
Abstract
IP3 receptor (IP3R)-mediated Ca2+ transfer at the mitochondria-endoplasmic reticulum (ER) contact sites (MERCS) drives mitochondrial Ca2+ uptake and oxidative metabolism and is linked to different pathologies, including Parkinson’s disease (PD). The dependence of Ca2+ transfer efficiency on the ER-mitochondria distance remains unexplored. Employing molecular rulers that stabilize ER-mitochondrial distances at 5 nm resolution, and using genetically encoded Ca2+ indicators targeting the ER lumen and the sub-mitochondrial compartments, we now show that a distance of ~20 nm is optimal for Ca2+ transfer and mitochondrial oxidative metabolism due to enrichment of IP3R at MERCS. In human iPSC-derived astrocytes from PD patients, 20 nm MERCS were specifically reduced, which correlated with a reduction of mitochondrial Ca2+ uptake. Stabilization of the ER-mitochondrial interaction at 20 nm, but not at 10 nm, fully rescued mitochondrial Ca2+ uptake in PD astrocytes. Our work determines with precision the optimal distance for Ca2+ flux between ER and mitochondria and suggests a new paradigm for fine control over mitochondrial function.
Similar content being viewed by others

Introduction
Ca2+ signals are required to drive mitochondrial bioenergetics through the activation of enzymes in the mitochondrial matrix1,2, and deficiency or excessive mitochondrial Ca2+ signals have been associated with cellular dysfunction and disease pathogenesis3,4,5,6. Mitochondria take up Ca2+ through a low-affinity mitochondrial Ca2+ uniporter complex (mtCU)7. Close apposition of the endoplasmic reticulum (ER) membrane and the outer mitochondrial membrane (OMM) at the mitochondria-ER contact sites (MERCS) leads to high [Ca2+] hot spots and a direct transfer of Ca2+ through the ER-located inositol-1,4,5-trisphosphate receptors (IP3Rs) and OMM-located porin/voltage-dependent cation channel 1 (VDAC1)8,9,10,11. The distance between the ER and the OMM, at which Ca2+ transfer from the ER to the mitochondrial matrix occurs, has been proposed to lay within the 10–25 nm range12,13,14,15. However, it is not known whether ER-mitochondrial Ca2+ transfer operates with the same efficiency in a range of ER-OMM distances or if a specific distance exists for optimal Ca2+ flux through IP3R to mitochondria (Fig. 1a).

a A scheme depicting question addressed in this study: what is the distance between ER membrane and OMM for optimal Ca2+ flux? Lipid bilayer, IP3R and VDAC are represented in scale. b Scheme illustrating the design of ER-mitochondrial linkers. TM transmembrane domain, FL flexible linker, RS rigid spacer, ER endoplasmic reticulum, OMM outer mitochondrial membrane, mRFP monomeric red fluorescent protein. c TEM validation of EMLs overexpressed in HeLa cells. Arrows indicate ER membrane adjacent to mitochondria. Scale bar = 500 nm. d Quantification of ER-OMM distance on TEM images. Inset scales up quantified distances and shows the average distance per EML. e Quantification of the length of the interface between ER and mitochondria. One-way ANOVA, Tukey post hoc test, n = 31–178 contacts from at least 15 cells per condition. *p < 0.05, **p < 0.01, ****p < 0.0001.
To address this question, we employed a palette of existent and newly generated synthetic ER-mitochondrial linkers (EML) designed to maintain the distance between ER and OMM in a range from 5 to 30 nm with a spatial resolution of 5 nm and genetically encoded Ca2+ indicators targeting the ER lumen or the sub-mitochondrial compartments.
Results
Generation of the extended palette of ER-mitochondrial linkers
We used five EMLs fixing ER-OMM distance at ≤5 nm (denominated as 5nm-EML), ≤10–12 nm (10nm-EML), 15 nm, 20 nm and 30 nm (15nm-EML, 20nm-EML and 30nm-EML, respectively). 5nm-EML and 10nm-EML, kindly provided by Gyorgy Hajnoczky, Thomas Jefferson University, were published elsewhere12,16. 15nm-EML, 20nm-EML, and 30nm-EML were generated de novo (Fig. 1b, Supplementary Fig. 1) and were composed of monomeric red fluorescent protein (mRFP) flanked by a rigid α-helical spacer of a defined length, derived from the structure of myosin-VI known for its exceptional rigidity17 (PDB code: 6OBI). Structural prediction, using Robetta service18, was used to select the correct number of amino acids resulting in the desired ruler lengths. At the N- and C-termini, targeting sequences to OMM (AKAP1) and to the ER membrane (Ubc6)16, were attached, respectively (Supplementary Fig. 1). All linkers were validated by electron microscopy (Fig. 1c), and their ability to impose the defined distance has been confirmed (Fig. 1d). Moreover, all linkers significantly increased the length of the interaction between the two organelles (Fig. 1e). Importantly, none of the EMLs affected cell viability up to 72 h post-transfection (Supplementary Fig. 2a). Similarly, no changes of total levels of proteins, implicated in mitochondrial dynamics, such as dynamin-related protein (DRP1), p-DRP1, mitofusin 1 (MFN1) and MFN219 (Supplementary Fig. 2b) or in Ca2+ uptake (mitochondrial Ca2+ uniporter, MCU)20,21 (Supplementary Fig. 2c) were observed. Localization of EMLs was confirmed using confocal microscopy (Supplementary Fig. 3). Analysis of mitochondrial morphology on TEM images showed no significant changes in mitochondrial perimeter, total area, or Feret’s diameter (Supplementary Fig. 4). To better validate the robustness of EMLs in stabilizing the distance between ER and mitochondria, we co-expressed 20nm-EML and SPLICS-Short, a split-GFP contact site sensor reconstituting bright GFP fluorescence at a distance of 8–10 nm22. We found that the cells overexpressing 20nm-EML, do not form 8–10 nm contacts (filled arrows in Supplementary Fig. 5). Interestingly, the cells with a low 20nm-EML expression level form 8–10 nm ER-mitochondrial contacts, reconstituting SPLICS-Short fluorescence (empty arrow in Supplementary Fig. 5), although, expectedly, 20nm-EML and SPLICS-Short signals did not co-locolize. These data demonstrate that EML overexpression in HeLa cells is a suitable model to study the correlation between ER-mitochondrial distance and Ca2+ transfer.
A bell-shaped ER-OMM distance-dependence of the ER-mitochondrial Ca2+ flux with a maximum at ∼20 nm
Assessment of mitochondrial calcium uptake, using 4mtD3cpv probe targeted to the mitochondrial matrix23 48 h after co-transfection with EMLs, revealed that the amplitude of ATP-induced Ca2+ transients in the mitochondrial matrix ([Ca2+]M) in 5nm- and 10nm-EML-expressing cells was significantly reduced compared with control cells expressing ER-mRFP (Fig. 2a). Overexpression of 15nm-EML had no effect, while 20nm-EML strongly enhanced ATP-evoked [Ca2+]M transient. Overexpression of 30nm-EML resulted in a drastic reduction of ATP-evoked [Ca2+]M signals (Fig. 2a, b). These results confirm previous observations and predictions that when the ER-OMM distance is too short (5nm-EML) or too long (30nm-EML), the Ca2+ transfer is inefficient14,15,16. Intriguingly, 10nm-EML strongly suppressed, while 15nm-EML did not change ER-mitochondrial Ca2+ flux compared with control cells (Fig. 2a, b) in spite of significant increase of the interface length between the membranes (Fig. 1e). Stimulation with histamine, acting on a different class of metabotropic receptors, resulted in similar results: 10nm-EML strongly reduced histamine-induced Ca2+ transient in the mitochondrial matrix, while 20nm-EML significantly potentiated the response (Supplementary Fig. 6a).

Representative traces and quantifications of Ca2+ signals in the mitochondrial matrix (a, b), cytosol (c, d), ER lumen (e, f and g), and mitochondrial cristae lumen (h, i). Whisker plots of data collected from 88–150 (b), 130–492 (d), 91–260 (f, g), 75–212 (i) cells from at least three independent coverslips analyzed from 3 independent experiments. One-way ANOVA, Tukey post hoc test. **p < 0.01, ***p < 0.001.
