
Abstract
The kidney is vulnerable to ischemia and reperfusion (I/R) injury that can be fatal after major surgery. Currently, there are no effective treatments for I/R-induced kidney injury. Trimethylamine N-oxide (TMAO) is a gut-derived metabolite linked to many diseases, but its role in I/R-induced kidney injury remains unclear. Here, our clinical data reveals an association between preoperative systemic TMAO levels and postoperative kidney injury in patients after post-cardiopulmonary bypass surgery. By genetic deletion of TMAO-producing enzyme flavin-containing monooxygenase 3 (FMO3) and dietary supplementation of choline to modulate TMAO levels, we found that TMAO aggravated acute kidney injury through the triggering of endoplasmic reticulum (ER) stress and worsened subsequent renal fibrosis through TGFβ/Smad signaling activation. Together, our study underscores the negative role of TMAO in I/R-induced kidney injury and highlights the therapeutic potential through the modulation of TMAO levels by targeting FMO3, thereby mitigating acute kidney injury and preventing subsequent renal fibrosis.
Similar content being viewed by others
Introduction
Approximately 10–15% of hospitalized adults and more than 50% of patients in intensive care units experience acute kidney injury (AKI)1,2,3. This injury is commonly prompted by ischemia and reperfusion (I/R) injury, and is still without effective therapies4,5. Moreover, AKI is a major risk factor for progressive renal insufficiency. The severity and number of AKI episodes can predict progressive and long-term renal dysfunction6,7. Additionally, there is a considerable reduction in the quality of life experienced by patients suffering from AKI and subsequent renal dysfunction. Increased metabolic by-products of intestinal bacteria have contributed to the increased burden on these patients8.
Trimethylamine N-oxide (TMAO) is a metabolic byproduct generated by the oxidation of trimethylamine (TMA) in the gut microbiota9. It is produced from precursors such as choline, betaine, L-carnitine, ergothioneine, and gamma-butyrobetaine obtained from dairy products10. In the conversion of TMA into TMAO, flavin-containing monooxygenase 3 (FMO3) plays an important role11. There is significant variability in the expression of FMO3 in the adult liver among individuals12, which potentially exerts a substantial influence on the production of TMAO. Studies have reported that serum TMAO concentration in patients with end-stage renal disease is 30- to 100-fold higher than that of people with normal renal function13,14,15. However, there is lacking research on the role of TMAO in the context of I/R-induced kidney injury.
In this study, we evaluated the association of circulating TMAO levels and the incidence of AKI in postoperative patients, and then investigated the effects and potential mechanisms of TMAO on I/R-induced kidney injury by genetic deletion of the TMAO-producing enzyme FMO3 and the supplementation of choline in the diet to elevate TMAO levels. Our data revealed that elevated TMAO levels aggravated AKI by activating endoplasmic reticulum (ER) stress and exacerbated subsequent renal fibrosis by upregulating the TGFβ/Smad pathway. These findings demonstrate the detrimental impact of TMAO on kidney injury induced by I/R. Modulating TMAO levels by targeting FMO3 may offer a therapeutic approach to mitigate AKI and prevent subsequent renal fibrosis.
Results
Systemic levels of TMAO are associated with AKI after CPB
Cardiopulmonary bypass (CPB) may cause hemodynamic changes, imbalance of oxygen supply and demand, inflammation, and oxidative stress, all of which may lead to postoperative AKI16,17,18. To investigate the association between circulating TMAO levels and the incidence of AKI, we recruited 9 AKI patients and 18 non-AKI controls after cardiac surgery under CPB. The 18 controls matched according to age and gender to the AKI group. Clinical, demographic, and laboratory characteristics for AKI and non-AKI patients were summarized in Table 1. All patients survived the surgery, and none died in hospital or within 30 days afterward. There are no significant differences in these clinical data between the two groups tested by the Mann–Whitney U-test or Student’s t-test. Compared with non-AKI patients, preoperative TMAO levels were significantly higher in AKI patients (Fig. 1a). By Pearson correlation analysis, we found that higher serum TMAO level is correlated to increased serum creatine (Fig.1b), increased creatinine clearance (Fig. 1c) and estimated glomerular filtration rate (Fig. 1d). Collectively, these data indicated that serum TMAO is associated with postoperative kidney injury.

a Serum TMAO levels of AKI and non-AKI patients at baseline (before the surgery). Data are presented as mean ± SEM. ***P < 0.001 (Student’s t-test). non-AKI group, n = 18; AKI group, n = 9. b Pearson correlation analysis of serum TMAO level and increased serum creatinine on Day 1 after surgery, r = 0.6521, P = 0.0002. c Pearson correlation analysis of serum TMAO level and decreased creatinine clearance after surgery, r = 0.4609, P = 0.0155. d Pearson correlation analysis of serum TMAO level and decreased estimated glomerular filtration rate after surgery, r = 0.4571, P = 0.0165. △ Creatinine, increased serum creatinine after surgery; △ Ccr, decreased creatinine clearance; △ eGFR, decreased estimated glomerular filtration rate after surgery.
FMO3 knockout confers protection against I/R-induced AKI in mice
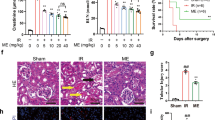
Since FMO3 is an important enzyme that converts TMA to TMAO19, we introduced a stop codon at residue 98 of FMO3 using CRISPR/Cas9 technology to produce Fmo3−/− mice for investigation. We first confirmed Fmo3 deficiency in kidney tissues by RT-qPCR. Compared with the WT mice, Fmo3 at mRNA level in Fmo3−/− mice was significantly lower (Fig. 2a). Similarly, FMO3 protein levels were significantly lower in kidney and liver tissues of Fmo3−/− mice (Fig. 2b). We further verified that Fmo3 deficiency inhibited the oxidation of TMA by measuring the baseline serum levels of TMAO in WT and Fmo3−/− mice. Compared with the control group, TMAO levels at baseline were lower in Fmo3−/− mice (Fig. 2c). Since I/R injury could partially mimic kidney injury after CPB20, we performed renal I/R surgeries on 52 control (n = 32) and Fmo3−/− mice (n = 20) (Fig. 2d). All Fmo3−/− mice survived after I/R, whereas only 15 (46.9%) mice in the control group survived for eight days (Fig. 2e). Taken together, these data indicate that higher TMAO levels are associated with poorer 7-day survival.

