
Abstract
Current treatments for ischemic stroke focus on removing neurovascular-occluding clots but overlook the resulting neuroinflammation. Clinical trials targeting brain-infiltrating peripheral immune cells have failed to improved outcomes, leaving mechanisms of neuroinflammation poorly understood. Whilst many studies have examined the inflammatory cells and cytokine profile of the post-stroke brain at days and weeks following injury onset, we propose that interventions at these timepoints are too late to limit brain damage. In this study, we examined brain immune cell composition, cytokine levels, and neurological dysfunction at hyperacute (3 h) and acute (24 h) stages following a preclinical mouse model of ischemic stroke. Interestingly, we detected elevated inflammatory cytokines in brain tissue as early as 3 h, notably before infiltrating neutrophil and monocyte arrival at 24 h. Depletion of peripheral immune cells by antibodies or genetic alteration did not dampen neuroinflammation, nor improve sensorimotor function. Intravital imaging of Cx3cr1gfp/+ mice showed that microglia, the brain-resident immune cell, display rapidly altered morphology, and swiftly upregulated cytokine production hyperacutely post-stroke. Altogether, our study provides evidence that microglia are key drivers of early neuroinflammation and modulating their function at clinically-relevant timepoints will improve stroke recovery.
Similar content being viewed by others

Introduction
Occlusion of cerebral arteries during ischemic stroke starves the brain of oxygen and nutrients, initiating a cascade of events that result in neuronal cell death, neuroinflammation and brain damage1,2,3. The existing therapeutic strategy of ischemic stroke aims to remove the occlusive embolus or clot and restore neurovascular perfusion4,5, yet there remains no therapeutic strategy that targets neuroinflammation to reduce its detrimental impact on the “salvageable” penumbra surrounding the ischemic core6. Multiple studies have separately reported various types of peripheral immune cells infiltrate the brain at days and weeks after stroke7,8, however, the lack of effective therapeutic is indicative of our limited understanding on the development of neuroinflammation after ischemic stroke. To this end, we propose that a characterization of the entire immune composition and cytokine levels in the brain during the hyperacute stage of stroke onset would provide insights into initiating the inflammatory response.
During homeostasis, entry of circulating materials into the brain is regulated by the blood-brain barrier (BBB), a neurovascular unit composed of endothelial cells, pericytes and astrocytes. However, BBB integrity is disrupted in response to local trauma or infection-driven inflammation, enabling both vascular solute permeability and cell trafficking9. Numerous studies have reported that neutrophils infiltrate brain tissue in both patients and experimental models of stroke10,11,12,13, and their inflammatory activity has been shown to be detrimental to the brain. In animal models of stroke, neutrophils have been shown to accumulate in the perivascular space and adjacent parenchyma within 12–24 h post-stroke, with more robust parenchymal infiltration typically evident at 1–3 days10,14. Monocyte-derived macrophages tend to infiltrate more slowly, generally becoming prominent after 48 h8,15. In humans, while imaging and CSF data suggest a similar delayed infiltration, definitive timing remains difficult to resolve due to variability in sampling16. Therapeutic strategies to block peripheral myeloid cell recruitment to the post-stroke brain reduced neuroinflammation in mice12,13,17. However, in clinical trials, interventions aimed at reducing neutrophil infiltration in the reperfusion phase have thrice failed (EAST, AUSTIN, and HALT trials)18,19,20. In fact, studies have shown a necessity for neutrophils infiltrating the brain to be phagocytosed by microglia, the brain-resident macrophage, to promote the resolution of inflammation21. To our knowledge, the brain-immune composition in the hyperacute (<6 h) phase has not been characterized. Similar to the acute (6–24 h) phase, we expect the immune composition of the brain at these timepoints to be largely dominated by microglia. Clearly, a greater understanding of neuroinflammation, specifically microglia function, in response to ischemic stroke is required.
As the tissue-resident immune cell in the brain, microglia are widely distributed and optimally positioned to detect and respond to brain injury. At steady state, microglia are mostly ramified to facilitate tissue surveillance, debris removal and neuronal pruning. They are highly dynamic cells that display multivariant morphological, metabolic and functional states in response to trauma22, capable of cytokine production and secretion. Cytokines play a pivotal role in driving neuroinflammation after stroke, however, the source and timing of their production in the post-stroke brain have been elusive. The aim of this study is to examine the cytokine profile of the post-stroke brain during hyperacute and acute phases of injury, and to investigate whether brain-infiltrating immune cells or brain-resident cells are key producers of these cytokines. This is a clinically relevant time point because it aligns with the accepted “therapeutic window” for effective treatment via tissue plasminogen activator. Intervention was originally approved based on the NINDS trial, which showed the most efficacy if administered <3 h of stroke onset23. Our study demonstrates that brain-infiltrating neutrophils and monocytes do not contribute to the initiation of neuroinflammation after ischemic stroke. In fact, a prominent inflammatory cytokine profile is detected in the brain markedly before the arrival of these brain-infiltrating myeloid cells. Our study provides evidence that microglia are the key producers of the major inflammatory cytokines (TNF-α, IL-6, and IL-1β) detected within the hyperacute phase of ischemic stroke, and modulating their function at a clinically relevant time point may improve stroke recovery.
Methodology
Mice
Adult male mice aged 7–10 weeks were used, and housed at a specific pathogen-free animal facility within the Monash Health Translational Precinct (MHTP-AF, Clayton, Australia). Conditions were maintained in a temperature- and humidity-controlled environment in a 12 h light/dark cycle, with access to food and water as required. Mice were acclimatized for ≥7 days before use. Wild-type C57BL/6J mice were obtained from the Monash Animal Research Platform (Clayton, Australia), whilst Cx3cr1gfp/+ mice were generated in-house by crossing Cx3cr1gfp/gfp mice24 with C57BL/6J mice. Neutrophil-reporter (CatchupIVM-red) mice25, and CCR2-deficient mice (Ccr2rfp/rfp) were also used26. All procedures were performed in accordance with Australian law (Victorian Prevention of Cruelty to Animals Act 1986) and were approved by the Monash Medical Center Animal Ethics Committee (MMCB/2021/32).
Stroke model
Photothrombotic (PT) strokes were performed as previously described27, under aseptic conditions. In brief, mice were intraperitoneally (IP) injected with rose bengal (20 mg/kg; Merck; Supplemental Table 1), before anesthesia induction by isoflurane (2–3%). Mice were positioned into a digital stereotactic frame (Stoelting Co.), fur on the head was removed, and a 1 cm incision was made over the parietal plane to expose the skull. 10 min post-rose bengal injection, a 1500 μm-diameter fiber optic cable was positioned over the sensorimotor cortex (2 mm right, 1 mm posterior of bregma)28 and the area was illuminated with a 532 nm cold laser (ThorLabs) for 15 min. After illumination, the wound was closed with surgical suture, and the mice were removed from isoflurane to quickly regain consciousness. Mice were allowed to recover on a heat pad. Sham mice were treated in an identical manner, but with no illumination.
VASCULAR INTEGRITY was determined at intervals post-stroke. Mice were anesthetized IP with a solution of ketamine (150 mg/kg) and xylazine (10 mg/kg), before an intravenous injection with Evans blue dye (80 mg/kg; Merck) or Dextran (70 kDa)—Rhodamine B (20 mg/kg; Invitrogen). The dye or dextran was allowed to circulate for 10 min before mice were perfused transcardially with ice-cold phosphate buffered saline (PBS). For vascular integrity visualization (Evans blue), the brain was removed from the skull, sectioned coronally at 2 mm, and images were captured using a Canon PowerShot SX730 HS mounted above a light box with Evans blue-positive tissue quantified using ImageJ v2.14. For volumetric quantification (70 kDa dextran), infarcted brain tissue was snap-frozen, before homogenization in 1 ml radioimmunoprecipitation buffer (#89900 ThermoFisher) in an “M-tube” (#130-093-236 Miltenyi Biotech) using a GentleMACS Octo Disassociator (Miltenyi Biotech). Lysate was centrifuged at 10,000 xg, and supernatant was collected, twice. Rhodamine was detected at 590 nm using an Infinite M Nano (Tecan) plate reader.
INFARCT SIZE was determined from 30 μm coronal sections taken every 210 μm and sections were stained with 0.1% thionin (Merck) to visualize tissue death. Images of thionin-stained brains were captured using a Canon PowerShot SX730 HS mounted above a light box and infarct volume was measured using ImageJ v2.14.
