iodide+ (PI+AnV-) LECs was determined after treatment with (g) PEVs or (h) rbEVs. The concentration of EVs produced from LECs in the culture supernatant was measured after treatment with (i) rbEVs or (j) PEVs. Each point (n) represents cells treated with PEVs produced from a single donor ± SEM. PEVs: platelet extracellular vesicles; rbEVs: red blood cell extracellular vesicles; WGA: Wheat Germ Agglutinin; ROS: reactive oxygen species; SOD: superoxide dismutase. AU: arbitrary unit.
EVs are released from BECs during activation and apoptosis34, and high concentrations of BEC-derived EVs in the blood circulation are associated with future cardiovascular events35. Given the negative clinical outcomes associated with the endothelium shedding, we asked whether similar processes can be triggered in LECs with implications for lymphatic dysfunction. Therefore, we tested whether exogenous PEVs could act on LECs by hampering the release of EVs. We measured the concentration of EVs produced by the lymphatic endothelium following a 24 h treatment with PEVs or rbEVs. MHCI+VEGFR-3+ EVs detected in the cell supernatant by a small particle option flow cytometer were identified as LEC-derived EVs (LEC-EVs). We observed that rbEVs increased the release of LEC-EVs upon incubation with the cell monolayer (Fig. 3i). In contrast, PEV treatment did not increase the production of EVs from LECs (Fig. 3j).
Finally, to better define the mechanisms by which PEVs might abrogate the loss in LEC integrity, we analysed the gene expression regulation in LECs upon incubation with PEVs. RNA sequencing revealed that specific genes were significantly down- or upregulated following PEV incubation (p < 0.05 and FC > 1.3; Fig. 4a, blue and red, respectively,Table 3 and supplementary data 2). IFIT1, MX2, MX1 and RSAD2 were downregulated in LECs treated for 24 h with PEVs. FZD7, ATAD3C, VWA7, and KLB genes were upregulated. Moreover, qPCR analysis highlighted that genes implicated in the cellular junction stability (F11R; Fig. 4b) and lymphatic integrity (PROX1 and FLT4; Fig. 4c, d, respectively) were upregulated upon incubation with PEVs. However, TJP1 and CDH5 expression were not modulated by PEV treatment (Fig. 4e, f, respectively). Nonetheless, rbEVs downregulated the expression of CDH5 and tended to downregulate F11R (p-value = 0.06), two proteins known to be implicated in the maintenance of cellular junctions (Fig. 4g, h, respectively). The gene expression of PROX1 and FLT4 was unchanged upon incubation with rbEVs (Fig. 4i, j, respectively). Thus, the interaction of LECs with PEVs triggers multiple events that may contribute to the ability of the lymphatic vasculature to face physiological and pathological challenges.

Human LECs were treated for 24 h with PEVs. a Volcano plot of transcriptomic analysis was performed in human LEC. Y axis = p-value (-LOG10) of the gene expression. X axis = fold change PEVs/Control (LOG2) of gene expression. Each gene is represented by a dot. Significant down- and upregulated genes in PEV-treated LECs are depicted in blue and red, respectively. mRNA expression in human LECs was also performed by qPCR analysis for (b) F11R, (c) PROX1, (d) FLT4, (e) TJP1 and (f) CDH5 mRNA. Human LECs were also treated for 24 h with rbEVs, and mRNA expression of (g) CDH5, (h) F11R, (i) PROX1 and (j) FLT4 was subsequently measured by qPCR analysis. Each point (n) represents cells treated with PEVs produced from a single donor ± SEM. PEVs: platelet extracellular vesicles; rbEVs: red blood cell extracellular vesicles; F11R: F11 Receptor; PROX1: Prospero homeobox protein 1; FLT4: Fms-related tyrosine kinase 4; TJP1: Tight junction protein-1; CDH5: cadherin 5.
Exogenous PEVs travel through lymph in vivo and interact with multiple cells
We next tested whether exogenous PEVs could also preferentially travel through lymph, bind to the interior of lymphatic vessels and enhance lymphatic contraction. Ten million human PEVs were stained with CellVue and injected into the dermis of the footpad of wt mice. Images obtained following whole mount immunofluorescence imaging revealed that PEVs were efficiently drained to regional lymph nodes (LNs; popliteal/inguinal), but not the non-draining LNs (axillary) at 120 min post-injection (Fig. 5a). Indeed, two hours post-injection, PEVs could be found within lymph node-derived professional phagocytic cells and in the LN extract following its enzymatic digestion (Fig. 5b, c). Moreover, while traveling through lymph, PEVs were also taken up by LECs, as depicted by the immunofluorescent imaging revealing CD41a staining within cells lining the draining popliteal lymphatic vessel compared to control mice (non-injected) (Fig. 5d).

CellVue+ human PEVs (1 × 107) or vehicle control were injected in the dermis of the footpad in wild-type mice. Lymph nodes (LNs) draining the site of injection (green boxes) and LNs located at a distal site (beige boxes) were collected 15 min and 2 h post-injection. Then, they were (a) visualized with a fluorescence imaging scanner (IVIS Lumina II) or (b) digested with collagenase D to determine the percentage of cells containing EVs. c The concentration of free EVs within the LNs were assessed by flow cytometry using anti-mouse MHCII and anti-human CD62P antibodies. d Two hours post-PEV injection, popliteal lymphatic vessels draining the injection site were also collected, harvested and stained with anti-human CD41a antibody (red) and DAPI (blue) to be imaged with a confocal microscope (white scale bar, 20 µm). The lymphatic contraction capacity was measured in vivo 48 h after the injection of (e) PEVs or (f) rbEVs in Ldlr-/-, and (g) after injection of PEVs in wild-type mice. h The lymph nodes (N-D, beige; D, green) and (i) the aorta were also collected at 48 h and visualized with fluorescent imaging. Each point represents a mouse ± SEM. N-D: non-draining; D: draining; LN: lymph nodes; EVs: extracellular vesicles. PEVs: platelet extracellular vesicles. rbEVs: red blood cell extracellular vesicles. Created with Biorender.com.
PEVs improve lymphatic contraction capacity in vivo
Given the apparent multiplicity of interactions between exogenous PEVs and cells within the lymphatic network, we investigated the resulting functional consequences. One disease model in which lymphatic function is known to be impaired is atherosclerosis, particularly in association with deficiency of the Ldlr11,13,15. Therefore, we asked whether PEVs would exert any modulating effects on lymphatic vessel function in this setting. The injection of PEVs significantly improved lymphatic contractions after 48 h in Ldlr-/- mice, which are known to have impaired lymphatic function at 3 months of age13 (Fig. 5e, Supplementary Movie 1a, b), whereas rbEVs did not enhance the lymphatic contractions (Fig. 5f). When injected into wt, healthy animals, PEVs had no detectable effect on the contraction frequency (Fig. 5g). After 2 days following the intradermal injection, the CellVue-positive EVs were still detectable by immunofluorescence in the draining lymph nodes (Fig. 5h). Exogenous PEVs could also be found in the aorta and its surrounding adipose tissue (Fig. 5i). These observations suggest that as PEVs travel through the lymphatic system, they may exert functional (reparative) effects on LECs, through a presently unknown mechanism.
Discussion
Plasma EVs can penetrate into lymph
Our study brings forward several observations. Notably, we report that PEVs could permeate extravascular and lymphatic compartments thereby extending the regulatory effects of platelets. Plasma contains EVs that mostly originate from RBC and CD41+ cells (platelets)36, and we have reported that these EVs also travel through lymph26,27. This is not surprising as lymph is mostly formed by plasma ultrafiltrate, but the ability of particulate elements (such as EVs) to partake in this trafficking is of considerable interest23. Under physiological conditions, EVs enter into the lymphatics via the lymphatic capillaries. Under inflammatory conditions, this could also occur via the collecting lymphatic vessels, which are embedded in fat22. What has not been widely appreciated is that this filtrate creates an access route of RBC- and platelet-EVs to the lymphatic system whereby they can extend their regulatory influences. There is a growing appreciation for the role of EVs as important cell-cell messengers capable of transporting messenger RNA, microRNA, proteins and lipids37 between cells, but specific intercellular pathways are still to be mapped.