To investigate if alterations of Ca2+ handling in cytosol or ER, upon EMLs overexpression, could affect [Ca2+]M response, we first assessed ATP-induced IP3R-mediated Ca2+ dynamics in the cytosol ([Ca2+]C) using Fura-2 probe24. As shown in Fig. 2c, d, presenting baseline-normalized Ca2+ traces, in 5nm- and 20nm-EML-expressing cells there were no differences in ATP-evoked [Ca2+]C transient. In 10nm-EML, 15nm- and 30nm-EML-expressing cells [Ca2+]C response was enhanced. There was no difference in resting [Ca2+]C (Supplementary Fig. 6c–e). Resting Steady-state luminal ER Ca2+ levels ([Ca2+]L) and the ER releasable Ca2+ pool (Δ[Ca2+]L) were measured using a green fluorescent protein (GFP)-aequorin fusion protein (GAP3) probe25. The GAP3-transduced HeLa cells were stimulated with a cocktail, containing ATP (100 µM) and tert-butyl hydroquinone (TBHQ, 100 µM) in a Ca2+-free KRB solution supplemented with 500 µM EGTA, to induce rapid and complete ER depletion (Fig. 2e). No significant differences in [Ca2+]L and Δ[Ca2+]L were observed upon EML overexpression, with the exception of 20nm-EML, which resulted in a modest but significant reduction of the steady-state [Ca2+]L compared with control cells expressing ER-mRFP (Fig. 2f). Nevertheless, the Δ[Ca2+]L in 20nm-EML-expressing HeLa was not different from control and no significant differences were found in the decay kinetics between samples (Fig. 2g). These results suggest that the changes of ATP-evoked [Ca2+]M transients in EMLs-expressing cells were not due to alterations of ER Ca2+ content and/or Ca2+ release capacity.
Potentially, alterations in mtCU activity could influence Ca2+ transients in the mitochondrial matrix. To confirm that the observed effects of EMLs (Fig. 2a) were independent of mtCU, we measured Ca2+ in the mitochondrial cristae lumen (CL) ([Ca2+]CL). Employing a recently developed ratiometric probe exploiting the targeting sequence from Reactive Oxygen Species Modulator 1 protein (denominated as ROMO-GemGeCO)26, we show that, quantitatively, [Ca2+]CL signals detected by ROMO-GemGeCO (Fig. 2h, i) faithfully resemble those of [Ca2+]M measured by 4mtD3cpv (see Fig. 2a).
We also checked if the 20 nm ER-mitochondrial distance enhances Ca2+ transfer in other cellular types. We found that ATP-induced [Ca2+]M was significantly potentiated in the hepatocellular carcinoma Huh-7 cell line (Supplementary Fig. 7) and in primary murine embryonic fibroblasts (MEFs) transduced with 20nm-EML (see below). These results demonstrate that (1) EMLs, based on rigid α-helical structures, represent a unique tool for the control of ER-OMM distance, allowing discrimination of events with ER-OMM distance resolution of 5 nm; (2) ER-mitochondrial Ca2+ flux critically depends on transversal ER-OMM distance and (3) the dependence is bell-shaped with ~20 nm representing an optimal distance for both ER-mitochondria Ca2+ transfer and mitochondrial Ca2+ uptake.
Enrichment of IP3R in 20 nm ER-OMM space with formation of functional IP3R-VDAC Ca2+ transferring units
To investigate if IP3R and VDAC were implicated in the increase of Ca2+ transfer in 20nm-EML-expressing cells27, we used a number of complementary approaches focusing on 5nm-, 10nm- and 20nm-EMLs because they produced clear opposite effects on ER-mitochondrial Ca2+ flux. First, we performed a proximity ligation assay (PLA), widely exploited to study IP3R-VDAC1 juxtaposition (Fig. 3a). As shown in Fig. 3b, in control ER-mRFP-expressing cells, PLA signals show diffuse dotty patterns corresponding to juxtaposed (≤40 nm) IP3R and VDAC1/3 proteins using anti-pan-IP3R and anti-VDAC1/3 antibodies, detecting putative close apposition of IP3R and VDAC1/3 at MERCS. Overexpression of 5nm-EML suppressed PLA signal, suggesting that at this distance, the juxtaposition of IP3R and VDAC1/3 is inhibited. Overexpression of 10nm-EML resulted in a significant increase of PLA signal, suggesting an enhanced juxtaposition of IP3R and VDAC1/3. However, a higher magnification examination shows only partial colocalization of PLA signal with 10nm-EML. Considering the reduced ATP-induced [Ca2+]M and [Ca2+]CL transients (Fig. 2), this result indicates that, although juxtaposed, IP3R and VDAC1/3 are unable to form functional Ca2+-transferring complexes. Strikingly, overexpression of 20nm-EML resulted in a strong increase of PLA signal (Fig. 3c) with a significantly higher colocalization between PLA signal and 20nm-EML (Fig. 3d), suggesting an enrichment of 20 nm MERCS with functional IP3R-VDAC complexes. Next, we performed immunofluorescence (IF) analysis using anti-pan-IP3R antibody, recognizing all three IP3R isoforms, which, in HeLa cells, form hetero-tetrameric IP3Rs28, to analyze the distribution of IP3Rs in EMLs-expressing cells (Fig. 3e). Overexpression of 5nm-EML did not significantly alter the wide-spread distribution of IP3R in the cell, while 10nm- and 20nm-EMLs produced an enrichment of IP3Rs in the proximity of EMLs (Fig. 3f). In line with PLA data, Pearson correlation coefficient quantification showed a significantly higher colocalization between IP3R and 20nm-EML compared with 10nm-EML (Fig. 3g).

a–d Proximity ligation assay (PLA) using anti-pan-IP3R, and anti-VDAC1/3 antibody (green) of HeLa cells expressing control plasmid (ER-mRFP) and ER-mitochondrial linkers (EMLs) (red). a Cartoon illustrating that only IP3Rs and VDAC1/3 juxtaposed at ≤40 nm are labeled. Representative images (b), quantification of the PLA-labeled area intensity (c) and PLA-EML Pearson colocalization coefficient (d). Scale bars = 20 µM. e–g Immunofluorescence analysis using anti-pan-IP3R antibody (green) in ER-mRFP and EML-expressing HeLa cells (red). e Cartoon illustrating that all IP3Rs present in the cells are labeled. Representative images (f) and quantification of IP3R-EML Pearson colocalization coefficient (g). Scale bars = 20 µM. h–j Localization of endogenous IP3R1 tagged with EGFP in HeLa cells (endogenous IP3R1-EGFP, green) upon expression of ER-EBFP, and 10nm- and 20nm-EMLs containing EBFP (magenta). h Cartoon illustrating that all endogenous IP3R1 receptors are tagged with EGFP. Representative images (i) and quantification of EGFP-EML Pearson colocalization coefficient (j). Scale bars = 20 µM. Whisker plots report data collected from 54–106 (c), 25–30 (d), 8–18 (g), 16–18 (j) cells from at least 3 independent coverslips analyzed from 3 independent cultures. One-way ANOVA, Tukey post hoc test. **p < 0.01, ***p < 0.001, ****p < 0.0001.
Enrichment of IP3R fluorescent signal in the EML region and its strong reduction in the resting part of the cytosol in 20nm-EML-expressing cells suggest relocation of IP3R into 20 nm ER-mitochondrial space. To confirm this hypothesis in an unbiased manner, independent of the enhancement of fluorescent signal during immunodecoration with first and second antibodies, we expressed 10nm- and 20nm-EMLs in a recently generated HeLa cell line, in which endogenous IP3R1 was tagged with EGFP using CRISPR/Cas9 technology (HeLa-IP3R1-EGFP)28 (Fig. 3h). To avoid minimal possible contamination of EGFP signal by fluorescence deriving from mRFP-containing linker variants, we generated and validated EMLs expressing enhanced blue fluorescent protein (EBFP) (Supplementary Fig. 8). Overexpression of the control (ER-EBFP) construct in HeLa-IP3R1-EGFP cells did not alter the distribution of endogenous IP3R1-EGFP signal (Fig. 3i, ER-EBFP). However, overexpression of 10nm- and 20nm-EML-EBFP constructs 48 h after transfection resulted in strong re-localization of fluorescent signals in the area of EMLs. In line with PLA and IF analyses, colocalization between IP3R1-EGFP and EMLs was significantly higher for 20nm-EML compared with 10nm-EML (Fig. 3j). A residual signal remained in some intracellular areas or in correspondence to the edge of the cell. Western blot analysis of total lysates showed that the enrichment of IP3R in 20 nm MERCS was not due to altered expression of IP3Rs (Fig. 4a, b and Supplementary Fig. 9a). Furthermore, in confirmation of the IP3R relocation hypothesis, IP3R protein was significantly increased in MERCS isolated from HeLa cells stably expressing 20nm-EML compared with cells expressing control ER-mRFP construct (Fig. 4a–c). Interestingly, the amount of VDAC1/3 or GRP75 proteins was not different from control cells either in total cell lysates (Fig. 4b and Supplementary Fig. 9b) or in MERCS isolated from 20nm-EML-overexpressing cells (Fig. 4c) suggesting that enrichment of IP3Rs in 20 nm MERCS was independent of VDAC1/3 localization. No significant changes were found in MERCS, upon overexpression of 10nm-EML, in IP3R, VDAC1/3, or GRP75 levels, although a tendency to increase IP3R was observed (Supplementary Fig. 9a–c).