a In WT and Fmo3−/− mice, the mRNA levels of Fmo3 in kidney tissues were analyzed by RT-qPCR, n = 10 per group. Data are presented as mean ± SEM. ***P < 0.001 (Student’s t-test). b Representative immunoblotting images showing protein levels of FMO3 in WT and Fmo3−/− mice kidneys (upper panel) and livers (lower panel). c Serum TMAO levels of WT and Fmo3−/− mice tested before I/R, n = 6 per group. Data are presented as mean ± SEM. ***P < 0.001 (Student’s t-test). d Schematics of the experiment. Renal I/R surgery including the removal of the contralateral kidney was performed in WT (C57BL/6 N) and Fmo3−/− mice. Serum samples were collected at 24 h, 48 h, and 72 h after I/R. Renal tissue samples were collected at 48 h after I/R for histology of AKI and 4 weeks after I/R for observation of chronic injury. The image was created with figdraw.com and released under a license: PPTTY74a57. e Kaplan–Meier curve of WT and Fmo3−/− mice after renal I/R. All the Fmo3−/− mice survived one month after I/R surgery, whereas only 15 (46.9%) mice in the WT group survived 8 days. WT group, n = 32; Fmo3−/− group, n = 20, P < 0.001 (Log-rank (Mantel–Cox) test). f Renal function (serum creatinine and BUN) of WT and Fmo3−/− mice was measured at 24 h, 48 h, and 72 h after I/R, n = 4–8 per group. Data are represented as mean ± SEM. Fmo3−/− I/R vs WT I/R, **P < 0.01; ***P < 0.001 (Student’s t-test). g Relative mRNA level of KIM-1 (Havcr1) at 48 h post-I/R, n = 5 per group. Data are presented as mean ± SEM. **P < 0.01; ***P < 0.001 (one-way ANOVA with post hoc Tukey test). h Representative histopathological images of Periodic acid-Schiff (PAS) staining of kidneys from mice sacrificed at 48 h post-reperfusion and from sham-operated animals. Black arrows indicate the disappeared brush edge of renal tubules, tubular epithelial cell necrosis, and detachment from the basement membranes in WT mice after I/R injury. Red arrows indicate swollen tubules and renal casts. Scale bar, 50 μm. i Renal tubular injury score evaluated at 48 h post-I/R in mice. The tubular injury was defined as tubular dilation, tubular atrophy, tubular atrophy, tubular cast formation, sloughing of tubular epithelial cells, or loss of the brush border. Tubular injury scores (0–5) were valued and quantified according to histopathological images of kidneys from four groups, n = 5 per group. Data are presented as mean ± SEM. **P < 0.01; ***P < 0.001 (one-way ANOVA with post hoc Tukey test). j Evaluation of apoptotic nephrocytes in kidneys stained with TUNEL staining (red) at 48 h. Scale bar, 100 μm. k Quantification of the average number of apoptotic cells per field at 48 h after I/R, n = 5 per group. ***P < 0.001 (one-way ANOVA with post hoc Tukey test). WT wild type, I/R ischemia-reperfusion injury, BUN blood urea nitrogen, TUNEL TdT-mediated dUTP-biotin nick end labeling.
To determine whether the silencing of Fmo3 protects renal function after I/R, we assessed kidney function 24 h, 48 h, and 72 h after I/R in WT and Fmo3−/− mice. WT mice developed kidney dysfunction, as reflected by elevated serum creatinine and blood urea nitrogen (BUN) concentrations. In contrast, creatinine and BUN levels were significantly lower in Fmo3−/− mice than in WT mice after I/R (Fig. 2f). In addition, the serum creatinine and BUN levels increased significantly at 24 h after I/R in WT mice, and remained high at 48 h, while the serum creatinine and BUN levels in Fmo3−/− group returned to normal earlier, suggesting that Fmo3 knockout protects renal function after I/R.
To investigate the acute stage of kidney injury after I/R, a series of animals were harvested 48 h after I/R. Kidney injury molecule 1 (KIM-1, also as Havcr1, an established biomarker for renal proximal tubule injury21,22) mRNA levels were lower in Fmo3−/− mice 48 h after I/R compared to that of the control group (Fig. 2g). Histological examination of renal injury was performed in the kidneys of WT and Fmo3−/− mice subjected to I/R. Consistent with the functional deterioration, kidneys of WT mice subjected to I/R showed remarkable tubular injury, as reflected by tubular dilation, detachment of necrotic epithelial cells from basement membranes, and filling of tubule lumens by casts comprising necrotic cell debris by Periodic Acid-Schiff (PAS) stain. In contrast, the kidneys of Fmo3−/− mice subjected to I/R showed considerably less injury than those of WT mice (Fig. 2h). Likewise, tubular injury scores were calculated at 48 h after I/R and Fmo3−/− mice had a significantly lower injury score (Fig. 2i).
To evaluate the effect of FMO3 deficiency on renal cell death in I/R-injured kidneys, we performed the triphosphate nick-end labeling (TUNEL) assay and observed a significant increase in the number of TUNEL-positive cells in the kidneys of I/R-injured mice compared to that in sham-treated mice. Moreover, Fmo3 knockout significantly reduced the number of TUNEL-positive cells in I/R-injured kidneys (Fig. 2j, k). Taken together, these data indicate that FMO3 deficiency reduces apoptosis and protects renal function from renal I/R injury.
FMO3 knockout represses ER stress at the acute phase after I/R
To gain insights into the mechanism of Fmo3 deficiency-induced renal protection against I/R injury, we collected the kidney tissues from the WT I/R group and Fmo3−/− I/R group and performed RNA-seq analysis (Fig. 3a–e). Compared to the WT I/R group, 642 upregulated genes and 629 downregulated genes were identified in Fmo3−/− I/R group (Fig. 3b), suggesting transcriptional changes induced by Fmo3 deficiency. Further, we performed gene set enrichment analysis (GSEA) to investigate the mechanism underlying the renal protective effects by Fmo3 knockout and showed that the upregulated genes in Fmo3−/− I/R group vs WT I/R group were primarily enriched in ER-related pathways such as ER unfolded protein response (Fig. 3e), along with metabolism-related pathways including ATP biosynthetic process, electron transport chain, and oxidative phosphorylation (Fig. S1).

a Principal component analysis (PCA) using all the identified genes from RNA sequencing data for WT (pink) and Fmo3−/− (blue) after I/R reveals in-group clusters with minimal overlap. b Volcano plots illustrating differentially expressed genes identified in Fmo3−/− vs WT groups. The blue dots denote downregulated genes, the red dots denote upregulated genes and the gray dots denote genes without significantly differential expression. Compared to the WT I/R group, 642 upregulated genes and 629 downregulated genes were identified in the Fmo3−/− I/R group. c Heatmap of significantly differential genes identified in kidney tissue between WT and Fmo3−/− groups. d The top ten enriched gene ontology biological process (GO-BP) terms of genes that exhibited an upregulation in the Fmo3−/− group (pink) and genes that exhibited a downregulation in the Fmo3−/− group (green) are presented below. e GSEA showing enrichment of gene signatures associated with response to ER stress between Fmo3−/− and WT group. The normalized enrichment scores (NES) and P values are shown in the plot. NES normalized enrichment score.
Accordingly, we performed qPCR analysis to detect the changes in the mRNA levels of ER stress-related genes in WT and Fmo3−/− kidneys after sham surgery or I/R injury. We found that Atf4, Ddit3, Xbp1, and Ern1 were highly expressed in WT mice after I/R surgery compared with WT Sham group, while this upregulation was significantly suppressed in the Fmo3−/− group after I/R (Fig. 4a). Similarly, immunoblotting analysis revealed that the protein levels of p-PERK/PERK, p-eIF2α/eIF2α, ATF4, XBP1s, and CHOP significantly increased in the WT kidneys after I/R when compared to the Sham group, which were also suppressed in the Fmo3−/− group after I/R (Fig. 4b). By comparison, ATF6 protein level were not significantly altered in all the groups detected. Taken together, these data indicate that Fmo3 knockout leads to repressed ER stress at the acute phase after I/R.