We observed that EV trafficking between plasma and lymph is not random. For example, the white blood cell (WBC)-derived EVs were more concentrated in lymph than in plasma. No such difference was observed in TER-119+ EVs. Increased levels in leukocyte-derived EVs in lymph could be attributable to the fact that leukocytes are generally more numerous in efferent lymph than in plasma38. WBC-EVs contained in lymph could thus originate from the WBC present in both the plasma ultrafiltrate and lymph per se. On the other hand, lymph is normally devoid of RBCs, suggesting that rbEVs in lymph would solely be taken up into lymphatics from the interstitial fluid space containing plasma filtrate.
CLEC-2 expression on lymph PEVs grants them unique functional properties
It has been demonstrated that most of the circulating CD41+CD42b+ EVs found in the blood circulation are derived, primarily, from megakaryocytes rather than from platelets39,40. In lymph, however, we propose that CD41+ EVs might be more abundant in lymph than in plasma, as two sources of cells could be involved: megakaryocytes and platelets. Upon their adhesion to the external junctions between blood and lymphatic vessels, platelets inevitably activate and thus produce PEVs, that could in turn enter the lymphatic circulation along with the CD41+ EVs already present in the plasma ultrafiltrate. CD41+ EVs in lymph might have a distinct role than in plasma. We have recently published that, albeit at a lower level than plasma, lymph can generate thrombin26, which supports the fact that coagulation can occur in lymph41. However, in a mouse model of rheumatoid arthritis, we showed that the specific removal of CD41+ EVs from lymph and plasma reduced thrombin generation in plasma, but not in lymph26. This points to a distinct function of CD41+ EVs between both fluids.
Like plasma, lymph contains EVs that are double-positive for CD41 and CLEC-2. Of interest, no ligand for CLEC-2 has been identified yet in blood42, suggesting that CLEC-2+ EVs in lymph might further bind to the interior of the lymphatics. Given the potential for beneficial effects of platelets on lymphatic endothelial function via the interaction between CLEC-2 receptors and podoplanin on LECs, we propose that the in vivo execution of this process relies on PEVs. Thus, PEVs represent a unique mechanism whereby the beneficial effect of platelets can be extended into lymphatics by releasing encapsulated platelet content that can travel through lymph and access LECs in situ. We hypothesize that PEVs can also bind to LECs via a CLEC-2/podoplanin interaction to exert their function, but the exact mechanism depicting the effect of PEVs on lymphatics remains to be elucidated.
Extracellular vesicles as carriers of lipids into lymph
The lymphatic system plays important roles in lipid transport from the intestine to the blood circulation1. Practically all dietary lipids packaged in chylomicrons are absorbed by the small intestine and transported to the blood stream via the lymphatic system. As chylomicrons are triglyceride-carrying particles, it is not surprising to observe more TG in lymph than in plasma, as also described in the litterature43. Beside the 16 validated TG entities, DG(16:0_18:2) was also identified as an upregulated lipid in lymph compared to plasma. Diacylglycerols are intermediates in TG metabolism second messengers that activate proteins involved in a variety of signaling cascades44. LPC, a major class of glycerophospholipids in human plasma that is implicated in inflammation45 was rather downregulated in whole lymph compared to plasma. Altogether, our results confirm that lymph actively transports lipid to in turn modulate lipid metabolism46.
In addition to lipoproteins, EVs may serve as carriers of lipids between fluid compartments and tissues. Like cells, EVs are characterized by lipid bilayer, and lipids are essential elements within extracellular vesicles47,48. Lipids are essentials for various cellular functions, and the set of lipids needed in each cell differs from one cell type to another depending on the cell function or pathological state49. Along these lines, we have recently reported that LECs are sensitive to membrane lipid changes and composition of their lipid rafts modulates the expression of key regulators of lymphatic function, such as vascular endothelial growth factor (VEGF) receptor-3 (VEGFR-3) and VEGF-C mRNA14. However, our understanding of both the lipid composition and their functions within EVs remains constrained. Recently, PEVs have been identified as carriers of specific lipids and metabolites in severe allergic inflammation28. The lipid content of these PEVs varies depending on the degree of inflammation in the patients. It is known that the membrane lipids comprised in EVs might act as signaling molecules capable of stabilizing the vesicles in their new environments, thus facilitating their binding to recipient cells50. Therefore, we sought to determine whether specialized lipids could seek to specifically reach LECs by traveling through lymph EVs when lymphatic dysregulation is present.
While the lipidomics profile of EVs contained in plasma and lymph did not differ extensively, the only annotated lipid significantly increased in lymph EVs was the PC (36:4). Of note, no such discrepancy was noted between the global levels of these lipids in whole fluids. This observation may suggest that enhancing PC in lymph EVs may signifiy mechanisms of selective trafficking of EV subsets. In addition, it cannot be excluded that PC could also contribute to interactions between EVs and LECs. PC has been studied in lymphedema and lipidema, two inflammatory conditions characterized by fluid and fat accumulation51. Despite having distinct molecular and genetic regulators, those two inflammatory and complex conditions are both still undiagnosed and mismanaged, leaving affected patients physical and emotional distress. Hence, subcuteanous injections of phosphatidylcholine helped patients to manage their pain and decreased fat deposition52. Phosphatidylcholine and deoxycholate (PC-DC) injections have been studied in a randomized, controlled albeit small trial, and have been described as successful in decreasing abdominal fat volume and thickness by inducing adipocyte necrosis53. Whereas these observations do not allow to pinpoint a specific role of the lymphatics in the process, it can be suggested that the lymphatic clearance capacity got enhanced. A more recent study found 33% more endogenous phosphatidylcholines in serum than in lymphedema-associated adipose tissue54. These observations implies that PC is not readily accessible to lymph. A direct access of PC in lymph to specifically reach the LECs through PEVs could optimize the effect of lymphatic therapy while limiting the systemic adverse effect.
PEVs preserve the healthy state of lymphatic endothelial cells in vitro
The trafficking of endogenous PEVs into the lymphatic system suggests the potential for intralymphatic EV therapy. We first used an in vitro model to test whether and how exogenous PEVs can exert beneficial effects on lymphatic endothelial cells. We report that PEVs restrict the production of EVs from the lymphatic endothelium. This is in contrast to EVs derived from RBC that induce massive EV release from LECs. In addition, PEVs decreased the necrosis rate of LECs and reduced LEC-ROS production. Our data pinpoint the potential protective role of PEVs on the maintenance of the integrity of the lymphatic endothelium. PEVs have been extensively studied in various settings including vascular inflammation, atherosclerosis and hemostasis55,56,57,58,59,60,61. They can be detrimental to endothelial function but they have also been shown to protect the microvasculature from harmful factors, such as thrombin, capable of disrupting the control of endothelial permeability62. Our results converge on multiple roles of PEVs as guardians of lymphatic integrity.
PEVs downregulate the expression of interferon-stimulated genes
Transcriptomic analysis revealed that IFIT1, MX2, MX1 and RSAD2 were downregulated in LECs treated for 24 h with PEVs. Interferon-induced protein with tetratricopeptide repeats 1 (IFIT1), like IFIT2 and IFIT3, may inhibit the translation and replication of viruses63. Intriguingly also Radical S-adenosyl methionine domain-containing protein 2 (RSAD2) has underlying anti-viral egress and replication effects64. Antiviral response pathway is also implicated by changes in Interferon-induced GTP-binding protein MX (MX1) along with MX2, both having a high level of homology65. MX1 is mainly induced after type I IFN (IFNα/β) stimulation during the antiviral response and has a potentially suppressive effect on the activity of viral ribonucleoprotein complex and its GTPase. It also plays a role in some solid tumors as high MX1 protein expression was associated with features of cancer aggressiveness66. It was associated with large tumor size, high tumor grade, hormone receptor negativity and high Ki67 expression66. On the other hand, MX2 can be expressed at significant levels even in the absence of IFN while still being effective in repressing viral replication, transcription, and nucleocapsid shuttling67. MX2 has been shown to display additional, viral-independent cellular functions including regulation of cell-cycle progression.
The downregulation of these interferon-stimulated genes (ISGs) have also been shown to have modulatory effects on the immune system and promote autoimmunity in chronic inflammatory diseases, such as systemic lupus erythematosus (SLE)68. An elevated type I IFN activity negatively affects endothelial function and increases prevalence of atherosclerosis in patients with an autoimmune disease69,70. Consequently, Tydén et al. propose that activation of type I IFN may have direct impact on the cardiovascular risk for patients with a chronic autoimmune disease like SLE71. In their work, the authors also observed a concomitant deposition of complement factors on platelets, a measure of platelet activation, in patients with endothelial dysfunction. Of interest, endothelial dysfunction was assessed using surrogate markers of endothelial activation, and endothelial microparticles71. Collectively, our observations may suggest that a part of the LEC-protective effect of PEVs may stem from tuning down the ISG expression.
