
Abstract
The essential l,d-transpeptidase of Mycobacterium tuberculosis (LdtMt2) catalyses the formation of 3\(\to\)3 cross-links in cell wall peptidoglycan and is a target for development of antituberculosis therapeutics. Efforts to inhibit LdtMt2 have been hampered by lack of knowledge of how it binds its substrate. To address this gap, we optimised the isolation of natural disaccharide tetrapeptide monomers from the Corynebacterium jeikeium bacterial cell wall through overproduction of the peptidoglycan sacculus. The tetrapeptides were used in binding / turnover assays and biophysical studies on LdtMt2. We determined a crystal structure of wild-type LdtMt2 reacted with its natural substrate, the tetrapeptide monomer of the peptidoglycan layer. This structure shows formation of a thioester linking the catalytic cysteine and the donor substrate, reflecting an intermediate in the transpeptidase reaction; it informs on the mode of entrance of the donor substrate into the LdtMt2 active site. The results will be useful in design of LdtMt2 inhibitors, including those based on substrate binding interactions, a strategy successfully employed for other nucleophilic cysteine enzymes.

Similar content being viewed by others


Introduction
Tuberculosis (TB) imposes a major impact on global health and causes an estimated 1.6 million deaths per year1. There is a limited set of drugs for TB treatment; however, the prolonged treatment regimens required for efficacy and increasing resistance mean there is a clear need for new therapeutics targeting Mycobacterium tuberculosis (Mtb), the causative agent of TB2,3.
Disruption of cell wall biosynthesis is arguably the most effective current approach for treatment of bacterial infections, with the cell wall biosynthesis targeting β-lactams being amongst the most successful of antibacterial drug classes4. β-Lactams are efficient and normally safe inhibitors of the penicillin binding proteins (PBPs), a class of nucleophilic serine enzymes that are involved in several of the final steps of peptidoglycan synthesis; PBPs include d,d-transpeptidases (some with bifunctional glycosyltransferase activity), d,d-carboxypeptidases, and endopeptidases (Fig. 1A)5. To date, the application of β-lactams for the treatment of TB has been limited, reflecting challenges including β-lactamase mediated resistance6. A further issue with β-lactam based TB treatment is that in addition to the nucleophilic serine PBPs, nucleophilic cysteine enzymes play a key role in Mtb cell wall biosynthesis7.

A Penicillin binding proteins (PBPs) with glycosyltransferase activity link lipid II to peptidoglycan, PBPs with carboxypeptidase activity cleave peptide strands from pentapeptides to give tetrapeptides and PBPs with transpeptidase activity form 4\(\to\)3 cross-links between pentapeptide strands. The l,d-transpeptidases (Ldts) form 3\(\to\)3 cross-links between tetrapeptide strands. Endopeptidases reverse the transpeptidase activity, cleaving the peptide bonds between cross-links. Lcps cleave peptide strands from tetrapeptides to tripeptides. The figure was created using BioRender.com. GlcNAc is in green, MurNAc is in teal, amino acids are in light blue, cross-linked amino acids are in dark blue. B Mechanism for the cross-linking reaction between the disaccharide tetrapeptide monomers catalysed by LdtMt2 (adapted from de Munnik et al.26).
Whereas the d,d-transpeptidases catalyse the formation of 4\(\to\)3 cross-links between peptidoglycan pentapeptide chains, the functionally related l,d-transpeptidases (Ldts) catalyse the formation of 3\(\to\)3 cross-links between tetrapeptide chains (Fig. 1)8,9. Despite being well conserved between bacterial species10, the low abundancy of 3\(\to\)3 cross-links is proposed to render Ldts redundant in most bacteria, at least in controlled laboratory environments11. However, Mtb displays up to 80% of 3\(\to\)3 cross-links during the stationary phase7. Hence, the Ldts have been identified as attractive targets for the development of anti-TB treatment12. In particular LdtMt2, one of the five Ldts of Mtb, is recognised as being important for virulence13,14,15.
Whereas the d,d-carboxypeptidases and d,d-transpeptidases are nucleophilic serine enzymes (Fig. S1)16,17, the Ldts employ a catalytic triad involving the sidechains of a nucleophilic cysteine and a histidine residue, and the backbone carbonyl of a third residue in a characteristic Hxx14–17(S/T)HGCZN motif, in which Z represents a hydrophobic residue18. The Ldts, which do not manifest structural homology with the PBPs and likely originate from a different evolutionary precursor18, share a common YkuD fold (named after the first identified member of this class), distinguished by a β-sandwich with two mixed β-sheets and one helix, and a set of four loops (Fig. S1)18,19,20. One of these loops borders the LdtMt2 active site19, creating two entrances towards the nucleophilic cysteine: the inner cavity and the outer cavity (Fig. S1)19.
The established substrate for LdtMt2 is the disaccharide tetrapeptide monomer 1 (GlcNAc-MurNAc-l-Ala-d-iGluNH2-m-DAPNH2-d-Ala; Fig. 2A)7,13. The formation of 3\(\to\)3 crosslinks is proposed to occur through covalent reaction of the amide bond between the 3rd and 4th residue with the nucleophilic cysteine of LdtMt2, i.e., Cys354, to give an acyl-enzyme complex21. The 3rd residue of a second strand of the tetrapeptide chain then reacts with the thioester intermediate to give the 3\(\to\)3 crosslinked product (Fig. 1B)22.

A Structures of the tetrapeptide substrates of LdtMt2 1-3, obtained through isolation from the cell wall of C. jeikeium. B Structure of the pentapeptide monomer of peptidoglycan from Mtb and C. jeikeium 4, obtained through isolation from the cell wall of C. jeikeium. C The structures of the products of transpeptidase activity of LdtMt2 5-7, obtained through cross-linking of 1, 2 and 3, respectively. D Peptidoglycan fragments 8-13 that do not act as substrates for LdtMt2, were obtained through solid-phase peptide synthesis (9 and 11) or commercial sources (8, 10, 12 and 13). GlcNAc stands for N-acetylglucosamine, MurNAc stands for N-acetylmuramic acid.
Though traditionally viewed as PBP inhibitors, selected β-lactams also inhibit the Ldts. In particular, carbapenems have been shown to inhibit Ldts23,24,25, and recent work has shown the potential of cephalosporins to inhibit LdtMt226. Several other classes of covalently reacting electrophilic compounds, e.g., ebselen, nitriles and acrylamides, have also been reported to inhibit LdtMt226,27. Structure-based design of Ldt inhibitors has, however, been limited by a lack of detailed knowledge of enzyme-substrate interactions.
Studies of LdtMt2 in complex with its substrate or acyl-enzyme intermediate have been hindered by the restricted availability of 1. Reported studies of the LdtMt2-substrate complex have thus far relied on unnatural substrate analogues, e.g., a crystal structure of the peptidoglycan fragment d-γ-Glu-m-DAP bound to the LdtMt2 active site22. Efforts to chemically synthesise substrate 3 (lactoyl-l-Ala-d-iGluNH2-m-DAPNH2-d-Ala) have yielded only low amounts of material28. The peptidoglycan substrates 1, 2 (GlcNAc-Muramitol-l-Ala-d-iGluNH2-m-DAPNH2-d-Ala), and 3 can, however, be obtained through isolation from the cell well of Corynebacterium jeikeium, which shares an identical peptidoglycan monomer structure with Mtb7,13,29. Using a variation of this method, we obtained 1-3 from the cell wall of C. jeikeium in significantly improved yields, enabling detailed studies on their interaction with LdtMt2. Kinetic and binding studies suggest that isolated LdtMt2 has a low affinity for the isolated tetrapeptide peptidoglycan monomer. X-ray crystallographic studies of LdtMt2 and substrate 3 yielded a structure of the proposed thioester intermediate of the transpeptidase reaction, which suggests the donor substrate enters through the inner cavity, but subsequently interacts with both the inner and outer cavities.