a RT-qPCR results showing mRNA expression of ER-Stress related genes Atf4, Ddit3, Xbp1, Ern1 detected in WT and Fmo3−/− kidney tissues 48 h after I/R. All normalized to Gapdh, n = 5 per group. *P < 0.05; **P < 0.01; ***P < 0.001 (one-way ANOVA with post hoc Tukey test). b Representative immunoblotting images showing protein levels of p-PERK, PERK, p-eIF2α, eIF2α, ATF4, CHOP, XBP1s, ATF6, and HSP90 in WT Sham, Fmo3−/− Sham, WT I/R and Fmo3−/− I/R kidney tissues (left panel). Quantification of protein levels of p-PERK and p-eIF2α are normalized by PERK and eIF2α, respectively (right panel). Quantification of protein levels of ATF4, CHOP, XBP1s, and ATF6 are normalized by HSP90 (right panel), n = 3 per group. ns not significant; *P < 0.05; **P < 0.01 (one-way ANOVA with post hoc Tukey test).
FMO3 knockout suppresses the activation of TGFβ/Smad signaling and reduces renal fibrosis after I/R
The response of renal tubular cells following AKI is initially adaptive but tends towards subsequent renal fibrosis with time23,24,25. Accordingly, we investigated whether Fmo3 deficiency affects I/R-induced renal fibrosis. We first assessed kidney function in WT and Fmo3−/− mice at 4wk after I/R. Consistent with observations during the acute stage, creatinine and BUN levels in Fmo3−/− mice were significantly lower than those in WT mice following I/R at the chronic stage (Fig. 5a, b). Serum levels of TMAO were measured in WT and Fmo3−/− mice. At 4 weeks, TMAO levels remained lower in Fmo3−/− mice compared to the WT group (Fig. 5c). Furthermore, chronic renal atrophy was observed in the WT group represented by a significantly lower ratio of kidney weight to body weight (KW/BW) (Fig. 5d) when compared with the Sham groups. While Fmo3−/− mice had a relatively normal KW/BW ratio compared with WT I/R group (Fig. 5d). In addition, substantial renal injury and fibrosis were observed in WT mice at 4 weeks after I/R, as revealed by marked glomerular atrophy and extensive renal interstitial fibrosis by HE staining and Masson’s Trichrome staining, which was ameliorated in Fmo3−/− mice after I/R (Fig. 5e, f).

a, b Renal function (serum creatinine and BUN) of WT and Fmo3−/− mice was measured at 4wk after I/R, n = 6 per group. Data are presented as mean ± SEM, **P < 0.01; ***P < 0.001 (Student’s t-test). c Serum TMAO levels of WT and Fmo3−/− mice were measured at 4 week after I/R, n = 6 per group. Data are presented as mean ± SEM. ***P < 0.001 (Student’s t-test). d Ratio of kidney weight to body weight (KW/BW) in WT and Fmo3−/− mice. Data are shown as the mean ± SEMs, n = 6 per group. ***P < 0.001 (Student’s t-test). e Representative histopathological images of HE-stained (upper panel) and Masson’s trichrome-stained (lower panel) sections of kidneys from mice sacrificed at 4 weeks post-I/R and from sham-operated animals. The scale bar represents 100 μm. f Quantification of fibrotic area ratio as the percentage of area, n = 6 per group. Data are presented as mean ± SEM. ***P < 0.001 (one-way ANOVA with post hoc Tukey test). g The relative mRNA levels of fibrosis-related gene including Tgfb1, Tgfbr1, Tgfbr2, Acta2 in Fmo3−/− and WT mice, n = 5 per group. Data are presented as mean ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001 (one-way ANOVA with post-Dunnett’s test). h Representative immunoblotting images showing protein levels of TGF-β1, TGFBR1, TGFBR2, phosphorylated to total SMAD2/3, α-SMA and HSP90 detected in WT Sham, Fmo3−/− Sham, WT I/R and Fmo3−/− I/R kidney tissue 4 weeks after I/R (left panel). Quantification of protein levels of TGF-β1, TGFBR1, TGFBR2, SMAD2/3, and α-SMA were normalized by HSP90, and phosphorylated SMAD2/3 were normalized by total SMAD2/3 (right panel), n = 3 per group. Data are presented as mean ± SEM. ns not significant; *P < 0.05; **P < 0.01; ***P < 0.001 (one-way ANOVA with post hoc Tukey test).
Previous studies have suggested that the activation of TGFβ/Smad pathway is implicated in renal fibrosis26. Accordingly, we evaluated whether the TGFβ/Smad pathway was differentially regulated in WT and Fmo3−/− mice at 4 wk after I/R. We showed that several genes related to TGFβ/Smad signaling pathway, such as Tgfb1, Tgfbr1, Tgfbr2, and Acta2, were significantly upregulated in WT kidneys when compared to the sham groups by qPCR analysis, which was significantly suppressed in the Fmo3−/− group at 4 wk after I/R (Fig. 5g). Similarly, the immunoblotting analysis revealed that the protein levels of TGF-β1, TGFBR1, TGFBR2, phosphorylated to total SMAD2/3, and α-SMA significantly increased in WT kidneys at 4 weeks after I/R when compared to the sham groups, which were also suppressed in Fmo3−/− mice (Fig. 5h), suggesting that FMO3 deficiency suppresses the activation of TGFβ/Smad signaling at the chronic phase after renal I/R. To investigate whether the activation of TGFβ/Smad signaling may be associated with ER stress, we examined the mRNA levels of ER stress-related genes including Atf4, Ern1, Xbp1, and Atf6 in kidney tissues at 4 weeks after I/R. We found that, in contrast to the aforementioned significant downregulation by FMO3 deficiency at the acute phase after I/R, they were no longer downregulated in Fmo3−/− mice at 4 wk after I/R (Fig. S2a). Taken together, these data suggest that FMO3 deficiency suppresses the activation of TGFβ/Smad signaling and reduces renal fibrosis after I/R.
Increased TMAO concentration aggravates acute and chronic renal injury after I/R
We next examined whether the effects of FMO3 on I/R-induced acute and chronic kidney injuries are mediated via TMAO. WT mice were fed a 1% choline diet to increase the serum levels of TMAO before subjected to kidney I/R surgery (Fig. 6a). Compared with mice fed standard chow diet, serum TMAO level was significantly higher in mice with choline diet (Fig. 6b). We performed renal I/R surgeries on these mice to assess the effect of increased circulating TMAO levels on kidney function and injury at the acute and chronic phases after I/R.

a Schematics of the experiment. Renal I/R surgery including the removal of the contralateral kidney was performed in mice after 2 weeks of chow or 1% choline feeding. Kidney samples and serum were collected at 48 h after I/R for observation of AKI. 1% choline was needed to increase the levels of TMAO in mice. Mice fed a chow diet were set as control. The image was created with figdraw.com and released under a license: PPTTY74a57. b Serum TMAO levels were measured after 1% choline feeding, n = 8 per group. Data are presented as mean ± SEM. **P < 0.01 (Student’s t-test). c, d Renal function (serum creatinine and BUN) of mice was measured at 48 h after I/R, n = 4 per group. Data are presented as mean ± SEM. *P < 0.05 (Student’s t-test). e Representative histopathological images of HE staining of kidneys from mice sacrificed at 48 h post-I/R (left panel). Red arrows indicate swollen tubules and renal casts. Scale bar, 100 μm. Renal tubular injury score evaluated at 48 h post-I/R in mice (right panel), n = 4 per group. Data are presented as mean ± SEM. *P < 0.05(Student’s t-test). f Evaluation of apoptotic nephrocytes in kidneys stained with TUNEL staining (red) at 48 h. Scale bar, 100 μm, n = 4 per group. Data are presented as mean ± SEM. *P < 0.05 (Student’s t-test). g Representative immunoblotting images showing protein levels of p-PERK, PERK, p-eIF2α, eIF2α, ATF4, CHOP, and HSP90 in chow I/R and choline I/R kidney tissues (left panel). Quantification of protein levels of p-PERK and p-eIF2α are normalized by PERK and eIF-2α, respectively (right panel). Quantification of protein levels of ATF4, CHOP, and XBP1s is normalized by HSP90 (right panel), n = 4 per group. Data are presented as mean ± SEM. *P < 0.05, **P < 0.01 (Student’s t-test).