EDEMA SIZE was observed by magnetic resonance imaging (MRI). A Bruker 9.4 T animal MRI Scanner (Bruker BioSpin GmbH) with a Bruker 86 mm volumetric transmitter coil and a Bruker single-channel mouse head receiver coil was used to generate T2-weighted scans of the brain. Animals were anesthetized using 1% isoflurane prior to and for the duration of the scan, whilst respiration rate and body temperature were monitored throughout. The Bruker 2D T2-weighted RARE imaging sequence was used with the following parameters: TE/TR = 36 ms/7000 ms; RARE factor = 8; Field of view (FOV) = 16.8 × 16.8 mm2; number of slices = 26; slice thickness = 0.5 mm; matrix = 168 × 168; averages = 4; orientation = Axial. Images were processed using Mango v4.1 (University of Texas).
Neurological assessment
ADHESIVE TAPE REMOVAL TEST. At experimental end points, mice were moved from their housing cage and allowed to acclimatize in a clear testing container (22.5 cm × 16 cm x 10.5 cm) for 5 min. Mice were restrained as adhesive squares (3 mm; Implant media) were securely adhered onto the palm region of each forepaw—a red sticker on the right paw and green on the left. A recording device below the testing container was used to record the behavior of the mouse, and each animal was recorded for a maximum of 180 s to observe each adhesive being removed. All videos were compiled, and investigators were blinded to the identity of the animal during analysis. Investigators analyzed the time taken for the animal to perform the initial snout-to-paw contact (adhesive detection) and complete removal of each adhesive (adhesive removal).
WIRE HANG TEST. At experimental endpoints, mice were moved from their housing cage and allowed to grasp a metal wire that was fixed at a height of 40 cm above a thick layer of bedding. A maximum duration of each trial was set at 180 s, and the latency time of when each mouse fell was recorded. Mice were subject to a total of three trials with a 5-min break in between each trial, and the final latency time was calculated as the average of the three trials.
Peripheral myeloid cell depletion
Mice were treated with anti-Ly6C/Ly6G (RB6-8C5, #BE0075 BioXCell) or IgG2b isotype control (LTF-2, #BE0090 BioXCell) by IP injection at 10 mg/kg. Mice were dosed 24 h prior and 1 h prior to PT stroke induction. Myeloid depletion was confirmed by flow cytometry analysis of ammonium-chloride-potassium (ACK)-hemolyzed blood 24 h post-stroke, compared to isotype control. Depletion was further confirmed in whole blood using a VETSCAN HM5 Hematology Analyser (Zoetis; data not shown).
Brain inflammatory cytokine assessment
At 3 h and 24 h post-stroke, mice were anesthetized with ketamine/xylazine IP, then transcardially perfused with ice-cold PBS. The brain was removed from the skull (cerebellum removed), separated into target tissue locations, weighed and snap frozen. Frozen tissue was homogenized in 1 ml radioimmunoprecipitation buffer (#89900 ThermoFisher) containing complete, ethylenediaminetetraacetic acid (EDTA)-free protease inhibitor (#04693132001 Roche) in an “M-tube” (#130-093-236 Miltenyi Biotech) using a GentleMACS Octo Disassociator (Miltenyi Biotech). Lysate was centrifuged at 10,000 xg, and supernatant was collected twice before storage. Analytes were quantified using the LEGENDplex Mouse Inflammation Panel (740446 BioLegend) in accordance with the manufacturer’s instructions. LEGENDplex beads were acquired using an LSR X Fortessa (BD Biosciences) and analyzed using the LEGENDplex Analysis Software. Raw analyte concentration was normalized to the pre-determined tissue weight, and shown as pg/g.
Flow cytometry
At 3 h and 24 h post-stroke, mice were anesthetized with ketamine/xylazine IP. Blood was collected from the inferior vena cava into an EDTA-coated (0.5 M) 1 ml syringe and tube. Erythrocytes were lysed using ACK buffer for 10 min, before staining and analysis by flow cytometry. Then, the animal was transcardially perfused with iced-cold PBS, and the brain was harvested (cerebellum removed) and separated into target tissue locations. Brains were processed using an Adult Brain Dissociation Kit (#130-107-677, Miltenyi Biotech), modified from the manufacturer's instructions, using a “C tube” (#130-093-237, Miltenyi Biotech) with the 37C_ABDK_01 protocol of a GentleMACS Octo Disassociator with heaters. Upon dissociation, cells were isolated by centrifugation using a 30%/80% Percoll gradient (Bio-Strategy) at 1850 x g for 20 min without brake, at room temperature. Cells were collected from Percoll and washed twice with ice-cold PBS-EDTA (2 mM), before incubation with fixable viability stain (FVS)780 (1:1,000, #565388 BD Biosciences) for 20 min on ice. Cells were washed before blockade with PBS-fetal calf serum (FCS; 10%) with anti-CD16/32 (1:100, #553142 BD Biosciences) for 20 min on ice. Cells were then incubated with conjugated antibodies (Supplemental Table 1) against cell surface proteins in PBS-EDTA (2 mM) for 30 min on ice. Cells were fixed and permeabilized using a CytoPerm Solution Kit (#555028, BD Biosciences) for 20 min on ice, before incubation with conjugated antibodies against intracellular proteins in permeabilization wash buffer for 30 min on ice.
Cell cytokine production was assessed by isolating cells from the brain as described above. Isolated cells were cultured for 3 h in 2 ml complete Dulbecco’s Modified Eagle Medium (10% FCS, Penicillin-Streptomycin 100 U/ml and L-glutamine 0.5 mg/ml ThermoFisher) at 37 °C and 5% CO2 in a sterile, humidified atmosphere. Culture was performed in the presence of GolgiPlug (containing Brefeldin A, #555029 BD Biosciences) to retain cytokines in cells, and plates were agitated every 30 min to reduce adhesion. Cells were then stained for flow cytometry as above.
Cell acquisition was performed using an Aurora Spectral Flow Cytometer (Cytek Biosciences), and the entire sample was acquired. Cell count was calculated using a Colter Counter Analyzer (Beckman Coulter). Cell and UltraComp eBeads Plus (#01-2222-42 ThermoFisher) single color controls were used for spectral unmixing with SpectroFlo Software (Cytek Biosciences). Flow cytometry panel design was assisted by the Cytek Cloud software, and unmixed samples were processed using FlowJo v10.1.
Immunohistochemistry
At 3 h and 24 h post-stroke, mice were anesthetized with ketamine/xylazine IP, then transcardially perfused with iced-cold PBS and 4% paraformaldehyde (PFA). Upon harvesting, brains were incubated at 4 °C overnight in 4% PFA, and then 20% sucrose solution at 4 °C overnight, before snap freezing. 30 μm coronal sections were collected using a cryostat, and sections were washed with Tris-buffered saline (TBS)-T (0.3% Triton-X, 0.1% Tween20), before blockade with 1% bovine serum albumin (BSA) TBS-T for 1 h at room temperature. Sections were incubated with primary antibodies (Supplemental Table 1, 1:100) in 1% BSA-TBS-T overnight at 4 °C in a humidity box. Sections were thrice washed in TBS-T before incubation with fluorochrome-conjugated secondary antibodies (1:500) in 1% BSA-TBS-T for 1 h at room temperature. Sections were washed in TBS-T before incubation with DAPI (1:1,000, Sigma) in TBS-T for 10 min at room temperature. Sections were thrice washed in TBS-T, and mounted using Fluorescence Mounting Medium (#S302380-2 Dako). Tissue fluorescence was imaged using an Olympus VS120 slide scanner with a 20× lens, using DAPI as fine focus for automated acquisition.
Microscopy and image analysis
TISSUE TWO-PHOTON MICROSCOPY. Images of 30 µm thick, immunofluorescence-stained coronal brain sections taken from Cx3cr1gfp/+ mice were acquired using an Olympus FVMPE-RS multiphoton microscope. Three regions of brain tissue were identified in the same mouse in the ipsilateral (IL) tissue: the infarct core, the peri-infarct region, and a region distal to the infarct core. One FOV was acquired of the contralateral (CL) tissue, which was in a position mirroring the infarct location. Images were obtained using Galvano one-way scanning at 0.6 μm Z-intervals using a 20× water-immersion objective at a resolution of 512 × 512 pixels with 2-times frame averaging. Fluorescence images of GFP-positive microglia were obtained via scanning at 950 nm, and positive signals from Brilliant Violet 421-conjugated anti-CD49b (platelets) and Alexa Fluor 594-conjugated anti-laminin (vasculature) were obtained via scanning at 825 nm; both used 5% laser power. Images were first compiled and merged using ImageJ v2.14, and then morphological analysis was performed using Imaris v10.2.0 software (Oxford Instruments).