PEVs upregulate the expression of genes involved in the preservation of lymphatic integrity
In addition to downregulating ISGs, PEVs significantly upregulated the expression of specific genes implicated in lymphatic integrity. Among them ATPase Family AAA Domain Containing 3 C (ATAD3C), a truncated form of ATAD3, has been recently identified as important in the control of mitochondrial cristae in mouse muscle cells, influencing mitochondrial DNA replication and cholesterol levels72. Indeed, knocking down ATAD3 in mice resulted in a 30% decrease in the ratio of cholesterol esters (CEs) to total cholesterol in 5-month-old mice compared to controls72. Changes in the ratio of CEs/free cholesterol might reflect changes in lipid rafts, which could, in turn, affect the expression and signaling of key proteins involved in lymphatic function14.
We also observed upregulation of von Willebrand factor A domain containing 7 (VWA7) transcript encoding a protein regulated by a blood endothelial cell-specific transcription factor, SRY-Box Transcription Factor 7 (SOX7)73. While not expressed by LECs, SOX7 has very recently been identified as the first direct blood endothelial cell‐specific regulator of Vegf-c transcription74. This regulation of angiocrine signals is essential for correct lymphatic growth and patterning. We have also observe elevated transcripts for β-klotho (KLB), a single-pass transmembrane protein with a major function in longevity, metabolic regulation, glucose uptake, bile acid synthesis, and fatty acid metabolism75. β-Klotho is an essential receptor component for the endocrine fibroblast growth factors (FGFs), FGF-19 and FGF-2176. FGF-21 cannot directly bind to FGFR when KLB is absent, thereby positioning KLB as prerequisite in mediating the biological function of FGF-2177. Although no direct effect of FGF-21 has been associated to lymphatic function as yet, mice lacking Fgf-21 develop significant pancreatic periductal lymphocytic inflammation when fed with a high fat obesogenic diet78. Further, Fgf-21 has been shown to maintain vascular functions and exert an anti-atherosclerotic effect79.
The PEV induction of Frizzled-7 (FZD7) is of special interest as part of the mechanism regulating cellular growth, stemness and multiple functions. For example, FZD7 acts as Wnt receptor expressed in endothelial cells where it controls vascular permeability via the canonical Wnt/FZD pathway80. It has recently been evidenced that FZD7 controls blood vessel formation through Wnt/β-catenin canonical signaling81. Based on our transcriptomic analysis, we hypothesize that FZD7 upregulation may favor the maintenance of the integrity of the lymphatic endothelium. FDZ7 is known to accumulate at the sites of cell-cell contact in association with β-catenin and VE-cadherin80. VE-cadherin is an important adherent junction molecule involved in the regulation of mechanosignaling in the lymphatic vasculature82. It stabilizes β-catenin and in turn regulates two of the main genes involved in lymphatic valve formation and maintenance: PROX1 and FOXC283.
Finally, qPCR analysis revealed that F11R is upregulated in LECs after a 24 h PEV treatment. F11R is a tight junction transmembrane protein implicated in cell migration84, cellular permeability85 and immune cell transmigration86. Moreover, F11R regulates platelet adhesion to BECs87. Our results suggest that PEV treatment leads to the upregulation of adhesion molecules to facilitate their entry into LECs. All toghether, our data suggest that the upregulation of FZD7 increases VE-cadherin and/or β-catenin expression and affects PROX1 and FLT4 gene expression. We posit that PEVs might, at least in part, act through FZD7 upregulation to mediate its beneficial effect on lymphatic vessel integrity and permeability. However, this remains to be directly tested.
Exogenous PEVs reach lymphatic endothelial cells and draining lymph nodes
We next sought to investigate whether the beneficial effects of the exogenous PEVs on lymphatic endothelial cells in vitro could be extended in vivo to in turn peserve lymphatic transport. In this regard, we observed that PEVs were rapidly taken up (within 2 h) by the initial lymphatics, and traveled within the collecting lymphatic vessels. In the same timeframe, a subfraction of PEVs were found adhering to the interior of collecting lymphatic vessels and a proportion of PEVs reached the draining lymph nodes. Interestingly, not only free PEVs could be found in lymph node fluids, but PEV signal was present within immune cells contained in the lymph nodes. Srinivasan et al. studied the route taken by exogenous exosomes derived from HEY cell line to get to skin draining LNs, and identified macrophages and B-cells as key players in exosome uptake88. Our data suggest that free PEVs could be taken up by antigen-presenting cells before reaching the LNs, while CD41+CLEC-2+ EVs could be preferentially internalized by LECs thus serving two different biological processes.
PEV treatment improves lymphatic pumping and might has a systemic reach
Intraluminal valves within the collecting lymphatic vessels are essential to ensure the propelling of lymph forward through the lymphatic vasculature89. Our findings on the effect of PEVs on the lymphatic endothelial cell monolayer in culture combined with the distribution of the injected exogenous PEVs in vivo led us to suggest that PEVs might enhance lymphatic contraction capacity in vivo. We found that PEVs improve the contraction capacity in 3 month old Ldlr-/- mice fed with a regular chow diet 48 h following the intradermal injection in the footpad dermis. PEVs could be retrieved, but to a lesser extent than at 2 h, in the draining LN. Fluorescent imaging also revealed that PEVs might be reaching the adipose tissue weaving in and out the wall of mouse aorta, where lymphatics are embedded5. These mice were fed with a regular chow diet and thereby protected from formation of the overt atherosclerotic lesion, but regardless, PEVs were able to interact with LECs at remote location from the injection site. Of note, PEVs had no detectable effect on lymphatic contractions in wt mice.
We have previously published that Ldlr-/- mice first present a lymphatic dysfunction that resides at the level of the collecting lymphatics rather than in the lymphatic capillaries15. Indeed, in these mice, the uptake of the interstitial fluid and components by the lymphatic capillaries are normal, but there is reduced capacity to propel the lymph toward the draining lymph nodes and the blood circulation15. Our follow-up publication demonstrated that the optimization of lymphatic function with VEGF-C 152 S before the atherogenesis later continuously supports the contractile capacity of lymphatic vessels during the induction of atherogenesis. This boosting in lymphatic transport consequently limits the progression of atherosclerosis13. Herein, we cannot exclude that the beneficial effects of PEVs could be also mediated through the release of VEGF-C upon platelet activation90 and through the transduction of intracellular signals91. Among the signaling pathways that would be relevant figures VEGFR-3, one of the main receptors of VEGF-C, that is mostly located in cell membrane lipid rafts92 and we demonstrated here that it is upregulated after a 24 h treatment with PEVs.
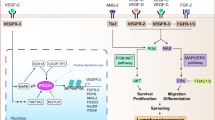
In summary, we present that PEVs might represent a new way in which platelets may act on lymphatic vessels. PEVs may become a new therapeutic option in the treatment of chronic inflammatory diseases where lymphatics are known to be defective (e.g. in atherosclerosis). We propose that PEVs can directly promote lymphatic pumping function and ameliorate dysfunctional lymphatic function. We suggest that selecting PEVs based on their CLEC-2 expression might enrich for these effects. As CLEC-2 supports platelet activation in mouse but not in human blood at arterial shear rate42, CLEC-2+ PEVs may present a promising and safe option as a potential lymphatic-targeting therapy in humans (Fig. 6).

CD41+ EVs travel from the blood circulation and enter the lymph mostly via a the lymphovenous junctions (crossing point between the lymphatic vessel and the subclavian vein) and b the lymphatic capillaries. CD41+ PEVs are also produced upon the adhesion and activation of platelets at the exterior of the lymphatic vessels before entering lymph. CD41+CLEC2+ EVs are mostly attached to the interior side of the lymphatic vessel. c Like platelets, CD41+ EVs bind to LECs via the interaction of their CLEC-2 receptor with podoplanin on LECs. d The specific signature of the EVs that bind LECs confers them the ability to enhance lymphatic contractions in mice known to display a defect in lymphatic pumping. Ultimately, enhancing lymphatic function with exogenous PEVs might become a new therapeutic option in the treatment of chronic inflammatory diseases where lymphatics are known to be defective, like atherosclerosis. Created with BioRender.com.
