
Abstract
Excessive signaling by various GPCRs underlies a variety of human disorders. Suppression of GPCRs by “enhanced” arrestin mutants was proposed as therapy. We hypothesized that GPCR binding of endogenous arrestins can be increased by small molecules stabilizing pre-activated conformation. Using molecular dynamics, we identified potentially druggable pockets in pre-activated conformation of arrestin-3 and discovered a compound targeting one of these pockets. Saturation-transfer difference NMR data showed that the compound binds at the back loop of arrestin-3. FRET- and NanoBiT-based assays in living cells showed that the compound increased in-cell arrestin-3, but not arrestin-2, binding to basal β2-adrenergic receptor and its phosphorylation-deficient mutant, but not to muscarinic M2 receptor. These experiments demonstrated the feasibility of enhancing the binding of endogenous wild type arrestin-3 to GPCRs in a receptor-specific and arrestin-subtype selective manner.
Similar content being viewed by others

Introduction
G protein-coupled receptors (GPCRs) are involved in various physiological and pathophysiological processes and targeted by about a third of clinically used drugs1. Agonist binding to GPCRs triggers a series of conformational rearrangements, which are propagated to the cytoplasmic tip of the receptor2. According to the canonical mechanism, the active GPCR conformation initiates G protein-mediated signaling. G protein-coupled receptor kinases (GRKs) phosphorylate certain serine and threonine residues on the cytoplasmic elements of the activated receptor3. Consequently, active and phosphorylated receptor recruits another cytosolic protein, arrestin, which terminates G protein-mediated signaling4,5,6 and activates alternative signaling pathways7,8,9.
Mammals express four arrestin subtypes10. Arrestin-1 and arrestin-4 are visual, expressed in photoreceptors, whereas arrestin-2 (β-arrestin-1) and arrestin-3 (β-arrestin-2) are non-visual arrestins expressed in virtually every cell in the body. All arrestins are elongated molecules consisting of an N- and a C-domain11. In the basal state, the carboxy-terminal tail (C-tail) of arrestin folds back and is anchored to the N-domain of the protein, thereby stabilizing the basal conformation. Arrestin binding to the GPCR requires receptor attached phosphates and an active receptor conformation12,13,14,15. The requirement for GPCR phosphorylation among arrestin proteins differs. Arrestin-1 absolutely requires rhodopsin phosphorylation for tight binding, whereas in case of arrestin-2 and arrestin-3 phosphorylation requirement depends on the receptor type3.
Receptor binding induces several conformational rearrangements in arrestin, the most prominent being the release of the C-tail and twisting of the two domains relative to each other by ~20o16,17,18,19,20,21,22. These are triggered by the phosphorylated parts of the receptor that bind to the N-domain of arrestin, displacing the C-tail and releasing the structural constraints that stabilize its basal conformation. The interdomain rotation assists rearrangement of the central-crest loops, the gate, back, lariat and finger loop, each of which is engaged in the formation of the high-affinity complex with the receptor16,19,20,22.
Arrestin mutants with destabilized basal conformation bind GPCRs, including their non-phosphorylated forms, better than parental wild type (WT) proteins. Therefore, these mutants have been suggested as therapeutic tools for to correct defects of phosphorylation-deficient receptors, which cause a variety of human disorders, including retinitis pigmentosa23, certain forms of cancer24, diabetes25, and others26. However, these mutants usually demonstrate reduced thermal stability and receptor selectivity27, limiting their therapeutic potential.
An alternative approach is the stabilization of receptor-bound-like arrestin conformations by small molecules. Arrestin-3 is intrinsically flexible, transiently sampling these conformations28. Comparison of structures of arrestins in the basal and receptor-bound state has revealed remarkable rearrangements in certain regions. As compared to the basal, the receptor-bound state features a more exposed C loop (residues 249–254 in arrestin-2 and 244–249 in arrestin-3), rearranged gate loop (residues 295–306 in arrestin-2 and 290–299 in arrestin-3)29 and unfolded back loop (residues 313–317 in arrestin-3, 312–316 in arrestin-2; 318–322 in arrestin-1)30,31,32. Intermediate state of arrestin-1 (PDB ID:4ZRG &3UGU) features structural properties that fall between basal and receptor-bound states.
Here, we tested whether stabilization of receptor bound-like conformation enhances arrestin-3 recruitment to a non-phosphorylated GPCR. A computational drug discovery approach, based on targeting these conformations of arrestin-3 extracted from molecular dynamics (MD) simulations, identified a small molecule predicted to bind arrestin-3. The binding of the molecule was verified by saturation-transfer difference (STD) NMR. We showed that this compound enhanced arrestin-3 binding to incative WT and activated phosphorylation-deficient β2-adrenergic receptor (β2AR), but not to muscarinic M2 receptor. No impact on arrestin-2 recruitment to either receptor was observed, indicating selectivity of the compound for the arrestin-3.
Results
Basal arrestin-3 samples receptor bound-like conformations
MD simulations of basal arrestin-3 revealed a marked flexibility of the interdomain interface30. To explore spontaneously adopted GPCR bound-like states for in silico drug discovery, we ran three ~1 μs MD trajectories in explicit water (Supplementary Fig. 1 for RMSDs of the replicates). Conformation of the back loop, the rearrangement of the gate loop16,17,18,19,20,21,22,33 and inter-domain rotation were monitored as indicators of global conformational rearrangements. We selected two reaction coordinates to classify the state of arrestin-3 along the MD trajectories. These are the pair-distances between: 1) the Cα atoms of residues K313 and G317 located at the N- and C-terminus of the back loop, respectively, (blue in Fig. 1A), 2) the Cα atoms of residues D291 and H296 on the gate loop (red in Fig. 1A), which report on conformational changes occurring upon binding of the receptor. Simulation snapshots were classified as basal/pre-activated/active based on the reference values of these two parameters in the corresponding structures (See Methods for definitions of the values and Supplementary Table 1 for a list of structures). Importantly, these reference values are well separated in basal and receptor-bound arrestins (Fig. 1B), which enabled us to link the conformation of arrestin-3 to its functional state. Arrestin-3 sampled basal/pre-activated/active states of the back loop in (68/29/3)%, (59/40/1)%, and (15/54/31)% of the three independent simulations. Also, pre-activated, albeit not fully active, states of the gate loop were sampled in 72, 60, and 50% of them, respectively. The population of active and pre-activated states was confirmed in a separate accelerated molecular dynamics (aMD) simulation: here, the back loop showed a distribution of basal/pre-activated/active states (61/33/6)%, respectively, whereas the gate loop was in pre-activated states in 78% of the trajectory. The destabilization of the aromatic core at the interdomain interface was also proposed as an activation-related change30. We found that it populates basal/pre-activated/active states in (10/79/11)%, (12/41/46)% and (17/82/1)% of the three arrestin-3 trajectories. However, we did not consider targeting this region as it is involved in GPCR binding, interacting with the intracellular loop 2 of the receptor16.

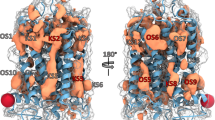
A Residue positions K313 and G317 at the termini of the back loop (blue), and D291 and H296 for gate loop (pink) are shown on the 3D structure of arrestin-3 (PDB ID: 3P2D)28 together with the residues of the polar core (green) and three-element region (yellow) as well as inter-domain aromatic core (purple). B Distribution of the distances used to identify the conformational state of the gate loop and the back loop in arrestin-3 trajectories and those measured in active/pre-activated and inactive structures of arrestins (See Supplementary Table 1 for the complete list of structures). Values for inactive and receptor-bound structures are shown in red and green dots, respectively. Notably, inositol hexakisphosphate-bound arrestin-3 (PDB ID:5TV1)16 adopts an extremely long distance (See green dot outlier) at the gate loop, due to the perturbation induced by the IP6 molecule near the polar core. C Superposition of traces of two extreme conformations from the principal component analysis of arrestin-3 structures, aligned on the N terminal domain, showing the rotation axis and direction (See Methods). D Table summarizing the occurrence of rotated states in arrestin-3-compound complexes and the average rotation angle.