Results
Isolation of the tetrapeptide monomer
Initially, we isolated the reduced disaccharide tetrapeptide monomer 2 from the cell wall of C. jeikeium using reported methods29,30. C. jeikeium cells were lysed via high-pressure homogenesis, boiled in 8% sodium dodecyl sulphate, then treated with trypsin, pronase, mutanolysin, and lysozyme to yield soluble peptidoglycan monomers (Figure S2). MurNAc moieties in peptidoglycan monomers were then reduced to muramitol using sodium borohydride, to improve resolution during HPLC separation (Fig. S3A)30. This method was scaled to a dried cell mass of 94.6 g C. jeikeium from 21.6 L of culture, which yielded ∼4.5 mg of 2 (Fig. S4A). Aiming to improve the yield, we attempted to bypass the reduction of MurNAc in 1. Decreased separation of peaks was observed by HPLC (Fig. S3B); however, highly purified (>95%) disaccharide-tetrapeptide 1 (Fig. S4B) was obtained (∼3.6 mg from 61.3 g dried cell mass).
As described29, we observed that the distribution between tetrapeptide monomer 1 and pentapeptide monomer 4 favours 1 in crude samples. Addition of ampicillin in apparently subinhibitory concentrations (2 μg/mL)29 increased the abundance of 4 (Fig. S3C), likely due to ampicillin-mediated inhibition of d,d-carboxypeptidase and d,d-transpeptidase activities, leading to accumulation of their substrate (4)5,17. Subsequent incubation of isolated 4 with the purified recombinant Escherichia coli d,d-carboxypeptidase DacA gave 1 (Fig. S5). This procedure substantially (∼10 fold) improved the overall yield of 1 (∼20.5 mg from 55.9 g dried cell mass). The disaccharide moiety of 1 was cleaved by reaction with ammonium hydroxide29,30, giving lactoyl-tetrapeptide 3 (Fig. S6). The identities and purities of 1, 2 and 3 were confirmed by liquid chromatography mass spectrometry (LC-MS; Fig. S4) and 1H and 13C NMR characterisation (Figs. S7 and 8).
Studies of LdtMt2 activity with peptidoglycan monomers
We assessed LdtMt2 catalysis via LC-MS analysis of reaction mixtures of recombinant LdtMt2 with the three variants of l-Ala-d-iGluNH2-m-DAPNH2-d-Ala: the peptide containing GlcNAc-MurNAc 1, the peptide containing GlcNAc-Muramitol 2, and the lactoyl peptide 3. Incubation of LdtMt2 (5 µM) with 1, 2, or 3 (100 µM) led to formation of products 5, 6 and 7, as evidenced by the observation of species with masses of 1786 Da, 1790 Da, and 974 Da, respectively, (Fig. S9), as reported for 2 and 313,28.
Next, we assessed the structural requirements of the tetrapeptide residues that enable reaction with LdtMt2. A previous study identified the importance of sidechain amidation of both the d-iGlu and m-DAP residues for turnover28. To further investigate the structural requirements for LdtMt2 substrates, we incubated LdtMt2 with tetrapeptide 10 (l-Ala-d-iGlu-l-Lys-d-Ala), a peptidoglycan monomer that is often observed in Gram-positive bacteria11. We observed no evidence for turnover of 10. The tetrapeptide 11 (l-Ala-d-iGluNH2-l-Lys-d-Ala), which contains an amidated d-iGlu residue, but wherein m-DAPNH2 is replaced with l-Lys, was also not a substrate (over 24 h), evidencing the importance of the m-DAPNH2 residue in peptidoglycan fragments for turnover with isolated LdtMt2.
It is reported that the reduced pentapeptide monomer of Mtb (GlcNAc-muramitol-l-Ala-d-iGluNH2-m-DAPNH2-d-Ala-d-Ala) is not an LdtMt2 substrate13. Consistent with this, incubation of LdtMt2 with the pentapeptide monomer of Mtb 4 (GlcNAc-MurNAc-l-Ala-d-iGluNH2-m-DAPNH2-d-Ala-d-Ala), or pentapeptide 8 (l-Ala-d-iGlu-l-Lys-d-Ala-d-Ala), did not manifest evidence for reaction, further evidencing that pentapeptide monomers are not LdtMt2 substrates.
We were interested in assessing the transpeptidase activity of the active site cysteine to serine mutant of LdtMt2 (LdtMt2C354S), in part because previous reports have shown that in contrast to wild-type LdtMt213,21, LdtMt2C354S is unable to form a covalent complex with several penem and carbapenem derivatives23,31. When LdtMt2C354S was incubated with 1, 2, 3 or 4, no product formation was observed, suggesting the active site cysteine is essential for transpeptidase activity, and cannot be replaced by a potentially nucleophilic serine residue.
Differential scanning fluorometric thermal shift assays32 (Fig. S10 and Table S1) with a 100-fold excess (500 µM) of peptidoglycan fragments showed pentapeptide 4 induced a modest stabilising effect, as evidenced by a shift in the melting temperature (Tm) of ≥2 °C. With a 200-fold excess (1 mM), stabilisation was observed with tetrapeptides 1, 3 and 9, and pentapeptides 4 and 8, all manifesting a Tm shift of 2-3 °C; no significant shift in Tm was observed with 10 and 11.
Protein-observed solid-phase extraction mass spectrometric (SPE-MS) studies were performed with LdtMt2 (1 µM) in the presence of 1 and 3 (100 µM). While transpeptidase activity was confirmed with 1 and 3, as evidenced by observation of protein unbound species with masses of 1786 Da, and 974 Da, (correlating to 5 and 7, respectively), no covalent adducts of LdtMt2 were observed, suggesting that in solution the lifetime of the covalent thioester (predicted mass increase of 849 Da in the case of 1 and 443 Da in the case of 3, relative to unmodified LdtMt2) intermediate is short-lived (Fig. S11).
Kinetics of the LdtMt2 transpeptidase reaction with peptidoglycan monomers
Thus far, LdtMt2 transpeptidase activity has been measured by LC-MS analysis13, and LdtMt2 inhibition assays have relied on use of unnatural chromogenic or fluorogenic probe substrates23,33,34, as employed in an assay suitable for high-throughput screening26. These assays are, however, likely poor mimics of the natural reaction. We therefore developed an efficient, quantitative, and biologically relevant method of determining LdtMt2 activity and inhibition based on turnover of its natural substrate. A microtiter plate-based assay that determines PBP activity has been reported, based on detection of d-Ala, which is released on d,d-transpeptidase and d,d-carboxypeptidase activity (Fig. 3A)35,36,37. d-Ala is converted to pyruvate, ammonia and hydrogen peroxide by d-amino acid oxidase (DAAO). Hydrogen peroxide is then used by horseradish peroxidase (HRP) to oxidise Amplex Red (AR) forming fluorescent resorufin35. We considered that the transpeptidase assay could be extended to monitor LdtMt2 catalysis.

A Principle of the LdtMt2 transpeptidase assay, employing d-amino acid oxidase (DAAO) and horseradish peroxidase (HRP) to report on the release of d-Ala. B Correlation between pIC50 values obtained with the transpeptidase activity assay and fluorogenic thiol probe assay26. Individual data points are shown. C Michaelis-Menten graph of LdtMt2 in reaction with substrate 1. D Michaelis-Menten graph of LdtMt2 in reaction with substrate 3.