After 48 h of I/R, the mice fed a choline diet demonstrated worse kidney function compared to the standard chow group, as evidenced by significantly elevated serum creatinine (Fig. 6c) and BUN (Fig. 6d) concentrations. Consistent with functional deterioration, histological analysis demonstrated a more significant tubular injury in the kidneys of mice subjected to a choline diet compared to those maintained on a standard chow diet after I/R. This was evidenced by a greater extent of necrotic epithelial cell detachment from the basement membranes and filling of tubule lumens by casts comprising necrotic cell debris along with a significantly higher injury score (Fig. 6e). To evaluate the effect of higher TMAO levels on renal cell death in I/R-injured kidneys, we performed TUNEL assay and observed a significant increase in the number of TUNEL-positive cells in the I/R-injured kidneys of mice fed choline diet as compared to those fed standard chow diet (Fig. 6f).
Molecular examination of renal injury at the acute phase after I/R by qPCR analysis revealed that the mRNA level of Kim-1 was significantly higher in mice fed a choline diet after I/R than in mice fed a standard chow diet (Fig. S2b). In addition, ER stress-related genes including Atf4, Ddit3, and Xbp1 were significantly upregulated in mice fed a choline diet at 48 h after I/R when compared to those fed a standard chow diet after I/R, whereas the mRNA levels of Atf6 were comparable between the two groups (Fig. S2c). Further, immunoblotting analysis revealed that the levels of p-PERK/PERK, p-eIF2α/eIF2α, ATF4, and CHOP significantly increased in the kidneys of mice fed choline diet at 48 h after I/R as compared to those fed standard chow diet (Fig. 6g). Taken together, these data suggest that TMAO aggravates ER stress and AKI after renal I/R.
Lastly, we investigated whether TMAO may aggravate renal injury at the chronic phase after renal I/R. Masson’s trichrome staining revealed that there were significantly more fibrotic areas in the kidneys of mice fed a choline diet when compared to thosefed standard chow diet at 4 wk after I/R (Fig. 7a). Likewise, immunoblotting analysis revealed that the protein levels of TGF-β1, TGFBR1, TGFBR2, phosphorylated to total SMAD2/3, and α-SMA significantly increased areas in the kidneys of mice fed choline diet when compared to those with standard chow diet at 4 wk after I/R (Fig. 7b). Taken together, these data suggest that TMAO activates TGF-β/SMAD signaling and aggravates renal fibrosis after renal I/R.

a Representative histopathological images of Masson’s trichrome-stained (left panel) sections of kidneys from mice sacrificed at 4 weeks post-I/R. The scale bar represents 100 μm. Quantification of the fibrotic area ratio is shown in the right panel, n = 4 per group. Data are presented as mean ± SEM. ***P < 0.001. b Representative immunoblotting images showing protein levels of TGF-β1, TGFBR1, TGFBR2, phosphorylated SMAD2/3, total SMAD2/3, α-SMA, and HSP90 detected in chow Sham, chow I/R, choline Sham and choline I/R kidney tissues 4 weeks after I/R (upper panel). Quantification of protein levels of TGF-β1, TGFBR1, TGFBR2, SMAD2/3, and α-SMA were normalized by HSP90, and phosphorylated SMAD2/3 was normalized by total SMAD2/3 (lower panel), n = 4 per group. Data are presented as mean ± SEM. ns not significant; *P < 0.05; **P < 0.01; ***P < 0.001 (one-way ANOVA with post hoc Tukey test).
Discussion
Accumulating evidence indicates that I/R is a prevalent etiology for AKI and the subsequent development of chronic kidney disease. Previous studies showed that elevated choline and TMAO levels were associated with an increased risk of developing chronic kidney diseases27 and tubulointerstitial renal fibrosis in mice28. However, the contribution of TMAO to I/R-induced kidney injury is largely unstudied. Since major surgery (especially under CPB) is the leading cause of I/R-induced kidney injury, we first investigated patients after CPB and found an association between higher baseline serum TMAO and worse renal function after surgery. Since FMO is an important enzyme for the synthesis of TMAO, we then constructed Fmo3−/− mice to further investigate if deficiency of FMO3 could protect renal function after I/R. We found that Fmo3−/− mice had repressed ER stress and preserved renal function at the acute phase after I/R. In addition, we found that deficiency of FMO3 suppressed the activation of TGFβ/Smad signaling and reduced subsequent renal fibrosis after I/R. Lastly, we showed that the effects of FMO3 on I/R-induced acute and chronic kidney injuries were at least in part mediated via TMAO in mice with increased TMAO circulating levels from a 1% choline diet. Taken together, our findings uncover a bidirectional relationship between circulating TMAO levels and kidney functions and injuries after I/R, thus pointing to therapeutic strategies to reduce TMAO levels to protect kidney function and alleviate kidney injury at both acute and chronic stages after I/R.
Previous studies indicate that TMAO activates NF-κB signaling to impact inflammation pathways, with G-protein coupled receptor (GPCR) playing a crucial role in its pro-inflammatory effects on endothelial cells29. TMAO has also been reported to have the potential to trigger ER stress and increase inflammation in several disease models, such as diabetes30, aortic valve fibrosis31,32, and Alzheimer’s disease33,34. FMO3 is found as a transmembrane protein in the ER of different tissues, including the kidneys35. Previous studies have shown that kidney injury caused by ischemia can lead to ER stress, which can cause apoptosis and contribute to kidney diseases36. In our I/R-induced AKI models, we found that lowering circulating TMAO levels by Fmo3 knockout could alleviate I/R-induced renal injury along with suppressed ER stress at the acute stage after I/R, thus pointing to a potential new gene therapy option for treating AKI.
It has been revealed that the initial response of renal tubular cells following AKI is adaptive. However, over time, it tends to progress towards the development of chronic renal fibrosis23,24,25. Given that previous studies have shown that TMAO accelerates fibroblast-myofibroblast differentiation and contributes to cardiac fibrosis37,38, here we investigated whether lowering circulating TMAO levels by Fmo3 knockout could alleviate I/R-induced renal fibrosis at the chronic stage. Consistent with a previous report39 showing the activation of TGFβ/Smad signaling in the development of renal fibrosis after AKI, we found that FMO3 deficiency suppresses the activation of TGFβ/Smad signaling and reduces renal fibrosis after I/R, shedding light on the therapeutic intervention of renal fibrosis after I/R.