INTRAVITAL TWO-PHOTON MICROSCOPY. At 3 h and 24 h post-stroke, Cx3cr1gfp/+ mice were anesthetized with ketamine/xylazine IP, and their tail veins were cannulated for the addition of fluorescent antibodies or additional anesthetic, if required. The animal was then mounted in a stereotactic frame. Fur on the head was shaved, and a 1 cm incision was made in the skin to reveal the skull. The skull was thinned over the parietal plane in the identical region in each mouse, being the same area which had been illuminated previously to excite the Rose Bengal, creating a 3 mm x 3 mm square using a Microtorque dental drill (SDR Scientific), ensuring the skull remained intact. Focus was determined using a fluorescence-enabled eyepiece, and images were acquired using an Olympus FVMPE-RS multiphoton microscope. Imaging location was determined by the presence of vessel-occlusive thrombus in post-stroke mice, in the center of the thinned square. Images were obtained using Galvano one-way scanning at 1 μm Z-intervals using a 20× water-immersion objective at a resolution of 512 × 512 pixels with no averaging. Z-intervals began imaging below the dura, a structure which is easily distinguishable by its unique vessel shapes. Laser power increased with Z-intervals, beginning at 12%, and reaching 40% when imaging was 100 µm into the brain. Fluorescence images of GFP-positive microglia were obtained via scanning at 950 nm, and positive signals from Brilliant Violet 421-conjugated anti-CD49b (platelets) were obtained via scanning at 825 nm. Images were first compiled and merged using ImageJ v2.14, and then morphological analysis was performed using Imaris v10.2.0 software (Oxford Instruments).
RENDERING AND IMAGE ANALYSIS. Using Imaris v10.2.0, reconstruction of microglial dendrites was performed using the Filament Tracer tool, while reconstruction of microglial cell bodies was performed using the Surfaces tool. Cells were manually removed and discounted from analysis using IMARIS if the cell body extended beyond the XY plane. Cell counts, body sphericity and volume were determined using the surfaces tool, while measures of dendrites and dendrite branch number were determined using the filament tracer tool: starting point 18 µm; “Use surface as markers”; Seed point start 1.5 µm; seed threshold automatically defined; terminal ends 3 µm; cylinder 1.5 µm with detail 1 µm.
Statistical analysis
All statistical analyses were performed using Prism 10 (GraphPad Software Inc.). Data normality was tested using a Shapiro-Wilk normality test and analyzed as required. Outliers were excluded using Grubb’s test α ≤ 0.05. All values are presented as mean ± SD, unless otherwise stated. Statistical significance was represented by stars: *p < 0.05 **p < 0.01 ***p < 0.001 ****p < 0.0001, or “ns” for no significance.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Results
Stroke induces a rapid loss of cerebral vascular integrity and impairs motor function
The photothrombotic (PT) stroke model generates a consistent and reproducible cerebral ischemia injury in the cortical microvasculature, mimicking a lacunar-like infarct29. Mice were injected with photosensitive rose bengal (Fig. 1ai) and challenged with a cold laser (ii) to induce an infarct (iii) within the primary motor cortex (iv), but also covering the secondary motor and primary somatosensory regions. Whilst the stroke-induced infarct was notable at 3 h (2.7 ± 0.6 mm3), it developed 5-fold within 24 h (15.2 ± 4.0 mm3) (Fig. 1b). This is associated with a loss of cerebral vascular integrity within just 30 min of stroke induction, as demonstrated by leakage of Evans blue and Rhodamine B dextran (70 kDa) into the brain parenchyma (Figs. 1c and S1). This developed over the subsequent 3 h, and to a lesser extent, from 6 h onwards due to the occlusion of microvasculature. In parallel, brain weight increased by 10% 24 h post-stroke, as a result of the vascular leakage and edema (Fig. 1d). This is supported by intravital MRI of mice 24 h after stroke, identifying clearly demarcated volumes of localized edema in the brain surrounding the predicted stroke location (Fig. 1e and Video S1).

a Methodology to induce a photothrombotic (PT) stroke, (i) intraperitoneal injection of rose bengal (20 mg/kg), before (ii) 15 min excitation by a 532 nm cold laser, (iii) causing a sizeable, reproducible infarct above the (iv) primary motor cortex (outlined red), shown by Nissl stain (left) and anatomical annotations (right) from the Allen Mouse Brain Atlas [mouse.brain-map.org]. Image created with BioRender.com. b Representative image of thionin-stained 30 µm brain sections at 3 and 24 h post-PT stroke, and quantification of stroke infarct volume from stacked sections (sham n = 7, 3 h n = 7, 24 h n = 6). c. Representative images of 2 mm brain sections at intervals post-PT stroke, after intravenous injection of Evans blue (EB), and quantification of coverage (volume of EB-positive tissue; Sham n = 1/timepoint, PT n = 3/timepoint). d Weight of brains (minus cerebellum) at intervals post-stroke (n = 9–10). e A representative magnetic resonance imaging scan taken 24 h post-PT stroke in a live, isoflurane-anesthetized mouse. Red arrow indicates the area of edema (n = 4). Mice were assessed for sensory and motor function at 3 and 24 h post-PT stroke or sham control. Time taken to f detect or g remove an adhesive from the left and right paw was measured (sham n = 7, PT n = 9). Time mice were able to remain on a suspended wire at h 3 h and i 24 h post-stroke (n = 10). Graphs are presented as mean ± SD. A one-way ANOVA or Kruskal-Wallis test was performed for analysis of multiple groups, and a Mann–Whitney test was performed on the wire hang test. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, ns no significance.
Clinically, ischemic stroke induces severe neurological deficits amongst survivors, leaving up to 30% of patients with permanent, life-altering disabilities30. To determine whether neurological dysfunction is observed post-PT stroke, we performed the adhesive tape removal test, which assesses sensorimotor deficit31, and the wire hang test to assess muscle/motor function32. The time taken to detect and remove an adhesive from the left paw (CL to injury) did not differ from that on the right paw, or sham control, indicating that sensorimotor function was not impacted 24 h after stroke (Fig. 1f, g). In contrast, the wire hang test identified severe (73.8%) impairment of motor function within 3 h of stroke, compared to sham controls. This impairment was maintained (81.5% impairment vs sham) when tested 24 h post-stroke (Fig. 1h, i). These observations indicate that the severity of brain injury induced by the PT stroke used for these experiments is sufficient to result in significant motor dysfunction.
Inflammatory signals are detected in the brain prior to the infiltration of peripheral immune cells
To determine the degree of neuroinflammation in response to PT stroke over time, we quantified inflammation-associated cytokines in perfused brain tissue homogenates. Cytokine abundance was spatially quantified in each cerebral hemisphere, denoted as CL tissue and stroke-impacted IL tissue. The IL hemisphere was further dissected for to the “infarct core” and the tissue adjacent to the infarct (peri-infarct; Fig. 2a). A panel of inflammatory cytokines was quantified in the infarct core, peri-infarct and CL regions in mice at 3 or 24 h after PT stroke, and compared to sham controls (Fig. 2b). The concentration of inflammatory cytokines interleukin (IL)-1α, IL-1β, and tumor necrosis factor (TNF)-α were substantially increased in the infarct core within 3 h of a stroke, which was either maintained or further increased after 24 h, compared to controls (Fig. 2c–e). IL-6 was drastically increased in the infarct core by 24 h, but was not significantly different from sham controls within 3 h of stroke (Fig. 2f). Notably, minimal concentrations of cytokines were detected in peri-infarct tissue at 3 h or 24 h (Fig. S2a, b), suggesting the inflammation induced by the PT stroke does not penetrate the striatum. Granulocyte-colony stimulating factor and transforming growth factor-β1 were expressed at minimal levels in the infarct core after 24 h compared to controls, and interferon (IFN)-γ, IL-12p70, IL-23, granulocyte macrophage-CSF, IFN-β, IL-27, and M-CSF did not change significantly in the infarct core in response to stroke at 3 h or 24 h (Fig. S2c–k). IL-10, IL-17A, IL-18, VEGF, and CD40L were below the threshold of detection in all tissues, in all conditions (data not shown).