Representative images (a, d) and quantification of total cell lysates (b) and isolated MERCS (c, e) using antibodies for pan-IP3R, VDAC1/3, GRP75, MFN1, MFN2, DRP1 or TOM70. Anti-β-actin (a–c) or anti-GAPDH (d, e) were used for normalization of raw data. Data are expressed as mean ± SEM of n = 3–6 independent experiments. Unpaired two-tailed Student’s t-test. **p < 0.01.
The above-described experiments are based on a generally accepted paradigm that both IP3R and VDAC are required for ER-mitochondrial Ca2+ transfer29,30. However, forced membrane tethering could potentially predispose to an IP3R-independent Ca2+ release, e.g., via Ca2+ leak from the ER and/or activation of Ca2+-induced Ca2+ release mechanism31,32. On the other hand, it has been suggested that several β-barrel-forming OMM proteins can be permeable to ions33,34. To investigate whether IP3R and VDAC1/3 are the only ones responsible for the enhanced Ca2+ flux in 20nm-EML-expressing cells, we used HeLa cells with triple IP3R1,2,3 knock-out (HeLa-TKO)30 and MEFs derived from VDAC1/3-KO mice35. Control IF analysis showed no fluorescent signal either in WT HeLa probed with non-specific serum, or in HeLa-TKO probed with anti-pan-IP3R antibody (Supplementary Fig. 10). ATP-induced Ca2+ signals were monitored in mitochondrial CL using ROMO-GemGeCO probe. [Ca2+]CL signals were not detected in ER-mRFP-expressing HeLa-TKO cells, nor in cells overexpressing either 10nm- or 20nm-EML (Supplementary Fig. 11a, b). Similarly, [Ca2+]CL signals were completely absent in ATP-stimulated VDAC1/3-KO MEFs in either condition (Supplementary Fig. 11c, d). Notably, in both WT cells, HeLa and MEFs, [Ca2+]CL signals were detected, and, in line with that reported in Fig. 2, 10nm-EML reduced, while 20nm-EML significantly enhanced Ca2+ transients in CL. Therefore, both IP3R and VDAC1/3 are necessary for Ca2+ transfer at 20 nm MERCS.
10 nm ER-OMM distance inhibits, while 20 nm distance promotes mitochondrial respiration and ATP production
One of the most accredited functions of Ca2+ signals in mitochondria is the regulation of mitochondrial energetics through the activation of metabolic enzymes and ATP synthase2. Therefore, we investigated the effect of EMLs overexpression on mitochondrial respiration and ATP production using Oroboros oxygraphy (Fig. 5a). We found that 10nm-EML overexpression reduced, while the 20nm-EML one significantly increased both basal oxygen consumption (Fig. 5b), respiratory reserve (Fig. 5c) and ATP-linked respiration (Fig. 5d). These data were corroborated by quantification of total cellular ATP content using ATPlite assay (Fig. 4e). Potentially, variations of the mitochondrial membrane potential (ΔΨm), due to expression of EMLs, could influence respiration and ATP production. However, measurement of ΔΨm with JC-1 probe using flow cytometry, in cells expressing EBFP variant EMLs, did not reveal any difference in ΔΨm among ER-EBFP, 10nm-EML and 20nm-EML-expressing HeLa cells (Supplementary Fig. 12). Our results suggest that mitochondrial oxidative metabolism and ATP supply may be efficiently regulated, through regulation of Ca2+ flux, by modulating the distance between ER and mitochondria.

a Representative traces (ER-RFP), illustrating the protocol of high-resolution respirometry for quantification of respiration using Oroboros’ protocol for intact cells; blue line: oxygen concentration, red line: oxygen flux. b Oxygen flux in the routine state (R); in the leakage state (L) after the addition of oligomycin, an inhibitor of ATP synthase; after the addition of FCCP, an uncoupler of oxidative phosphorylation to induce maximum respiratory capacity (E). All data are expressed as specific flux, i.e., oxygen consumption normalized to the sample protein content and after subtraction of non-mitochondrial oxygen flux (ROX). c Reserve respiratory capacity obtained by the subtraction of R from E. d Oxygen consumption linked to ATP production, i.e., oligomycin-sensitive respiration obtained by the subtraction of L from R. Data are expressed as mean ± SEM of fold changes above the control of n = 4 (ER-RFP and 10nm-EML) or n = 3 (ER-RFP and 20nm-EML) independent experiments. Unpaired two-tailed t-test 10nm-EML or 20nm-EML vs ER-RFP. *p-value < 0.05; **p-value < 0.01. e Total cellular ATP. Data are expressed as mean ± SEM of % of control (ER-RFP) of n = 6 independent experiments. One-way ANOVA, Tukey post hoc test, **p-value < 0.01; ****p-value < 0.0001.
MERCS are disrupted in Parkinson’s disease astrocytes: rescue by overexpression of 20nm-EML
The data, obtained using overexpression of artificial ER-OMM tethers, suggest that 20 nm distance specifically promotes Ca2+ transfer between ER and mitochondria through preferential localization of IP3Rs at 20 nm MERCS. Estimation of physiological abundance of MERCS on TEM images suggests that 18–22 nm MERCS account for ~18% of all MERCS ranging from 5 to 80 nm, indicating that almost 1/5 of MERCS are potentially deputed to Ca2+ transfer, although this percentage likely depends on cell type and condition14.
Increasing evidence suggests that MERCS are altered in neurodegeneration, including Alzheimer’s (AD) and Parkinson’s (PD) diseases15,36,37,38. To investigate whether specific alteration of 20 nm MERCS may mediate mitochondrial Ca2+ deficiency in pathological conditions, we took advantage of a recently reported split-GFP contact site sensors (SPLICS)22,39, which we adapted to reconstitute bright GFP fluorescence specifically at 20 nm between ER and OMM (Supplementary Fig. 13). Transfection of 20nm-SPLICS resulted in the appearance of bright fluorescent dots distributed throughout the cell in sites of the higher ER and mitochondrial density (Supplementary Fig. 14a). Co-expression of 20nm-SPLICS probe with 20nm-EML resulted in a drastic increase of SPLICS signal and its complete colocalization with 20nm-EML (Supplementary Fig. 14b), confirming proper functioning of the probe, while immunodecoration of IP3Rs showed close juxtaposition of 20nm-SPLICS with a fraction of IP3Rs, although, expectedly, SPLICS-free IP3Rs were also detected (Supplementary Fig. 14c).
As a model of pathological condition that explores the potential alteration of MERCS-mediated Ca2+ transfer, we capitalized on our recent characterization of hiPSC-differentiated astrocytes (hiA) from human subjects bearing a Parkinson’s disease (PD)-causing LRRKG2019S mutation40 (referred as to PD-hiA)41. PD-hiA displayed no differences in basal or evoked cytosolic Ca2+ signals while presenting severe defects of mitochondrial respiratory activity and significant reduction of ΔΨm41. Therefore, PD-hiA are suitable to explore if defects of mitochondrial Ca2+ handling are caused by the deficiency of the ER-mitochondrial Ca2+ transfer and distance. We employed 4mtD3cpv probe to demonstrate that [Ca2+]M signals were dramatically reduced in PD-hiA compared with control cells prepared from aged-paired non-PD subjects (Ctr-hiA) (Fig. 6a–d), suggesting impaired ER-mitochondrial Ca2+ transfer. To assess ER-mitochondria interaction, we used three available SPLICS sensors: SPLICS-Short, detecting distances at 8–10 nm; the newly generated 20nm-SPLICS and SPLICS-Long, detecting contact sites at 40–50 nm22,39. As shown in Fig. 6e, no differences were found in MERCS at 8–10 nm distance (SPLICS-Short) between Ctr-hiA and PD-hiA; however, a dramatic decrease was observed in PD-hiA in the abundance of contact sites detected by 20nm-SPLICS (Fig. 6f). Instead, there was a significant increase of contact sites detected by SPLICS-Long in PD-hiA compared with Ctr-hiA (Fig. 6g), suggesting an enlargement of the distance between ER and mitochondria. To validate SPLICS data we quantified ER-mitochondrial distance on TEM images in a range from 5 to 100 nm. As shown in Fig. 6h, the average distance between ER and mitochondria increased from ~20 to ~38 nm, corroborating SPLICS quantification.