Given the complexity of the process leading to I/R-induced renal fibrosis and the observed downregulation of TGFβ/Smad signaling in the Fmo3−/− group40, further research should focus on identifying the specific molecule that interacts with TMAO to activate TGFβ/Smad signaling. Recent findings have suggested that TMAO can directly stimulate the ERK1/2 pathway in human renal fibroblasts, leading to their differentiation into myofibroblasts and concurrent activation of the TGFβ1 pathway41. Additional investigations are required to determine if TMAO directly modulates ERK1/2 signaling. In addition, the involvement of the PERK/eIF2α pathway in fibrosis has been documented in various organs, including the liver, lung, and heart42,43,44. A recent study demonstrated that inhibition of PERK decreased TGFβ1-induced autophagy in kidney proximal tubular cells, resulting in reduced fibrogenic response and apoptosis45. Nevertheless, the impact of ER stress on renal fibrosis varies across different experimental models. Therefore, further evidence is required to determine whether ER stress directly induces renal fibrosis.
The expression of FMO3, the primary FMO gene observed in adult livers, exhibits considerable interindividual variability, with potential fluctuations of up to a 20-fold difference12. These genetic differences in population lead to differences in plasma TMAO levels, suggesting targeting FMO3 may serve as a potential strategy for lowering TMAO levels and subsequently ameliorating kidney injury. Further investigation is necessary to ascertain whether dietary and pharmaceutical interventions or disruption of the meta-organismal pathway associated with TMAO production can potentially impede the progression of renal dysfunction following I/R injury or the advancement of renal functional impairment in individuals with pre-existing renal diseases.
In conclusion, the present data indicate that a higher level of circulating TMAO is associated with a higher risk of acute injury, functional impairment, progressive renal fibrosis, and poorer long-term survival after I/R. Lowering TMAO levels by Fmo3 knockout protects the kidney from I/R injury by suppressing ER stress at the acute phase and by inhibiting TGFβ/Smad signaling activation at the chronic phase. Our results pave the way for a better understanding of the mechanisms underlying the effects of TMAO and the development of novel therapeutic strategies in the context of I/R-induced kidney disease.
Materials and methods
Clinical study population
Patients undergoing major cardiac surgery (on CPB) admitted to Anzhen Hospital, Capital Medical University from June 2023 to October 2023 were enrolled. Specifically, participants were from the non-intervention group of the study “The Application of Enhanced Recovery After Surgery for Cardiovascular Surgery in Adults (NCT 05914090, ongoing)”. The exclusion criteria, in addition to the criteria initially set by the clinical trial, also include: emergency surgery; renal dysfunction (eGFR <60 mL/min/1.73 m²); preoperative anemia (hemoglobin < 130 g/L in males and <120 g/L in females); preoperative use of nephrotoxic drugs such as nonsteroidal anti-inflammatory drugs, diuretics, beta-lactams, aminoglycosides, and/or amphotericin antibiotics; active-phase endocarditis; congenital heart disease; infections expected to affect postoperative recovery; persistent heart failure. Patients were divided to AKI and non-AKI groups after surgery based on the KDIGO (the kidney disease: improving global outcomes) classifications46. Clinical, demographic, and laboratory characteristics for AKI and non-AKI patients were collected and summarized. Serum samples were collected before cardiac surgery and the level of TMAO was measured. The human study was performed in accordance with the Declaration of Helsinki and was approved by the Institutional Ethics Committee of Beijing Anzhen Hospital (approval no. KS2023064). The protocol of the clinical study was registered at Clinical Trial (NCT05914090). Informed consent was obtained from each study participant. All ethical regulations relevant to human research participants were followed.
Animals
Ten- to 12-week-old male Fmo3−/− mice (C57BL/6N) were produced using CRISPR/Cas9 technology (Cyagen Biosciences, Suzhou, China). The Cas9 protein and two gRNA molecules (gRNA1: TCACATGTGATGCGTATTGC, gRNA2: TGAAGCCAGTTCTAGGGTAA) were administered into C57BL/6N mouse embryos. These embryos were then placed into the uteruses of pseudo-pregnant foster mothers. Mice with the following characteristics were identified and backcrossed at least six times onto the C57BL/6N background: (i) carrying a 7-base deletion resulting in a frameshift, and (ii) carrying a prematurely truncated FMO3 protein with a stop codon at residue 98. The genotype of the knockout mice was confirmed through a polymerase chain reaction (PCR) using two pairs of primers (F1: TCTCACCATCATCTAATTCCTCCTC, R1: ATCCTCTGTAACACTCTTTCCTAG; F1: TCTCACCATCATCTAATTCCTCCTC, R2: CTATATGGTCCTGTGAACGACAGT). To directly test for a potential contribution of increased TMAO level to the promotion of kidney injury after I/R, WT mice were fed either a standard chow (SPF-F02-001, spfbiotech, China) (total choline, 0.07%) or the same diet supplemented to 1.0% choline (C875195-1g, Maclin, China).
All animal experiments were performed with the approval of the Institutional Animal Care and Use Committee of the Chinese Academy of Sciences. We have complied with all relevant ethical regulations for animal use. All mice were raised in a sound-attenuated room with a 12-h light–dark cycle.
Renal ischemia–reperfusion model
Renal I/R surgery was performed in mice using the uIRx model, as previously described47,48. Mice were anesthetized using isoflurane (Veteasy, R510-22-10, Guang Dong, China) and placed on a heating pad (37 °C). Body temperature and respiration were monitored to ensure adequate depth. Following a midline laparotomy of approximately 1.5 cm, the bilateral renal pedicle was exposed. Subsequently, a right renal arteriovenous ligation and nephrectomy were performed. To induce left renal ischemia, the left renal artery was occluded by clamping it with an atraumatic vascular clip (Scanlan, Saint Paul, Minnesota, USA), resulting in a gradual color change of the kidney from red to dark purple within 1–2 min. After a duration of 30 min, the vascular clip was released to initiate reperfusion. Once the kidney color changed from dark purple to red, the muscle layer and skin were closed using Vicryl 4-0 sutures. Moreover, the left kidneys from the age and gender-matched mice that underwent the same surgical procedure but without renal pedicle clamping were used as the sham group.
Measurement of serum creatinine, BUN, and TMAO levels
Serum BUN and creatinine levels were measured using an INTEGRA 400 plus bioanalyzer (Roche Diagnostics, Indianapolis, IN, USA).
Mouse serum TMAO levels were quantified through high-performance liquid chromatography with a triple quadrupole mass spectrometer (AB SCIEX QTRAP 6500 System, USA) as previously described49. Briefly, a solution of 150 μL acetonitrile was added to 50 μL serum in 1.5 mL Eppendorf tubes prior to HPLC-MS analysis for protein precipitation. The mixture was vortexed for 1 min and centrifuged at 13,000 rpm for 15 min at 4 °C. The resulting supernatant was filtered through a 0.22 μm filter and analyzed using Liquid chromatography separation was performed on a C18 5 μm 4.6 × 250 mm column using an ExionLC LC system (Applied Biosystems/Sciex). The injection volume was 10 μL. Separation proceeded using a linear gradient of 0.1% formic acid (A) in water and 100% acetonitrile (B) as follows: 0–1 min, 35% B; 1–7 min 35–95% B; 7–8 min, 95% B; 8–8.8 min 95–30% B; 8.8–12 min 30% B. Flow rate was maintained at 0.35 mL min−1 at 40 °C. The HPLC instrument was coupled to a QTRAP triple-quadruple mass spectrometer (Applied Biosystems/Sciex) with electrospray ionization and multiple reaction monitoring scans. The concentrations of the calibration standards were 0.05–200 μM TMAO. The amount of TMAO in the sample was calculated by plotting the TMAO calibration curve.