Optimisation initially involved variation of DAAO and HRP concentrations, aiming to minimise delay in conversion of d-Ala. When the assay mixture contained an excess of DAAO (2 U/mL) and HRP (2 U/mL), conversion of d-Ala was apparently near instantaneous (Fig. S12A, B). The assay performed better in sodium phosphate compared to Tris or HEPES buffers (Fig. S12C), and in the presence of 100 mM NaCl (Fig. S12D). A higher pH manifested increased LdtMt2 activity (tested pH range 7.0-8.5; Fig. S12E) and activity was higher at 37 °C than room temperature (Fig. S12F). Assays were therefore performed at 37 °C in 50 mM sodium phosphate pH 8.0, 100 mM NaCl and 0.01% (v/v) Triton X-100, with an LdtMt2 concentration of 500 nM and substrate (3) concentration of 35 µM (Fig. S12G). Under these conditions, a Z′ value of 0.88 and a signal to background value of 5.0 was obtained. The hydrogen peroxide produced has potential to oxidise the LdtMt2 catalytic cysteine, however, dose-response studies34 manifested no evidence for interference with assay relevant concentrations (Fig. S12H).
The assay was applied to investigate the inhibition of reported LdtMt2 inhibitors (Fig. S13 and Table S2)26,27,33. The results showed good correlation with the reported LdtMt2 ligand binding assay utilising a fluorogenic thiol reactive probe (Fig. 3B)34. The DAAO/HRP method presents an improved assay for LdtMt2 inhibitor screening, as the assay more closely resembles the biological process. The possibility for interference through inhibition of DAAO and HRP necessitates use of appropriate controls; indeed, the nonspecific inhibitors ebselen 24 and ebsulfur analogue 23 were found to cause interference (Fig. S14 and Table S2). Note that the assay could be adapted to use l-lactate dehydrogenase to monitor reduction of NAD+ in the presence of l-lactate.
With fixed concentrations of 1 and 3 (35 µM), a substantial difference in reaction rates was observed (120.0 ± 1.7 RFU min−1 and 224.2 ± 2.6 RFU min−1 for 1 and 3, respectively; Fig. S15), suggesting that interaction of the GlcNAc-MurNAc moiety of the substrate, at least with isolated components, has the potential to hinder the reaction rate. Increasing concentrations of 1 did not saturate the reaction (up to 20 mM; Fig. 3C and Table S3), hence the Km of 1 could not be determined. With 3, a Km of 6.2 mM was obtained, indicating low affinity; kcat was 2.6 × 102 min−1, providing a kcat/Km of 4.2 min−1 M−1 (Fig. 3D and Table S3). Note, that the effective incorporation of fluorescent amino acids and peptidoglycan fragments into the Mtb cell wall suggests more efficient turnover occurs in a cellular context38. The difference between isolated activity and cellular activity may reflect a need for complex formation, missing cofactors, or for binding of full-length peptidoglycan for optimal LdtMt2 activity.
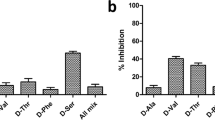
The non-substrate peptidoglycan fragments 4, 8, 10, 12 and 13 showed modest LdtMt2 inhibition (Fig. S16). Pentapeptides 8 and 10 and tripeptide 12 were the most potent (pIC50 4.8), while dipeptide 13 manifested the lowest potency (pIC50 4.0). The ability of structurally related non-substrate peptides to displace the substrate from the active site shows potential for the development of substrate-like inhibitors, although optimisation to improve affinity will be required.
X-ray crystallographic studies of the LdtMt2-substrate complex
To investigate the structural interactions of LdtMt2 with its substrate, we carried out X-ray crystallographic studies with the peptidoglycan fragments, aiming to obtain a structure of an enzyme-substrate complex; we obtained structures of LdtMt2 in complex with 1 and 3.
While in the absence of 3, LdtMt2 crystallised in the reported P1211 space group26,27, in the presence of 3 LdtMt2 crystallised in the P21212 space group with a single protein chain in the asymmetric unit (1.98 Å, Fig. 4; PDB 8PXZ), using precipitation solutions with a pH between 7.0–7.5, yielding structures of LdtMt2 with a molecule of 3 bound to the LdtMt2 immunoglobulin-like domain 2 (IgD2). Notably, however, when LdtMt2 and 3 were co-crystallised using a precipitation solution at pH 7.0, we observed additional electron density extending from the sidechain of the nucleophilic residue Cys354. The results are in agreement with formation of the dipeptide (d-iGluNH2-m-DAPNH2), covalently linked to LdtMt2 via a thioester with Cys354, apparently reflecting a key intermediate of the transpeptidase reaction with the donor peptide (Fig. 4A). The thioester is observed in the (Z)-stereochemistry, consistent with prior studies on nucleophilic serine- and cysteine-enzymes, including the SARS-CoV-2 main protease (Mpro; Fig. S17A)39,40 and porcine pancreatic elastase41.

A A structure of LdtMt2 reacted with 3 (PDB 8PXZ, 1.98 Å resolution) shows the covalent thioester intermediate structure, and a second molecule of 3 bound to the IgD2 of LdtMt2. The mF0 − DFc polder OMIT map is contoured at 3.0 σ and is carved around the 3-derived thioester; 3 is in grey mesh. Polar interactions are shown in grey dashes. B View of the inner cavity, which interacts with the d-iGluNH2 residue. C View of the outer cavity, which interacts with the m-DAPNH2 residue.
In the LdtMt2-3 complex structure, the m-DAPNH2 residue sidechain protrudes into the outer cavity, while the d-iGluNH2 sidechain is located in the inner cavity, suggesting that the donor substrate enters from the inner cavity (Fig. 4B, C). Note that no electron density was observed corresponding to the l-Ala residue of 3, which has previously been suggested not to play a significant role in substrate binding42. Additional electron density at the entrance of the outer cavity was observed, in which d-Ala, released during the formation of the thioester intermediate, could be modelled (Fig. 4A). We did not observe density that could reflect binding of an acceptor substrate.
The overall fold of the LdtMt2-3 complex strongly resembles that observed for unmodified LdtMt2 (PDB 6RLG27; main chain RMSD 0.65 Å). Similarly to unmodified LdtMt227,43, the Sγ atom of Cys354 is positioned 3.9 Å from the Nε2 atom of His336, and the Nδ1 atom of His336 is positioned for polar interactions with the main-chain carbonyl O of Ser337 at a distance of 2.7 Å. Notably, no conformational changes relative to unmodified LdtMt2 are observed in loop II (Fig. S18), which encloses the active site (residues 300-323) and which has previously been observed to undergo conformational changes upon binding of various LdtMt2 inhibitors, and which has been proposed to undergo extensive conformational changes upon substrate binding26,33,43.
The thioester acyl group, as well as the m-DAPNH2 residue and the d-iGluNH2 residue of 3 are positioned to engage in interactions with elements in the LdtMt2-3 complex active site (Fig. 4, S19). The thioester acyl group is located in the proposed oxyanion hole (the backbone NH groups of His352, Gly353 and Cys354) at respective distances of 3.8 Å, 3.3 Å and 2.3 Å, suggesting that, at least in the crystal structure obtained, no strong interactions are made with the backbone NH groups of His352. Polar interactions of m-DAPNH2 with His336 (3.0 Å), Asn356 (3.4 Å) and Thr320 (2.8 Å), as well as water-mediated polar interactions with His352 (3.3 Å to water, 3.4 Å to His352) are observed. The m-DAPNH2 carbon chain is positioned for hydrophobic interactions with the sidechain of Tyr318. The d-iGluNH2 residue is observed to engage in water-mediated polar interactions with His352 (2.7 Å to water and 3.3 Å to His352) and Ser331 (2.9 Å to water and 2.8 Å to Ser331), as well as hydrophobic interactions with Gly332.