Representative traces and quantifications of raw (a, b) and baseline-normalized (c, d) ATP (100 µM)-induced Ca2+ transients in hiA from healthy subjects (Ctr) and from LRRK2G2019S patients (PD). Data are expressed as mean ± SEM of basal 530/475 ratio (Basal) and peak of the response (Peak) from 56–63 cells from three independent experiments. Representative images and quantification of fluorescence SPLICS-Short (e), 20nm-SPLICS (f) and SPLICS-Long (g) transfected in hiA from healthy subjects (Ctr) and from LRRK2G2019S patients (PD). Data are expressed as mean ± SEM of GFP area normalized to cell area from n = 19–22 cells from at least three independent transfections. Unpaired two-tailed Student’s t-test, ***p-value < 0.001; ****p-value < 0.0001. h Representative TEM micrographs and quantifications of the ER-OMM distance of hiA from healthy subjects (Ctr) and from LRRK2G2019S patients (PD). Arrows indicate ER membrane adjacent to mitochondria. Scale bar = 500 nm. Data are mean ± SEM from 72–88 individual contact sites from 28 (Ctr) or 20 (PD) slices per genotype. Unpaired two-tailed Student’s t-test, ****p-value < 0.0001.
In order to investigate if mitochondrial Ca2+ uptake in PD-hiA could be rescued by stabilizing the ER-mitochondrial interaction at the 20 nm distance, optimal for Ca2+ transfer, we expressed control (ER-mRFP), 10nm-EML and 20nm-EML together with 4mtD3cpv probe in PD-hiA and stimulated cells with ATP. As shown in Fig. 7a–d, 10nm-EML did not rescue mitochondrial Ca2+ uptake, moreover, both basal Ca2+ level in the mitochondrial matrix and the ATP-induced Ca2+ transient in 10nm-EML-expressing PD-hiA were lower than in ER-mRFP-expressing PD-hiA cells. Strikingly, 20nm-EML overexpression fully rescued both the basal Ca2+ level in the matrix and the Ca2+ transient, which were similar to those observed in Ctr-hiA expressing control ER-mRFP construct (Fig. 7a–d). These results (1) validate the importance of Ca2+ transfer at 20 nm MERCS suggesting that, contrarily to Alzheimer’s disease astrocytes, in which a shortening of the ER-mitochondrial distance is found15,42,43 in PD-hiA bearing LRRKG2019S mutation, impairment of mitochondrial bioenergetics41 is due to enlargement of ER-mitochondrial distance, loss of 20 nm MERCS and inability of Ca2+, released from the ER, to reach mitochondria; (2) the defect of the mitochondrial Ca2+ uptake can be efficiently rescued by stabilizing the membranes at the distance of 20 nm but not 10 nm. Figure 7e schematically depicts the proposed hypothesis. Interestingly, in line with the absence of the effect of EMLs expression on ΔΨm in HeLa cells, overexpression of 20nm-EML in PD-hiA failed to normalize depolarization of ΔΨm41 (Fig. 7f), suggesting that the alterations of mitochondrial Ca2+ uptake and of ΔΨm might be caused by independent mechanisms.

Representative traces and quantifications of raw (a, b) and baseline-normalized (c, d) ATP (100 µM)-induced Ca2+ transients in Ctr-hiA PD-hiA transfected with ER-mRFP, 10nm-EML and 20nm-EML. Box plot from 44–54 cells from three independent experiments. One-way ANOVA with Tukey post hoc test, ****p-value < 0.0001. e Cartoon illustrating MERCS alterations in hiA from LRRK2G2019S patients (PD) compared with healthy subjects (Ctr). Enlargement of ER-OMM distance with specific reduction of 20 nm MERCS leads to failure of high [Ca2+] hot spots to reach mitochondria. The alterations are rescued by the expression of 20nm-EML but not by 10nm-EML. f Quantification of G-aggregates/monomers ratio of JC-1 by flow cytometry in Ctr-hiA and PD-hiA transfected with ER-EBFP and EBFP-expressing 10nm-EML and 20nm-EML. Data are expressed mean ± SEM of percent of 105 gated cells from three independent experiments. One-way ANOVA, Tukey post hoc test. ****p-value < 0.0001.
Discussion
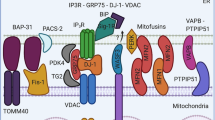
Since the discovery of the close apposition between ER and mitochondrial membranes to warrant a low-affinity mitochondrial Ca2+ uptake8,9, attempts have been made over the last three decades to estimate how ER-mitochondria interaction affects Ca2+ transfer. A range of ER-mitochondria distances between 10 and 25 nm has been hypothesized based on theoretical considerations14,15. A more narrow range of 10–15 nm was considered for the design of ER-mitochondria linking probes to quantify [Ca2+] in the ER-OMM cleft during IP3-mediated Ca2+ release12. However, our results do not support this scenario, suggesting that a larger, ∼20 nm space is required to maximize ER-mitochondrial Ca2+ transfer. Although, at 10 nm, IP3Rs may be recruited into MERCS, the Ca2+ transfer is significantly less efficient compared with control, suggesting that at 10 nm, the formation of functional IP3R-VDAC1 Ca2+ transferring units is inhibited. This conclusion is supported by the reduction of [Ca2+]CL and [Ca2+]M Ca2+ transients in 10nm-EML-expressing cells in spite of significant increase of the length of ER-OMM interface. Currently, it is unclear how 20 nm MERCS are organized and what is the mechanism of relocation of IP3Rs to MERCS. IP3Rs have several dozens of protein partners, many of which have been shown to promote IP3R-mediated Ca2+ signals at MERCS. Plausible candidates may include Sigma-1 receptor44 and IRE1α45 on the ER membrane, and Tom70 on the OMM46. A hypothetical ∼7–10 nm gap between IP3R and VDAC might accommodate bridging proteins such as GRP75, suggesting a stoichiometric formation of IP3R-GRP75-VDAC complex27. However, even if we show that both IP3R and VDAC1/3 are required for ER-mitochondrial Ca2+ transfer, our data do not support the hypothesis of physical interaction because we do not observe enrichment of both GRP75 and VDAC in 20 nm MERCS. Instead, our data are in line with a recent report suggesting that IP3R does not require physical linkage with a pore-forming protein in the OMM to warrant ER-mitochondrial Ca2+ flux47. However, it should be acknowledged that VDAC and GRP75 are highly abundant proteins, and their availability may not be a limiting factor for the formation of IP3R-GRP75-VDAC complex and the IP3R-mediated linkage to the OMM.
Impairment of mitochondrial oxidative metabolism and reduction of ATP production are common hallmarks of neurodegenerative diseases48. Recently, we reported that in both AD and PD astrocytes, basal respiration, respiratory reserve capacity, and ATP synthesis are impaired41,42. In AD astrocytes, bioenergetic deficit was associated with a reduction of mitochondrial Ca2+ uptake due to the shortening of the distance between ER and mitochondria with the specific increase of the interaction at 8–10 nm42,43, i.e., the distance at which Ca2+ transfer is impaired, according to our current data. This prompted us to investigate if PD astrocytes had alterations of the ER-mitochondrial interaction similar to those found in AD astrocytes. Strikingly, and contrarily to AD astrocytes, we found a dramatic enlargement of the space between organelles with a reduction of 20 nm contacts associated with severe impairment of mitochondrial Ca2+ uptake. This finding suggests that, in pathological conditions, impairment of mitochondrial bioenergetics, as a consequence of the deficit of Ca2+ signals, may result from both shortening (in the case of AD) and enlargement (in the case of PD) of the distance between ER and mitochondria. Therefore, normalization of the ER-mitochondrial Ca2+ transfer through modulation of the ER-mitochondrial interaction may represent a universal therapeutic approach to normalize cellular homeostasis in different pathological conditions characterized by the impairment of mitochondrial function.
Taken together, in this study we demonstrate that the ER-OMM distance is a critical parameter for ER-mitochondrial Ca2+ transfer and OXPHOS. We show that a narrow range of distances close to 20 nm is optimal for Ca2+ flux between the organelles; the shortening of the distance below 20 nm dramatically affects both mitochondrial Ca2+ uptake and OXPHOS. We provide a framework and extended molecular tool for manipulating mitochondrial function through precise control over ER-mitochondrial distance.
Methods
DNA constructs
ER-mitochondrial linkers
5nm-EML and 10nm-EML were a kind gift from Georgy Hajnoszky (Jefferson University, USA)12,16. Sequences of other linkers are reported in Supplementary Fig. 1.
Linkers are composed of targeting sequences for OMM (mouse AKAP1, residues 34–63) and ER (yeast Ucb6, residues 233–250)16; flexible linkers, rigid α-helical spacers (yellow), and mRFP1 sequence (https://www.fpbase.org/protein/mrfp1/). EMLs were synthesized and cloned in pcDNA3.1 using Nhe1 and Xho1 restriction sites by GenScript (https://www.genscript.com/). EBFP-containing EMLs were synthesized by substituting mRFP1 sequence with EBFP2 sequence (https://www.fpbase.org/protein/ebfp2/).