Histological assessment
The kidneys were fixed in 4% paraformaldehyde (Leagene Biotech, Beijing, China) for 24 h, embedded in paraffin, and cut into 5 mm sections. These sections were then stained with hematoxylin (ZSJQ Biotechnology Co., Beijing, China), eosin (Leagene Biotech, Beijing, China), and PAS reagent (Leagene Biotech, Beijing, China). Based on the severity of tubular cell swelling, loss of brush border, and nuclear condensation, the tubular injury was scored from 0 to 4 (0, no changes; 1, changes affecting 25%; 2, changes affecting 25% to 50%; 3, changes affecting 50–75%; 4, changes affecting 75–100% of the section), as previously described50. Masson’s trichrome staining (Scytek, Logan, Utah, USA) was performed according to the manufacturer’s instructions. The collagen-enriched area was quantified using the ImageJ software (v1.8.0) based on our previous description51.
TUNEL apoptosis detection
Following the manufacturer’s instructions, we performed the terminal deoxynucleotidyl transferase-mediated digoxigenin–deoxyuridine TUNEL assay using the TUNEL BrightRed Apoptosis Detection Kit (Vazyme Biotech Co, A113-01, Nanjing, China). The samples were imaged using confocal microscopy (Leica, Wetzlar, Germany, TCS SP5). To quantify the fluorescence density of the apoptotic cells, ten random optical fields were analyzed from three distinct kidneys per group using the ImageJ software (v1.8.0) based on our previous description52.
Immunoblotting
Kidney tissue homogenates were prepared by homogenizing fresh or liquid nitrogen snap-frozen kidney biopsies in Tissue Extraction Reagent II (Thermo Fisher Scientific, FNN0071) with protease inhibitors (Roche, 05892970001) and phosphatase inhibitors (Roche, 04906837001). Kidney tissue homogenates were harvested from the supernatant after centrifugation (3800 × g) at 4 °C for 10 min and quantified using a Bradford assay. Protein lysates (30 μg) were subjected to 10% SDS-PAGE and electro-transferred to a 0.22 μm PVDF membrane (Pall, BSP0161). The membrane was blocked using 5% skim milk (Coolaber, CN311911100, Beijing, China) for 1 h at room temperature, incubated with a primary antibody overnight at 4 °C and a secondary antibody for 1 h at room temperature, and visualized using an ECL chemiluminescence reagent (Tanon, 180-501, Shanghai, China).
The antibodies used were:
Anti-FMO3 antibody 1:2000 (ab126711, Abcam, USA), Phospho-PERK (Thr982) antibody 1:1000 (DF7576, Affinity, China), PERK/EIF2AK3 Polyclonal antibody 1:1000 (24390-1-AP, Proteintech, China), Phospho-eIF2α-S51 Rabbit mAb 1:1000 (AP0692, Abclonal, China), eIF2α Rabbit pAb 1:1000 (A0764, Abclonal, China), ATF4 (D4B4) Rabbit mAb 1:1000 (#11815, CST, USA), CHOP (L63F7) Mouse mAb 1:1000 (2895S, CST, USA), XBP1S-specific Polyclonal antibody 1:500 (24868-1-AP, Proteintech, China), ATF6 Rabbit pAb 1:1000 (A0202, Abclonal, China), HSP90 (C45G5) Rabbit mAb 1:1000 (4877S, CST, USA), Phospho-ERK1/2 (Thr202/Tyr204) Antibody 1:1000 (TA1015S, Abmart, China), ERK1/2 Antibody 1:1000 (TA0155, Abmart, China), TGF beta 1 Rabbit pAb 1:1000 (A2124, Abclonal, China), TGFBR1 Rabbit pAb 1:1000 (A16983, Abclonal, China), TGFBR2 Monoclonal antibody 1:1000 (66636-1-Ig, Proteintech, China), Phospho-Smad2-S465/467+Smad3-S423/425 Rabbit pAb 1:1000 (AP0548, Abclonal, China), SMAD2/SMAD3 Rabbit pAb 1:1000 (A7536, Abclonal, China), and ACTA2/smooth muscle actin Rabbit Polyclonal Antibody 1:2000 (14395-1-AP, Proteintech, China).
RNA extraction and quantitative PCR
Total RNA was extracted from wild-type (WT, C57BL/6N) and Fmo3−/− mouse kidneys using the TRIzol Reagent (Thermo Fisher Scientific, 15596018) and reverse transcribed into cDNA using RevertAid Master Mix (Thermo Fisher Scientific, M1631), according to the manufacturer’s instructions. The TB Green Premix Ex Taq (Tli RNaseH Plus; Takara, RR420D) was used to amplify the PCR products. The mRNA levels of the genes were normalized to those of Gapdh. The primers used in the qPCR procedure are as follows:
Fmo3 F:5′-CCTTCATTGGTGCAAAGCCC-3′, R:5′-CCACTGTGTTAGGATGGCGT-3′;
Atf4 F:5′-AATGGCCGGCTATGGATGAT-3′, R:5′-CAATCTGTCCCGGAAAAGGC-3′;
Ddit3 F:5′-CCCCAGGAAACGAAGAGGAA-3′, R:5′-TGACCTCTGTTGGCCCTG-3′;
Xbp1 F:5′-ACACGTTTGGGAATGGACAC-3′, R:5′-CCATGGGAAGATGTTCTGGG-3′;
Ern1 F:5′- GACCGGCAGTTCCAGTACAT-3′, R:5′-TTGAGAGAATGCAGGTGTGC-3′;
Tgfb1 F:5′-CCACCTGCAAGACCATCGAC-3′, R:5′-CTGGCGAGCCTTAGTTTGGAC-3′;
Tgfbr1 F:5′-ATTGCTGGTCCAGTCTGCTT-3′, R:5′-TTTTAAGGTGGTGCCCTCTG-3′;
Tgfbr2 F:5′- AGCTGTTGGCGAGGAGTTTC-3′, R:5′-ACAGCTTAGGTGGATGGATGC-3′;
Acta2 F:5′-TGCTGACAGAGGCACCACTGAA-3′, R: 5′-CAGTTGTACGTCCAGAGGCATAG-3′;
Gapdh F:5′-GGAGAAACCTGCCAAGTATGA-3′, R:5′-TTGAAGTCACAGGAGACAACC-3′.
RNA sequencing analysis
As an input material for the RNA sample preparation, tissues were homogenized in TRIzol (Thermo Fisher Scientific, USA) after I/R. RNA sequencing libraries were prepared according to the manufacturer’s recommendations using the NEBNext Ultra RNA Library Prep Kit for Illumina (E7530L, NEB, USA). Index codes were added to attribute sequences to samples. On Illumina platforms, qualified libraries were pooled and sequenced using the PE150 strategy (Novogene Bioinformatics Technology, Beijing, China) based on effective library concentrations. DESeq2 (version 1.40.2) was used to identify differential expression genes (DEGs) with absolute log2FC (fold change) ≥ 0.5 and adjusted P value < 0.05 (Benjamini–Hochberg method). First, principal component analysis (PCA) was performed on the full set of processed detected genes. PCA was performed using the prcomp function of the R Stats Package (version 4.3.0), which stabilized the variance of the FPKM (Fragments Per Kilobase of exon model per Million mapped fragments). Visualization of the volcano plot and heatmap was performed using the R packages ggplot2 (version 3.4.2) and ComplexHeatmap (version 2.16.0). GSEA was applied to identify underlying pathways and a P value < 0.05 was considered statistically significant.