Methods
Experimental animals
This study was conducted in 10–16 week-old wt and Ldlr-/- mice on a C57BL/6 background that were purchased from Jackson Laboratory. Animals were housed in a pathogen-free environment under a 12 h light-dark cycles with free access to water and a standard chow diet. All experiments were performed in accordance with the Canadian Council on Animal Care guidelines and received the ethical approval by the Montreal Heart Institute (MHI) Animal Care Committee (protocol #2019-14-03).
Lymph and plasma collection
Mouse blood and lymph was collected on ethylenediaminetetraacetic acid (EDTA) as previously described27. Lymph was collected from the thoracic duct of anesthetized mice for an average of 45 min to 1 h. The collected lymph and plasma were centrifuged at 1,200 g and 2,400 g, respectively, for 10 min at 4 °C. Samples were aliquoted and stored at -80 °C until further use.
Production of human platelet and red blood cell extracellular vesicles
All ethical regulations relevant to human research participants were followed and approved by the MHI Ethic Committee (protocol #2016-2117). Every subject gave written informed consent. Human blood from healthy donors was collected with a syringe containing citrate-dextrose solution (ACD, Sigma cat. #C3821). Blood was transferred into 50 mL tubes and centrifuged at 280 g for 10 min. Unless otherwise specified, all centrifugations were performed at room temperature (RT). Then, the red blood cell fraction was kept to produce rbEVs and the PRP fraction remove for the production of PEVs.
PEVs were produced as described previously93. Briefly, the PRP was harvested and 1/5 volume of ACD and 1/50 volume of EDTA were added. The PRP was then centrifuged at 400 g for 5 min and the supernatant was harvested and centrifuged for 5 min at 1,300 g. The supernatant was discarded, and the pellet was resuspended in 2 mL of Tyrode’s buffer pH 6.5 and 13 mL of Tyrode’s buffer pH 7.4 were added to the homogeneous preparation of platelets. Platelets were counted and diluted at 1 × 108 platelets/mL with Tyrode’s buffer pH 7.4 containing 5 mM of CaCl2. Next, platelets were stimulated for 2 h with 0.5 Unit/mL of thrombin (from bovine serum, Sigma cat. #T4648) at 37 °C. Platelet activation was stopped by the addition of 20 mM of EDTA and centrifuged twice for 10 min at 2,000 g to eliminate any platelet remnants. The supernatant was collected and centrifuged for 90 min at 18,000 g without brakes. The supernatant was discarded and the pellet containing PEVs was resuspended in Tyrode’s buffer containing 5 mM of CaCl2 (pH 7.4). PEVs were aliquoted and stored at -80 °C until use.
When fluorescent PEVs were required, human PEVs were incubated with CellVue™ Claret Far Red Fluorescent (Sigma cat. #Minclaret) according to the manufacturer’s protocol, and centrifuged twice at 18,000 g for 90 min. The pellet was resuspended in 0.22 μm-filtered phosphate buffer saline (PBS) and immediately used for the in vivo experiments.
RbEVs were produced as previously described93. A total of 200 µL of the red blood cell fraction was incubated for 10 min with 50 mL of filtered double-distilled water. A volume of 5.5 mL of filtered-PBS 10X (Growcells cat. #75801-000) was added to stop the hypotonic reaction. RbEVs were then aliquoted and stored at -80 °C until further use.
Flow cytometry for the characterization of extracellular vesicles
EVs were analyzed in mouse plasma and lymph samples following a 30 min incubation at 37 °C in annexin V buffer (10 mM Hepes pH 7.4, 140 mM NaCl, and 2.5 mM CaCl2; BD Biosciences cat. #556454) containing 1 μM carboxyfluorescein diacetate succinimidyl ester (CFSE, Selleck chemicals cat. #S8269) and 10 μM D-Phe-Pro-Arg-chloromethylketone (PPACK, Cayman Chemical cat. #15160-1), and one or more of the following mouse antibodies: anti-MHCI (BV711, BD Biosciences cat. #743539), anti-CD45 (APC, BioLegend cat. #103112), anti-CD41 (AF700, BioLegend cat. #133925), anti-CLEC-2 (PE, BioLegend cat. #146104), annexin V (PerCP Cy5.5, BioLegend cat. #640936), anti-TER-119 (BV510, BD Biosciences cat. #640936), anti-CD62P (BV605, BD Biosciences cat. #740358), anti-CD11c (Pe-Cy7, Tonbo Biosciences cat. #600114), anti-CD3e (BV650, BD Biosciences cat. #564378), anti-CD19 (BV510, BD Biosciences cat. #562956), anti-LY6G (PerCP Cy5.5, BD Biosciences cat. #560602) and anti-CD11b (AF700, BioLegend cat. #101222). The flow cytometer was calibrated for EV detection using an ApogeeMix (Apogee Flow Systems cat. #1493), a mixture of non-fluorescent silica beads (180, 240, 300, 590, 880, and 1300 nm) and FITC-fluorescent latex beads (110 and 500 nm). Count beads (Apogee Flow System cat. #1426) were added to each mix to determine the concentration of EVs per sample. The EV gate was set using the ApogeeMix and absolute concentrations were determined for each sample with count beads. Acquisitions were made using our custom-made small particle option FACS Aria Fusion cell sorter (BD Biosciences) equipped with a FSC-PMT and 1 mm-thick magnetron sputtered 405/10 bandpass filter (Chroma Technology), which is referred throughout the manuscript as V-SSC.
The quantification and purity of the produced human EVs was assessed with a BD FACSCelesta (BD Biosciences) also customized with 405/10 bandpass filter (V-SSC). PEVs and rbEVs produced from human blood, as well as LEC-EVs produced from lymphatic endothelial cells in vitro, were stained for 30 min at 37 °C with human anti-CD62P (PE, Biolegend cat. #304906), anti-CD235a (BV786, BD Biosciences cat. #740984) and anti-VEGFR-3 (PE, Biolegend cat. #356204) antibodies, respectively, in a 0.22 µm-filtered annexin V buffer containing 1 µM CFSE and 10 µM PPACK. Like for the mouse EVs, the EV gate was set using an Apogee Mix (Supplementary Fig. 2a). EVs were also first identified based on their CFSE expression (Supplementary Fig. 2b and c) before being specifically categorized according to their membrane marker expression (Supplementary Fig. 2d; Supplementary Fig. 1a; CD235a for rbEVs and CD62P for PEVs). To confirm that the identified events were of cellular origin, 0.1% Triton X-100 (0.05% final concentration) was added to a sample tube to disrupt the vesicle membranes, and the sample was further analyzed by flow cytometry (Supplementary Fig. 2e and f). The drastic decrease of events confirmed the presence of PEVs or rbEVs. Count beads were added (Supplementary Fig. 2g) to determine the concentration of EVs according to the following calculation: (# of events in EV gate / # of events in count beads gate) x (total number of beads in sample/sample volume) x dilution factor. FACS plots are showing all parameters in height (indicated as –H), as recommended for EV detection94. For EV analysis, events were acquired at a flow rate of 12 µL/min, which is the lowest flow rate on the BD FACSCelesta. The flow rate during acquisition was kept to the minimum to avoid swarming effects and coincident detection95. After acquisition, data analysis was performed with FlowJo v10.5.
Sorting of mouse plasma and lymph extracellular vesicles
Plasma and lymph samples were incubated with 1 μM CFSE, 10 μM PPACK and anti-mouse MHCI antibody (BV711) in 0.22 μm-filtered Annexin V binding buffer 1X. Samples were analyzed with a custom-made small particle option BD FACSAria Fusion cell sorter (BD Biosciences). As described above, the flow cytometer was calibrated for EV detection. The EV gate was set using the ApogeeMix and absolute concentrations were determined for each sample with count beads. CFSE+MHCI+ events were sorted, collected in 5 mL tubes, aliquoted and kept in -80 °C for further analysis. To confirm the cellular origin of the vesicles detected, 0.1% Triton X-100 (0.05% final concentration) was added to the samples for 30 min, and the decrease in EV count in the region of interest determined.