Split-GFP contact site sensors (SPLICS)
Generation of SPLICS-Short and SPLICS-Long was described elsewhere22,39. 20nm-SPLICS was synthetized by GenScript (https://www.genscript.com/) and cloned in pcDNA3.1. Sequence of 20nm-SPLICS is reported in Supplementary Fig. 13.
We deposited all original constructs in Addgene.
Cell lines
HeLa (https://www.atcc.org/products/ccl-2), Huh-7 (https://huh7.com/) cells, HeLa-IP3R1-EGFP cells28, HeLa-IP3R-TKO30, MEF-VDAC1,3-KO35, and their respective WT lines were maintained in complete culture media containing Dulbecco’s modified Eagle’s medium (DMEM; Sigma-Aldrich, Cat. D5671) supplemented with 10% fetal bovine serum (FBS, Gibco, Cat. 10270), 2 mM L-glutamine (Sigma-Aldrich, Cat. G7513), and 1% penicillin/streptomycin solution (Sigma-Aldrich, Cat. P0781).
Cell transfection
In all experiments, except MERCS fractionation, transient transfection of linkers and other plasmids was used. In this, 3 × 104 cells/well were resuspended in 250 µl of complete DMEM and 250 µl of transfection mix and plated onto 13 mm glass coverslips in 24-well plates. For the transfection mix, unless otherwise specified, 1 µl Lipofectamine 2000 (Thermo Fisher Scientific, Cat. 11668-019) and 1 µg plasmid DNA were mixed in Optimem (Gibco, Cat. 11058-021). After 3 h, transfection medium was replaced with complete medium. After 48 h, cells were used for experiments. For transfection of 20nm-SPLICS probe, 0.1 µg plasmid DNA per well was used for 24-well plate.
Cell viability assay
Crystal violet is a viability assay that discriminates between alive and dead cells in culture by employing a blue/violet dye exclusively binding to DNA and proteins in well-adherent, viable cells. HeLa and Huh-7 were seeded, respectively, at a density of 7.5 × 103 and 15 × 103 cells/well and transfected with 5nm-EML, 20nm-EML, and ER-RFP on 96-well plates. Forty-eight hours post-transfection, media was removed, and cells were fixed in methanol at 4 °C. After incubation for 10–20 min with 50 µl/well of 0.1% crystal violet, the dye was carefully removed, and each well was washed with phosphate-buffered saline solution (PBS). Then, plates were allowed to dry for 12 h and crystal violet was solubilized in 50 µl/well of 30% acetic acid. Lastly, absorbance at 595 nm was measured using Victor3V 1420 multilabel counter (Perkin Elmer).
Transmission electron microscopy
For transmission electron microscopy (TEM) analysis, following trypsinization, 1 × 106 cells were centrifuged at 900 rpm for 5 min and then fixed with 2.5% glutaraldehyde in culture medium, for 2 h at room temperature. The pellet was then rinsed in PBS, post-fixed in 1% aqueous OsO4 for 2 h at room temperature, and rinsed in H2O. Cells were pre-embedded in 2% agarose in water, dehydrated in a graded acetone scale, and then embedded in epoxy resin (Electron Microscopy Sciences, EM-bed812). Ultrathin sections (60–80 nm) were cut on a Reichert OM-U3 ultramicrotome, collected on nickel grids, and then stained with uranyl acetate and lead citrate. The specimens were observed with a JEM 1200 EX II (JEOL, Peabody, MA, USA) electron microscope operating at 100 kV and equipped with a MegaView G2 CCD camera (Olympus OSIS, Tokyo, Japan)49. Images were analyzed via ImageJ 1.54F.
For the ER-mitochondrial distance/interface analysis, the regions of juxtaposition between ER membrane and OMM were identified with a gap from 0 to 100 nm. These regions were considered as MERCS and the length of their extension was quantified as “MERCS interface” or “length of the ER-mitochondria interaction”. Transversal distance between ER and OMM was quantified manually using ImageJ “measure” tool. Within a single contact site with an extended interface, the transversal distances were measured along the interface with ∼20 nm intervals. A single contact of the same distance, defined by the continuous interface between ER and OMM, was considered as one point for statistical analysis.
Mitochondrial morphology was assessed by manually tracing OMM perimeter and morphological parameters were quantified using ImageJ “measure” tool.
Western blot
Forty-eight hours post-transfection, cells were lysed with lysis buffer (50 mM Tris-HCl (pH 7.4), sodium dodecyl sulfate (SDS) 0.5%, 5 mM EDTA), complemented with protease inhibitors cocktail (PIC, Millipore, Cat. 539133) and phosphatase inhibitor cocktail (Thermo Fisher Scientific, Cat. 78428), and collected in a 1.5 ml tube. Lysates were quantified with QuantiPro BCA Assay Kit (Sigma, Cat. SLBF3463). According to the relative abundance of the protein of interest, 20–40 µg of proteins were mixed with the right amount of Laemmli Sample Buffer 4X (Bio-Rad) and boiled. Then samples were loaded on a 6–12% polyacrylamide-sodium dodecyl sulfate gel for SDS-PAGE. Proteins were transferred onto nitrocellulose membrane, using Mini Transfer Packs or Midi Transfer Packs, with Trans-Blot® Turbo TM (Bio-Rad) according to the manufacturer’s instructions (Bio-Rad). The membranes were blocked in 5% skim milk (Sigma, Cat. 70166) for 45 min at room temperature. Subsequently, membranes were incubated with the indicated primary antibody overnight at 4 °C. Anti-β-Actin was used to normalize protein loading. A list of primary antibodies is provided in Table 1.
Goat anti-mouse IgG (H+L) horseradish peroxidase-conjugated (Bio-Rad, 1:5000; Cat. 170-6516) and Goat anti-rabbit IgG (H+L) horseradish peroxidase-conjugated secondary antibodies (Bio-Rad, 1:5000; Cat. 170-6515,) were used. Detection was carried out with SuperSignalTM West Pico/femto PLUS Chemiluminescent Substrate (Thermo Scientific, Cat. 34578), based on the chemiluminescence of luminol and developed using ChemiDocTM Imaging System (Bio-Rad).
Time-lapse ratiometric fluorescent imaging
Imaging of Fura-2, GAP3, 4mtD3cpv, and ROMO-GemGeCO Ca2+ probes was performed using an epifluorescent Leica DMI6000B microscope equipped with an S Fluor 40×/1.3 objective, a Polychrome V monochromator (Till Photonics, Munich, Germany), a Photometrics DV2 dual-imager (Teledyne Photometrics, Tucson, US). For imaging of mitochondria, an internal lens with a 1.6 optical increment was used. Images were acquired by a Hamamatsu cooled CCD camera (Hamamatsu Photonics, Hamamatsu City, Japan) and registered using MetaFluor software (Molecular Devices, Sunnyvale, CA, USA). Microsoft Excel and GraphPad Prism were used for offline analysis and figure preparation.
Mitochondrial Ca2+ imaging
Mitochondrial Ca2+ dynamics were monitored with 4mtD3cpv sensor, a genetically encoded Ca2+ indicator targeted to the mitochondrial matrix23. Forty-eight hours post-transfection, expression of 4mtD3cpv was checked, and mitochondrial matrix calcium dynamics were monitored. Coverslips were washed with Ca2+-containing KRB solution (125 mM NaCl, 5 mM KCl, 1 mM Na3PO4, 1 mM MgSO4, 5.5 mM glucose, 20 mM HEPES, pH 7.4, supplemented with 2 mM CaCl2), transferred to an acquisition chamber, and mounted on the stage of the microscope. Samples were illuminated at 420 nm and simultaneously acquired at 475 nm (donor, ECFP) and 530 nm (acceptor, circularly permuted (cp) Venus). CpVenus/ECFP ratio was calculated online using MetaFluor software. After acquisition of basal Ca2+ levels (first 30 s of acquisition), the cells were stimulated with 100 μM ATP. Regions of interest (ROIs) were defined around individual mitochondria.
Fura-2 Ca2+ imaging
Cells were plated onto 24 mm round coverslips (3 × 104 cell/coverslip), and loaded with 2.5 μM Fura-2/AM (Cat. No. F1201, Life Technologies, Milan, Italy) in the presence of 0.005% Pluronic F-127 (Cat. No. P6867, Life Technologies) and 10 μM sulfinpyrazone (Cat. S9509, Sigma) in Ca2+-containing KRB solution. After loading (30 min in the dark at RT), cells were washed once with KRB solution and allowed to de-esterify for 30 min. After this, the coverslips were mounted in an acquisition chamber and placed on the stage of the microscope, and cells were alternately excited at 340 and 380 nm; the fluorescent signal was collected through a 510/20 nm bandpass filter. The cells were stimulated with 100 μM ATP. For comparison of Ca2+ dynamics, measured as an amplitude of Ca2+ increase from the baseline level, Fura-2 ratio values were normalized using the formula (Fi-F0)/F0 [referred to as Normalized (Norm.) Fura Ratio].