A All atom structure of basal arrestin-3 highlighting the two interface regions targeted by drug discovery: the back loop characterized by the short helix (red box) and the gate loop (blue box). B Structures of the candidate compounds selected after virtual screening and close-up of complexes: from left to right, LSH-1, LSH-2, LSH-3, LGL.
Targeting the back loop stabilizes receptor- bound-like inter-domain rotation in arrestin-3
We set out to find small molecules that stabilize receptor bound-like conformations sampled in MD simulations. These conformational states, identified by the reaction coordinates defined above, were sufficiently populated in the MD trajectory dataset to extract snapshots for in silico drug discovery. We chose seven structures (Supplementary Fig. 2), characterized by a diverse set of conformational states of the back loop, the gate loop, and interdomain rotation (Supplementary Table 2). In three of the selected snapshots the back loop was in the basal state and the gate loop was in the pre-activated state; in two the back loop was in the activated state and the gate loop was in the basal state, in the other two, the back loop was in the activated state and the gate loop was in the pre-activated state (Supplementary Table 2). In these selected conformations, the inter-domain rotation angle fluctuates between active and basal values (Supplementary Table 2).
We then searched for available binding pockets on these selected structures focusing on the interdomain interface. Two consensus high-ranking pockets (see Methods, and Supplementary Table 3 for the properties of pockets identified) were found in this region. One of them is located near the back loop and the other one at the gate loop and both consistently emerge in multiple detection methods (see Methods and Supplementary Table 4).
Virtual screening on these pockets using a dataset of compounds from ZINC purchasable small molecules (see Methods and Supplementary Table 5) yielded ~2000 hits, which were further analyzed by docking and MD simulations of the predicted complexes with arrestin-3. One compound, referred to as LGL, bound stably to the gate loop throughout the course of 1 μs simulation (Supplementary Fig. 3). On the back loop pocket, we first selected 22 highest ranking candidate compounds (LSH series) based on their docking scores (Supplementary Table 6), ADME predictors (Methods), and binding poses (Methods and Fig. 2C, D). Among them, 10 compounds stably bound to the back loop (Methods for the list of compounds that left the target site within 50 ns or shorter) in a 1 μs MD trajectory. To test whether these compounds stabilize the binding competent conformation of arrestin-3, we measured the inter-domain rotation angle in the MD simulations of complexes. Within the LSH series, we observed a pre-activated rotation angle for three of them (Supplementary Table 6, highlighted green). The LSH-3 complex adopted a rotated state in 46% of the trajectories with an average value of 12.90, whereas LSH-1 and LSH-2 (Fig. 2C, D), stabilized a rotation angle ~6.50 (Fig. 1D). Despite stabilizing a lower inter-domain rotation angle, we also included LGL in experimental studies to compare the effect of a compound that is bound to the gate loop. We also examined the dynamics of the back loop and the gate loop in the arrestin-3/compound complexes. The gate loop parameters (RMSD and the distance between Cα atoms of D291 and H296, both of which were expected to increase upon activation, Supplementary Fig. 5), are more perturbed with bound LSH-3 than in the other complexes, in agreement with the trend observed for the inter-domain rotation angle (Fig. 1D). Also, the distance between the gate loop and the polar core34, in arrestin-3/compound trajectories, monitored by the side chains of D291 and R170 (D296 and R175 in arrestin-1) increases (5.0 Å) with LSH-3 relative to the crystal of basal arrestin-3 (PDB ID: 3P2D).
Interestingly, the perturbation of the back loop induced by the compounds is not always associated with the stabilization of pre-activated inter-domain rotation. For instance, LSH-1 stabilized the back loop in the unfolded state (Supplementary Fig. 5A), but this did not perturb the gate loop, leading to sampling lower inter-domain rotation angles (Supplementary Fig. 5B). This suggests that both stabilization of the back loop in the unfolded state and perturbation of the gate loop toward pre-activated state are required to sample active-like inter-domain rotation angles.
LSH-3 binds arrestin-3 at the interdomain interface
To validate the interaction between arrestin-3 and LSH-3, STD NMR was employed. STD-NMR is a powerful biophysical method for identifying binding between small molecules and larger molecules, facilitating the screening of compounds35. The STD-NMR spectrum and the binding epitope (the protons in close contact with the protein) are shown in Fig. 3. Protons H14 and H15 of the piperazine ring showed the highest absolute STD%, suggesting that this moiety is the closest group to the protein. A high STD value is also displayed by the methyl group (H17), while proton H16 could not be considered due to proximity of its signal to dissolved in dimethyl sulfoxide (DMSO) peak. Thus, we conclude that piperazine ring and its short aliphatic chain effectively engage the binding pocket in arrestin-3. The aromatic (H7, H8, and H9) and cyclohexene (H1, H2, H3, H4, and H5) protons demonstrate lower intensity (0.3–.5 STD%), indicating weaker interactions with the protein surface. Thus, the molecule is arranged in the binding pocket of arrestin-3 so that the piperazine moiety has the strongest interaction with the protein.

A Epitope map of LSH-3 with relative STD percentages at initial slope conveyed by color code, acquired at saturation time of 2 s, irradiating at 0 ppm. Dark blue dots indicate the most intense signal (100% relative STD), light blue dots between 20% and 80% and green dots under 20%. B 1H spectrum of LSH-3 C. STD spectrum of LSH-3 in the presence of arrestin-3.
While STD experiments identify the functional groups of the compound critical for target binding, they do not identify the protein binding site. To evaluate which site of arrestin-3 is involved, differential epitope mapping by STD-NMR spectroscopy (DEEP-STD NMR)36 was employed. This approach requires protein saturation at different irradiation frequencies to highlight parts of the compound contacting protein in the bound state. The analysis was carried out irradiating at 0–8.8 ppm, centered on the methyl group of the aliphatic chains and the aromatic or NH protons, respectively. These two STD-NMR experiments (Supplementary Fig. 6) showed different compound epitopes under different irradiations, suggesting that the aromatic protons of the compound are more intense upon the irradiation of the protein at 8.8, while the piperazine ring is surrounded by aliphatic amino acids. The two main putative binding sites emerging from the computational site mapping, SH-back loop (from the helix turn present in the inactive back loop) and GL-gate loop (Fig. 2), differ in the amino acid composition: higher number of hydrophobic residues is present in the SH binding site, while charged residues are mainly present at the GL site (Supplementary Fig. 4). To discriminate among them, the total compound saturation was calculated for both STD spectra. A larger total saturation was observed in the STD spectrum irradiated at 0 ppm (Supplementary Table 8), indicating that the compound preferentially occupies the site lined by hydrophobic residues.
Finally, the binding constant (KD) of the compound was determined using titration STD experiments. The curves were built with four concentrations (from 0.3 to 5 mM) and the KD values were determined from the binding isotherm with STD-AF measured at saturation times of 2 s. (Supplementary Fig. 7). To calculate the STD amplification factor (STD-AF), which relates the protons’ proximity to the protein surface, protons H14, H15, and H17 were selected. The experimental data were fitted with an exponential function. The KD obtained by STD-NMR is 0.8 mM, indicating that LSH-3 has sufficient affinity for arrestin-3 to be used as the chemical scaffold.