a Diagram outlining the regions of the brain, relative to infarct location: contralateral (CL), ipsilateral (IL), peri-infarct tissue and core-infarct tissue. Image created with BioRender.com. b–f Mice were subjected to photothrombotic (PT) stroke for 3 or 24 h, and perfused brains were homogenized, and supernatant was analyzed using a LEGENDplex cytokine panel. b Analytes taken from infarct core homogenates were measured for cytokines and presented as log2 fold-change over sham control. Concentration (pg/g of brain tissue) of c IL-1α d IL-1β, e TNF-α and f IL-6 in regions of the brain at 3 and 24 h post-stroke is shown (sham n = 10, 3 h PT n = 9, 24 h PT n = 10). g–l. Mice were subjected to PT Stroke for 3 or 24 h, perfused brains were dissociated, and cells were stained for analysis by spectral flow cytometry. g Gating strategy of single, viable cells (see also Fig. S2) to identify neutrophils, monocytes/monocyte-derived macrophages and microglia. Total count of h neutrophils (CD45hiCD11b+Ly6G+), i monocytes (CD45hiCD11b+Ly6G−F4/80loLy6C+), j monocyte-derived macrophages (CD45hiCD11b+Ly6G−F4/80midLy6C+) and k microglia (CD45midCD11b+Ly6G−F4/80hiLy6C−) in contralateral (CL) or ipsilateral (IL) regions of the brain (Sham n = 4, PT n = 6). l Representative fluorescent image of a 30 µm section of a CatchupIVM-red (neutrophil reporter; red) perfused mouse brain at 24 h post-stroke, with DAPI (blue) and anti-GPIbβ (platelets; cyan), imaged using an Olympus VS120 slide scanner. Arrows point to extravasated neutrophils, and the dotted line indicates the infarct core region (n = 3). Graphs are presented as mean ± SD. A one-way ANOVA was performed for the analysis of multiple groups. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, ns no significance.
The cellular origins of the cytokines produced in the brain during the hyperacute (3 h) post-stroke phase are unclear. Although it has been proposed that circulating neutrophils releases cytokines to drive neuroinflammation after stroke, the earliest detection of infiltrating neutrophils in the brain was at 24 h following stroke onset10,11,12,13,33. Thus, they are unlikely to be a major producer of inflammatory cytokines in the brain at these early time points. To address this, perfused brains were digested and analyzed by spectral flow cytometry. A gating strategy was used to identify viable neutrophils (CD45hiCD11b+Ly6G+), monocytes (CD45hiCD11b+Ly6G−Ly6C+F4/80lo), monocyte-derived macrophages (mono-macs; CD45hiCD11b+Ly6G−Ly6C+F4/80hi) and microglia (CD45midCD11b+Ly6G−Ly6C−F4/80hi) in the brain (Fig. 2g). Whilst we observed a small increase in neutrophil numbers in the IL tissue at 3 h post-stroke compared to CL or sham controls; this was negligible when compared to the 10-fold increase at 24 h post-injury (Fig. 2h). This is mirrored when assessing the stroke-induced brain infiltration of monocytes and mono-macs, both of which mildly increased at 3 h, but increased 5- and 8-fold after 24 h, respectively (Fig. 2i, j). The number of brain-resident macrophage cells, microglia, was unchanged in response to stroke (Fig. 2k), in agreeance with other pre-clinical stroke models11. The extravasation of immune cells was observed using the CatchupIVM-red (neutrophil reporter; red) mouse, whereby neutrophils were observed at very low levels in infarcted tissue 3 h post-stroke, but both within the infarct core and in the peri-infarct region 24 h post-stroke (Figs. 2l and S3a). Neutrophils were not observed in the CL tissue of post-stroke mice, nor in either hemisphere of the sham control (Fig. S3b, c). A limited number of lymphocytes (B cells CD45hiCD11b−CD19+; T cells CD45hiCD11b−CD3+) were identified in the brain at 3 h and 24 h post-stroke (Fig. S4a–e). These observations demonstrate that the arrival of peripheral immune infiltration occurs >3 h post-stroke, after the detection of inflammatory signals.
Manipulation of peripheral immune cells does not improve acute stroke outcome
To further investigate if brain-infiltrating peripheral myeloid cells mediate neuroinflammation after stroke, we depleted this population using anti-Ly6C/Ly6G (anti-Gr-1). This strategy successfully depleted peripheral blood neutrophils by 99.9% and monocytes by 79.1% compared to mice treated with IgG2b isotype control (Fig. 3a–c). Depletion was further confirmed using a hematology analyzer (data not shown). We then examined the effect of this intervention on stroke-induced inflammation-associated cytokines 24 h post-stroke. Mirroring previous observations, the concentration of IL-1α, IL-1β, IL-6, and TNF-α was increased in the infarct core, compared to CL tissue from the same mouse. However, there was no difference in inflammatory profile between peripheral myeloid-competent and -depleted mice (Figs. 3d and S5a–l). Moreover, there was no improvement in motor function in peripheral myeloid-depleted mice, compared to competent controls (Fig. 3e). These findings demonstrate that removal of peripheral myeloid cells does not modify stroke outcome in this experimental setting.

a–e Peripheral myeloid cells were depleted by the intraperitoneal injection of anti-Ly6C/Ly6G antibody (10 µg/g) on day -1 and day 0 of photothrombotic (PT) stroke induction. Myeloid-competent mice received an equivalent dose of IgG2b (IgG) control. a Gating strategy used for flow cytometry analyses of red blood cell-lysed blood to confirm myeloid cell depletion. Blood immune populations were further analyzed for b neutrophils (CD45hiCD11b+Ly6G+) and c monocytes (CD45hiCD11b+Ly6G−Ly6C+). d Perfused brains were homogenized, and tissue supernatant was analyzed using a LEGENDplex inflammatory cytokine panel. Analytes taken from infarct core homogenates were measured for cytokines and presented as log2 fold-change over contralateral (CL) control (IgG n = 6, Ly6C/G n = 8). e Time taken for mice to fall from a suspended wire. Graphs are presented as mean ± SD. An unpaired t test was used when comparing 2 groups. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, ns no significance.
Depleting myeloid cells by anti-Ly6C/Ly6G did not allow distinction between the effects of neutrophils and monocytes/monocyte-derived macrophages. This is critical in that mono-mac subpopulations have been shown to be both host-protective and damaging during neuroinflammatory events34,35, although little is known regarding their phenotype and function in response to ischemic stroke. Therefore, we next assessed the expression of activation markers on mono-macs in the brains of wild-type mice by flow cytometry at intervals after PT stroke. At 3 h after stroke, the activation markers on brain mono-macs were comparable to sham control, however, this was drastically altered by 24 h, as a result of the abundant infiltration of mono-macs. Relative to sham controls, 24 h post-stroke brains were enriched for mono-macs expressing CD80, CD86, MHC-II, Arginase-1, and EGR2, while expression of CD206 and iNOS was reduced in post-stroke mono-macs (Fig. 4a–g). Notably, these markers are characteristic of mono-macs of both inflammatory and inflammation-resolving phenotypes. Indeed, Arg1+CD80+MHC-II+ macrophages have previously been described in response to traumatic brain injury36, although their function is unknown.

a–g Mice were subjected to photothrombotic (PT) stroke or sham control for 3 or 24 h, perfused brains were dissociated, and cells were stained for analysis by spectral flow cytometry. Single, viable cells (also see Fig. S2) were used to identify monocyte-derived macrophages (mono-mac; CD45hiCD11b+Ly6G−F4/80midLy6C+). Mean fluorescence intensity of a CD206, b CD80 c CD86, d MHC-II, e Arg1, f EGR2 and g iNOS are displayed as fold-change versus sham control of the same region (IL—Ipsilateral, CL—contralateral; sham n = 4, PT n = 6). h–n C57BL/6 (WT—wild type) or Ccr2rfp/rfp (Ccr2−/−) mice were subjected to a PT stroke 24 h. h Deficiency of functional mouse CCR2 in Ccr2rpf/rfp mice. Image created with BioRender.com. Perfused brains were dissociated, and cell populations were analyzed by flow cytometry for i microglia (CD45midCD11b+Ly6G−F4/80hiLy6C−), j neutrophils (CD45hiCD11b+Ly6G+), k monocytes (CD45hiCD11b+Ly6G−F4/80loLy6C+) and l monocyte-derived macrophages (CD45hiCD11b+Ly6G−F4/80midLy6C+). m Perfused brains were homogenized, and the supernatant was analyzed using a LEGENDplex cytokine panel. Analytes taken from infarct core homogenate were assessed and compared to contralateral (CL) tissue, presented as log2 fold-change (C57BL/6 n = 6, ccr2−/− n = 8). n Time mice were able to remain on a suspended wire. Dotted line represents mean sham hang time (n = 6). A one-way ANOVA was used for analysis of multiple groups, and a Mann–Whitney test was performed when analyzing the wire hang test. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, ns no significance.