Crystallographic studies of LdtMt2 in the presence of 1 did not manifest evidence for the presence of substrate at the active site; similarly to 3, however, 1 was observed to bind to the surface of the IgD2 domain through various polar interactions, as observed with both wild-type LdtMt2 and with LdtMt2C354S (PDB 8PXY, 2.15 Å; Fig. S20). The GlcNAc moiety of 1, which is absent in 3, was observed to interact with the surface the catalytic domain of a second symmetry related molecule of LdtMt2, manifesting polar interactions with Asp367 (2.5 Å and 2.8 Å) and with Gln363 (3.0 Å), located ~20 Å away from the nucleophilic cysteine (or serine, in the case of LdtMt2C354S), potentially preventing binding of the second molecule of 1 at the LdtMt2 active side as observed in the structure of LdtMt2 co-crystallised with 3.
Discussion
Targeting bacterial cell wall transpeptidases is a validated method for treating bacterial infections, in particular via β-lactam mediated PBP inhibition5. Though a role for β-lactams in the widespread treatment of TB has not been established, promising activity against Mtb has been observed, particularly with carbapenems, which inhibit both PBPs and Ldts44. Ldts, and specifically LdtMt2, are essential for Mtb13, but have been considerably less intensively studied than PBPs. Whilst several early-stage studies on inhibitors for LdtMt2 are reported23,26, the search for potent and clinically useful inhibitors is ongoing. Despite its biological and medicinal importance, the mechanism of the transpeptidase reaction of LdtMt2 has been the subject of limited studies, in part likely due to the lack of availability of the peptidoglycan monomers. Reported studies on LdtMt2 have thus mainly relied on the use of relatively simple substrate analogues22,45. Our modified procedure for the isolation of peptidoglycan monomers from the C. jeikeium cell wall enabled us to obtain a crystal structure of LdtMt2 reacted with 3 and to obtain insights into the binding and reaction kinetics of LdtMt2.
Exposure of C. jeikeium cells to subinhibitory concentrations of ampicillin during growth, results in accumulation of peptidoglycan pentapeptide monomer 4. LdtMt2 substrate 1 can subsequently be obtained from 4 via enzymatic cleavage of the terminal d-Ala. Via this method, the tetrapeptide monomer was obtained in a yield of ∼21 mg from 56 g of dried cell mass. Presently, this approach is substantially more efficient than reported synthetic procedures28.
X-ray crystallographic studies of LdtMt2 with substrate 3 yielded a structure of the covalently linked thioester acyl-enzyme complex, likely substantially reflecting the transient intermediate during the LdtMt2 transpeptidation reaction. Many enzymes employing nucleophilic cysteine- or serine-residues proceed through covalent acyl-enzyme intermediates; commonly these intermediates are unstable, and hence structures of them in complex with substrates, including at the acyl-enzyme complex stage, can be challenging to obtain under catalytically relevant conditions46. Crystallographic studies of the (thio)ester intermediates frequently rely on active site mutations (e.g., ref. 47), the use of substrate mimics (e.g., ref. 48), the use of rate-reducing conditions, for example, non-optimal pH (e.g., refs. 41,49), or use of an excess of a reversibly reacting product41. There is a particular lack of reported thioester, compared to ester, intermediate structures, perhaps reflecting the increased reactivity of thioesters with nucleophiles. To our knowledge, the only other nucleophilic cysteine enzyme where structural analysis of a thioester derived from a natural substrate has been achieved is SARS-CoV-2 Mpro39,40, where a thioester was observed to form in crystallo after being presented with and excess of an acid product, resulting in reverse reaction.
The observation of a thioester in our LdtMt2-3 complex acyl-enzyme crystal structure could in part be due to the inability of the acceptor substrate to enter the active site in the crystalline state due to reduced flexibility that may be required for acceptor substrate binding. In addition to the use of an excess of substrate (and hence product) and lattice interactions, other factors that may affect the persistence of the thioester in crystallo are the reduced pH (7.0, compared to 7.5) and temperature (4 °C, compared to rt) with respect to the solution-based studies, both of which reduce the rate of the LdtMt2 reaction (Fig. S12D-E). It is also important to note that transpeptidation reactions are fundamentally different from hydrolysis reactions, in that during transpeptidation the acyl-enzyme complex is required to be sufficiently stable in order to allow for the m-DAP residue of the acceptor substrate to react with the (thio)ester of the donating substrate.
The relative positions of the catalytic residues and of the oxyanion hole residues in the LdtMt2-3 complex are similar to those observed in the Mpro thioester intermediate structure, and in structures of Ldts in complex β-lactams, e.g., in a structure of the Enterococcus faecium Ldtfm complexed with ertapenem50, and in structures of LdtMt2 complexed with meropenem or biapenem (Fig. S17A–D)21,51. In all cases the thioester link has the (Z)-stereochemistry that is commonly associated with acyl-enzyme intermediates46,52, including in a structure of PBP4a of B. subtilis in complex with a peptidoglycan mimetic (Fig. S17E)53.
The structure reported here informs on how LdtMt2 interacts with a substantial part of its donor substrate. In a structure of a non-substrate peptidoglycan fragment (13, d-γ-Glu-m-DAP) bound to the LdtMt2 active site22, the m-DAP residue faces into the inner cavity, contrasting with our structure, suggesting entrance from the outer cavity (Figs. S21A, B and S22A). Molecular dynamics studies have suggested that entrance of the substrate from the inner cavity of LdtMt2 will not lead to acylation42. Our results suggest that during the initial steps of productive binding via the inner cavity, the substrate may project deeper into the active site than predicted by the reported calculations. In agreement with this proposal, a solid-state NMR structure of the B. subtilis l,d-transpeptidase (LdtBs) in complex with intact peptidoglycan suggests binding of the donor substrate in the analogous inner cavity (Figs. S21C, S22B)54.
An allosteric binding pocket of LdtMt2, named the S-pocket, is proposed to bind the sugar-moieties (Fig. S21E)45. We did not observe interaction of GlcNAc-MurNAc of 1 in this pocket. Nevertheless, extrapolation of the studies on LdtBs bound to intact peptidoglycan suggests that intact peptidoglycan likely binds in the S-pocket associated with the IgD2 domain of LdtMt254.
A coupled assay for detecting d-Ala was optimised for assessing LdtMt2 activity. The assay represents an increased throughput method of assessing turnover in comparison with previous LC-MS studies13,28, and provides increased biologically relevance over the reported high-throughput assay for inhibition of LdtMt226. However, due to the relatively low enzymatic activity, high concentrations of substrate are required, making the use of this assay in a high-throughput setting challenging without a well-established synthesis route. Additionally, the coupled enzymes DAAO and HRP increase the risk of assay interference. Based on these observations, we recommend that the fluorescence-based transpeptidase assay is perhaps most suited for use as a secondary assay following more high-throughput assays for LdtMt2, testing potential inhibitors in a more biologically relevant manner.
Although our studies suggest that the isolated recombinant LdtMt2 has a relatively low affinity and catalytic activity with 1 and 3, under our conditions 3 is a somewhat more efficient substrate. Interestingly, evidence for efficient transpeptidase activity by Ldts has been observed in Mtb cells in a previous report38, suggesting the need for increased interactions than present in our assays; potentially these will be achieved with use of full-length peptidoglycan. Alternatively, complex formation with related proteins / enzymes may increase activity, as observed with the functionally related d,d-transpeptidases and glycosyltransferases PBP1A and PBP2B, which require interactions with lipoprotein cofactors LpoA and LpoB, both in isolated enzyme and cellular systems55,56,57,58. In E. coli, the Ldt YcbB was found to associate with PBP1b and PBP5 revealing links between the two types of peptidoglycan transpeptidases59.