LSH-3 enhances recruitment of arrestin-3 to phosphorylation-deficient β2AR in cells
β2AR C-tail contains four residues which can be phosphorylated by GRK2: Ser355,356,364 and Thr360. We replaced these residues with alanines to generate a phosphorylation-deficient mutant, as described37. We visualized arrestin-3 recruitment to WT and phosphorylation-deficient β2AR by fluorescent microscopy in response to an β2AR- agonist isoproterenol (Iso)38. Since this interaction requires the phosphorylation of the receptor, GRK2 was also added. Arrestin-3 was recruited to WT, but not phosphorylation-deficient β2AR (Fig. 4F, I).

A Taken from mCh. channel after 30 s Iso stimulation. B Taken from EGFP channel after 30 s Iso stimulation. C Overlay of A and B. D Taken from mCh. channel after 5 min Iso stimulation E Taken from EGFP channel After 5 min Iso stimulation. F Overlay of (D) and (E). (G–L) Spinning disc confocal microscopy images of N2a cells transfected with β2AR- mCh., arrestin-3-177 mEGFP, and wild-type GRK2. Spinning disc confocal microscopy images of N2a cells transfected with β2AR- mCh., arrestin-3-177 mEGFP, and GRK2. G Taken from mCh. channel after 30 s Iso stimulation. H Taken from EGFP channel after 30 s Iso stimulation. I Overlay of (G) and (H). J Taken from mCh. channel after 5 min Iso stimulation K Taken from EGFP channel After 5 min Iso stimulation. Yellow arrows show arrestin-3 colocalizations with β2AR. L Overlay of J and K. Magnification 63X, scale bars are 10 µm.
Next, arrestin-3 recruitment to WT and phosphorylation-deficient β2AR with or without test compounds or Iso were analyzed by FRET39 using membrane-targeted Gap43-mCh.-meGFP as a positive control. After assessing cytotoxicity of LSH-1, LSH-2, and LSH-3 by succinate dehydrogenase activity (MTT) assay40 in N2a and HEK293 cells (Supplementary Fig. 9), experiments were carried out using 100 µM of each test compound.
To perform FRET experiments in a phospho-deficient system using a plate reader, we validated that the FRET/Donor ratio calculated from the plate reader correlates with % FRET efficiency measured by confocal fluorescence microscopy, analyzed with the ImageJ PixFRET plug-in. N2a cells overexpressing the control FRET construct, Gap43-mCh.-mEGFP, were first imaged by microscopy, resulting in a 10–15% FRET efficiency. Subsequently, the same experiment was conducted using the plate reader, yielding a FRET/Donor ratio of 0.12, which was close to the microscopy measurements. This consistency is largely due to the minimal bleed-through of these fluorescent proteins into each other’s channels, which was negligible under our imaging conditions (data not shown).
For further experiments, FRET/Donor ratios calculated from plate reader measurements were used. Basal FRET/Donor ratios (prior to stimulation) and response to Iso (20 µM) were measured in N2a cells expressing either wild-type (WT) β2AR or phosphorylation-deficient β2AR together with arrestin-3 (Fig. 5). N2a cells expressing WT β2AR showed approximately 75% higher FRET/Donor signal compared to those expressing the phosphorylation-deficient β2AR (Fig. 5A). Iso treatment further increased the FRET/Donor ratio in the WT system, while no increase was observed in the phospho-deficient system (Fig. 5B). Thus, arrestin-3 recruitment to the phosphorylation-deficient β2AR was significantly reduced.

Upper left panel: Basal FRET levels in N2a cells expressing phosphorylation-deficient β2AR-mCherry, arrestin-3-177-mEGFP, GRK2, or β2AR-mCherry, arrestin-3-177-mEGFP, GRK2. Upper right panel: Calculated FRET levels in N2a cell before and after 20 µM Iso stimulation. Means ± s.e.m. are shown. **p < 0005, ****p < 0.0001 statistically significant differences. ns: no significant differences. Lower left panel: LSH-3 chemical (1H-Indole-3-acetic acid, 6-[[(6-carboxy-3-cyclohexen-1-yl) carbonyl] amino] - α- (4-ethyl- 1- piperazinyl) increases FRET between b2AR phosphorylation-deficient–mCh., and arrestin-3-177-mEGFP. Each chemical was tested in two separate experiments n = 6. Means ± s.e.m. are shown. **p < 0005, ***p < 0001, statistically significant differences. ns: no significant differences. Lower right panel: Chemical interaction with membrane targeting sequence fused mCh. and mEGFP transfected N2a cells. Means ± s.e.m. are shown. ***p < 0.001, statistically significant differences, ns: no significant differences. Chemical-treated samples were compared with Gap 43 vehicle (1% DMSO).
Next, we used this system to test whether selected compounds promote interaction between phosphorylation-deficient β2AR and arrestin-3. 30-minute measurements were recorded following 30-minute incubation (Fig. 5C). FRET/Donor values for phospho-deficient β2AR and with and without 1% DMSO (negative control) were calculated as 0.038 and 0.044, respectively. FRET/Donor values for LSH-1, LSH-2, and LSH-3 were found to be 0.033, 0.030, and 0.064, respectively. Compound LSH-3 increased FRET by nearly 50%, which suggested that LSH-3 facilitates interaction between phosphorylation-deficient β2AR and arrestin-3.
To evaluate whether the compounds interfered with mEGFP or mCh fluorescense, FRET experiment was performed in the presence of test compounds using the positive control Gap43-mCh.-mEGFP. Results indicate that LSH-1 and LSH-3 do not alter the FRET in this construct, while LSH-2 decreases FRET by around 25%. This suggests that the impact of LSH-1 and LSH-3 on FRET in phosphorylation-deficient β2AR-arrestin-3 system is not due to non-specific interference, confirming that LSH-3 increased FRET by bringing phosphorylation-deficient β2AR and arrestin-3 close to each other.
LSH-3 effect is receptor- and subtype- specific
To investigate the effect of LSH-3 treatment on arrestin recruitment to β2AR and muscarinic M2 receptor (M2R), we performed a bioluminescence-based protein-protein interaction assay (NanoBiT) in HEK293 cells, where endogenous arrestin-2 and arrestin-3 were knocked out41. Cells were co-transfected with arrestin-3 or arrestin-2 with N-terminally fused SmBiT and β2AR or M2R with C-terminally fused LgBiT. 48-h post-transfection, the cells were treated with 100 μM LSH-3 or DMSO (vehicle control) for 30 min prior to the addition of luciferase substrate.
In case of arrestin-3, LSH-3 treatment significantly enhanced recruitment to the basal β2AR, particularly prominent in the initial 15 min of the assay (Fig. 6C). Notably, the maximum arrestin-3 recruitment to the active β2AR remained unchanged (Fig. 6A). In contrast, LSH-3 treatment showed no significant influence on arrestin-3 recruitment to M2R, regardless of the receptor activation state or agonist (carbachol) concentration (Figs. 6B, D).

HEK293 arrestin2/3 KO cells were co-transfected with plasmids encoding arrestin-3 (A–D) or arrestin-2 (E–H), both with N-terminal SmBiT and β2AR (A, C, E, and G) or M2R (B, D, F, and H) with C-terminal LgBiT. The cells were treated with 100 mM LSH3 or DMSO (vehicle control) for 30 min prior to the addition of luciferase substrate (time point 0). Agonist (10 mM isoproterenol (ISO) for β2AR or 10 mM carbachol for M2R) was added at 30 min. A Arrestin-3 recruitment to β2AR. B Arrestin-3 recruitment to M2R. C Arrestin-3 recruitment to inactive β2AR (no agonist). D Arrestin-3 recruitment to inactive M2R. E Arrestin-2 recruitment to β2AR. F Arrestin-2 recruitment to M2R. G Arrestin-2 recruitment to inactive β2AR. H Arrestin-2 recruitment to inactive M2R. Data are presented as means ± SEM (N = 6). Statistical significance was determined by two-way repeat measures ANOVA followed by Fisher’s LSD post hoc test and indicated, as follows: *, p < 0.05.