To investigate if the recruitment of mono-mac populations into the brain mediates post-stroke neuroinflammation, we utilized the Ccr2rfp/rfp (Ccr2−/−) mouse, which does not express functional CCR2 (Fig. 4h), allowing for the investigation of the contribution CCR2-expressing monocytes to neuroinflammation37. Peripheral immune cell populations were unaltered in Ccr2−/− mice compared to wild-type controls (Fig. S6a–d). Similarly, microglia count in the brain was unaltered in Ccr2−/− mice compared to controls after stroke (Fig. 4i). Consistent with previous observations, CCR2-competent mice displayed significant elevation of neutrophils (Fig. 4j), monocytes (Fig. 4k) and mono-macs (Fig. 4l) in the IL hemisphere of post-stroke brain at 24 h. In contrast, stroke-induced infiltration of these myeloid subsets was absent in Ccr2−/− mice (Fig. 4i–l). Perfused brain homogenate from Ccr2−/− and wild type mice was compared 24 h post-stroke, and no difference in inflammatory cytokine profile was observed (Figs. 4m and S7a–l). Moreover, Ccr2−/− mice displayed no improvement in motor function compared to wild-type controls (Fig. 4n). Taken together, these data suggest that neutrophils and mono-macs do not contribute to neuroinflammation or neurological deficits in the first 24 h following experimental PT stroke.
Microglia rapidly respond to stroke through morphological rearrangement
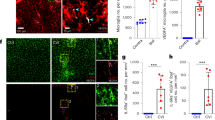
Our results indicate that infiltrating myeloid cells are not the contributors of inflammatory cytokines in post-stroke brain at hyperacute stages following injury. This raised the possibility that microglia, the brain-resident myeloid immune cell, was contributing to the neuroinflammation observed at this time point. Therefore, we examined the phenotype and function of microglia at 3 and 24 h post-stroke. In Initial assessments of phenotype by flow cytometry, microglia in the IL of post-stroke brains displayed minimal changes in classical pro-inflammatory (CD80, CD86, MHC-II, iNOS) or resolving (CD206, Arg1, EGR2) markers, as compared to sham or CL control (Fig. S8a–g). We next used Cx3cr1gfp/+ mice to analyze microglia morphology in coronal sections of post-stroke brain, stained with endothelial (anti-laminin; red) and platelet (GPIbβ; cyan) markers (Fig. S9a, b). Microglia in CL tissue have small, spherical cell bodies with numerous dendrites and as many as 8 branch levels (complexities). In the IL stroke-affected tissue, this was found to be altered within just 3 h of ischemic injury, when the number of microglial cells in the infarct core was significantly reduced (61.2% ± 4.2) compared to CL counterparts. Additionally, the microglia in IL had elevated cell body volume, whilst their dendrite number and complexity were reduced compared to CL controls (Fig. 5a–f). These stroke-induced changes in microglia morphology were exacerbated at 24 h post-stroke, at which we also observed microglia morphological changes in peri-infarct regions (Fig. 5g–k). Furthermore, of the microglia that remained in the infarct core at 24 h following stroke onset, there was a marked reduction in their dendrite complexity (Fig. 5l). Importantly, microglia were morphologically indifferent across these same regions in sham control mice (Fig. S10a–f). Interestingly, microglia in the infarct core and peri-infarct regions appear to be co-localized with thrombus-rich (GPIBβ, cyan) vasculature (Laminin, red) at hyperacute and acute timepoints. We next used two-photon intravital microscopy to investigate microglia morphology in vivo at 3 and 24 h post-stroke. The brains of Cx3cr1gfp/+ mice were imaged through a thinned skull over the stroke infarct location. Similar to histological imaging, microglia were found to have notably reduced dendrites and larger cell bodies within 3 h of stroke, with large blebbed cell bodies compared to naïve control (Fig. 6a, b, and Video S2). Moreover, at 24 h post-stroke, microglia dendrites were almost completely ablated (Fig. 6c). These observations demonstrate that microglial cells display severely altered morphology in the post-stroke brain.

Cx3cr1gfp/+ mice were subjected to a photothrombotic (PT) stroke for a–f 3 or g–l 24 h. Brain sections (30 µm) were immunofluorescently stained with anti-GPIbβ (platelets; cyan) and anti-laminin (vasculature; red) to complement the microglia green fluorescent protein (GFP)-reporter (green) and Z-stack images (0.5 µm intervals) were acquired using an Olympus FVMPE-RS two-photon microscope. a, g Representative images were taken at regions across the brain, relative to stroke location. Raw Z-compressed images were 3-D rendered (using IMARIS v10) for morphological analysis. CX3CR1+ microglia were assessed for b, h count per field of view (FOV), c, i cell body volume, d, j cell body sphericity, e, k dendrites per cell and f, l dendrite complexity (n = 5 mice; individual cells are plotted for volume and sphericity [≈n = 500/region]). A repeated measures one-way ANOVA was used for analysis of multiple groups. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, ns no significance.

Cx3cr1gfp/+ mice were intravenously administered with anti-CD49d-BV421 (platelets; cyan) to complement microglia-gfp (green) and were either a naive control or subjected to photothrombotic (PT) stroke for b 3 or c 24 h. The skulls of anesthetized mice were thinned over the parietal plane, and brains were imaged using a two-photon microscope. 100 µm of brain was imaged with 1 µm Z-stack steps. Images shown display (left) 10 µm of Z-compressed images and (right) 100 µm of 3-D rendered images using IMARIS v10. Images are representative of 3 mice/condition.
Microglia and neurons are responsible for inflammatory cytokine production in the hyperacute response to stroke
Given that we have demonstrated peripheral myeloid cells play a minimal role in the production of inflammatory cytokines at hyperacute stages of stroke, but at the same time we detected dramatic changes in microglial morphology, we next examined if brain-resident cells are critical players for initiating post-stroke neuroinflammation. To do this, we isolated and assessed the inflammatory cytokine production of brain-resident populations at 3 h after PT stroke, including microglia (CD45midCX3CR1+), astrocytes (CD45loCX3CR1-GLAST-1+), endothelial cells (CD45loCX3CR1-CD31+), pericytes (CD45loCX3CR1-PDGFRβ+) and neurons (CD45loCX3CR1-NeuN+) (Fig. 7a), which accounted for >85% of cells in the brain. We were particularly interested in which cell types were accountable for the stroke-induced production of IL-1α, IL-1β, IL-6, and TNF-α at 3 h post-stroke (Fig. 2b–f). We found astrocytes, endothelial cells, and pericytes had negligible alterations in production of these cytokines in the core tissue, compared to CL control (Fig. S11a–l). Interestingly, neurons displayed a mild inflammatory profile in the core tissue, compared to CL control, as shown by increases in production of IL-1β (34% increase), IL-6 (20% increase), and TNF-α (31% increase) following stroke (Fig. 7b–e). However, microglia markedly upregulated production of IL-1β (50% increase), IL-6 (44% increase) and TNF-α (30% increase) (Fig. 7f–i). IL-1α expression was reduced in both neurons (46%) and microglia (60%) in core tissue compared to CL control (Fig. 7d, h). This was not surprising, as IL-1α is a canonical alarmin which is pre-stored and released in response to trauma. Thus, IL-1α was likely released in situ in the brain, prior to cell culture38,39. Together with the knowledge that peripheral immune cells are mostly absent at 3 h post-stroke, these findings indicate that microglia are the most notable source of inflammatory cytokines IL-1β, IL-6 and TNF-α in the hyperacute post-stroke brain. Given these findings, we conclude that microglia are critical in the instigation of the neuroinflammatory response to ischemic stroke.

Cx3cr1gfp/+ mice were subjected to photothrombotic (PT) stroke for 3 h, perfused brains were dissociated, and brain cells were cultured in the presence of GolgiPlug for 3 h, before staining for analysis by spectral flow cytometry. a Single, viable cells (strategy shown in Fig. S2) were used to identify microglia (CD45midCX3CR1+), astrocytes (CD45loGLAST-1+), endothelial cells (CD45loGLAST-1−CD31+), pericytes (CD45loGLAST-1−PDGFRβ+) and neurons (CD45loGLAST-1−NeuN+). Neurons isolated from contralateral (CL), ipsilateral (IL) and core brain regions were assessed for the expression of b IL-6, c TNF-α, d IL-1α, and e IL-1β (n = 6). Microglia isolated from CL, IL and core brain regions were assessed for the expression of f IL-6, g TNF-α, h IL-1α, and i IL-1β (n = 6). Data is presented as fold-change in median fluorescence intensity (MFI) versus the CL tissue. A repeated measures one-way ANOVA was used for analysis of multiple groups. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, ns no significance.