Untargeted lipidomic by liquid chromatography quadrupole time-of-flight mass spectrometry (LC-QTOF-MS)
Untargeted lipidomic analyses were performed at the MHI as previously described96. Lipids were extracted from plasma, lymph or sorted EVs spiked with the following internal standards used as quality controls for the reproducibility of the measures (Avanti Polar Lipids Inc): (monoacylglycerosphosphocholine (LPC) 13:0, diacylglycerophosphocholine (PC) 14:0/14:0 and 19:0/19:0, phosphatidylserine (PS) 12:0/12:0, diacylglycerophosphoethanolamine (PE) 17:0/17:0, and diacylglycerophosphoglycerol (PG) 15:0/15:0). Samples were injected in a high-performance liquid chromatograph (1290 Infinity HPLC, Agilent Technologies Inc) coupled to a quadrupole time-of-flight mass spectrometry (6550 accurate system, Agilent Technologies Inc.) equipped with a dual electrospray ionization source and analyzed in both positive and negative mode. The lipid elution was performed with a Zorbax Eclipse plus column (C18, 2.1 mm × 100 mm, particle size 1.8 μm, Agilent Technologies Inc.) for 83 min at 40 °C using a gradient of two different solvents (0.2% formic acid and 10 mM ammonium formate in water and 0.2% formic acid and 5 mM ammonium formate in methanol/acetonitrile/methyl tert-butyl ether [MTBE], 55:35:10 [v/v/v]). Mass spectrometry data analysis was performed with the Mass Hunter Qualitative Analysis software package (version B.07) and lipids of interest were identified using MS/MS analysis or by manual alignment with our previously acquired human database for which the plasma lipid entities have been already validated using MSMS analysis for which spectra were manually interpreted. Student’s t-test followed by Benjamini-Hochberg correction (lymph vs plasma) or unpaired Student’s t-test without correction (lymph EVs vs plasma EVs) were performed with Mass Professional Pro version 12.6.1 (Agilent Technologies Inc.).
Nanoparticle tracking analysis
To determine the size of the produced human PEVs and rbEVs, NTA was performed using a NanoSight LM10 with a 642 nm laser (Malvern Instruments Ltd), using software version 2.3, screen gain 4 and camera level 10. Three, 1 min videos were recorded per sample diluted 1:20 in PBS with analysis screen gain 10 and detection threshold 4. Temperature ranged from 20 to 23.5 °C.
Transmission electron microscopy
Ten (10) µL of 1 × 107 human PEVs or rbEVs were incubated during 2 min onto a 200 mesh cooper grids with formvar/carbon support film (Electron Microscopy Sciences, EMS). Samples on grids were stained twice with 10 µL of freshly prepared 2% uranyl acetate (EMS). The excess liquid was removed by blotting after each step. Images were acquired using a FEI Tecnai T12 transmission electron microscope operated at 80 kV at 52,000x of magnification.
Immunoblotting
EV samples were prepared by diluting 1 × 107 EVs in 2X Laemmli buffer and heating the mixture at 95˚C for 5 min. Samples were then resolved by electrophoresis under reducing conditions on a 10% SDS-PAGE gel and proteins were transferred onto a PVDF membrane. The membrane was blocked with 5% skim milk powder (BioShop cat. #SKI400) in Tris-buffered saline (TBST, 0.1% Tween 20) for 1 h at RT and washed twice with TBST. Then, the membrane was incubated overnight at 4 °C with an anti-flotillin-1 (Abcam cat. #ab133497) and an anti-calnexin antibody (Invitrogen cat. #PA534665). After three washes in TBST, the membrane was incubated for 1 h at RT with a horseradish peroxidase-conjugated anti-rabbit secondary antibody (Elabscience cat. #E-AB-1003) and washed three times with TBST. Chemiluminescence was detected with the EasySee Western Blot Kit (Transgen Biotech) and captured on Western blotting film (Ultident).
Human lymphatic endothelial cell culture
Human dermal microvascular lymphatic endothelial cells isolated from adult donors (HMVEC-dLyAd, Lonza cat. #CA10064-286) were seeded in complete Endothelial Cell Growth Media MV2 (Promocell cat. #10175-256, P5 to P7). Twenty-four hours prior to the treatment, the media was replaced with an EV-depleted complete media. Then, PEVs (1 × 106/mL) or rbEVs (2 × 106/mL) were added in EV-depleted complete media for 24 h. Following treatments, the supernatant was collected and centrifuged for 5 min at 430 g, aliquoted and stored at -80 °C for further analysis. Cells were washed with media, harvested using Tryspin-EDTA (Fisher cat. #MT25052CI) and immediately processed for flow cytometry, functional assays, or immunofluorescence analysis, as described below.
Confocal microscopy
Following a 2- and 24 h treatment with CellVue+ PEVs, cultured human LECs were washed with phenol-free and EV-depleted medium and cytoplasmic membranes were stained with WGA (AF555, Thermo Fisher cat. #W32464) and bisBenzimide H 33342 trihydrochloride (Hoechst, Sigma cat. #14533) for nuclei staining for 7.5 min at 37 °C. Live cells were washed three times with phenol-free and EV-depleted medium before confocal microscopy imaging (Z-stacks) using an LSM 710 Confocal Microscope (Zeiss) equipped with a 63/1.4 oil DIC objective. The quantification of the volume occupied by PEVs per cell on the deconvolved (Theoritical PSF) Z-stacked images was performed using the Huygens Professional software (Scientific Volume Imaging).
Measurement of cell apoptosis and necrosis
Lymphatic endothelial cells were harvested and stained with PI (VWR cat. #89139-066) and annexin V (V450, BD Biosciences cat. #560506) in annexin V binding buffer, centrifuged at 430 g for 5 min at 4 °C and resuspended in FACS buffer. Cells were analyzed with a BD FACSCelesta (BD Biosciences).
Quantification of reactive oxygen species production
LECs were seeded in 96 well-plates, treated with PEVs (1 × 106/mL) or rbEVs (2 × 106/mL) for 24 h and stained with 10 µM CM-H2DCFDA (Invitrogen cat. #C6827) for 30 min at 37 °C as described by Mahmoud et al.97. Upon cleavage by intracellular esterase and oxidation by ROS, the molecule becomes highly fluorescent. Its fluorescence intensity was measured using Synergy 2 plate reader every 1.13 min for 1 h. Superoxide dismutase (100 U/mL) (Alfa Aesar cat. #AAJ630038PL) was used as a negative control, as it protects cells against oxidative stress and blocks ROS production.
RNA sequencing
Following RNA extraction, EV-treated LECs were also analyzed with Illumina next-generation sequencing at the Institute for Research in Immunology and Cancer (IRIC) Genomics Platform at the University of Montreal (Montreal, Quebec, Canada). Adapter sequences and low-quality bases in the resulting FASTQ files were trimmed using Trimmomatic version 0.35 and genome alignments were conducted using STAR version 2.7.1a98,99. The sequences were aligned to the human genome version GRCh38, with gene annotations from Gencode version 37 based on Ensembl 103. As part of quality control, the sequences were aligned to several different genomes to verify that there was no sample contamination. Gene expressions were obtained both as raw readcount directly from STAR as well as computed using RSEM in order to obtain gene and transcript level expression in reads per transcripts per million (TPM) for these stranded RNA libraries100. DESeq2 version 1.22.2 was then used to normalize gene read counts and compute differential expression between the various experimental conditions. Sample clustering based on normalized log read counts produces a hierarchy of samples. A principal component analysis (PCA) is also used to validate that samples correlate as expected101.
qPCR analysis
LECs were harvested after a 24 h treatment and lysed in Trizol. RNA was extracted according to the manufacturer’s protocol (Invitrogen cat. # 12183025) and stored at -80 °C. Reverse transcription of RNA was performed using the High-Capacity cDNA Reverse Transcription Kit (ThermoFisher Scientific, cat. #4368814). Quantitative PCR was performed on the QuantStudio™ 3 (ThermoFisher Scientific) using 10 ng of complementary DNA mixed with Itaq™ Universal SYBR® Green Supermix (Biorad, cat. #1725121). The amplification cycles were performed at 94 °C for 10 s and at 60 °C for 45 s for 40 cycles. The relative expression was calculated by the comparative method of (2-ΔΔCT) and normalized to the housekeeping gene ACTB as previously described102. All samples were then normalized to the control. The primers used are displayed in Table 4.
In vivo injection of platelet extracellular vesicles in mice
Mice were first anesthetized with 2.0% isoflurane and 0.5 L/min O2. Then, a total of 20 µL containing 1 × 107 CellVue+ PEVs (or control PBS) were injected with a 10 µL Hamilton syringe in the dermis on both sides of the right footpad. Mice were sacrificed 15 min, 120 min or 48 h after the injection. Popliteal lymphatic vessels, draining LNs (popliteal and inguinal), non-draining LNs (axillary) and the aorta were harvested.