To determine whether this effect was specific for arrestin-3, we performed parallel experiments with arrestin-2. LSH-3 treatment had no significant effect on arrestin-2 recruitment to either β2AR or M2R, with or without the agonist (Fig. 6E–H). Thus, LSH3 selectively enhances arrestin-3, but not arrestin-2, recruitment to basal β2AR, demonstrating subtype- and receptor-specific action of the compound. This selectivity was consistent with our computational prediction: Induced Fit Docking of compound LSH-3 was performed on representative arrestin-3 and arrestin-2 structures from MD simulations (see Methods), as well as on arrestin-1 active and pre-activated crystal structures. Results showed that only arrestin-3 yields low-energy binding poses inserted in the back loop pocket (Supplementary Fig. 10), compatible with the binding mode in Fig. 2.
Discussion
GPCRs are involved in numerous signaling pathways in the body. Precise regulation of initiation and termination of the signal is crucial for the homeostasis of the cell. One of the mechanisms that cells use for terminating the GPCR-mediated signaling is the phosphorylation of certain cytoplasmic receptor residues. Recognition of phosphoresidues by arrestin stabilizes the pre-activated conformation and contributes to GPCR affinity. Dysregulation of phosphorylation prevents arrestin binding and leads to various diseases24,42.
Enhanced arrestin mutants were designed to increase their affinity for phosphorylation-deficient GPCRs, but their use requires gene therapy. The effect of phosphorylation in inducing arrestin pre-activation can be recapitulated by phosphopeptides mimicking the C-tail of the receptor43, as shown by crystal structures of phosphopeptide-bound arrestin complexes. Other negatively charged molecules like heparin or IP6 mimic the effects of phosphopeptides as well44. However, these compounds cannot be a viable therapeutic option because they do not cross the plasma membrane. Here, we demonstrated that the conformational equilibrium of arrestin can be perturbed by small molecules.
We focused on arrestin-3, since available evidence indicates that it is the most conformationally flexible non-visual subtype28,30. We confirmed its flexibility by MD simulations and identified a potential binding pocket that emerges upon its transition to pre-activated state. We discovered in silico compounds that fit one of these pockets and demonstrated experimentally that one of the predicted compounds, LSH-3, binds arrestin-3 (Fig. 3). In MD simulations LSH-3, predicted to stably bind to the back loop, stabilizes its unfolded conformation, and transmits the perturbation from the back loop to the gate loop, triggering its translocation from the polar core towards the lariat loop, facilitating inter-domain rotation. Importantly, the back loop was shown to be perturbed upon binding PIP2 which led to arrestin-3 pre-activation45, and upon arrestin-2 pre-activation46. Interestingly, these conformational rearrangements occurred without full dislocation of the C-tail of arrestin-3, which is consistent with our MD simulations, but opposed to the common model of phosphorylation-dependent activation, where the C-tail is displaced upon binding to the phosphoreceptor. Interestingly, it was previously shown that the C-tail of arrestin-3 can be localized close to its N-domain upon binding to the phosphorylated receptor47.
We demonstrated experimentally that in the presence of LSH-3 arrestin-3 binds phosphorylation-deficient, as well as unstimulated (and therefore not phosphorylated) β2AR. It should be noted that “unstimulated” does not necessarily refer to the basal conformation, as β2AR samples active conformations even in the absence of an agonist48.
Our data suggests that LSH-3 acts as a selective facilitator of arrestin-3 binding to β2AR. The finding that LSH-3 does not enhance arrestin-3 binding to M2 muscarinic receptor suggests that arrestin-3 bound to different GPCRs assumes distinct conformations. Indeed, the back loop is helical in the structure of arrestin-2/M2R complex19, while it is unfolded in the arrestin-2/β1-adrenergic receptor complex18, suggesting a receptor-specific modulation. It is tempting to speculate that LSH-3 might modulate receptor-specific binding of arrestin-3 by stabilizing the back loop in the unfolded state.
4950In summary, our results indicate that arrestin-GPCR interaction can be enhanced by small molecules in a receptor- and arrestin subtype-specific manner. Mutations in GPCRs are at the root of numerous congenital disorders (reviewed in refs. 24,26,49,50,51,52,53). Excessive signaling by gain-of-function mutants of various GPCRs was shown to cause retinal degeneration (rhodopsin42,54), hyperthyroidism or toxic thyroid adenomas (thyrotropin receptor55,56,57), male precocious puberty (LH receptor58), obesity (melanocortin-4 receptor59), and various forms of cancer60, to name just a few disorders. Therefore, enhanced endogenous arrestins stabilized by small molecules to increase their binding to overactive GPCRs, appear to be an alternative therapeutic approach to the treatment of such diseases. Further studies are necessary to explore the uses and limitations of this approach.
Materials and methods
Modeling
We used the crystal structure of bovine arrestin-3 (PDB-ID:3P2D, chain B) as a template to model human arrestin-3 (sequence identity: 95.2%) since the number of C-tail residues resolved is higher than in other crystal structures. The missing residues were built using Swiss-Model61 while the C-terminal tail was modeled using Modeler software62 as it contained a higher number of missing residues than could be handled by Swiss-Model. The remaining C-tail residues were modeled as coil, without any secondary structure, being consistently disordered and missing in crystal structures. The lowest DOPE energy conformation was selected as the starting structure. The modeled C tail residues were rather mobile along the MD simulations as expected. The same modeling protocol was applied for human arrestin-2, starting from the crystal structure of bovine arrestin-2 (PDB-ID:1JSY).
The protonation states of residues were determined using the PROPKA63 methodology, which computes pKa values of ionizable residues by accounting for the effect of the protein environment at pH = 7. The parametrization of small molecules, which were used in simulations of complexes with arrestin-3, was done using CHARMM-GUI64 .The output files were examined to check the penalty values associated with parametrization. CHARMM-GUI was also used for solvation and ionization of the systems studied, which was done by neutralizing the systems with 0.15 M KCl. We used CHARMM36m (Chemistry at Harvard Macromolecular Mechanics force field)65 and TIP3P66 to model protein and water molecules, respectively.
MD simulations
Atomistic MD simulations were performed on both arrestin-3 and arrestin-3-compound complexes using GROMACS 4.5.0.167 package and the following settings. The particle mesh Ewald68 method was used for electrostatics with a real space cut-off of 1.0 nm and a grid spacing of 0.13 nm. The Lennard-Jones interactions were determined with a twin-range cut-off scheme of 1.0 and 1.4 nm with the long-range interactions updated every ten steps. The time step was 2 fs. The temperature and pressure were set at 310 K and 1 atm using the Nose-Hoover thermostat69 and Parrinello-Rahman barostat70, respectively. We performed three replicates for apo arrestin-3 and two for arrestin-3-compound complexes, each of which started with a different velocity distribution. All simulations were performed around 1 μs. Arrestin-2 was subjected to the same simulation protocol for a total simulation time of 3 μs. Clustering of MD trajectories for Induced Fit Docking was carried out using GROMACS, the GROMOS method71 and a cutoff of 0.25 nm focusing on Cα atoms of residues 7-348, 385-391.
aMD simulations
aMD was used as an independent validation to check whether the system might be exploring more conformational states than the ones observed in standard MD. One aMD simulation72 of length 1 μs was produced using the dual boost option, where independent boost potentials were applied to the dihedral and potential energy of the system, as implemented in NAMD package73. We used values of 4455 kcal/mol, 81, −544,852 kcal/mol, and −35,198 for describing the threshold energy for dihedral, dihedral acceleration factor, threshold energy for the potential, and acceleration factor in the dual boost mode, respectively, which were calculated according to the equation in ref. 74. in aMD simulations. Vavg_dihedral and Vavg_potential values were obtained from classical MD simulations.