Statistics and reproducibility
Continuous variables are expressed as the means ± standard deviations or as medians with interquartile ranges; categorical data were expressed as a percentage. The Student’s t-test or the Mann–Whitney U-test was used to compare continuous variables, as appropriate. Pearson’s chi-square test or Fisher’s exact test was used for categorical variables. Pearson test was used for correlation analysis. Additionally, Kaplan–Meier survival plots were used to show 95% confidence intervals (95% CI) for all-cause mortality. A one-way analysis of variance (ANOVA) followed by a post-hoc Tukey test is used when comparing more than two groups. All analyses were performed using GraphPad Prism 8.4 software (Prism, San Diego, CA, USA). Statistical significance was set at *P < 0.05, **P < 0.01, and ***P < 0.001.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The data that support the findings of this study are available as Supplementary Information. Uncropped and unedited blot/gel images are provided in Fig. s3. Supplementary Data 1 represents the numerical source data in this paper. All the sequencing data have been deposited in NCBI under submission number SUB14589014, BioProject accession number PRJNA1132774.
References
Susantitaphong, P. et al. World incidence of AKI: a meta-analysis. Clin. J. Am. Soc. Nephrol. 8, 1482–1493 (2013).
Bedford, M., Farmer, C., Levin, A., Ali, T. & Stevens, P. Acute kidney injury and CKD: chicken or egg? Am. J. Kidney Dis. 59, 485–491 (2012).
Ronco, C., Bellomo, R. & Kellum, J. A. Acute kidney injury. Lancet 394, 1949–1964 (2019).
Molitoris, B. A. Therapeutic translation in acute kidney injury: the epithelial/endothelial axis. J. Clin. Invest. 124, 2355–2363 (2014).
Oh, C. J. et al. Inhibition of pyruvate dehydrogenase kinase 4 ameliorates kidney ischemia–reperfusion injury by reducing succinate accumulation during ischemia and preserving mitochondrial function during reperfusion. Kidney Int. 104, 724–739 (2023).
Ishani, A. et al. Acute kidney injury increases risk of ESRD among elderly. J. Am. Soc. Nephrol. 20, 223–228 (2009).
Amdur, R. L., Chawla, L. S., Amodeo, S., Kimmel, P. L. & Palant, C. E. Outcomes following diagnosis of acute renal failure in U.S. veterans: focus on acute tubular necrosis. Kidney Int. 76, 1089–1097 (2009).
Ramezani, A. et al. Role of the gut microbiome in uremia: a potential therapeutic target. Am. J. Kidney Dis. J. Natl Kidney Found. 67, 483–498 (2016).
Tang, W. H. et al. Gut microbiota-dependent trimethylamine N-oxide (TMAO) pathway contributes to both development of renal insufficiency and mortality risk in chronic kidney disease. Circ. Res. 116, 448–455 (2015).
Koeth, R. A. et al. Intestinal microbiota metabolism of L-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat. Med 19, 576–585 (2013).
Gatarek, P. & Kaluzna-Czaplinska, J. Trimethylamine N-oxide (TMAO) in human health. EXCLI J. 20, 301–319 (2021).
Koukouritaki, S. B., Simpson, P., Yeung, C. K., Rettie, A. E. & Hines, R. N. Human hepatic flavin-containing monooxygenases 1 (FMO1) and 3 (FMO3) developmental expression. Pediatr. Res. 51, 236–243 (2002).
Kaysen, G. A. et al. Associations of trimethylamine N-oxide with nutritional and inflammatory biomarkers and cardiovascular outcomes in patients new to dialysis. J. Ren. Nutr. 25, 351–356 (2015).
Stubbs, J. R. et al. Serum triethylamine N-oxide is elevated in CKD and correlates with coronary atherosclerosis burden. J. Am. Soc. Nephrol. 27, 305–313 (2016).
Shafi, T. et al. Trimethylamine N-oxide and cardiovascular events in hemodialysis patients. J. Am. Soc. Nephrol. 28, 321–331 (2017).
Massoth, C., Zarbock, A. & Meersch, M. Acute kidney injury in cardiac surgery. Crit. Care Clin. 37, 267–278 (2021).
Lannemyr, L. et al. Effects of cardiopulmonary bypass on renal perfusion, filtration, and oxygenation in patients undergoing cardiac surgery. Anesthesiology 126, 205–213 (2017).
Ranucci, M. et al. Oxygen delivery during cardiopulmonary bypass and acute renal failure after coronary operations. Ann. Thorac. Surg. 80, 2213–2220 (2005).
Bennett, B. J. et al. Trimethylamine N-oxide, a metabolite associated with atherosclerosis, exhibits complex genetic and dietary regulation. Cell Metab. 17, 49–60 (2013).
Cheruku, S. R., Raphael, J., Neyra, J. A. & Fox, A. A. Acute kidney injury after cardiac surgery: prediction, prevention, and management. Anesthesiology 139, 880–898 (2023).
Chaturvedi, S., Farmer, T. & Kapke, G. F. Assay validation for KIM-1: human urinary renal dysfunction biomarker. Int J. Biol. Sci. 5, 128–134 (2009).
Du, H. et al. Establishment of epithelial inflammatory injury model using adult kidney organoids. Life Med. https://doi.org/10.1093/lifemedi/lnae022 (2024).
Xu, L., Sharkey, D. & Cantley, L. G. Tubular GM-CSF promotes late MCP-1/CCR2-mediated fibrosis and inflammation after ischemia/reperfusion injury. J. Am. Soc. Nephrol. 30, 1825–1840 (2019).
Yang, B. et al. Caspase-3 is a pivotal regulator of microvascular rarefaction and renal fibrosis after ischemia–reperfusion injury. J. Am. Soc. Nephrol. 29, 1900–1916 (2018).
Zhou, X. et al. Tubular cell-derived exosomal miR-150-5p contributes to renal fibrosis following unilateral ischemia–reperfusion injury by activating fibroblast in vitro and in vivo. Int J. Biol. Sci. 17, 4021–4033 (2021).
Meng, X. M., Nikolic-Paterson, D. J. & Lan, H. Y. TGF-beta: the master regulator of fibrosis. Nat. Rev. Nephrol. 12, 325–338 (2016).
Rhee, E. P. et al. A combined epidemiologic and metabolomic approach improves CKD prediction. J. Am. Soc. Nephrol. 24, 1330–1338 (2013).
Kapetanaki, S., Kumawat, A. K., Persson, K. & Demirel, I. The fibrotic effects of TMAO on human renal fibroblasts is mediated by NLRP3, caspase-1 and the PERK/Akt/mTOR pathway. Int. J. Mol. Sci. https://doi.org/10.3390/ijms222111864 (2021).
Seldin, M. M. et al. Trimethylamine N-oxide promotes vascular inflammation through signaling of mitogen-activated protein kinase and nuclear factor-kappaB. J. Am. Heart Assoc. https://doi.org/10.1161/JAHA.115.002767 (2016).
Kong, L. et al. Trimethylamine N-oxide impairs beta-cell function and glucose tolerance. Nat. Commun. 15, 2526 (2024).