Discussion
Infarcted tissue in the brain post-stroke is irreparable. However, the ischemic tissue surrounding the infarct core, the penumbra, can be salvaged if stroke-driven neuroinflammation is sufficiently controlled. Based on the results of pre-clinical studies, it was believed that brain-infiltrating peripheral immune cells were major contributors to neuroinflammation after stroke. However, clinical trials of interventions aimed at blocking myeloid cell infiltration into the brain failed to improve patient outcomes, and even worsened symptoms in some patients18,19,20. In this study, we demonstrate that peripheral myeloid leukocytes do not contribute to the development of a neuroinflammatory response during the hyperacute stage of ischemic stroke. This is supported by our observation that inflammatory cytokines are present in the brain in abundance 3 h following stroke onset, notably before the arrival of significant numbers of infiltrating myeloid cells, and depletion of these cells fails to modify stroke outcomes over the subsequent 24 h. These observations provide evidence why therapeutically targeting these infiltrating leukocytes is likely to have minimal protective effects in patients. In contrast, at this post-stroke timepoint, microglia show evidence of activation and produce inflammatory cytokines in and around the stroke-affected tissue in a spatially discriminated manner. Consistent with the literature, neurons also participate in the development of neuroinflammation40, although to a lesser extent. In light of this, our findings provide evidence that microglia-initiated neuroinflammation is an important contributor to brain injury and neurological deficits post-stroke.
There is a growing body of evidence to suggest the function of microglia during inflammation is determined by the cause, location and timing of injury. During infection-driven neuroinflammation, microglia have host-protective functions in pathogen phagocytosis, astrocyte activation and peripheral immune cell recruitment35,41,42. However, during sterile injury such as traumatic brain injury or stroke, the function of microglia is controversial. The role of microglia in ischemic stroke has been investigated in experimental stroke models, which target different brain regions and cause brain injury of diverse severity. The permanent middle cerebral artery occlusion (MCAO) model of stroke causes a large infarct (≈60 mm3) in the brain striatum27, while the PT model induces a smaller infarct (≈15 mm3) in the brain parenchyma. Depletion of microglia pre-MCAO model increases infarct size, indicating microglia are crucial for post-stroke neuroprotection21,43,44. However, in the PT model, infarct size was reduced in the absence of microglia45. The latter finding is in agreeance with data in the present study, which demonstrates microglia as a major source of inflammatory cytokines, and drivers of hyperacute neuroinflammation. In line with these data, it is logical to surmise that the net effect of microglia in the PT model is exacerbation of brain injury. In contrast, it is likely that the severity of the damage caused by the MCAO model potentially disables the ability of microglia to respond to the stimuli expressed in the ischemic brain. Instead, we speculate that microglia uptake MCAO-driven damage-associated molecular patterns (DAMPs) to protect neurons, and then undergo apoptosis, limiting an exaggerated inflammatory response. Which of these two mechanisms translates to ischemic stroke in humans is unclear, but as with mice, this is likely dependent on the location, severity and longevity of ischemia.
Our data indicate that microglia undergo early activation in the hyperacute phase of ischemic stroke, as evidenced by significant morphological remodeling and increased cytokine secretion. Interestingly, these functional and structural changes occur in the absence of significant alterations in microglial number or in the expression of classical activation markers, such as CD80, MHC-II, or CD206. This apparent discrepancy underscores the limitations of binary M1/M2 classification schemes22, particularly at very early timepoints. Previous studies have demonstrated that morphological and secretory changes can precede transcriptional shifts or surface marker upregulation46,47. Our findings suggest that during the hyperacute window, microglia may adopt a transitional activation state that is not well captured by canonical marker panels, highlighting the need for more temporally resolved and functionally nuanced definitions of microglial activation.
The BBB is integral to maintaining brain homeostasis and blocking DAMP-brain crosstalk. A loss of BBB integrity is observed in response to stroke, both clinically and in pre-clinical models48,49,50. Whilst microglia form part of the neurovascular unit51, microglia do not contribute to BBB integrity in homeostatic conditions49, nor does their depletion worsen BBB leakage during ischemic stroke43. However, the abundant and uniform distribution of microglia across the brain perfectly positions them to robustly respond to BBB breakdown. The release and/or extravasation of DAMPs, including S100A8, hemin, HMGB1, and fibrinogen during stroke is detrimental to tissue survival52,53,54,55,56. Consistent with this, microglia express an abundance of receptors for DAMPs on their cell membranes, including purine receptors, “receptor for advanced glycation end products”, Mac-1 (CD11b/CD18), toll-like receptors. We demonstrated a loss of vascular integrity within 30 min of PT stroke onset, which facilitates the extravasation of circulating DAMPs to exacerbate neuroinflammation. The impact of blood-DAMPs on microglia has been extensively researched in the context of neurodegenerative disease, where the generation/extravasation of fibrin has been shown to promote microglia clustering and axonal damage57,58. In contrast, little is known regarding the role of fibrin in hyperacute neuroinflammation. Blocking fibrin generation in patients after stroke improved neurological function, although it is unclear whether this benefit is microglia-dependent59. The two-hit of ischemia (tissue hypoxia) and the resultant DAMP generation and extravasation are detrimental to microglia, and the current reperfusion strategy only targets ischemia. Targeting the brain-vascular-immune interface may be the best strategy to dampen neuroinflammation60, subsequently salvaging the penumbra, and improving patient prognosis after stroke.
In this study, we have demonstrated that neutrophils nor CCR2-expressing monocytes are required for the development of hyperacute neuroinflammation following ischemic stroke. This is in agreeance with the previous negative clinical trials that tested interventions aimed at inhibiting leukocyte recruitment into the brain18,19,20. The hyperacute abundance of inflammatory cytokines observed in the present study, notably before the acute infiltration of peripheral immune cells, demonstrates that this recruitment is a consequence, rather than an initiator of neuroinflammation. We are yet to study the function of brain-infiltrating immune cells beyond 24 h in the PT model. However, learnings from other experimental models of stroke indicate that peripheral myeloid cells have neuro-protective function in the later stages of the response, via promotion of the resolution of neuroinflammation. It was demonstrated that neutrophils extravasate and interact with microglia in the cerebral infarct and surrounding tissue61,62. Microglia actively phagocytose both live and dead extravasated neutrophils in the stroke infarct core63,64,65, subsequently promoting the resolution of inflammation. It is unclear whether the resultant resolution of inflammation is a result of the neutrophil clearance or if microglia need to phagocytose neutrophils to re-polarize and return to their homeostatic, pro-resolving state. The interplay between microglia and neutrophils, and their potential resolving function, may explain why targeting the infiltration of peripheral immune cells into the brain failed to benefit patients after stroke.
We acknowledge the limitations of this study. Elucidation of microglia function is complicated by caveats regarding the techniques for their depletion. Microglia greatly depend on the signaling of colony-stimulating factor-1 receptor (CSF-1R) for development and survival. Thus, these cells can be pharmacologically depleted from the brain using CSF-1R inhibitors66. Variants of the CSF-1R inhibitor, PLX, are used across the field, which have a range of microglia-depleting efficiency. However, the shared properties of microglia with CSF-1R-expressing peripheral immune cells and other tissue-resident macrophages mean that any depletion strategies would have the undesired effect of also removing these subsets, meaning that any result observed from this intervention cannot be definitively ascribed to microglia67,68. Adding to this complexity, single-cell RNA-seq studies have identified as many as 16 distinct clusters of microglial cells following experimental ischemic stroke69. Our study has provided evidence that microglia are dominant producers of inflammatory cytokines in the hyperacute phase following ischemic stroke. We used an ex vivo culture approach to quantify the production of cytokines, but have not discriminated for microglia subtype contribution. Furthermore, astrocytes have been shown to rapidly respond to ischemia, notably through reactive gliosis (and later glial scar formation)70, which was not assessed with our experimental design. Future studies implementing a single cell “omics” approach will delineate heterogeneous cell populations and further characterize the roles glial cells play in the early response to ischemia. Evidently, there is a need for new techniques for specific depletion of microglia for experimental study, as a means to better understand their function. Furthermore, we accept that expression of CX3CR1 is not limited to microglia, and is known to be presented on a sub-population of non-classical monocytes and monocyte-derived macrophages. However, these cell types are not common in the brain at the acute timepoints here chosen following stroke onset, and the Cx3cr1gfp/+ mouse is a well-accepted tool for microglia morphological and functional analysis in the community22.