Reaction coordinates and structure classification
Trajectories of arrestin-3 were analyzed using a set of local and global structural properties: the conformational state of the back loop and of the gate loop, and the inter-domain rotation angle, as detailed in the next Methods section. We measured the distance between Cα atoms of residues F76 and F245 (F75 and F244 in arrestin-2; F79 and Y250 in arrestin-1) for the aromatic core, K313 and G317 (R312 and G316 in arrestin-2; K318 and G322 in arrestin-1) for the back loop, and D291 and H296 (D290 and H295 in arrestin-2; D296 and H301 in arrestin-1) for the gate loop. We compared these values to those measured in the structures of arrestin-1, arrestin-2, and arrestin-3 in their basal and receptor-bound states (Supplementary Table 1 for the list of the crystal structures, PDB IDs: 7DF9, 2WTR, 1G4M,1G4R, 3GC3, 5W0P, 3GD1, 1SUJ, 1CF1, 4ZRG, 3UGU, 1AYR, 4J2Q, 3UGX, 3P2D, 1ZSH, 1JSY, 4ZWJ, 6U1N, 6TKO, 6K3F, 7JTB, and 7F1W). The gate loop and back loop distances discriminate receptor-bound and basal experimental structures, so they were selected for the classification of the conformations. In detail, crystal basal systems show a folded α-helix at the back loop and a characteristic distance below 8.5 Å for the pair given above. Basal conformations also show a gate loop distance below 10.1 Å. In contrast, receptor-bound experimental structures have distances above 7.7 and 11.5 Å, for back loop and gate loop, respectively. Arrestin-3 MD snapshots mostly fall within the basal range or in between basal and receptor-bound: we classified these as pre-activated states.
Analysis of inter-domain rotation
The interdomain rotation axis was defined as follows: first, we superimposed the N-terminal domain of a set of structures of arrestin-1, arrestin-2, and arrestin-3 in the basal and receptor-bound state (the same PDB IDs given above were used, See Supplementary Table 1 for the list of structures used), obtaining a pseudo-trajectory which was then subjected to PCA analysis using the gmx covar tool of GROMACS67. The first PC projection of this pseudo-trajectory was then analyzed with the hingefind algorithm75 to identify the rotation axis of the C-domain relative to the N-domain. Finally, the rotation angle of the center of mass of the C-domain around this axis was calculated for every MD trajectory after superimposing the N-domain using an in-house program adapted from ref. 76. We classified an interdomain rotation angle as active when positive, i.e., rotated counterclockwise relative to the basal state.
Virtual screening on selected conformations
The SiteMap tool77 from Schrodinger software was used to identify binding pockets on the selected snapshots. The pocket candidates were evaluated using the following parameters: the number of site points, hydrophobic and hydrophilic, enclosure, exposure, donor/acceptor character of the pocket, volume, and druggability score (Dscore) (Supplementary Table 3). The pocket ranking was cross validated using the web server-based prediction tool fpocket78, as well as the AI based tool deepsite79. Both confirm a high score for the best SiteMap result (Supplementary Table 4). Hence, this pocket and the second best, both located at the N-C domain interface, referred to as back loop and gate loop, were used to build a receptor-based pharmacophore using the Phase module of Schrodinger80: a pharmacophore consists of a geometrical representation of the required chemical functionalities such as hydrophobicity, the capability of acting as hydrogen donors/acceptors, the aromaticity of the target region (See Supplementary Table 5). Pharmacophore-based virtual screening was carried out on a subset of the latest available ZINC purchasable compounds Database on ZincPharmer81.
Docking
The set of 2055 compound hits obtained from the virtual screening step was docked to the binding pocket. Glide docking tool82 was employed for this purpose, following standard preparation protocols. The candidate molecules were docked flexibly to the target site by SP docking, where only the trans conformers of amino acids were allowed. We used residues 123-128/306-308/316 and 168-172/291-299 to dock to the back loop and the gate loop, respectively. We then filtered them by means of a combined strategy. First, we preliminarily ranked the compounds considering their binding energies (Supplementary Table 6), ADME predictors (number of metabolites, human oral absorption value, molecular weight, QplogHERG, and QplogKhsa) and binding poses (those that block interaction between the finger loop and the back loop, which stabilizes basal conformation, were prioritized).
The 22 docking complexes with the highest scores were ranked for subsequent MD-based evaluation. Among them, 12 compounds dissociated from arrestin-3 within 50 ns. The ZINC IDs of these compounds were: 67755739, 55211759, 06864823, 03871402, 13546985, 68235734, 57778648, 47248657, 77319466, 79523936, 48397525, and 41124261. Ten compounds stably bound to the back loop, but only three of them perturbed the inter-domain rotation angle.
To computationally predict ligand selectivity, comparative induced fit docking of compound LSH3 on arrestin-2 and arrestin-3 was carried out using Schrodinger glide-based induced fit docking protocol82, with default settings, on representative structures of the most populated clusters obtained from corresponding MD simulations (see above), after preparing the structures in Maestro. Using the same protocol, induced fit docking of compound LSH3 on arrestin-1 was performed on the crystal structures of pre-activated arrestin-1 (PDB ID: 4ZRG) and of rhodopsin bound arrestin-1 (PDB:5W0P).
Reagents
LSH-1 compound (Benzamide, 2- chloro- N- [3-[(cyclopropylcarbonyl) amino] phenyl] - 5- (methylthio)-) was purchased from Enamine, (ZINC ID:31135800), CAS Registry Number: 1386112-75-9. LSH-2 compound (1H- Pyrazolo[4, 3- c] pyridine- 3- carboxylic acid, 4, 5, 6, 7- tetrahydro- 5- [(2- methyl- 1H- imidazol- 5- yl) carbonyl] - 1- (3- phenylpropyl) -) was purchased from ChemBridge, (ZINC ID:67490967), CAS Registry Number: 1309319-17-2. LSH-3 compound (1H- Indole- 3- acetic acid, 6- [[(6- carboxy- 3- cyclohexen- 1- yl) carbonyl] amino] - α- (4- ethyl- 1- piperazinyl) -) was purchased from InterBioScreen (ZINC ID:36365956), CAS Registry Number: 1214645-87-0. Lgl1 compound was purchased from Enamine, (ZINC ID:12915185). All the chemicals were DMSO at 10 mM concentration, then aliquoted at 10 µL in PCR tubes, and kept in a −80 °C freezer for further use.
N2a cell culture
N2a (mouse neuroblastoma) cells were cultured in 44.5% v/v DMEM with L-glutamine (Gibco, REF 41966-029), 44.5% v/v Optimem® reduced serum medium with L-glutamine (Gibco, REF 11058-021), 10% v/v heat-inactivated Fetal Bovine Serum (Biological Industries REF 04-127-1B), 1% v/v Penicillin/streptomycin solution (Biological Industries REF 03-031-1B). HEK293 cells were cultured in the same reagents with DMEM ratio 90%, and no Optimem. Both cell lines were grown at 37 °C in a humidified 95% air/5% CO2 atmosphere. The cells were routinely subcultured twice a week.