Xiong, Z. et al. The gut microbe-derived metabolite trimethylamine-N-oxide induces aortic valve fibrosis via PERK/ATF-4 and IRE-1alpha/XBP-1s signaling in vitro and in vivo. Atherosclerosis 391, 117431 (2024).
Li, J. et al. Trimethylamine N-oxide induces osteogenic responses in human aortic valve interstitial cells in vitro and aggravates aortic valve lesions in mice. Cardiovasc. Res. 118, 2018–2030 (2022).
Wang, F. et al. Transplantation of fecal microbiota from APP/PS1 mice and Alzheimer’s disease patients enhanced endoplasmic reticulum stress in the cerebral cortex of wild-type mice. Front. Aging Neurosci. 14, 858130 (2022).
Govindarajulu, M. et al. Gut metabolite TMAO induces synaptic plasticity deficits by promoting endoplasmic reticulum stress. Front. Mol. Neurosci. 13, 138 (2020).
Lattard, V., Lachuer, J., Buronfosse, T., Garnier, F. & Benoit, E. Physiological factors affecting the expression of FMO1 and FMO3 in the rat liver and kidney. Biochem. Pharm. 63, 1453–1464 (2002).
Qiu, L. et al. Beyond UPR: cell-specific roles of ER stress sensor IRE1alpha in kidney ischemic injury and transplant rejection. Kidney Int. 104, 463–469 (2023).
Yang, W. et al. Gut microbe-derived metabolite trimethylamine N-oxide accelerates fibroblast-myofibroblast differentiation and induces cardiac fibrosis. J. Mol. Cell Cardiol. 134, 119–130 (2019).
Consortium, A. B. et al. A biomarker framework for cardiac aging: the Aging Biomarker Consortium consensus statement. Life Med. https://doi.org/10.1093/lifemedi/lnad035 (2023).
Livingston, M. J. et al. Tubular cells produce FGF2 via autophagy after acute kidney injury leading to fibroblast activation and renal fibrosis. Autophagy 19, 256–277 (2023).
Humphreys, B. D. Mechanisms of renal fibrosis. Annu Rev. Physiol. 80, 309–326 (2018).
Andrikopoulos, P. et al. Evidence of a causal and modifiable relationship between kidney function and circulating trimethylamine N-oxide. Nat. Commun. 14, 5843 (2023).
Kim, H. J. et al. Carbon monoxide protects against hepatic steatosis in mice by inducing sestrin-2 via the PERK-eIF2alpha-ATF4 pathway. Free Radic. Biol. Med 110, 81–91 (2017).
Blohmke, C. J. et al. Atypical activation of the unfolded protein response in cystic fibrosis airway cells contributes to p38 MAPK-mediated innate immune responses. J. Immunol. 189, 5467–5475 (2012).
Zhao, Y. S. et al. Hydrogen and oxygen mixture to improve cardiac dysfunction and myocardial pathological changes induced by intermittent hypoxia in rats. Oxid. Med Cell Longev. 2019, 7415212 (2019).
Shu, S. et al. Reciprocal regulation between ER stress and autophagy in renal tubular fibrosis and apoptosis. Cell Death Dis. 12, 1016 (2021).
Khwaja, A. KDIGO clinical practice guidelines for acute kidney injury. Nephron Clin. Pr. 120, c179–184 (2012).
Fu, Y. et al. Rodent models of AKI-CKD transition. Am. J. Physiol. Ren. Physiol. 315, F1098–F1106 (2018).
Zager, R. A., Johnson, A. C. & Becker, K. Acute unilateral ischemic renal injury induces progressive renal inflammation, lipid accumulation, histone modification, and “end-stage” kidney disease. Am. J. Physiol. Ren. Physiol. 301, F1334–1345 (2011).
Ocque, A. J., Stubbs, J. R. & Nolin, T. D. Development and validation of a simple UHPLC-MS/MS method for the simultaneous determination of trimethylamine N-oxide, choline, and betaine in human plasma and urine. J. Pharm. Biomed. Anal. 109, 128–135 (2015).
Lee, P. Y. et al. Induced pluripotent stem cells without c-Myc attenuate acute kidney injury via downregulating the signaling of oxidative stress and inflammation in ischemia–reperfusion rats. Cell Transpl. 21, 2569–2585 (2012).
Wang, J. et al. Prophylactic supplementation with lactobacillus reuteri or its metabolite GABA protects against acute ischemic cardiac injury. Adv. Sci. 11, e2307233 (2024).
Zhang, H. et al. Prophylactic supplementation with Bifidobacterium infantis or its metabolite inosine attenuates cardiac ischemia/reperfusion injury. iMeta https://doi.org/10.1002/imt2.220 (2024).
Acknowledgements
The authors would like to acknowledge Dr. Weilin Li (Institutional Center for Shared Technologies and Facilities of Institute of Microbiology, Chinese Academy of Sciences), for their work on targeted metabolomics analysis of TMAO via a 6500 QTRAP triple quadrupole mass spectrometer (Applied Biosystems/Sciex) in this manuscript. This work was supported by the National Key Research and Development Program of China (2020YFA0113400), Beijing Natural Science Foundation (JQ22017), National Natural Science Foundation of China (92368112, 81921006), CAS Project for Young Scientists in Basic Research (YSBR-076), Initiative Scientific Research Program of the Institute of Zoology, Chinese Academy of Sciences (202310Z0202), State Key Laboratory of Membrane Biology, and Key Laboratory of Organ Regeneration and Reconstruction of the Chinese Academy of Sciences.
Author information
Authors and Affiliations
Contributions
Conceptualization: Moshi Song, Jun Wang, and Jiawan Wang. Methodology: Wei Wang, Jiandong Zhang, and Haozhou Wang. Formal analysis: Jiawan Wang, Siqi Chen, and Zeyu Gao. Investigation: Jiawan Wang, Jiandong Zhang, Zeya Li, Heng Du, Siqi Liu, Jun Wang, and Moshi Song. Omics: Pengfei Xu and Xuan Zhang. Clinical data: Fei Xiao, Zeya Li, Huili Li, and Sheng Wang. Original draft: Jiawan Wang and Wei Wang. Review and editing: Jun Wang and Moshi Song. Funding acquisition: Jun Wang and Moshi Song.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Peer review
Peer review information
Communications Biology thanks the anonymous reviewers for their contribution to the peer review of this work. Primary Handling Editor: Dario Ummarino.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wang, J., Wang, W., Zhang, J. et al. Deficiency of flavin-containing monooxygenase 3 protects kidney function after ischemia–reperfusion in mice. Commun Biol 7, 1054 (2024). https://doi.org/10.1038/s42003-024-06718-0
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s42003-024-06718-0
This article is cited by
-
Targeting gut–liver–kidney axis: microbiota-derived metabolites and therapeutic implications
Cell Communication and Signaling (2026)
-
CUGBP Elav-like family member 4 promotes cardiac remodeling through Inhibition of FMO2
BMC Cardiovascular Disorders (2025)
-
Medium from human iPSC-derived primitive macrophages promotes adult cardiomyocyte proliferation and cardiac regeneration
Nature Communications (2025)
-
Adipocyte FMO3-derived TMAO induces WAT dysfunction and metabolic disorders by promoting inflammasome activation in ageing
Nature Communications (2025)