A recent perspective by Paolicelli et al. has defined numerous subpopulations of microglia, including disease-associated, IFN-responsive, glioma-associated, lipid droplet-accumulating and proliferation-region-associated22, with many of the recent discoveries made through the innovative use of brain intravital microscopy coupled with “omics” analysis71. Here, our study demonstrates that microglia spatially located adjacent to the infarct core robustly modify their morphology and are key producers of inflammatory cytokines, with the potential to drive the development of neuroinflammation. Experimentally targeting microglia to limit neuroinflammation, such as blocking purinergic signaling or inflammasome activation, has had preclinical success. However, therapeutically modulating microglia inflammatory activity, without impacting immune cells globally, remains a key translational challenge. Emerging techniques such as targeted nanoparticles72, coupled with receptor-targeted peptides, may allow for more precise and successful intervention in the future.
Data availability
All raw data are available in supplemental information. Any data that support the findings of this study which are not shown here are available from the corresponding author upon reasonable request.
References
DiSabato, D. J., Quan, N. & Godbout, J. P. Neuroinflammation: the devil is in the details. J. Neurochem 139, 136–153 (2016).
Gauberti, M. et al. Ischemia-reperfusion injury after endovascular thrombectomy for ischemic stroke. Stroke 49, 3071–3074 (2018).
Wu, L. et al. Targeting oxidative stress and inflammation to prevent ischemia-reperfusion injury. Front. Mol. Neurosci. 13, 28 (2020).
Tissue plasminogen activator for acute ischemic stroke. N. Engl. J. Med. 333, 1581–1587 (1995).
Berkhemer, O. A. et al. A randomized trial of intraarterial treatment for acute ischemic stroke. N. Engl. J. Med. 372, 11–20 (2015).
Ramos-Cabrer, P., Campos, F., Sobrino, T. & Castillo, J. Targeting the ischemic penumbra. Stroke 42, S7–S11 (2011).
Cai, W. et al. Functional dynamics of neutrophils after ischemic stroke. Transl. Stroke Res. 11, 108–121 (2020).
Wattananit, S. et al. Monocyte-derived macrophages contribute to spontaneous long-term functional recovery after stroke in mice. J. Neurosci. 36, 4182–4195 (2016).
Varatharaj, A. & Galea, I. The blood-brain barrier in systemic inflammation. Brain Behav. Immun. 60, 1–12 (2017).
Akopov, S. E., Simonian, N. A. & Grigorian, G. S. Dynamics of polymorphonuclear leukocyte accumulation in acute cerebral infarction and their correlation with brain tissue damage. Stroke 27, 1739–1743 (1996).
Wanrooy, B. J. et al. Brain-associated innate leukocytes display diverse inflammatory states following experimental stroke. Immunol. Cell Biol. 100, 482–496 (2022).
Matsuo, Y. et al. Role of cell adhesion molecules in brain injury after transient middle cerebral artery occlusion in the rat. Brain Res. 656, 344–352 (1994).
Shibuya, N., Itokazu, T., Ueda, T. & Yamashita, T. Intravital imaging reveals the ameliorating effect of colchicine in a photothrombotic stroke model via inhibition of neutrophil recruitment. Transl. Stroke Res. 14, 100–110 (2023).
Enzmann, G. et al. The neurovascular unit as a selective barrier to polymorphonuclear granulocyte (PMN) infiltration into the brain after ischemic injury. Acta Neuropathol. 125, 395–412 (2013).
Gliem, M. et al. Macrophages prevent hemorrhagic infarct transformation in murine stroke models. Ann. Neurol. 71, 743–752 (2012).
Beuker, C. et al. Immune cell infiltration into the brain after ischemic stroke in humans compared to mice and rats: a systematic review and meta-analysis. Transl. Stroke Res. 12, 976–990 (2021).
Dimitrijevic, O. B., Stamatovic, S. M., Keep, R. F. & Andjelkovic, A. V. Absence of the chemokine receptor CCR2 protects against cerebral ischemia/reperfusion injury in mice. Stroke 38, 1345–1353 (2007).
Use of anti-ICAM-1 therapy in ischemic stroke: results of the Enlimomab Acute Stroke Trial. Neurology 57, 1428–1434 (2001).
Krams, M. et al. Acute stroke therapy by inhibition of neutrophils (ASTIN): an adaptive dose-response study of UK-279,276 in acute ischemic stroke. Stroke 34, 2543–2548 (2003).
Veltkamp, R. & Gill, D. Clinical trials of immunomodulation in ischemic stroke. Neurotherapeutics 13, 791–800 (2016).
Otxoa-de-Amezaga, A. et al. Microglial cell loss after ischemic stroke favors brain neutrophil accumulation. Acta Neuropathol. 137, 321–341 (2019).
Paolicelli, R. C. et al. Microglia states and nomenclature: a field at its crossroads. Neuron 110, 3458–3483 (2022).
Group, N. I. o. N. D. a. S. r.-P. S. S. Tissue plasminogen activator for acute ischemic stroke. N. Engl. J. Med. 333, 1581–1587 (1995).
Jung, S. et al. Analysis of fractalkine receptor CX(3)CR1 function by targeted deletion and green fluorescent protein reporter gene insertion. Mol. Cell Biol. 20, 4106–4114 (2000).
Hasenberg, A. et al. Catchup: a mouse model for imaging-based tracking and modulation of neutrophil granulocytes. Nat. Methods 12, 445–452 (2015).
Saederup, N. et al. Selective chemokine receptor usage by central nervous system myeloid cells in CCR2-red fluorescent protein knock-in mice. PLoS ONE 5, e13693 (2010).
Shim, R. et al. Stroke severity, and not cerebral infarct location, increases the risk of infection. Transl. Stroke Res. 11, 387–401 (2020).
Wang, Q. et al. The allen mouse brain common coordinate framework: a 3D reference atlas. Cell 181, 936–953.e920 (2020).
Yaghi, S. et al. Lacunar stroke: mechanisms and therapeutic implications. J. Neurol. Neurosurg. Psychiatry https://doi.org/10.1136/jnnp-2021-326308 (2021).
Dimyan, M. A. & Cohen, L. G. Neuroplasticity in the context of motor rehabilitation after stroke. Nat. Rev. Neurol. 7, 76–85 (2011).
Bouet, V. et al. The adhesive removal test: a sensitive method to assess sensorimotor deficits in mice. Nat. Protoc. 4, 1560–1564 (2009).
Hoffman, E. & Winder, S. J. A. Modified wire hanging apparatus for small animal muscle function testing. PLoS Curr. 8 https://doi.org/10.1371/currents.md.1e2bec4e78697b7b0ff80ea25a1d38be (2016).
Otxoa-de-Amezaga, A. et al. Location of neutrophils in different compartments of the damaged mouse brain after severe ischemia/reperfusion. Stroke 50, 1548–1557 (2019).
Russo, M. V., Latour, L. L. & McGavern, D. B. Distinct myeloid cell subsets promote meningeal remodeling and vascular repair after mild traumatic brain injury. Nat. Immunol. 19, 442–452 (2018).
De Vlaminck, K. et al. Differential plasticity and fate of brain-resident and recruited macrophages during the onset and resolution of neuroinflammation. Immunity 55, 2085–2102.e2089 (2022).
Hsieh, C. L. et al. Traumatic brain injury induces macrophage subsets in the brain. Eur. J. Immunol. 43, 2010–2022 (2013).
Kuziel, W. A. et al. Severe reduction in leukocyte adhesion and monocyte extravasation in mice deficient in CC chemokine receptor 2. Proc. Natl. Acad. Sci. USA 94, 12053–12058 (1997).
Kim, B. et al. The interleukin-1α precursor is biologically active and is likely a key alarmin in the IL-1 family of cytokines. Front. Immunol. 4, 391 (2013).
Batista, S. J. et al. Gasdermin-D-dependent IL-1α release from microglia promotes protective immunity during chronic Toxoplasma gondii infection. Nat. Commun. 11, 3687 (2020).