Molecular cloning
Tagging the arrestin-3 gene from the 177th position was performed by Restriction Free Cloning, which involves two consecutive PCRs. In the first PCR, the mEGFP (monomeric enhanced green fluorescent protein) gene was amplified with primers that carry complementary to mEGFP gene sequences and 25–30 overhanging base pairs from the arrestin-3 sequence from the 177–178th position. The product of this “first” PCR, which carries flanking regions that are homologous to the arrestin-3 gene, was used as a double-stranded “mega primer” in a second PCR to amplify the whole arrestin-3 carrying plasmid. After the second PCR, 1 µL Dpn1 restriction enzyme was added directly into the PCR mix to digest the parental plasmid (arrestin-3). 2 µL of the final mix was utilized for the transformation of E. coli XL 1 Blue. Final constructs were verified by Sanger sequencing and subcloned into an empty pcDNA 3.1(-) for eukaryotic expression.
Confocal fluorescence microscopy
To examine the arrestin-3 colocalization with β2AR at the cell membrane, 80,000 N2a cells were seeded on 35 mm glass-bottom petri dishes (In vitro scientific, CA, USA). Twenty-four hours later, cells were transfected with 400 ng of pcDNA3.1 or pcDNA3-based plasmids encoding arrestin-3-177-mEGFP, β2AR–mCherry (mCh.)/ β2AR P.D.-mCh., and GRK2. Confocal microscope imaging was performed 48 h post-transfection. Before imaging, the media was aspirated, cells were washed with PBS, and 1 mL of PBS was added. Leica DMI 4000 equipped with Andor DsD2 spinning disc confocal microscopy with Leica 63x/1,32 HCX PL APO oil DIC objective was used for imaging. Cells were excited at 480 ± 10 nm and emission was collected at 525 ± 15 nm (mEGFP channel), excitation at 585 ± 15 nm and emission collected at 630 ± 20 nm (mCherry channel). FRET analysis of Gap43-mCh.-mEGFP was performed using ImageJ pix-FRET plug-in. First, 60,000 N2a cells were seeded in three 35 mm glass-bottom petri dishes. The next day, samples were transfected with 400 ng of plasmids encoding Gap43-mCh.-mEGFP, Gap43-mCh, or Gap43-mEGFP. At 48 h post-transfection, media was aspirated, cells were washed with 1 mL PBS, and 1 mL PBS was added to each plate before imaging. Donor spectral bleedthrough was calculated from the Gap43-mEGFP transfected sample by exciting the cells from the donor channel, 480 ± 10 nm emission collected at 525 ± 15 nm and FRET channel, excitation at 480 ± 10, and emission at 630 ± 20 nm. Acceptor bleedthrough calculated from Gap43-mCh. by exciting the cells from the FRET channel, excitation at 480 ± 10 and emission at 630 ± 20 nm, and acceptor channel, excitation at 585 ± 15 nm. and emission collected at 630 ± 20 nm. FRET sample (Gap43-mCh.-mEGFP) image was taken from all three channels. The images were processed using ImageJ Pix-FRET plug-in that uses an algorithm that calculates the total bleed through pixel by pixel in three stacks of microscope images. The resulting final image gives the %FRET efficiency map.
FRET measurements
N2a cells (125,000) were seeded on poly D-lysine (PDL) coated 35 mm plastic petri dishes 24 h prior to transfection. In the control plate, in each experiment, cells were transfected with an empty vector, and sample plates that were transfected with β2AR-P.D-mCh., arrestin-3-177-mEGFP, and GRK2 encoding plasmids (total DNA amounts of the controls and samples were 1200 ng). After 24 h, transfected cells were washed with PBS and then lifted by 140 µL TrypL-E™ Express (Gibco, Cat # 12605028). Cells were resuspended in a 1.8 mL complete medium and counted. 20,000 N2a cells were seeded in each well of PDL-coated F-bottom opaque black 96-well plates (SPL Life Sciences F bottom 96-well immunoplate #31496). After 24 h, cells were washed twice with 200 µL Hanks’ buffered saline solution (HBSS), and the final volume was adjusted to 150 µL with HBSS containing either 100 µM of tested chemical or 1% v/v DMSO (control). Each chemical was also added to each chemical’s blank group. After adding the chemicals, 96-well plates were incubated at 37 °C for 30 min in the cell culture incubator and then measured for 30 min at the wavelengths as follows; mEGFP; 470/510, FRET; 470/610, mCh.; 570/610 (excitation bandwidth: 15 nm, emission bandwidth: 25 nm) FRET was calculated as the ratio of FRET/Donor. Molecular Device Spectramax id3 was used for measurement.
The %FRET efficiency was found to be around 12% (data not shown). The same construct was used for FRET calculation in the microplate reader. To determine the cell density to use in further experiments, 10,000, 15,000, and 20,000 cells were seeded in each well, and FRET was calculated as FRET intensity/Donor intensity. According to the results, increased cell density increases the intensity of each channel (mEGFP, FRET, and mCh.), but as expected the FRET/Donor ratio was not affected by the cell density. The FRET/Donor ratio was calculated as 0.125, which correlates with the calculated FRET efficiency in a confocal microscope (around 12%).
To assess possible interference between the compounds and fluorescence of mEGFP and mCh, N2a cells were transfected with 400 ng of the positive FRET construct, before FRET measurement in a microplate reader. A 96-well plate was incubated for an hour with each compound (100 µM) in a cell culture incubator and then measured by Molecular Devices Spectramax id3 plate reader using the same settings as for the other experiments.
MTT toxicity assay
To test the toxicity of the compounds, the MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide) assay was used. N2a and HEK293 cells were seeded in clear F-bottom plastic 96 well microplates (Sarstedt AG&Co. KG, Germany, REF: 83.3924) in 100 µL final volume in complete medium, at a density of 10,000/well and 20,000/well for each cell line, respectively, and cultured at 37 °C in 5% Co2 incubator for 20 h. The test compounds were added at 50 and 100 µM and incubated for another 4 h. 10 µL MTT solution (5 mg/mL dissolved in PBS) was added to each well and incubated for another 4 h. After that, 100 µL of detergent solution (10% w/v SDS in 0.01 M HCl) was added to each well to dissolve the formed formazan crystals. The plates were shaken for 5 min and incubated at room temperature for 16 h. Absorbance was measured at 570 nm with Thermo Scientific Varioskan Lux microplate reader.
Saturation transfer difference (STD) – NMR
For the STD-NMR experiments, arrestin-3 was produced as 6xHis-tagged recombinant protein in E. coli using vector pET28a from Twist Bioscience (South San Francisco, California, USA), as described83,84. The LSH-3 compound was obtained by InterBioScreen with a non-stereoselective synthesis. It contains three stereocenters and thus can in principle exist as eight distinct stereoisomers. The commercially acquired compound was first characterized and a set of signals, corresponding to four of the possible diasteroisomers, were identified, with one of them accounting for the 50%. Therefore, to assess the ability of LSH-3 to bind arrestin-3, multiple STD spectra were acquired using different compound/protein samples, prepared to better characterize the binding mode. Reported STD data were calculated considering the main isomer.
The protein-compound samples were prepared in a 200:1, 500:1, 1500:1 and 3000:1 compound/protein ratio. The final concentration of the protein was 5 μM, and the final volume was 180 μL. The buffer used is a 20 mM deuterated phosphate buffer pH 7.4. A percentage (less than 5%) of DMSO‑d6 was added when necessary to improve the solubility of the compound.
1H-STD NMR experiments were performed on a 600 MHz Bruker Avance spectrometer. The probe temperature was maintained at 298 K. In the STD experiments, water suppression was achieved by the excitation sculpting pulse sequence.