Endoplasmic reticulum Ca2+ imaging
ER Ca2+ dynamics were monitored with ER-GAP3, a genetically encoded Ca2+ sensor, targeted to the ER lumen (referred to as GAP3)25. Forty-eight hours post-transfection, expression of GAP3 was checked, and ER calcium dynamics were monitored. Coverslips were mounted in a chamber in KRB solution and placed on the stage of the microscope. Cells were alternately excited at 405 and 470 nm, and the fluorescent signal was acquired using a 510/20 nm bandpass filter. After recording basal signal for 30 s, KRB solution was removed and replaced with a Ca2+-free solution (KRB + 500 µM EGTA). After allowing the signal to stabilize for an additional 30 s, cells were stimulated with 100 µM ATP and 100 µM Tert-butylhydroquinone (TBHQ), and the response was recorded for 300 s.
Ca2+ imaging in mitochondrial cristae lumen
Ca2+ dynamics in the mitochondrial cristae lumen (CL) were monitored with ROMO-GemGeCO, a genetically encoded Ca2+ indicator localized to the cristae lumen space, a kind gift from Wolfgang Graier (Medical University of Graz, Austria)26. Forty-eight hours post-transfection, coverslips were washed with Ca2+-containing KRB solution, transferred to the acquisition chamber and mounted on the stage of the microscope. Samples were illuminated at 420 nm and simultaneously acquired at 475 nm and 530 nm. Then, 530/475 nm ratio was calculated online using MetaFluor software. After acquisition of basal Ca2+ levels (first 30 s of acquisition), the cells were stimulated with 100 μM ATP. Regions of interest (ROIs) were defined around individual mitochondria.
Immunofluorescence (IF)
Cells, transfected or not according to the experimental design, were grown on 13 mm glass coverslips, fixed with 4% formaldehyde, permeabilized (7 min in 0.1% Triton X-100 in PBS), blocked in 1% gelatin, and immunoprobed with an appropriate primary antibody (diluted in PBS supplemented with 1% gelatin) overnight at 4 °C. After 3 times washing in PBS, an Alexa-conjugated secondary antibody (1:300 in PBS supplemented with 1% gelatin) was applied for 1 h at room temperature (RT). The following primary antibodies were used: anti-IP3R (rabbit, 1:500, Abcam, Cat. AB108517) and anti-VDAC1/3 (mouse, 1:250, Abcam, Cat. AB14734). Secondary antibodies were as follows: Alexa Fluor 488 anti-mouse IgG, Alexa Fluor 555 anti-rabbit IgG (all secondary antibodies were from Molecular Probes, Life Technologies, Monza, Italy). Nuclei were counter-stained with 4′,6-diamidino-2-phenylindole (DAPI). Images were acquired by Zeiss 710 confocal laser scanning microscope equipped with EC Plan-Neofluar 40×/1.30 Oil DIC M27 objective and Zen software or with a Leica SP8 LSCM equipped with a white light laser, and HCX PL APO 40×/1.25-075 OIL CL objective and LAS X software.
Proximity ligation assay (PLA)
In this, 1.75 × 104 cells/well were plated in 8-well chamber (IBIDI, Cat: 80806) and transfected with ER-mRFP, 5nm-EML, 10nm-EML and 20nm-EML. After 40 h, PLA was performed according to the manufacturer’s instructions (Duolink® Proximity Ligation Assay, Sigma). Briefly, cells were fixed in 4% paraformaldehyde, and incubated with primary antibodies anti-IP3R (1:500) and anti-VDAC1/3 (1:100) for 16 h at 4 °C. Duolink PLA probe incubation, according to the primary antibody species, was carried on for 1 h at 37 °C, and then the development of the signal was obtained by Duolink green fluorescence detection reagent by ligation and amplification reactions. Duolink in situ mounting media with DAPI was used. Images were acquired by Zeiss 710 laser scanning confocal microscope (LSCM) equipped with EC Plan-Neofluar 40×/1.30 Oil DIC M27 objective and Zen software.
Confocal images acquisition and processing
For both IF and PLA analyses, image stacks were acquired using nonsaturating parameters throughout the entire volume of the cell using a software-suggested (LAS X for Leica and Zen for Zeiss) resolution, inter-plane interval and number of images in the stack. For comparison, different conditions were acquired with fixed parameters of resolution, laser power and gain. Images were flattened using maximum intensity projection tool, exported as full-resolution TIFF images and assembled in figures using either Adobe Photoshop CS6 or Microsoft Office PowerPoint 2021.
Post-image analysis was performed using Fiji ImageJ 1.52p software. For colocalization analysis, data are expressed as fluorescence intensity/cell area ratio. For measuring colocalization of two fluorophores, we used an ImageJ JACoP toolbox for subcellular colocalization analysis50. Intracellular fluorescence was calculated for the entire cell area (CTCF) = Integrated Density − (Area of selected cell × Mean background fluorescence).
Generation of stable lines expressing ER-mRFP and 20nm-EML
To isolate MERCS-enriched cellular fraction, stable HeLa cell lines expressing ER-mRFP, 10nm-EML and 20nm-EML were generated. To generate ER-mRFP, 10nm-EML and 20nm-EML-expressing lentiviral backbones, ORFs were excised from pcDNA3.1 using Nhe1 and Xho1 restriction sites and cloned into Xba1 and Sal1 restriction sites, respectively, of pRRLSIN.cPPT.hCMV-GFP.WPRE vector. Lentiviral particles were produced and concentrated using the ultracentrifugation protocol described elsewhere51. For generation of stable lines, 24 h after plating (104 cells/well in 24-well plates), HeLa cells were infected with lentiviral particles at MOI from 5 to 20. The wells presenting more than 50% of infected cells, as detected by fluorescence of reporter proteins, were further processed. Cells were expanded, and ER-mRFP, 10nm-EML and 20nm-EML-expressing cells were enriched using fluorescence-activated cell sorting (S3e Cell Sorter, Bio-Rad, Segrate, Milano). Sorted cells were expanded, cryopreserved and stored in liquid nitrogen until needed.
Isolation of MERCS
HeLa stably expressing ER-mRFP (also referred to as ER-RFP), 10nm-EML and 20nm-EML were plated at a concentration of 0.5 × 106 cells/dish in 10 cm Petri dishes (50 dishes per line). Forty-eight hours later, cells were washed twice with PBS, detached with trypsin, and then collected in 50 ml centrifuge tubes. Cells were pelleted, and the pellets were processed according to the protocol described elsewhere52, using an Eppendorf CR30NX ultracentrifuge equipped with an R25ST rotor.
ATPlite™ assay
For the determination of total cell ATP using ATPlite™ assay (PerkinElmer, Cat. ATPLT-0415), cells were seeded at a density of 7.5 × 103 cell/well and transfected with control and EML-expressing plasmids in 96-well plates. Forty-eight hours post-transfection, 30 µl/well of mammalian cell lysis solution was added, and lysis was favored by shaking the plate at 400/500 rpm for 5 min. Then, 30 µl/well of substrate buffer solution (containing Luciferase and D-Luciferin) was added, and the plate was put again at 400/500 rpm for 5 min, protected from light according to the manufacturer’s instructions. After another 10 min of incubation, luminescence was measured using Victor3V 1420 multilabel counter (Perkin Elmer).
Mitochondrial membrane potential determination
JC-1 dye (Cayman, Ann Arbor, Michigan, USA, Cat.15003) was used according to the manufacturer’s instructions to assess mitochondrial membrane potential (ΔΨm). Control and EMLs expressing cells were resuspended in complete media to a final cell density of 1 × 106 cells. JC-1 dye was added to the cell suspension at a final concentration of 1 µg/µl and incubated for 15 min at 37 °C and 5% CO2. A negative control sample was prepared by adding an equal volume of vehicle to the cell suspension. After washing with warm PBS, cells were resuspended in PBS+EDTA (500 µM) and immediately acquired with a flow cytometer (Accuri C6 Plus BD 660517). The fluorescence was measured with excitation wavelength at 485 nm, dual emission filters at 529 and 590 nm, and cut-off at 515 nm. 105 events were acquired for each sample using the following parameters: forward scatter (FSC), side scatter (SSC), PE (red, JC-1 aggregate), and FITC (green, JC-1 monomer) fluorescence channels. Gating strategies were applied to exclude cell debris and doublets based on FSC and SSC properties. The mitochondrial membrane potential was determined as a ratio of JC-1 aggregates (red fluorescence) to monomers (green fluorescence). As a positive control, cells were treated with FCCP 10 µM (Tocris, Cat. 0453), for 5 min. Data analysis was performed using FlowJo software v10. Gated events were plotted on a bivariate dot plot to analyze the distribution of JC-1 fluorescence.