Amidation of both d-iGlu and m-DAP residues is reported to be required for LdtMt2 catalysis, as well as for growth of Mtb28. We observed that 11, a close analogue of 3, containing d-iGluNH2 but with the m-DAPNH2 substituted with l-Lys, is not a substrate for LdtMt2. The LdtMt2-3 complex structure suggests that the multiple polar interactions m-DAPNH2 makes with Asn356, His336, Thr320, and His352 are important, as l-Lys is likely only able to (directly) interact with Asn356 and His336. By contrast with the high substrate selectivity observed for LdtMt2, PBPs have been observed to react with d-Ala-d-Ala peptidoglycan mimetics, suggesting that modified features of the second and third peptide chain residues may not be as important for PBP catalysis as they appear to be for LdtMt260,61.
Most reported LdtMt2 inhibitors interact with the catalytic cysteine (Cys354), and may fill either the inner or the outer active site cavities, e.g., in the LdtMt2:ertapenem complex the inhibitor occupies the inner cavity, while in the LdtMt2:biapenem complex the inhibitor occupies the outer cavity (Fig. S17C-D)21,43,45,51. Based on the structure of LdtMt2 reacted with 3, elements of which occupy both the inner- and outer-cavities, improvement of inhibitor potency might be achieved through interactions with both the inner- and outer-cavities. Design of inhibitors achieving this may be based on our tetrapeptide structure, but optimisation will be necessary, especially in light of the low affinity of LdtMt2 for 3 in solution. The structure of LdtMt2 reacted with 3 suggests that a focus for future optimisation studies may be on the d-iGluNH2-m-DAPNH2-d-Ala residues, in particular the d-iGluNH2-m-DAPNH2 elements which are observed to interact with LdtMt2 in the intermediate structure. The addition of an electrophilic group to the m-DAPNH2 residue in lieu of the peptide bond with d-Ala may increase reactivity, e.g., a nitrile group, which has been shown to react with Cys354 of LdtMt226. Optimisation of the substrate for inhibition, including the addition of a reversibly covalently reacting nitrile group has been a successful method in the inhibitor development of Mpro, with one resulting inhibitor, nirmatrelvir, being approved for COVID-19 treatment62,63. Polar interactions with Tyr308 may be achieved through incorporation of a heteroatom in the d-iGluNH2 carbon chain. While the carbon chain of the m-DAPNH2 does not undergo direct interactions with the active site of LdtMt2, the flexibility of this residue may be required to allow for entrance into the active site and the polar interactions observed with His336, Tyr320, and Asn356, as well as the water-mediated interactions with His352.
Conclusion
LdtMt2 is a promising drug target for TB treatment, though little has been reported on the mechanism of the transpeptidase reaction that it catalyses. We optimised the isolation of the natural disaccharide tetrapeptide monomers 1-4 from the C. jeikeium bacterial cell wall through overproduction of peptidoglycan sacculus. We explored the interactions of LdtMt2 with the isolated tetrapeptide monomers 1 and 3 via X-ray crystallographic studies, obtaining a structure of the proposed thioester intermediate in LdtMt2 catalysis. The structure informs on the mode of entrance of the donor substrate into the LdtMt2 active site and active site interactions made by it. The combined results will be useful in the design of novel LdtMt2 inhibitors, including those based on substrate binding interactions, a strategy that has been successfully employed for other nucleophilic cysteine enzymes, including Mpro62,63.
Methods
Materials
Faropenem was from Fluorochem Ltd., FAD was from Alfa Aesar, AR was from Cambridge Bioscience Ltd., Fmoc- d-iGln-OH was from Ambeed, 12 and 13 were from InvivoGen. 16-23 were obtained from the GSK HTS compound library. All other compounds were from Merck.
Protein production and purification
Recombinant LdtMt2 was produced in Escherichia coli BL21(DE3) cells transformed with pNIC28-Bsa4-LdtMt2 Δ1-55, in the presence of 50 μg/mL kanamycin31. Expression was induced with isopropyl β-d-thiogalactopyranoside (IPTG, 0.5 mM) at OD600 of 0.6, with subsequent incubation overnight at 18 °C. Cells were lysed by sonication (SONIC Vibra-Cell, 60% amplitude) in the presence of DNase I; lysates were loaded onto a 5 mL HisTrap column (GE Life Sciences), pre-equilibrated in HisTrap buffer A (50 mM Tris, pH 8, 500 mM NaCl, 20 mM imidazole). The protein was eluted with HisTrap buffer B (50 mM this, pH 8, 500 mM NaCl, 500 mM imidazole) using a gradient from 0% to 100% buffer B. Protein-containing fractions were combined and the buffer was exchanged for HisTrap buffer A. The HisTag was cleaved using the TEV protease at 4 °C, over 12 h. The HisTag cleaved LdtMt2 was passed through a 5 mL HisTrap Column, and loaded onto a 300 mL Superdex 75 column (GE Life Sciences) pre-equilibrated in buffer C (50 mM Tris, pH 8, 500 mM NaCl). LdtMt2 was eluted with buffer C. The purity and identity of LdtMt2 was confirmed by SDS-PAGE (>95% purity) and MS (calculated 37975 Da, observed 37978 Da) analyses. Site-directed mutagenesis was carried out using the partial overlapping primer design method64, with primer sequences shown in Table 1 being used to achieve the C354S mutation of LdtMt2. PCR was performed using Q5 High-Fidelity DNA Polymerase (New England Biolabs) following standard protocols. LdtMt2C354S was produced and purified as described for the wild-type enzyme31. Purity and identity of LdtMt2C354S was confirmed by SDS-PAGE (>95% purity) and MS (calculated 37959 Da, observed 37962 Da). Recombinant E. coli DacA was produced and purified as reported65.
Peptidoglycan fragment isolation
C. jeikeium (National Collection of Type Cultures: NCTC 11913) was grown overnight at 37 °C on Columbia agar supplemented with 5% sheep blood (Scientific Laboratory Supplies Ltd). Cultures were used to inoculate brain heart infusion broth supplemented with 1% (v/v) Tween 80 and 2 μg/mL ampicillin29. The culture was incubated at 37 °C for 16 hours with shaking.
Cells were collected by centrifugation (15,000×g, 20 min, 4 °C). The cell pellet was resuspended (1 mL/g) in 10 mM sodium phosphate pH 7.0 and lysed using a high-pressure cell disruptor (CF1, Constant Systems, 40 kpsi, 3x). The cell wall was collected by centrifugation (15,000×g, 15 min, 4 °C) and resuspended in 25 mL of 10 mM sodium phosphate pH 7.0. The resulting suspension was added dropwise to 25 mL of 10 mM sodium phosphate pH 7.0 with 8% (w/v) SDS and incubated for 30 min at 100 °C. Peptidoglycan was pelleted by centrifugation (45,000×g, 30 min, 15 °C), washed with 10 mM sodium phosphate pH 7.0 (5x), and treated with Pronase E (200 µg/mL, overnight at 37 °C with shaking, in 10 mM Tris-HCl pH 7.4). Next, the peptidoglycan was pelleted by centrifugation (45,000×g, 30 min, 15 °C) and was washed with 20 mM sodium phosphate pH 7.8, before treatment with trypsin (200 µg/mL, overnight at 37 °C with shaking, in 20 mM sodium phosphate pH 7.8). Peptidoglycan was washed twice with water and dissolved in 25 mM sodium phosphate pH 6.0 with 0.1 mM MgCl2 and treated with mutanolysin (200 µg/mL) and lysozyme (200 µg/mL) and incubated overnight at 37 °C with shaking. Insoluble peptidoglycan was pelleted by centrifugation (4000×g, 30 min, 4 °C). The supernatant containing soluble disaccharide peptides was collected.