Popliteal lymphatic vessels were fixed with 4% paraformaldehyde for 24 h. Vessels were incubated overnight in donkey serum (5%, Jackson Immunoresearch cat #017-000-121) and stained with anti-human CD41a antibody (BD Biosciences cat. #555465) for 4 days at 4 °C. Vessels were then washed twice with PBS and incubated with a secondary antibody (Cy3, Jackson Immunoresearch cat. #115-165-003) and DAPI (BioShop cat. #DAP444.5) at RT. Z-Stacks were acquired using an LSM 710 Confocal Microscope (Zeiss) equipped with a 63/1.4 oil dic objective.
Non-draining and draining LNs were digested with collagenase D (2.68 mg/mL, Roche cat. #11088882001) for 25 min at 37 °C. Cells were filtered through a 70 µm cell strainer (Falcon cat. #352350) in Hank’s Balanced Salt Solution (HBSS,Corning cat. #CA-45000-458) and centrifuged at 430 g for 10 min at 4 °C. To identify the proportion of LN cells positive in PEVs, cells were resuspended in HBSS buffer supplemented with 0.5 M EDTA and 30% bovine serum albumin (BSA) and stained for 30 min at 4 °C with anti-mouse CD45 (FITC, Tonbo Biosciences cat. #350451), anti-mouse MHCII (V450, Tonbo Biosciences cat. #755321) and anti-human CD62P (PE, BioLegends cat. #304905) antibodies (Supplementary Fig. 3). To assess the proportion of PEVs that was not contained within LN cells, supernatant collected from LNs following tissue digestion was stained with anti-human CD62P antibody (AF700, Labome cat. #304932) and CFSE for 30 min at 37 °C. All FACS analyses were performed by flow cytometry as described above (BD FACSCelesta, BD Biosciences).
Furthermore, in a final set of mice, non-draining LNs, draining LNs and the aorta were harvested and imaged individually in a fluorescence imaging scanner (IVIS Lumina II, Caliper Life Sciences). To visualize the uptake of CellVue+ PEVs in each tissue, excitation and emission wavelengths were set to 640 nm and 700 nm, respectively. The exposure time was 10 s and no binning was applied.
Measurement of lymphatic vessel contraction
Lymphatic vessel contractions were measured 48 h following the injection of human PEVs or rbEVs, as described previously13. Briefly, an incision was made all around the upper hind limb of anesthetized mice, and the skin was then carefully removed. Warm saline was administered on the paw to keep the limb tissue hydrated throughout the imaging session. Two injections of ovalbumin-alexa fluor 647 (2 mg/mL in 10 µL, Fisher cat. #O34781) were performed in the dermis of each side of the footpad of the mouse103. The paw was then manually contracted three times to allow the fluorescent molecule to move up through lymph and enable the visualization of the collecting lymphatic vessels. After a 10 min stabilization, the contractions were recorded for 10 min with an Axiozoom V.16 microscope (Zeiss). A total of 326 pictures per imaging session were taken with an exposure time of 0.8 s. Images were stabilized using the Template Matching Imagej™ plugin and analyzed with the LymphPulse 3.0 Matlab™ based software. Three to five regions of interest (ROI) were delineated manually along each lymphatic vessel visualized. The resulting curves exhibited peaks and valleys at distinct fluorescent intensities (measured in arbitrary units, AU), to which the background intensity was subtracted. Contraction frequencies were calculated from each curve using a Matlab algorithm according to the formula (Np+Nv)/(2*dt), where Np and Nv represent the number of peaks and valleys, respectively, and dt denotes the analysis time (illustrated in Fig. 6d).
Statistics
Measurements were all taken from distinct samples. All results are expressed as the mean ± SEM, with n representing the number of mice or cell culture experiment. Statistics and graphs were performed using Prism™ 10.0 statistical software. Normality was tested with Shapiro-Wilk test, followed by either parametric (t-tests for only two experimental groups or two-way ANOVA with Tuckey’s post-hoc test for multiple comparisons) or non-parametric (Mann-Whitney for only two experimental groups) analysis. In all analysis, p ≤ 0.05 was required for statistical significance. All attempts at replication were successful.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Datasets generated during and/or analyzed during the current study are available in the supplementary data 1 for the lipidomic data, in the supplementary data 2 for RNA-sequencing as well as deposited on NIH sequence read archive (BioProject ID: PRJNA1133037 and submission code: SUB14590723) and in the supplementary data 3. Supplementary movie 1 is available on FigShare: https://doi.org/10.6084/m9.figshare.26180867.
References
Iqbal, J. & Hussain, M. M. Intestinal lipid absorption. Am. J. Physiol. Endocrinol. Metab. 296, E1183–E1194 (2009).
Miteva, D. O. et al. Transmural flow modulates cell and fluid transport functions of lymphatic endothelium. Circ. Res. 106, 920–931 (2010).
Hoggan, G. & Hoggan, F. E. The lymphatics of the walls of the larger blood-vessels and lymphatics. J. Anat. Physiol. 17, 1–23 (1882).
Lemole, G. M. The role of lymphstasis in atherogenesis. Ann. Thorac. Surg. 31, 290–293 (1981).
Martel, C. et al. Lymphatic vasculature mediates macrophage reverse cholesterol transport in mice. J. Clin. Invest 123, 1571–1579 (2013).
Lim, H. Y. et al. Lymphatic vessels are essential for the removal of cholesterol from peripheral tissues by SR-BI-mediated transport of HDL. Cell Metab. 17, 671–684 (2013).
Houssari, M. et al. Lymphatic and immune cell cross-talk regulates cardiac recovery after experimental myocardial infarction. Arterioscler Thromb. Vasc. Biol. 40, 1722–1737 (2020).
Wada, H. et al. Distinct characteristics of VEGF-D and VEGF-C to predict mortality in patients with suspected or known coronary artery disease. J. Am. Heart Assoc. 9, e015761 (2020).
Wada, H. et al. VEGF-C and mortality in patients with suspected or known coronary artery disease. J. Am. Heart Assoc. 7, e010355 (2018).
Drosos, I. et al. Increased lymphangiogenesis and lymphangiogenic growth factor expression in perivascular adipose tissue of patients with coronary artery disease. J. Clin. Med. 8, 1000 (2019).
Milasan, A. et al. apolipoprotein A-I modulates atherosclerosis through lymphatic vessel-dependent mechanisms in mice. J. Am. Heart Assoc. 6, e006892 (2017)
Milasan, A., Ledoux, J. & Martel, C. Lymphatic network in atherosclerosis: the underestimated path. Future Sci. OA 1, FSO61 (2015).
Milasan, A., Smaani, A. & Martel, C. Early rescue of lymphatic function limits atherosclerosis progression in Ldlr(-/-) mice. Atherosclerosis 283, 106–119 (2019).
Vachon, L. et al. Downregulation of low-density lipoprotein receptor mRNA in lymphatic endothelial cells impairs lymphatic function through changes in intracellular lipids. Theranostics 12, 1440–1458 (2022).
Milasan, A., Dallaire, F., Mayer, G. & Martel, C. Effects of LDL receptor modulation on lymphatic function. Sci. Rep. 6, 27862 (2016).
To, K. H. T. et al. T-type, but not L-type, voltage-gated calcium channels are dispensable for lymphatic pacemaking and spontaneous contractions. Sci. Rep. 10, 70 (2020).
Telinius, N. et al. Human lymphatic vessel contractile activity is inhibited in vitro but not in vivo by the calcium channel blocker nifedipine. J. Physiol. 592, 4697–4714 (2014).
Sestito, L. F. et al. Lymphatic-draining nanoparticles deliver Bay K8644 payload to lymphatic vessels and enhance their pumping function. Sci. Adv. 9, eabq0435 (2023).
Bolger, G. T., Weissman, B. A. & Skolnick, P. The behavioral effects of the calcium agonist Bay K 8644 in the mouse: antagonism by the calcium antagonist nifedipine. Naunyn Schmiedebergs Arch. Pharm. 328, 373–377 (1985).
Osada, M. et al. Platelet activation receptor CLEC-2 regulates blood/lymphatic vessel separation by inhibiting proliferation, migration, and tube formation of lymphatic endothelial cells. J. Biol. Chem. 287, 22241–22252 (2012).
Hess, P. R. et al. Platelets mediate lymphovenous hemostasis to maintain blood-lymphatic separation throughout life. J. Clin. Invest. 124, 273–284 (2014).
Milasan, A., Farhat, M. & Martel, C. Extracellular vesicles as potential prognostic markers of lymphatic dysfunction. Front Physiol. 11, 476 (2020).
Levick, J. R. & Michel, C. C. Microvascular fluid exchange and the revised starling principle. Cardiovasc. Res. 87, 198–210 (2010).