Oroboros high-resolution respirometry
Cellular respiration rates in real-time of intact control and EMLs expressing cells were measured using an Oroboros oxygraph-2K high-resolution respirometer (Oroboros Instruments, Innsbruck, Austria). The “substrate, uncoupler, inhibitor, titration” (SUIT) protocol, specifically SUIT-003_O2_ce_D012, was employed following the guidelines recommended by the manufacturer. Cells were detached from the plate, using trypsin-EDTA (Gibco, Cat 25200056), counted, and resuspended in pre-warmed respiration medium MiR05 (0.5 mM EGTA, 3.0 mM MgCl2, 60 mM potassium lactobionate, 20 mM taurine, 10 mM KH2PO4, 20 mM HEPES, 110 mM sucrose, 1 g/L bovine serum albumin, pH 7.1) to achieve a final cell density of 1 × 105 cells/ml. Pairs of ER-mRFP + 10nm-EML or ER-mRFP + 20nm-EML-expressing cells were analyzed simultaneously in the neighboring chambers, and oxygen concentration and flux were recorded using DatLab software (Oroboros). Baseline oxygen consumption rates were identified for each chamber during the “Routine” phase in the presence or absence of pyruvate (5 mM) stimulation, used to sustain TCA-linked respiration in MiR05 medium. Subsequently, oligomycin (5 nM) was added to inhibit ATP synthase and assess the uncoupled respiration (“Leak” phase). The protonophore carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone (FCCP) was then titrated (0.05 μM increments) until plateau of oxygen flux, indicative of maximal respiration, was achieved [“Electron transport (ET)” phase]. Finally, 1 μl each of rotenone (0.5 μM) and antimycin A (2.5 μM) were sequentially added to inhibit ETC complexes I and III, respectively, and to identify ET-independent respiration (ROX phase). Rates of O2 consumption (flux) were normalized to total protein content. Briefly, at the end of the experimental procedure, the cellular suspension from the two chambers was centrifuged at 1000 × g for 5 min. The cellular pellets were lysed in 200 µl of lysis buffer (10 mM HEPES, 60 mM KCl, 1 mM EDTA, 0.075% NP40, 1 mM DTT) and then centrifuged at 15,000 × g for 15 min at 4 °C. Protein concentration in the supernatant was measured with Bradford Reagent (Sigma, Cat. B6916).
Human induced astrocytes (hiA) derived from fibroblast of healthy donors and Parkinson’s disease patients with LRRK2G2019S mutation
Human fibroblasts were obtained from two healthy donors (Ctrl1 was purchased from AXOL-#AX0019 and Ctrl 2 from the Coriell stem cell bank-#ND291914) and two Parkinson’s disease patients with LRRK2G2019S mutation (PD1 from the Coriell stem cell bank-#PD33878 and PD 2 provided by the BioDonostia Hospital, San Sebastian, Spain). Generation and characterization of hiA have been described elsewhere41. hiA were differentiated from embryoid bodies, obtained in Aggrewell 800 (STEMCELL) from human iPSCs. Differentiation of NPC to progenitor astrocytes was triggered using the astrocyte differentiation medium (STEMdiff astrocyte differentiation #100-0013, StemCell). To maintain the appropriate cell density (70% of confluence), cells were passed every week in the same coating mix (human laminins 50% LN211 and 50% LN111 from Biolamina) for 21 days. Finally, astrocyte progenitor cells were maturated in the Astrocyte Maturation Medium (STEMdiff astrocyte maturation #100-0016, StemCell) for 60 to 75 days before running the experiments.
In experiments presented in Fig. 6, pairs Ctr1-vs-PD1 and Ctr2-vs-PD2 were analyzed separately. Given that both pairs yielded essentially the same results, to increase statistical power, the data of Ctr1 were pooled with Ctr2 and the data of PD1 were pooled with PD2. In experiments presented in Fig. 7, Ctr1 and PD1 were used.
Statistics and reproducibility
Statistical analysis was performed with GraphPad Prism software (GraphPad Software Inc., La Jolla, CA). A two-tailed unpaired Student’s t-test was used to compare two samples. To compare three or more samples, one-way ANOVA was used, followed by Tukey post hoc test, unless otherwise specified. A p-value < 0.05 was considered statistically significant. A full report on sample size, statistical methods and results is provided in Supplementary Data 1.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data generated or analyzed during this study are included in this published article and its supplementary information files. Full statistical report is provided in Supplementary Data 1. Source data underlying the graphs presented in the main and supplementary figures are provided as Supplementary Data 2 and 3, respectively. Original uncropped Western blots are provided in Supplementary Data 4.
References
Denton, R. M. Regulation of mitochondrial dehydrogenases by calcium ions. Biochim. Biophys. Acta 1787, 1309–1316 (2009).
Lee, S. H., Duron, H. E. & Chaudhuri, D. Beyond the TCA cycle: new insights into mitochondrial calcium regulation of oxidative phosphorylation. Biochem. Soc. Trans. 51, 1661–1673 (2023).
Granatiero, V., De Stefani, D. & Rizzuto, R. Mitochondrial calcium handling in physiology and disease. Adv. Exp. Med. Biol. 982, 25–47 (2017).
Giorgi, C., Marchi, S. & Pinton, P. The machineries, regulation and cellular functions of mitochondrial calcium. Nat. Rev. Mol. Cell Biol. 19, 713–730 (2018).
Bustos, G. et al. The ER-mitochondria Ca2+ signaling in cancer progression: fueling the monster. Int. Rev. Cell Mol. Biol. 363, 49–121 (2021).
Garbincius, J. F. & Elrod, J. W. Mitochondrial calcium exchange in physiology and disease. Physiol. Rev. 102, 893–992 (2022).
De Stefani, D., Patron, M. & Rizzuto, R. Structure and function of the mitochondrial calcium uniporter complex. Biochim. Biophys. Acta 1853, 2006–2011 (2015).
Rizzuto, R., Brini, M., Murgia, M. & Pozzan, T. Microdomains with high Ca2+ close to IP3-sensitive channels that are sensed by neighboring mitochondria. Science 262, 744–747 (1993).
Rizzuto, R. et al. Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science 280, 1763–1766 (1998).
Gincel, D., Zaid, H. & Shoshan-Barmatz, V. Calcium binding and translocation by the voltage-dependent anion channel: a possible regulatory mechanism in mitochondrial function. Biochem. J. 358, 147–155 (2001).
Rapizzi, E. et al. Recombinant expression of the voltage-dependent anion channel enhances the transfer of Ca2+ microdomains to mitochondria. J. Cell Biol. 159, 613–624 (2002).
Csordás, G. et al. Imaging interorganelle contacts and local calcium dynamics at the ER-mitochondrial interface. Mol. Cell 39, 121–132 (2010).
Csordás, G., Weaver, D. & Hajnóczky, G. Endoplasmic reticulum-mitochondrial contactology: structure and signaling functions. Trends Cell Biol. 28, 523–540 (2018).
Giacomello, M. & Pellegrini, L. The coming of age of the mitochondria-ER contact: a matter of thickness. Cell Death Differ. 23, 1417–1427 (2016).
Lim, D. et al. Ca2+ handling at the mitochondria-ER contact sites in neurodegeneration. Cell Calcium 98, 102453 (2021).
Csordás, G. et al. Structural and functional features and significance of the physical linkage between ER and mitochondria. J. Cell Biol. 174, 915–921 (2006).
Barnes, C. A. et al. Remarkable rigidity of the single α-helical domain of myosin-VI as revealed by NMR spectroscopy. J. Am. Chem. Soc. 141, 9004–9017 (2019).
Baek, M. et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science 373, 871–876 (2021).
Pernas, L. & Scorrano, L. Mito-morphosis: mitochondrial fusion, fission, and cristae remodeling as key mediators of cellular function. Annu. Rev. Physiol. 78, 505–531 (2016).
Baughman, J. M. et al. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 476, 341–345 (2011).
De Stefani, D., Raffaello, A., Teardo, E., Szabò, I. & Rizzuto, R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 476, 336–340 (2011).
Cieri, D. et al. SPLICS: a split green fluorescent protein-based contact site sensor for narrow and wide heterotypic organelle juxtaposition. Cell Death Differ. 25, 1131–1145 (2018).
Palmer, A. E. et al. Ca2+ indicators based on computationally redesigned calmodulin-peptide pairs. Chem. Biol. 13, 521–530 (2006).
Grynkiewicz, G., Poenie, M. & Tsien, R. Y. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J. Biol. Chem. 260, 3440–3450 (1985).
Navas-Navarro, P. et al. GFP-aequorin protein sensor for ex vivo and in vivo imaging of Ca2+ dynamics in high-Ca2+ organelles. Cell Chem. Biol. 23, 738–745 (2016).