The disaccharide peptides were separated via reverse phase high-performance liquid chromatography (HPLC) on a C18 column (10 µm, 21,2 × 250 mm, Avantor ACE AQ) at a flowrate of 15 mL/min. A gradient of 1-20% (v/v) of solvent B was applied between 0 and 20 min (solvent A: 0.05% (v/v) trifluoroacetic acid in water; solvent B: 0.035% (v/v) trifluoroacetic acid in acetonitrile). Peaks containing 4 were lyophilised and dissolved in 50 mM tris pH 7.5 and DacA (20 µM) was added. The mixture was incubated for 5 h at 37 °C, lyophilised and purified by HPLC on a C18 column (10 µm, 21,2 × 250 mm, Avantor ACE AQ) at a flowrate of 15 mL/min. A gradient of 1-5% (v/v) of solvent B was applied between 0 and 7.5 min, followed by a gradient of 5–7% (v/v) of solvent B over 40 min to obtain 1 as a white solid.
To obtain 2, fragment 1 was dissolved in 0.5 M borate buffer pH 8.0 containing 10 mg/mL sodium borohydride. After 20 min the pH was adjusted to 2-4 with orthophosphoric acid. Purification by HPLC was performed as described above.
To obtain 3, fragment 1 was dissolved in 25% (v/v) NH4OH in H2O and stirred at 37 °C for 5 hours. The mixture was then neutralised via addition of acetic acid and the mixture was lyophilised. Purification by HPLC was performed as described above.
Syntheses of 9 and 11
9 and 11 were synthesised by solid phase peptide synthesis using a Liberty Blue Automated Microwave Peptide Synthesizer. d-Alanine-Wang resin (715 mg, 0.5 mmol) was coupled with Fmoc protected amino acids d-Ala (1.5 mmol, 3 eq.), l-Lys (2.5 mmol, 5 eq.), d-iGluNH2 (2.5 mmol, 5 eq.), and l-Ala (2.5 mmol, 5 eq.), respectively, in the case of 9, and l-Lys (2.5 mmol, 5 eq.), d-iGluNH2 (2.5 mmol, 5 eq.), and l-Ala (2.5 mmol, 5 eq.), respectively in the case of 11. Coupling using the standard procedure was performed with N,N-diisopropylethylamine (DIPEA) in ethyl cyanohydroxyiminoacetate (Oxyma, 1 M; DIPEA/Oxyma 1.5% (v/v)) and 7.8% (v/v) N,N′-diisopropylcarbodiimide (DIC) in DMF. After each coupling step, the resin was washed with DMF and the Fmoc group was deprotected with 20% (v/v) piperidine in DMF using the standard deprotection procedure. After coupling and deprotection steps were repeated as required, the resin-bound peptide was washed with dichloromethane (DCM), methanol and DCM, and treated with a 5 mL solution of 2:1 TFA/DCM (2 h, rt). Solvent was removed by evaporation in vacuo and purified by HPLC on a C18 column (5 µm, 10 × 150 mm; SunFire, Waters) at a flowrate of 3 mL/min in 98% (v/v) buffer A (0.1% (v/v) formic acid in H2O) and 2% buffer B (0.1% (v/v) formic acid in acetonitrile).
Mass spectrometry assays
LC-MS assays were performed using a 1290 Infinity II LC System (Agilent Technologies) equipped with a C18 column (3 µm, 50 × 2.1 mm; Ace equivalence) with a gradient of 5-20% (v/v) water:acetonitrile 0.1% (v/v) formic acid over 0 and 20 min, coupled to an Agilent 6550 iFunnel Q-TOF mass spectrometer (Agilent Technologies) operating in positive mode. Data were analysed using MassHunter Qualitative Analysis B.07.00 (Agilent Technologies).
Protein-observed SPE-MS assays were performed with a RapidFire200 integrated autosampler/solid phase extraction (SPE) system (Agilent Technologies) coupled to an Agilent 6550 iFunnel Q-TOF mass spectrometer (Agilent Technologies) operating in the positive ionisation mode26. LdtMt2 (1 µM) in 50 mM tris pH 7.5 was incubated with peptidoglycan fragments (100 µM), loaded onto a C4 cartridge (Aligent Technologies), and eluted with organic phate (85% (v/v) acetonitrile, 15% (v/v) water, 0.1% (v/v) formic acid. Data were analysed using MassHunter Qualitative Analysis B.07.00 (Agilent Technologies).
Transpeptidase assays
A transpeptidase assay master mixture was prepared in assay buffer (50 mM sodium phosphate, pH 8.0, 100 mM NaCl, 0.01% (v/v) Triton X-100) as described in Table 2. The specified master mixture (14 µL) was added to a black polystyrene, clear- and flat-bottomed, 384-well microplate (Greiner Bio-One, part number 781096) using a MultiDrop Combi dispenser (Thermo Fisher Scientific).
Compounds (in DMSO, with final concentrations ranging from 40 µM to 20.3 nM; 10 dilutions of factor 3) were added using a CyBio liquid handling system (Analytik Jena AG), with 4 replicates per inhibitor concentration. In the case of Control 1 (expected 0% inhibition; 100% fluorescence signal) and Control 2 (expected 100% inhibition; 0% fluorescence signal) the compound solution was substituted with neat DMSO. LdtMt2 (5 µL, 500 nM final concentration) was added to all wells using a MultiDrop Combi dispenser (Thermo Fisher Scientific). In the case of Control 2, LdtMt2 was substituted with the assay buffer (5 µL). This mixture was incubated for 30 minutes at rt. The substrate (1 or 3, as specified, 5 µL, 35 µM final concentration) was then added to all wells. The fluorescence signal was measured using a PHERAstar plate reader (BMG Labtech) with λex = 540 nm and λem = 590 nm. Data were analysed using Prism 10 (GraphPad).
The transpeptidase interference assay was performed as described above, with the modification that the LdtMt2 mixture was replaced by 5 µL assay buffer, and the substrate was replaced by d-Ala (500 nM final concentration). Data were analysed using Prism 10 (GraphPad).
Crystallography
LdtMt2 wild type or LdtMt2C354S (Δ1-55; with the N-terminal His Tag removed, in 50 mM tris, pH 8.0, 100 mM NaCl) was incubated with substrate 1 or 3 for 30 minutes on ice, and then crystallised using a well solution consisting of 0.1 M HEPES, pH 7.0, and 25% (v/v) Jeffamine ED-2001 pH 7.0 (identified from the JCSG PlusTM crystallisation screen, Hampton Research), using sitting drop vapour diffusion crystallisation plates (low reservoir Intelli-Plate 93-3), with 1 µL of protein-substrate solution and 1 µL of well solution. Note that crystallisation conditions previously reported by us26,27 did not yield crystals with additional electron density in the active site upon either soaking existing crystals with substrates 1-3 or cocrystallisation with 1-3, neither did we obtain complexes with LdtMt2C354S. Crystallisation plates were stored at 4 °C, and crystals grew over 24 h. The crystals were mounted on nylon loops, cryocooled and stored in liquid nitrogen. Datasets were collected using the MX beamline i03 at Diamond Light Source at 100 K, at a wavelength of 0.97625 Å. Datasets were processed using the automated processing pipeline at Diamond Light Source, using xia266. Structures were solved by molecular replacement with Phaser67, using PDB entry 6RLG27 as the search model. Ligand restrain files were created using Grade268. Alternating cycles of refinement using PHENIX69 and manual model building using COOT70 were performed until no further significant improvement of Rwork and Rfree was achieved. Data collection and refinement statistics are described in Table 3. There were no Ramachandran outliers for the LdtMt2C354S-1 (PDB 8PXY) and LdtMt2-3 (PDB 8PXZ) structures, with 98% and 97% Ramachandran favoured, respectively.