Aspelund, A., Robciuc, M. R., Karaman, S., Makinen, T. & Alitalo, K. Lymphatic system in cardiovascular medicine. Circ. Res. 118, 515–530 (2016).
Milasan, A., Smaani, A. & Martel, C. Early rescue of lymphatic function limits atherosclerosis progression in Ldlr mice. Atherosclerosis 283, 106–119 (2019).
Tessandier, N. et al. Platelets disseminate extracellular vesicles in lymph in rheumatoid arthritis. Arterioscler Thromb. Vasc. Biol. 40, 929–942 (2020).
Milasan, A. et al. Extracellular vesicles are present in mouse lymph and their level differs in atherosclerosis. J. Extracell. Vesicles 5, 31427 (2016).
Couto-Rodriguez, A. et al. Platelet-derived extracellular vesicles as lipid carriers in severe allergic inflammation. Int. J. Mol.Sci. 24, 12714 (2023).
Hamid, M. A., Kunicki, T. J. & Aster, R. H. Lipid composition of freshly prepared and stored platelet concentrates. Blood 55, 124–130 (1980).
van der Veen, J. N. et al. The critical role of phosphatidylcholine and phosphatidylethanolamine metabolism in health and disease. Biochim. Biophys. Acta Biomembr. 1859, 1558–1572 (2017).
Welsh, J. A. et al. Minimal information for studies of extracellular vesicles (MISEV2023): From basic to advanced approaches. J. Extracell. Vesicles 13, e12404 (2024).
Camus, S. M. et al. Circulating cell membrane microparticles transfer heme to endothelial cells and trigger vasoocclusions in sickle cell disease. Blood 125, 3805–3814 (2015).
Pan, Y. et al. Platelet-secreted microRNA-223 promotes endothelial cell apoptosis induced by advanced glycation end products via targeting the insulin-like growth factor 1 receptor. J. Immunol. 192, 437–446 (2014).
Jimenez, J. J. et al. Endothelial cells release phenotypically and quantitatively distinct microparticles in activation and apoptosis. Thromb. Res. 109, 175–180 (2003).
Nozaki, T. et al. Significance of a multiple biomarkers strategy including endothelial dysfunction to improve risk stratification for cardiovascular events in patients at high risk for coronary heart disease. J. Am. Coll. Cardiol. 54, 601–608 (2009).
Freyssinet, J. M. Cellular microparticles: what are they bad or good for? J. Thromb. Haemost. 1, 1655–1662 (2003).
Yuana, Y., Sturk, A. & Nieuwland, R. Extracellular vesicles in physiological and pathological conditions. Blood Rev. 27, 31–39 (2013).
Mackay, C. R., Kimpton, W. G., Brandon, M. R. & Cahill, R. N. Lymphocyte subsets show marked differences in their distribution between blood and the afferent and efferent lymph of peripheral lymph nodes. J. Exp. Med. 167, 1755–1765 (1988).
Italiano, J. E. et al. Microvesicles, but not platelets, bud off from mouse bone marrow megakaryocytes. Blood 138, 1998–2001 (2021).
Flaumenhaft, R. et al. Megakaryocyte-derived microparticles: direct visualization and distinction from platelet-derived microparticles. Blood 113, 1112–1121 (2009).
Fantl, P. & Nelson, J. F. Coagulation in lymph. J. Physiol. 122, 33–37 (1953).
Bourne, J. H. et al. CLEC-2 supports platelet aggregation in mouse but not human blood at arterial sShear. Thromb. Haemost. 122, 1988–2000 (2022).
Casley-Smith, J. R. The identification of chylomicra and lipoproteins in tissue sections and their passage into jejunal lacteals. J. Cell Biol. 15, 259–277 (1962).
Goni, F. M. & Alonso, A. Structure and functional properties of diacylglycerols in membranes. Prog. Lipid Res. 38, 1–48 (1999).
Drzazga, A., Sowinska, A. & Koziolkiewicz, M. Lysophosphatidylcholine and lysophosphatidylinosiol–novel promissing signaling molecules and their possible therapeutic activity. Acta Pol. Pharm. 71, 887–899 (2014).
Dixon, J. B. Lymphatic lipid transport: sewer or subway? Trends Endocrinol. Metab. 21, 480–487 (2010).
Kavaliauskiene, S. et al. Cell density-induced changes in lipid composition and intracellular trafficking. Cell Mol. Life Sci. 71, 1097–1116 (2014).
Zabeo, D. et al. Exosomes purified from a single cell type have diverse morphology. J. Extracell. Vesicles 6, 1329476 (2017).
Muro, E., Atilla-Gokcumen, G. E. & Eggert, U. S. Lipids in cell biology: how can we understand them better? Mol. Biol. Cell 25, 1819–1823 (2014).
Skotland, T., Sagini, K., Sandvig, K. & Llorente, A. An emerging focus on lipids in extracellular vesicles. Adv. Drug Deliv. Rev. 159, 308–321 (2020).
Duhon, B. H., Phan, T. T., Taylor, S. L., Crescenzi, R. L. & Rutkowski, J. M. Current mechanistic understandings of lymphedema and lipedema: tales of fluid, fat, and fibrosis. Int. J. Mol. Sci. 23, 6621 (2022)
Rotunda, A. M. & Kolodney, M. S. Mesotherapy and phosphatidylcholine injections: historical clarification and review. Dermatol. Surg. 32, 465–480 (2006).
Reeds, D. N., Mohammed, B. S., Klein, S., Boswell, C. B. & Young, V. L. Metabolic and structural effects of phosphatidylcholine and deoxycholate injections on subcutaneous fat: a randomized, controlled trial. Aesthet. Surg. J. 33, 400–408 (2013).
Koc, M. et al. Lymphedema alters lipolytic, lipogenic, immune and angiogenic properties of adipose tissue: a hypothesis-generating study in breast cancer survivors. Sci. Rep. 11, 8171 (2021).
Mallat, Z. et al. Shed membrane microparticles with procoagulant potential in human atherosclerotic plaques: a role for apoptosis in plaque thrombogenicity. Circulation 99, 348–353 (1999).
Mackman, N., Tilley, R. E. & Key, N. S. Role of the extrinsic pathway of blood coagulation in hemostasis and thrombosis. Arterioscler Thromb. Vasc. Biol. 27, 1687–1693 (2007).
Marcos-Ramiro, B. et al. Microparticles in multiple sclerosis and clinically isolated syndrome: effect on endothelial barrier function. BMC Neurosci. 15, 110 (2014).
Boulanger, C. M., Loyer, X., Rautou, P. E. & Amabile, N. Extracellular vesicles in coronary artery disease. Nat. Rev. Cardiol. 14, 259–272 (2017).
Edrissi, H., Schock, S. C., Hakim, A. M. & Thompson, C. S. Microparticles generated during chronic cerebral ischemia increase the permeability of microvascular endothelial barriers in vitro. Brain Res. 1634, 83–93 (2016).
Barry, O. P., Praticò, D., Savani, R. C. & FitzGerald, G. A. Modulation of monocyte-endothelial cell interactions by platelet microparticles. J. Clin. Investig. 102, 136–144 (1998).
Weber, A., Koppen, H. O. & Schror, K. Platelet-derived microparticles stimulate coronary artery smooth muscle cell mitogenesis by a PDGF-independent mechanism. Thromb. Res. 98, 461–466 (2000).
Miyazawa, B. et al. Regulation of endothelial cell permeability by platelet-derived extracellular vesicles. J. Trauma Acute Care Surg. 86, 931–942 (2019).
Schoggins, J. W. & Rice, C. M. Interferon-stimulated genes and their antiviral effector functions. Curr. Opin. Virol. 1, 519–525 (2011).
Ghosh, S. & Marsh, E. N. G. Viperin: An ancient radical SAM enzyme finds its place in modern cellular metabolism and innate immunity. J. Biol. Chem. 295, 11513–11528 (2020).
Haller, O., Staeheli, P., Schwemmle, M. & Kochs, G. Mx GTPases: dynamin-like antiviral machines of innate immunity. Trends Microbiol. 23, 154–163 (2015).
Aljohani, A. I. et al. Myxovirus resistance 1 (MX1) is an independent predictor of poor outcome in invasive breast cancer. Breast Cancer Res. Treat. 181, 541–551 (2020).
Staeheli, P. & Haller, O. Human MX2/MxB: a potent interferon-induced postentry inhibitor of herpesviruses and HIV-1. J. Virol. 92, e00709-18(2018).