The on-resonance irradiation of the protein was performed at 0 and 8,8 ppm. Off-resonance irradiation was applied at 40 ppm, where no protein signals are visible. Selective presaturation of the protein was achieved by a train of Gauss-shaped pulses of 50 ms length each. The STD-NMR spectra were acquired with an optimized total length of saturation train of 0.5, 1, or 2 s. Blank experiments were conducted in absence of protein in order to avoid artefacts. The different signal intensities of the individual protons are best analyzed from the integral values in the reference and STD spectra, respectively. (I0 - Isat)/I0 is the fractional STD effect, expressing the signal intensity in the STD spectrum (ηSTD) as a fraction of the intensity of an unsaturated reference spectrum. In this equation, I0 is the intensity of one signal in the off-resonance or reference NMR spectrum, Isat is the intensity of a signal in the on-resonance NMR spectrum, and I0–Isat represents the intensity of the STD NMR spectrum. The STD NMR binding epitope was calculated based on the normalized STD values from the initial slope of each proton85, the initial growth rates approach consists in analyzing the protein–compound association curve using STD values at the limit of zero saturation time, when virtually no compound rebinding or relaxation takes place. The distance between the compound and the protein surface is expressed as absolute STD percentage (STD Abs) and the compound moieties interacting with the macromolecule are illustrated through a color-coded epitope map, obtained by normalizing all measured STD intensities against the most intense signal (to which is assigned an arbitrary value of 100%).
The STD amplification factor (STD-AF) is obtained multiplying ηSTD by the excess of compound and is proportional to the concentration of the protein–compound complex in solution. The curves were built with four concentrations (from 0.3 to 5 mM) and the KD values were extracted from the binding isotherm with STD-AF measured at saturation times of 2 s. To calculate the STD amplification factor (STD-AF), which relates the protons’ proximity to the protein surface, proton H14, H15, and H17 were selected as important moieties for the interaction. The experimental data were fitted with an exponential function.
NanoBiT assay
HEK293 cells with CRISPR/Cas9-mediated knockout of arrestin-2 and arrestin-341 were co-transfected with plasmids encoding arrestin-3 or arrestin-2 with N-terminal small-bit (SmBiT) and β2AR or muscarinic M2 receptor (M2R) with C-terminally fused large-bit (LgBiT) in a 24-well plate. Twenty-four hours post-transfection, cells were transferred to a 96-well plate in 100 μl serum-free DMEM and incubated for 16 h (serum starvation). Forty-eight hours post-transfection, cells were pre-treated with 100 μM LSH3 or DMSO (vehicle control) for 30 min at 37 °C in a CO2 incubator. Subsequently, 20 μl of nano-Glo substrate (Promega, #N2011) was added to each well, and total luminescence was recorded for 30 minutes until the signal became stable. Then 5 μl of agonist (isoproterenol for β2AR, carbachol for M2R) was added to a final concentration of 10 μM, and luminescence was immediately measured for 90 min.
Statistical analysis
All experiments were performed in triplicate, and the results were presented as mean ± S.E.M (Graphpad Prism version 8.0.1). Statical analyses were carried out using GraphPad Prism. The effect of LSH3 treatment was evaluated using two-way repeated measures ANOVA with time and treatment as factors, followed by Fisher’s LSD post hoc test with correction for multiple comparisons. p values < 0.05 were considered statistically significant.
Data availability
The trajectories generated during the current study are available in the https://zenodo.org/records/15503574.
References
Hauser, A. S., Attwood, M. M., Rask-Andersen, M., Schiöth, H. B. & Gloriam, D. E. Trends in GPCR drug discovery: new agents, targets and indications. Nat. Rev. Drug Discov. 16, 829–842 (2017).
Weis, W. I. & Kobilka, B. K. The molecular basis of G protein-coupled receptor activation. Annu. Rev. Biochem. 87, 897–919 (2018).
Gimenez, L. E. et al. Role of receptor-attached phosphates in binding of visual and non-visual arrestins to G Protein-coupled receptors. J. Biol. Chem. 287, 9028–9040 (2012).
Lohse, M. J., Benovic, J. L., Codina, J., Caron, M. G. & Lefkowitz, R. J. β-arrestin: a protein that regulates β-adrenergic receptor function. Science 248, 1547–1550 (1990).
Moore, C. A. C., Milano, S. K. & Benovic, J. L. Regulation of receptor trafficking by GRKs and arrestins. Annu. Rev. Physiol. 69, 451–482 (2007).
Carman, C. V. & Benovic, J. L. G-protein-coupled receptors: turn-ons and turn-offs. Curr. Opin. Neurobiol. 8, 335–344 (1998).
Chen, Q., Iverson, T. M. & Gurevich, V. V. Structural basis of arrestin-dependent signal transduction. Trends Biochem. Sci. 43, 412–423 (2018).
Peterson, Y. K. & Luttrell, L. M. The diverse roles of arrestin scaffolds in G protein–coupled receptor signaling. Pharm. Rev. 69, 256–297 (2017).
Wess, J., Oteng, A.-B., Rivera-Gonzalez, O., Gurevich, E. V. & Gurevich, V. V. β−arrestins: structure, function, physiology, and pharmacological perspectives. Pharm. Rev. 75, 854 (2023).
Indrischek, H., Prohaska, S. J., Gurevich, V. V., Gurevich, E. V. & Stadler, P. F. Uncovering missing pieces: Duplication and deletion history of arrestins in deuterostomes. BMC Evol. Biol. 17, 163 (2017).
Gurevich, V. V. Arrestins: a small family of multi-functional proteins. Int. J. Mol. Sci. 25, 6284 (2024).
Gurevich, V. V. et al. Arrestin interactions with G protein-coupled receptors: direct binding studies of wild type and mutant arrestins with rhodopsin, β2-adrenergic, and m2 muscarinic cholinergic receptors. J. Biol. Chem. 270, 720–731 (1995).
Pierce, K. L., Premont, R. T. & Lefkowitz, R. J. Seven-transmembrane receptors. Nat. Rev. Mol. Cell Biol. 3, 639–650 (2002).
Gainetdinov, R. R., Premont, R. T., Bohn, L. M., Lefkowitz, R. J. & Caron, M. G. Desensitization of G protein–coupled receptors and neuronal functions. Annu. Rev. Neurosci. 27, 107–144 (2004).
Kovoor, A., Celver, J., Abdryashitov, R. I., Chavkin, C. & Gurevich, V. V. Targeted construction of phosphorylation-independent β-arrestin mutants with constitutive activity in cells. J. Biol. Chem. 274, 6831–6834 (1999).
Kang, Y. et al. Crystal structure of rhodopsin bound to arrestin by femtosecond X-ray laser. Nature 523, 561–567 (2015).
Liao, Y. Y. et al. Snapshot of the cannabinoid receptor 1-arrestin complex unravels the biased signaling mechanism. Cell 186, 5784–5797 (2023).
Lee, Y. et al. Molecular basis of β-arrestin coupling to formoterol-bound β1-adrenoceptor. Nature 583, 862–866 (2020).
Staus, D. P. et al. Structure of the M2 muscarinic receptor–β-arrestin complex in a lipid nanodisc. Nature 579, 297–302 (2020).
Bous, J. et al. Structure of the vasopressin hormone-V2 receptor-β-arrestin1 ternary complex. Sci. Adv. 8, 7761 (2022).
Cao, C. et al. Signaling snapshots of a serotonin receptor activated by the prototypical psychedelic LSD. Neuron 110, 3154–3167 (2022).
Yin, W. et al. A complex structure of arrestin-2 bound to a G protein-coupled receptor. Cell Res. 29, 971–983 (2019).
Kristaponyte, I., Hong, Y., Lu, H. & Shieh, B.-H. Role of rhodopsin and arrestin phosphorylation in retinal degeneration of drosophila. J. Neurosci. 32, 10758 (2012).
Schöneberg, T. et al. Mutant G-protein-coupled receptors as a cause of human diseases. Pharm. Ther. 104, 173–206 (2004).