Joy, M. T. et al. CCR5 is a therapeutic target for recovery after stroke and traumatic brain injury. Cell 176, 1143–1157.e1113 (2019).
Drummond, R. A. et al. CARD9+ microglia promote antifungal immunity via IL-1β- and CXCL1-mediated neutrophil recruitment. Nat. Immunol. 20, 559–570 (2019).
Sun, H. et al. Bacteria reduce flagellin synthesis to evade microglia-astrocyte-driven immunity in the brain. Cell Rep. 40, 111033 (2022).
Szalay, G. et al. Microglia protect against brain injury and their selective elimination dysregulates neuronal network activity after stroke. Nat. Commun. 7, 11499 (2016).
Marino Lee, S., Hudobenko, J., McCullough, L. D. & Chauhan, A. Microglia depletion increase brain injury after acute ischemic stroke in aged mice. Exp. Neurol. 336, 113530 (2021).
Li, T. et al. Specific depletion of resident microglia in the early stage of stroke reduces cerebral ischemic damage. J. Neuroinflamm. 18, 81 (2021).
Ransohoff, R. M. A polarizing question: do M1 and M2 microglia exist? Nat. Neurosci. 19, 987–991 (2016).
Masuda, T. et al. Author correction: novel hexb-based tools for studying microglia in the CNS. Nat. Immunol. 21, 1302 (2020).
Ng, F. C. et al. Microvascular dysfunction in blood-brain barrier disruption and hypoperfusion within the infarct posttreatment are associated with cerebral edema. Stroke 53, 1597–1605 (2022).
Profaci, C. P. et al. Microglia are not necessary for maintenance of blood-brain barrier properties in health, but PLX5622 alters brain endothelial cholesterol metabolism. Neuron 112, 2910–2921.e2917 (2024).
Okada, T., Suzuki, H., Travis, Z. D. & Zhang, J. H. The stroke-induced blood-brain barrier disruption: current progress of inspection technique, mechanism, and therapeutic target. Curr. Neuropharmacol. 18, 1187–1212 (2020).
Haruwaka, K. et al. Dual microglia effects on blood brain barrier permeability induced by systemic inflammation. Nat. Commun. 10, 5816 (2019).
Gülke, E., Gelderblom, M. & Magnus, T. Danger signals in stroke and their role on microglia activation after ischemia. Ther. Adv. Neurol. Disord. 11, 1756286418774254 (2018).
Ha, J. S. et al. Hypoxia-induced S100A8 expression activates microglial inflammation and promotes neuronal apoptosis. Int. J. Mol. Sci. 22 https://doi.org/10.3390/ijms22031205 (2021).
Zille, M. et al. Hemin-induced death models hemorrhagic stroke and is a variant of classical neuronal ferroptosis. J. Neurosci. 42, 2065–2079 (2022).
Schuhmann, M. K. et al. Danger-associated molecular patterns are locally released during occlusion in hyper-acute stroke. Brain Behav. Immun. Health 15, 100270 (2021).
Han, L. et al. Fibrinogen deposition promotes neuroinflammation and fibrin-derived γ(377-395) peptide ameliorates neurological deficits after ischemic stroke. Int. Immunopharmacol. 131, 111831 (2024).
Davalos, D. et al. Fibrinogen-induced perivascular microglial clustering is required for the development of axonal damage in neuroinflammation. Nat. Commun. 3, 1227 (2012).
Mendiola, A. S. et al. Defining blood-induced microglia functions in neurodegeneration through multiomic profiling. Nat. Immunol. 24, 1173–1187 (2023).
Zhang, X. et al. Argatroban in patients with acute ischemic stroke with early neurological deterioration: a randomized Clinical Trial. JAMA Neurol. 81, 118–125 (2024).
Akassoglou, K. et al. Pioneering discovery and therapeutics at the brain-vascular-immune interface. Cell 187, 5871–5876 (2024).
McColl, B. W., Rothwell, N. J. & Allan, S. M. Systemic inflammatory stimulus potentiates the acute phase and CXC chemokine responses to experimental stroke and exacerbates brain damage via interleukin-1- and neutrophil-dependent mechanisms. J. Neurosci. 27, 4403–4412 (2007).
Neumann, J. et al. Very-late-antigen-4 (VLA-4)-mediated brain invasion by neutrophils leads to interactions with microglia, increased ischemic injury and impaired behavior in experimental stroke. Acta Neuropathol. 129, 259–277 (2015).
Hubert, V. et al. Multimodal imaging with NanoGd reveals spatiotemporal features of neuroinflammation after experimental stroke. Adv. Sci. 8, e2101433 (2021).
Neumann, J. et al. Beware the intruder: real time observation of infiltrated neutrophils and neutrophil-Microglia interaction during stroke in vivo. PLoS ONE 13, e0193970 (2018).
Li, L. et al. Resolvin D1 reprograms energy metabolism to promote microglia to phagocytize neutrophils after ischemic stroke. Cell Rep. 42, 112617 (2023).
Green, K. N., Crapser, J. D. & Hohsfield, L. A. To kill a microglia: a case for CSF1R inhibitors. Trends Immunol. 41, 771–784 (2020).
Basilico, B. et al. What microglia depletion approaches tell us about the role of microglia on synaptic function and behavior. Front. Cell Neurosci. 16, 1022431 (2022).
Dye, C. N., Franceschelli, D., Leuner, B. & Lenz, K. M. Microglia depletion facilitates the display of maternal behavior and alters activation of the maternal brain network in nulliparous female rats. Neuropsychopharmacology 48, 1869–1877 (2023).
Patir, A. et al. Phenotypic and spatial heterogeneity of brain myeloid cells after stroke is associated with cell ontogeny, tissue damage, and brain connectivity. Cell Rep. 43, 114250 (2024).
Shen, X.-Y. et al. Activation and role of astrocytes in ischemic stroke. Front. Cell. Neurosci. 15, 2021 (2021).
Suthya, A. R., Wong, C. H. Y. & Bourne, J. H. Diving head-first into brain intravital microscopy. Front. Immunol. 15, 1372996 (2024).
Ramírez-García, P. D. et al. A pH-responsive nanoparticle targets the neurokinin 1 receptor in endosomes to prevent chronic pain. Nat. Nanotechnol. 14, 1150–1159 (2019).
Acknowledgements
This work was supported by a Monash SCS Seed Grant and a CASS Foundation Grant awarded to J.H.B. (10310) and a Heart Foundation Vanguard Grant, awarded to C.H.Y.W. and J.H.B. (106921). C.H.Y.W. is supported by a Heart Foundation Future Leaders Fellowship (107214). Funding sources have no role in the research conceptualization or direction. The authors would like to thank Monash Biomedical Imaging Facility for the use of the MRI machine, Monash Animal Research Platform for assistance with mouse colony maintenance, Monash Histology Platform for processing brains for histology, and Monash Micro Imaging Facility for assistance with microscopy. We especially thank Michael Thomson and Dr Svenja Loering of the Monash Health Translational Precinct Flow Core facility for assistance with optimization of spectral flow cytometry.
Author information
Authors and Affiliations
Contributions
J.H.B. designed and performed research, collected, analyzed and interpreted data, and wrote the manuscript. C.H.Y.W. designed the research, interpreted the data, and wrote the manuscript. A.R.S. and B.J.W. performed research, collected and analyzed data. J.L.W., S.W.W., C.R.B. and G.Z. performed research. M.R. and M.J.H. interpreted data.
Corresponding author
Ethics declarations
Competing interests
C.H.Y.W. is an Editorial Board Member for Communications Biology, but was not involved in the editorial review of, nor the decision to publish this article. All other authors declare no competing interests.
Consent for publication
All authors have critically reviewed the manuscript and consent to publication.
Ethics approval and consent to participate
All procedures were performed in accordance with Australian law (Victorian Prevention of Cruelty to Animals Act 1986) and were approved by the Monash Medical Center Animal Ethics Committee (MMCB/2021/32).
Peer review
Peer review information
Communications Biology thanks Wei Gong and Todd Peterson for their contribution to the peer review of this work. Primary handling editors: Eliana Scemes and Benjamin Bessieres. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Bourne, J.H., Suthya, A.R., Wanrooy, B.J. et al. Microglia are prominent producers of inflammatory cytokines during the hyperacute phase of ischemic stroke. Commun Biol 8, 1193 (2025). https://doi.org/10.1038/s42003-025-08636-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-025-08636-1
This article is cited by
-
Microglia-astrocyte crosstalk following ischemic stroke
Molecular Brain (2025)