Statistics and reproducibility
All experiments were performed 2-4 times, as indicated. Individual data points are plotted, and average values and standard deviation are presented in corresponding tables.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The authors declare that the data that supports the findings of this study are available within the paper [and its supplementary files]. Source data for all figures and tables are available within the Supplementary Data file. Crystallographic data that supports the findings of this study have been deposited in the Protein Data Bank (PDB) with the accession codes “8PXY” and “8PXZ”. Requests for data should be sent to Christopher J. Schofield (christopher.schofield@chem.ox.ac.uk).
References
Global tuberculosis report 2022. Report No. Licence: CC BY-NC-SA 3.0 IGO (World Health Organization, 2022).
Sotgiu, G., Centis, R., D’Ambrosio, L. & Migliori, G. B. Tuberculosis treatment and drug regimens. Cold Spring Harb. Perspect. Med. 5, a017822 (2015).
Chakaya, J. et al. Global Tuberculosis Report 2020 – reflections on the Global TB burden, treatment and prevention efforts. Int. J. Infect. Dis. 113, S7–S12 (2021).
Lima, L. M., da Silva, B. N. M., Barbosa, G. & Barreiro, E. J. β-lactam antibiotics: an overview from a medicinal chemistry perspective. Eur. J. Med. Chem. 208, 112829 (2020).
Sauvage, E., Kerff, F., Terrak, M., Ayala, J. A. & Charlier, P. The penicillin-binding proteins: structure and role in peptidoglycan biosynthesis. FEMS Microbiol. Rev. 32, 234–258 (2008).
Kurz, S. G. & Bonomo, R. A. Reappraising the use of β-lactams to treat tuberculosis. Expert Rev. Anti-infect. Ther. 10, 999–1006 (2012).
Lavollay, M. et al. The peptidoglycan of stationary-phase Mycobacterium tuberculosis predominantly contains cross-links generated by L,D-transpeptidation. J. Bacteriol. 190, 4360–4366 (2008).
Glauner, B., Höltje, J. & Schwarz, U. The composition of the murein of Escherichia coli. J. Biol. Chem. 263, 10088–10095 (1988).
Aliashkevich, A. & Cava, F. LD‐transpeptidases: the great unknown among the peptidoglycan cross‐linkers. FEBS J. 289, 4718–4730 (2022).
Zandi, T. A., Marshburn, R. L., Stateler, P. K. & Brammer Basta, L. A. Phylogenetic and biochemical analyses of mycobacterial L,D-transpeptidases reveal a distinct enzyme class that is preferentially acylated by meropenem. ACS Infect. Dis. 5, 2047–2054 (2019).
Vollmer, W., Blanot, D. & De Pedro, M. A. Peptidoglycan structure and architecture. FEMS Microbiol. Rev. 32, 149–167 (2008).
Mainardi, J., Hugonnet, J., Gutmann, L. & Arthur, M. Fighting resistant tuberculosis with old compounds: the carbapenem paradigm. Clin. Microbiol. Infect. 17, 1755–1756 (2011).
Gupta, R. et al. The Mycobacterium tuberculosis protein LdtMt2 is a nonclassical transpeptidase required for virulence and resistance to amoxicillin. Nat. Med. 16, 466–469 (2010).
Schoonmaker, M. K., Bishai, W. R. & Lamichhane, G. Nonclassical transpeptidases of Mycobacterium tuberculosis alter cell size, morphology, the cytosolic matrix, protein localization, virulence, and resistance to β-lactams. J. Bacteriol. 196, 1394–1402 (2014).
Sanders, A. N., Wright, L. F. & Pavelka, M. S. Genetic characterization of mycobacterial L,D-transpeptidases. Microbiology 160, 1795–1806 (2014).
Andreeva, A. et al. SCOP database in 2004: refinements integrate structure and sequence family data. Nucleic Acids Res. 32, D226–D229 (2004).
Goffin, C. & Ghuysen, J.-M. Multimodular penicillin-binding proteins: an enigmatic family of orthologs and paralogs. Microbiol. Mol. Biol. Rev. 62, 1079–1093 (1998).
Biarrotte-Sorin, S. et al. Crystal structure of a novel β-lactam-insensitive peptidoglycan transpeptidase. J. Mol. Biol. 359, 533–538 (2006).
Böth, D. et al. Structure of LdtMt2, an L,D-transpeptidase from Mycobacterium tuberculosis. Acta Crystallogr. D 69, 432–441 (2013).
Bielnicki, J. et al. B. subtilis ykuD protein at 2.0 Å resolution: insights into the structure and function of a novel, ubiquitous family of bacterial enzymes. Proteins 62, 144–151 (2006).
Li, W. J. et al. Crystal structure of L,D-transpeptidase LdtMt2 in complex with meropenem reveals the mechanism of carbapenem against Mycobacterium tuberculosis. Cell Res. 23, 728–731 (2013).
Erdemli, S. B. et al. Targeting the cell wall of Mycobacterium tuberculosis: structure and mechanism of L,D-transpeptidase 2. Structure 20, 2103–2115 (2012).
Kumar, P. et al. Non-classical transpeptidases yield insight into new antibacterials. Nat. Chem. Biol. 13, 54–61 (2017).
Cordillot, M. et al. In vitro cross-linking of Mycobacterium tuberculosis peptidoglycan by L,D-transpeptidases and inactivation of these enzymes by carbapenems. Antimicrob. Agents Chemother. 57, 5940–5945 (2013).
Triboulet, S. et al. Inactivation kinetics of a new target of β-lactam antibiotics. J. Biol. Chem. 286, 22777–22784 (2011).
de Munnik, M. et al. High-throughput screen with the L,D-transpeptidase LdtMt2 of Mycobacterium tuberculosis reveals novel classes of covalently reacting inhibitors. Chem. Sci. 14, 7262–7278 (2023).
de Munnik, M. et al. Targeting the Mycobacterium tuberculosis transpeptidase LdtMt2 with cysteine-reactive inhibitors including ebselen. Chem. Commun. 55, 10214–10217 (2019).
Ngadjeua, F. et al. Critical impact of peptidoglycan precursor amidation on the activity of L,D-transpeptidases from Enterococcus faecium and Mycobacterium tuberculosis. Chem. Eur. J. 24, 5743–5747 (2018).
Lavollay, M. et al. The β-lactam-sensitive D,D-carboxypeptidase activity of Pbp4 controls the L,D and D,D transpeptidation pathways in Corynebacterium jeikeium. Mol. Microbiol. 74, 650–661 (2009).
Arbeloa, A. et al. Synthesis of mosaic peptidoglycan cross-bridges by hybrid peptidoglycan assembly pathways in gram-positive bacteria. J. Biol. Chem. 279, 41546–41556 (2004).
Lohans, C. T. et al. Non-hydrolytic β-lactam antibiotic fragmentation by L,D-transpeptidases and serine β-lactamase cysteine variants. Angew. Chem. Int. Ed. 58, 1990–1994 (2019).
Huynh, K. & Partch, C. L. Analysis of protein stability and ligand interactions by thermal shift assay. Curr. Protoc. Protein Sci. 79, 14–21 (2015).
Steiner, E. M., Schneider, G. & Schnell, R. Binding and processing of β‐lactam antibiotics by the transpeptidase LdtMt2 from Mycobacterium tuberculosis. FEBS J. 284, 725–741 (2017).
de Munnik, M. et al. A fluorescence‐based assay for screening β‐lactams targeting the mycobacterium tuberculosis transpeptidase LdtMt2. ChemBioChem 21, 368–372 (2020).
Gutheil, W. G., Stefanova, M. E. & Nicholas, R. A. Fluorescent coupled enzyme assays for D-alanine: application to penicillin-binding protein and vancomycin activity assays. Anal. Biochem. 287, 196–202 (2000).