Eloranta, M. L., Alm, G. V. & Ronnblom, L. Disease mechanisms in rheumatology–tools and pathways: plasmacytoid dendritic cells and their role in autoimmune rheumatic diseases. Arthritis Rheum. 65, 853–863 (2013).
Rajagopalan, S. et al. Endothelial cell apoptosis in systemic lupus erythematosus: a common pathway for abnormal vascular function and thrombosis propensity. Blood 103, 3677–3683 (2004).
Denny, M. F. et al. Interferon-alpha promotes abnormal vasculogenesis in lupus: a potential pathway for premature atherosclerosis. Blood 110, 2907–2915 (2007).
Tyden, H. et al. Endothelial dysfunction is associated with activation of the type I interferon system and platelets in patients with systemic lupus erythematosus. RMD Open 3, e000508 (2017).
Peralta, S. et al. ATAD3 controls mitochondrial cristae structure in mouse muscle, influencing mtDNA replication and cholesterol levels. J. Cell Sci. 131, jcs.217075 (2018).
Doyle, M. J. et al. Sox7 regulates lineage decisions in cardiovascular progenitor cells. Stem Cells Dev. 28, 1089–1103 (2019).
Chiang, I. K. N. et al. The blood vasculature instructs lymphatic patterning in a SOX7-dependent manner. EMBO J. 42, e109032 (2023).
Xu, Y. & Sun, Z. Molecular basis of Klotho: from gene to function in aging. Endocr. Rev. 36, 174–193 (2015).
Owen, B. M., Mangelsdorf, D. J. & Kliewer, S. A. Tissue-specific actions of the metabolic hormones FGF15/19 and FGF21. Trends Endocrinol. Metab. 26, 22–29 (2015).
Ogawa, Y. et al. BetaKlotho is required for metabolic activity of fibroblast growth factor 21. Proc. Natl Acad. Sci. USA 104, 7432–7437 (2007).
Singhal, G. et al. Fibroblast growth factor 21 (FGF21) protects against high fat diet induced inflammation and islet hyperplasia in pancreas. PLoS One 11, e0148252 (2016).
Lin, Z. et al. Fibroblast growth factor 21 prevents atherosclerosis by suppression of hepatic sterol regulatory element-binding protein-2 and induction of adiponectin in mice. Circulation 131, 1861–1871 (2015).
Ferreira Tojais, N. et al. Frizzled7 controls vascular permeability through the Wnt-canonical pathway and cross-talk with endothelial cell junction complexes. Cardiovasc Res. 103, 291–303 (2014).
Peghaire, C. et al. Fzd7 (Frizzled-7) expressed by endothelial cells controls blood vessel formation through Wnt/beta-catenin canonical signaling. Arterioscler Thromb. Vasc. Biol. 36, 2369–2380 (2016).
Hagerling, R. et al. Distinct roles of VE-cadherin for development and maintenance of specific lymph vessel beds. EMBO J. 37, e98271 (2018)
Yang, Y., Cha, B., Motawe, Z. Y., Srinivasan, R. S. & Scallan, J. P. VE-cadherin is required for lymphatic valve formation and maintenance. Cell Rep. 28, 2397–2412.e2394 (2019).
Huang, H., Cruz, F. & Bazzoni, G. Junctional adhesion molecule-A regulates cell migration and resistance to shear stress. J. Cell Physiol. 209, 122–130 (2006).
Kakogiannos, N. et al. JAM-A acts via C/EBP-alpha to promote claudin-5 expression and enhance endothelial barrier function. Circ. Res. 127, 1056–1073 (2020).
Ostermann, G., Weber, K. S., Zernecke, A., Schroder, A. & Weber, C. JAM-1 is a ligand of the beta(2) integrin LFA-1 involved in transendothelial migration of leukocytes. Nat. Immunol. 3, 151–158 (2002).
Babinska, A. et al. F11-receptor (F11R/JAM) mediates platelet adhesion to endothelial cells: role in inflammatory thrombosis. Thromb. Haemost. 88, 843–850 (2002).
Srinivasan, S., Vannberg, F. O. & Dixon, J. B. Lymphatic transport of exosomes as a rapid route of information dissemination to the lymph node. Sci. Rep. 6, 24436 (2016).
Breslin, J. W. et al. Lymphatic vessel network structure and physiology. Compr. Physiol. 9, 207–299 (2018).
Wartiovaara, U. et al. Peripheral blood platelets express VEGF-C and VEGF which are released during platelet activation. Thromb. Haemost. 80, 171–175 (1998).
Sunderland, N. et al. MicroRNA biomarkers and platelet reactivity: the clot thickens. Circ. Res. 120, 418–435 (2017).
Kukk, E. et al. VEGF-C receptor binding and pattern of expression with VEGFR-3 suggests a role in lymphatic vascular development. Development 122, 3829–3837 (1996).
Rousseau, M. et al. Detection and quantification of microparticles from different cellular lineages using flow cytometry. Evaluation of the impact of secreted phospholipase A2 on microparticle assessment. PLoS One 10, e0116812 (2015).
Poncelet, P. et al. Tips and tricks for flow cytometry-based analysis and counting of microparticles. Transfus. Apher. Sci. 53, 110–126 (2015).
Poncelet, P. et al. Standardized counting of circulating platelet microparticles using currently available flow cytometers and scatter-based triggering: forward or side scatter? Cytom. A 89, 148–158 (2016).
Forest, A. et al. Comprehensive and reproducible untargeted lipidomic workflow using LC-QTOF validated for human plasma analysis. J. Proteome Res. 17, 3657–3670 (2018).
Mahmoud, A. M. et al. Endothelial microparticles prevent lipid-induced endothelial damage via Akt/eNOS signaling and reduced oxidative stress. FASEB J. 31, 4636–4648 (2017).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2012).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Li, B. & Dewey, C. N. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinforma. 12, 323 (2011).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Amri, N. et al. Use of early donated COVID-19 convalescent plasma is optimal to preserve the integrity of lymphatic endothelial cells. Pharmaceuticals (Basel) 15, 365 (2022).
Bouta, E. M. et al. Lymphatic function measurements influenced by contrast agent volume and body position. JCI Insight 3, e96591 (2018)
Acknowledgements
The authors acknowledge the Institute for Research in Immunology and Cancer (IRIC) genomics core facility for RNA sequencing as well as Patrick Gendron at the IRIC bio-informatics core facility for sequencing analysis. The authors would also like to thank Carl Fortin for his technical support with flow cytometry analysis, Caroline Daneault and Isabelle Robillard Frayne from the MHI Metabolomics core facility for performing the untargeted lipidomic analysis, and Julianna Blagih for critical reading of the article. This project was supported by research grants from the Canadian Institutes for Health Research (PJT-148627; PRR-177917), the Natural Sciences and Engineering Research Council of Canada (RGPIN-2016-05331) and the Heart and Stroke Foundation of Canada (G-23-0034933) to CM.
Author information
Authors and Affiliations
Contributions
C.M., G.J. and A.M. conceived and designed the research. L.V., G.J., A.M., E.L., D.G.B., T.M.A., M.R. and N.T. performed experiments. C.M., L.V., G.J., M.R. and N.T. analyzed data. C.M., M.R., A.N., J.R. and E.B. interpreted the results. L.V., S.B., D.G.B., G.J., M.R. and N.T. prepared figures. L.V., G.J. and C.M. drafted the paper. V.F. developed the LymphPulse 3.0 software. L.V., G.J., A.M., L.V., J.R., S.B., A.N., J.C.T., M.R., E.B., N.T. and C.M. edited and revised the paper. L.V., G.J., A.M., E.L., D.G.B., L.V., J.R., A.N., S.B., T.M.A., J.C.T., V.F., M.R., E.B., N.T. and C.M. approved the final version of the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks Sathish Srinivasan and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editors: Ngan Huang and Dario Ummarino
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Vachon, L., Jean, G., Milasan, A. et al. Platelet extracellular vesicles preserve lymphatic endothelial cell integrity and enhance lymphatic vessel function. Commun Biol 7, 975 (2024). https://doi.org/10.1038/s42003-024-06675-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s42003-024-06675-8
This article is cited by
-
Exosomes derived from platelet-rich plasma alleviate synovial inflammation by enhancing synovial lymphatic function
Journal of Nanobiotechnology (2025)
-
Platelets as drivers of immunothrombosis in rheumatic diseases
Nature Reviews Rheumatology (2025)