Waldeck-Weiermair, M. et al. Development and application of sub-mitochondrial targeted Ca2+ biosensors. Front. Cell Neurosci. 13, 449 (2019).
Szabadkai, G. et al. Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J. Cell Biol. 175, 901–911 (2006).
Thillaiappan, N. B., Chavda, A. P., Tovey, S. C., Prole, D. L. & Taylor, C. W. Ca2+ signals initiate at immobile IP3 receptors adjacent to ER-plasma membrane junctions. Nat. Commun. 8, 1505 (2017).
Monaco, G. et al. The BH4 domain of anti-apoptotic Bcl-XL, but not that of the related Bcl-2, limits the voltage-dependent anion channel 1 (VDAC1)-mediated transfer of pro-apoptotic Ca2+ signals to mitochondria. J. Biol. Chem. 290, 9150–9161 (2015).
Ando, H., Hirose, M. & Mikoshiba, K. Aberrant IP3 receptor activities revealed by comprehensive analysis of pathological mutations causing spinocerebellar ataxia 29. Proc. Natl Acad. Sci. USA 115, 12259–12264 (2018).
Chami, M. et al. Role of SERCA1 truncated isoform in the proapoptotic calcium transfer from ER to mitochondria during ER stress. Mol. Cell 32, 641–651 (2008).
Lemos, F. O., Bultynck, G. & Parys, J. B. A comprehensive overview of the complex world of the endo- and sarcoplasmic reticulum Ca2+-leak channels. Biochim. Biophys. Acta Mol. Cell Res. 1868, 119020 (2021).
Hill, K. et al. Tom40 forms the hydrophilic channel of the mitochondrial import pore for preproteins [see comment]. Nature 395, 516–521 (1998).
Mazur, M., Kmita, H. & Wojtkowska, M. The diversity of the mitochondrial outer membrane protein import channels: emerging targets for modulation. Molecules 26, 4087 (2021).
Baines, C. P., Kaiser, R. A., Sheiko, T., Craigen, W. J. & Molkentin, J. D. Voltage-dependent anion channels are dispensable for mitochondrial-dependent cell death. Nat. Cell Biol. 9, 550–555 (2007).
Paillusson, S. et al. There’s something wrong with my MAM; the ER-mitochondria axis and neurodegenerative diseases. Trends Neurosci. 39, 146–157 (2016).
Area-Gomez, E. & Schon, E. A. On the pathogenesis of Alzheimer’s disease: the MAM hypothesis. FASEB J. 31, 864–867 (2017).
Vallese, F., Barazzuol, L., Maso, L., Brini, M. & Calì, T. ER-mitochondria calcium transfer, organelle contacts and neurodegenerative diseases. Adv. Exp. Med. Biol. 1131, 719–746 (2020).
Calì, T. & Brini, M. Quantification of organelle contact sites by split-GFP-based contact site sensors (SPLICS) in living cells. Nat. Protoc. 16, 5287–5308 (2021).
Tolosa, E., Vila, M., Klein, C. & Rascol, O. LRRK2 in Parkinson disease: challenges of clinical trials. Nat. Rev. Neurol. 16, 97–107 (2020).
Ramos-Gonzalez, P. et al. Astrocytic atrophy as a pathological feature of Parkinson’s disease with LRRK2 mutation. NPJ Parkinsons Dis. 7, 31 (2021).
Dematteis, G. et al. Proteomic analysis links alterations of bioenergetics, mitochondria-ER interactions and proteostasis in hippocampal astrocytes from 3xTg-AD mice. Cell Death Dis. 11, 645 (2020).
Tapella, L. et al. Protein synthesis inhibition and loss of homeostatic functions in astrocytes from an Alzheimer’s disease mouse model: a role for ER-mitochondria interaction. Cell Death Dis. 13, 878 (2022).
Hayashi, T. & Su, T.-P. Sigma-1 receptor chaperones at the ER-mitochondrion interface regulate Ca2+ signaling and cell survival. Cell 131, 596–610 (2007).
Carreras-Sureda, A. et al. Non-canonical function of IRE1α determines mitochondria-associated endoplasmic reticulum composition to control calcium transfer and bioenergetics. Nat. Cell Biol. 21, 755–767 (2019).
Filadi, R. et al. TOM70 sustains cell bioenergetics by promoting IP3R3-mediated ER to mitochondria Ca2+ transfer. Curr. Biol. 28, 369–382.e6 (2018).
Katona, M. et al. Capture at the ER-mitochondrial contacts licenses IP3 receptors to stimulate local Ca2+ transfer and oxidative metabolism. Nat. Commun. 13, 6779 (2022).
Area-Gomez, E., Guardia-Laguarta, C., Schon, E. A. & Przedborski, S. Mitochondria, OxPhos, and neurodegeneration: cells are not just running out of gas. J. Clin. Invest. 129, 34–45 (2019).
Casali, C. et al. Impact of heat and cold shock on epigenetics and chromatin structure. Eur. J. Cell Biol. 103, 151373 (2024).
Bolte, S. & Cordelières, F. P. A guided tour into subcellular colocalization analysis in light microscopy. J. Microsc. 224, 213–232 (2006).
Lim, D., Bertoli, A., Sorgato, M. C. & Moccia, F. Generation and usage of aequorin lentiviral vectors for Ca2+ measurement in sub-cellular compartments of hard-to-transfect cells. Cell Calcium 59, 228–239 (2016).
Wieckowski, M. R., Giorgi, C., Lebiedzinska, M., Duszynski, J. & Pinton, P. Isolation of mitochondria-associated membranes and mitochondria from animal tissues and cells. Nat. Protoc. 4, 1582–1590 (2009).
Acknowledgements
Centro Grandi Strumenti (CGS), Università di Pavia. Advanced microscopy facility, Center for Allergic and Autoimmune Diseases, CAAD, Università del Piemonte Orientale. EMBO short-term fellowship ASTF9854 (G.D.). CRT Foundation grant 1393-2017 (L.T.). Università del Piemonte Orientale grant FAR-2019 (D.L.). Next Generation EU, PRIN 2022 PNRR, grant P2022R43RA (D.L.). Royal Society Industry Fellowship grant NF\R2\212001 (G.L.). Biotechnology and Biological Sciences Research Council grants BB/T012986/1 and BB/W014831/1 (G.L.). CIBERNED grant CB06/05/0076 (C.M.). IKUR Strategy, Department of Education, Basque Government, Basque Country (P.R.G.). Italian Ministry of Education, University and Research (MIUR): Dipartimenti di Eccellenza Program (2018–2022)—Department of Biology and Biotechnology “L. Spallanzani”, University of Pavia (M.Bi.). This publication is part of the project MNESYS which has received funding from NextGeneration EU - MUR – M4C2 1.3 of PNRR with grant agreement no. PE00000006, CUP D33C22001340002 (L.T., M.G., D.L.).
Author information
Authors and Affiliations
Contributions
Conceptualization: G.D., L.T., A.A.G., D.L. Funding acquisition: G.D., L.T., D.L., G.L., P.R.G. Investigation: G.D., L.T., C.C., M.T., E.T., S.R., A.A., G.V., D.U., E.P., J.M., G.C., P.R.G., A.J., F.C., D.L. Methodology: G.D., G.L., K.M., T.C., M.Br., M.Bi., M.G., F.C., R.M., D.L. Project administration: G.D., D.L. Resources: G.D., L.T., M.T., C.D., M.G., G.L., N.F., L.G.F., K.M., C.M., F.C., A.A.G., D.L. Supervision: G.D., A.A.G., D.L. Writing—original draft: G.D., A.A.G., D.L. Writing—review & editing: G.D., L.T., C.C., M.T., E.T., S.R., M.G., G.L., N.F., L.G.F., C.M., A.J., T.C., M.Br., M.Bi., F.C., R.M., A.A.G., D.L.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks the anonymous reviewers for their contribution to the peer review of this work. Primary handling editors: Kaliya Georgieva. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Dematteis, G., Tapella, L., Casali, C. et al. ER-mitochondria distance is a critical parameter for efficient mitochondrial Ca2+ uptake and oxidative metabolism. Commun Biol 7, 1294 (2024). https://doi.org/10.1038/s42003-024-06933-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-024-06933-9
This article is cited by
-
Neurodegeneration models in Parkinson’s disease: cellular and molecular paths to neuron death
Behavioral and Brain Functions (2025)
-
Rescue of protein dyshomeostasis in hippocampal astrocytes from an Alzheimer’s disease mouse model by stabilizing ER-mitochondrial interactions at a 20 nm distance
Alzheimer's Research & Therapy (2025)
-
Calcium-mediated regulation of mitophagy: implications in neurodegenerative diseases
npj Metabolic Health and Disease (2025)