Frére, J.-M., Leyh-Bouille, M., Ghuysen, J.-M., Nieto, M. & Perkins, H. R. Exocellular dd-carboxypeptidases-transpeptidases from Streptomyces. Methods Enzymol. 45, 610-636 (1976).
Catherwood, A. C. et al. Substrate and stereochemical control of peptidoglycan cross-linking by transpeptidation by Escherichia coli PBP1B. J. Am. Chem. Soc. 142, 5034–5048 (2020).
Pidgeon, S. E. et al. L,D-Transpeptidase specific probe reveals spatial activity of peptidoglycan cross-linking. ACS Chem. Biol. 14, 2185–2196 (2019).
Lee, J. et al. Crystallographic structure of wild-type SARS-CoV-2 main protease acyl-enzyme intermediate with physiological C-terminal autoprocessing site. Nat. Commun. 11, 5877 (2020).
Lee, J. et al. X-ray crystallographic characterization of the SARS-CoV-2 main protease polyprotein cleavage sites essential for viral processing and maturation. Nat. Commun. 13, 5196 (2022).
Wilmouth, R. C. et al. Structure of a specific acyl-enzyme complex formed between β-casomorphin-7 and porcine pancreatic elastase. Nat. Struct. Biol. 4, 456–462 (1997).
Baldin, S., Misiura, N. & Švedas, V. Building a full-atom model of L,D-transpeptidase 2 from Mycobacterium tuberculosis for screening new inhibitors. Acta Nat. 9, 44–51 (2017).
Kim, H. S. et al. Structural basis for the inhibition of Mycobacterium tuberculosis L,D-transpeptidase by meropenem, a drug effective against extensively drug-resistant strains. Acta Crystallogr. D 69, 420–431 (2013).
Jaganath, D., Lamichhane, G. & Shah, M. Carbapenems against Mycobacterium tuberculosis: a review of the evidence. Int. J. Tuberc. Lung Dis. 20, 1436–1447 (2016).
Ahmad, N. et al. Allosteric cooperation in β-lactam binding to a non-classical transpeptidase. eLife 11, e73055 (2022).
Yang, W. & Drueckhammer, D. G. Understanding the relative acyl-transfer reactivity of oxoesters and thioesters: computational analysis of transition state delocalization effects. J. Am. Chem. Soc. 123, 11004–11009 (2001).
Huguenin-Dezot, N. et al. Trapping biosynthetic acyl-enzyme intermediates with encoded 2,3-diaminopropionic acid. Nature 565, 112–117 (2019).
Macheboeuf, P. et al. Trapping of an acyl–enzyme intermediate in a penicillin-binding protein (PBP)-catalyzed reaction. J. Mol. Biol. 376, 405–413 (2008).
Alber, T., Petsko, G. A. & Tsernoglou, D. Crystal structure of elastase–substrate complex at −55 °C. Nature 263, 297–300 (1976).
Lecoq, L. et al. Structure of Enterococcus faecium L,D-transpeptidase acylated by ertapenem provides insight into the inactivation mechanism. ACS Chem. Biol. 8, 1140–1146 (2013).
Bianchet, M. A. et al. Structural insight into the inactivation of Mycobacterium tuberculosis non-classical transpeptidase LdtMt2 by biapenem and tebipenem. BMC Biochem. 18, 8 (2017).
Liu, B., Schofield, C. J. & Wilmouth, R. C. Structural analyses on intermediates in serine protease catalysis. J. Biol. Chem. 281, 24024–24035 (2006).
Sauvage, E. et al. Crystal structure of the Bacillus subtilis penicillin-binding protein 4a, and its complex with a peptidoglycan mimetic peptide. J. Mol. Biol. 371, 528–539 (2007).
Schanda, P. et al. Atomic model of a cell-wall cross-linking enzyme in complex with an intact bacterial peptidoglycan. J. Am. Chem. Soc. 136, 17852–17860 (2014).
Paradis-Bleau, C. et al. Lipoprotein cofactors located in the outer membrane activate bacterial cell wall polymerases. Cell 143, 1110–1120 (2010).
Typas, A. et al. Regulation of peptidoglycan synthesis by outer-membrane proteins. Cell 143, 1097–1109 (2010).
Egan, A. J. et al. Outer-membrane lipoprotein LpoB spans the periplasm to stimulate the peptidoglycan synthase PBP1B. Proc. Natl Acad. Sci. USA 111, 8197–8202 (2014).
Lupoli, T. J. et al. Lipoprotein activators stimulate Escherichia coli penicillin-binding proteins by different mechanisms. J. Am. Chem. Soc. 136, 52–55 (2014).
Caveney, N. A. et al. Structural insight into YcbB-mediated beta-lactam resistance in Escherichia coli. Nat. Commun. 10, 1849 (2019).
Anderson, J. W. et al. On the substrate specificity of bacterial DD-peptidases: evidence from two series of peptidoglycan-mimetic peptides. Biochem. J. 373, 949–955 (2003).
Stefanova, M. E. et al. Neisseria gonorrhoeae penicillin-binding protein 3 exhibits exceptionally high carboxypeptidase and β-lactam binding activities. Biochemistry 42, 14614–14625 (2003).
Yang, H. & Yang, J. A review of the latest research on Mpro targeting SARS-COV inhibitors. RSC Med. Chem. 12, 1026–1036 (2021).
Owen, D. R. et al. An oral SARS-CoV-2 Mpro inhibitor clinical candidate for the treatment of COVID-19. Science 374, 1586–1593 (2021).
Zheng, L. An efficient one-step site-directed and site-saturation mutagenesis protocol. Nucleic Acids Res. 32, e115–e115 (2004).
Inglis, S. R., Strieker, M., Rydzik, A. M., Dessen, A. & Schofield, C. J. A boronic-acid-based probe for fluorescence polarization assays with penicillin binding proteins and β-lactamases. Anal. Biochem. 420, 41–47 (2012).
Winter, G. xia2: an expert system for macromolecular crystallography data reduction. J. Appl. Crystallogr. 43, 186–190 (2010).
McCoy, A. J. et al. Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 (2007).
Grade2 version 1.5.0. (Global Phasing Ltd., 2021).
Adams, P. D. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D 66, 213–221 (2010).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr. D 66, 486–501 (2010).
Acknowledgements
The project was co-funded by the Tres Cantos Open Lab Foundation (Project TC297). It was also supported by funding from the Biotechnology and Biological Sciences Research Council (BBSRC) [BB/M011224/1] and the Wellcome Trust (106244/Z/14/Z). P.A.L. thanks the National PhD Training Programme in Antimicrobial Resistance Research by the Medical Research Foundation (MRF-145-0004-TPG-AVISO) for a studentship. P.R. thanks the Wellcome Trust (227298/Z/23/Z).
Author information
Authors and Affiliations
Contributions
M.d.M., P.A.L., J.B. and C.J.S conceived the experiments; M.d.M. carried out experiments, with assistance from P.A.L., K.C.T. and P.R.; M.d.M. analysed the data, with assistance from P.A.L. and P.R.; M.d.M. drafted the manuscript with C.J.S, with input from all authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks the anonymous reviewers for their contribution to the peer review of this work. Primary Handling Editors: Ingrid Span and Laura Rodríguez Pérez.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
de Munnik, M., Lang, P.A., Calvopiña, K. et al. Biochemical and crystallographic studies of l,d-transpeptidase 2 from Mycobacterium tuberculosis with its natural monomer substrate. Commun Biol 7, 1173 (2024). https://doi.org/10.1038/s42003-024-06785-3
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s42003-024-06785-3
This article is cited by
-
Bacterial cell envelope-targeting antibiotics
Nature Reviews Microbiology (2025)