Schöneberg, T. & Liebscher, I. Mutations in G protein–coupled receptors: mechanisms, pathophysiology and potential therapeutic approaches. Pharm. Rev. 73, 89 (2021).
Stoy, H. & Gurevich, V. V. How genetic errors in GPCRs affect their function: Possible therapeutic strategies. Genes Dis. 2, 108–132 (2015).
Song, X. et al. Enhanced arrestin facilitates recovery and protects rods lacking rhodopsin phosphorylation. Curr. Biol. 19, 700–705 (2009).
Zhan, X., Gimenez, L. E., Gurevich, V. V. & Spiller, B. W. Crystal structure of arrestin-3 reveals the basis of the difference in receptor binding between two non-visual subtypes. J. Mol. Biol. 406, 467–478 (2011).
Scheerer, P. & Sommer, M. E. Structural mechanism of arrestin activation. Curr. Opin. Struct. Biol. 45, 160–169 (2017).
Sensoy, O., Moreira, I. S. & Morra, G. Understanding the differential selectivity of arrestins toward the phosphorylation state of the receptor. ACS Chem. Neurosci. 7, 1212–1224 (2016).
Granzin, J. et al. Crystal structure of p44, a constitutively active splice variant of visual arrestin. J. Mol. Biol. 416, 611–618 (2012).
Kim, Y. J. et al. Crystal structure of pre-activated arrestin p44. Nature 497, 142–146 (2013).
Zhou, X. E. et al. Identification of phosphorylation codes for arrestin recruitment by G protein-coupled receptors. Cell 170, 457–469 (2017).
Latorraca, N. R. et al. Molecular mechanism of GPCR-mediated arrestin activation. Nature 557, 452–456 (2018).
Meyer, B. & Peters, T. NMR spectroscopy techniques for screening and identifying ligand binding to protein receptors. Angew. Chem. - Int. Ed. 42, 864–890 (2003).
Monaco, S., Tailford, L. E., Juge, N. & Angulo, J. Differential epitope mapping by STD NMR spectroscopy to reveal the nature of protein–ligand contacts. Angew. Chem. 129, 15491–15495 (2017).
Krasel, C. et al. Dual role of the β2-adrenergic receptor C terminus for the binding of β-arrestin and receptor internalization. J. Biol. Chem. 283, 31840–31848 (2008).
Ruiz-Medina, B. E., Cadena-Medina, D. A., Esparza, E., Arrieta, A. J. & Kirken, R. A. Isoproterenol-induced beta-2 adrenergic receptor activation negatively regulates interleukin-2 signaling. Biochem. J. 475, 2907–2923 (2018).
Forster Th. energiewanderung und fluoreszenz. Naturwissenschaften 33, 166–175 (1946).
Morgan, D. M. L. Tetrazolium (MTT) Assay for cellular viability and activity. in polyamine protocols (ed. Morgan, D. M. L) 179–184 (Humana Press, 1998).
Grundmann, M. et al. Lack of beta-arrestin signaling in the absence of active G proteins. Nat Commun 9, 341 (2018).
Restagno, G. et al. A large deletion at the 3′ end of the rhodopsin gene in an Italian family with a diffuse form of autosomal dominant retinitis pigmentosa. Hum. Mol. Genet. 2, 207–208 (1993).
Abiko, L. A. et al. Biased agonism of carvedilol in the β 1-adrenergic receptor is governed by conformational exclusion. bioarXiv https://doi.org/10.1101/2024.07.19.604263.
Chen, Q. et al. Structural basis of arrestin-3 activation and signaling. Nat. Commun. 8, 1427 (2017).
Kim, K. & Chung, K. Y. Molecular mechanism of β-arrestin-2 pre-activation by phosphatidylinositol 4,5-bisphosphate. EMBO Rep. (2024) https://doi.org/10.1038/s44319-024-00239-x.
Asher, W. B. et al. GPCR-mediated β-arrestin activation deconvoluted with single-molecule precision. Cell 185, 1661–1675 (2022).
Zhuo, Y., Vishnivetskiy, S. A., Zhan, X., Gurevich, V. V. & Klug, C. S. Identification of receptor binding-induced conformational changes in non-visual arrestins. J. Biol. Chem. 289, 20991–21002 (2014).
Manglik, A. et al. Structural insights into the dynamic process of β2-adrenergic receptor signaling. Cell 161, 1101–1111 (2015).
Schöneberg, T. & Liebscher, I. Mutations in G protein–coupled receptors: mechanisms, pathophysiology and potential therapeutic approaches. Pharm. Rev. 73, 89–119 (2021).
Parnot, C., Miserey-Lenkei, S., Bardin, S., Corvol, P. & Clauser, E. Lessons from constitutively active mutants of G protein-coupled receptors. Trends Endocrinol. Metab. 13, 336–343 (2002).
Seifert, R. & Wenzel-Seifert, K. Constitutive activity of G-proteins-coupled receptors: cause of disease and common property of wild-type receptors. Naunyn Schmiedebergs Arch. Pharm. 366, 381–416 (2002).
Thompson, M. D., Hendy, G. N., Percy, M. E., Bichet, D. G. & Cole, D. E. C. G protein-coupled receptor mutations and human genetic disease. in Pharmacogenomics in Drug Discovery and Development (ed. Yan, Q.) 153–187 (Springer, 2014). https://doi.org/10.1007/978-1-4939-0956-8_8.
Vassart, G. & Costagliola, S. G protein-coupled receptors: mutations and endocrine diseases. Nat. Rev. Endocrinol. 7, 362–372 (2011).
Apfelstedt-Sylla, E. et al. Ocular findings in a family with autosomal dominant retinitis pigmentosa and a frameshift mutation altering the carboxyl terminal sequence of rhodopsin. Br. J. Ophthalmol. 77, 495 (1993).
Kosugi, S., Hai, N., Okamoto, H., Sugawa, H. & Mori, T. A novel activating mutation in the thyrotropin receptor gene in an autonomously functioning thyroid nodule developed by a Japanese patient. Eur. J. Endocrinol. 143, 471–477 (2000).
Parma, J. et al. Somatic mutations in the thyrotropin receptor gene cause hyperfunctioning thyroid adenomas. Nature 365, 649–651 (1993).
Parma, J. et al. Diversity and prevalence of somatic mutations in the thyrotropin receptor and Gsα genes as a cause of toxic thyroid adenomas1. J. Clin. Endocrinol. Metab. 82, 2695–2701 (1997).
Laue, L. et al. Genetic heterogeneity of constitutively activating mutations of the human luteinizing hormone receptor in familial male-limited precocious puberty communicated by Earl. Proc. Natl. Acad. Sci. USA 92, 1906–1910 (1995).
Tao, Y. X. Constitutive activity in melanocortin-4 Receptor: biased signaling of inverse agonists. in Advances in Pharmacology 70 135–154 (Academic Press Inc, 2014).
Arang, N. & Gutkind, J. S. G protein-coupled receptors and heterotrimeric G proteins as cancer drivers. FEBS Lett. 594, 4201–4232 (2020).
Waterhouse, A. et al. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 46, W296–W303 (2018).
Webb, B. & Sali, A. Comparative protein structure modeling using modeller. Curr. Protoc. Bioinforma. 54, 5.6.1–5.6.37 (2016).
Olsson, M. H. M., Søndergaard, C. R., Rostkowski, M. & Jensen, J. H. PROPKA3: consistent treatment of internal and surface residues in empirical pKa predictions. J. Chem. Theory Comput. 7, 525–537 (2011).
Jo, S., Kim, T., Iyer, V. G. & Im, W. CHARMM-GUI: a web-based graphical user interface for CHARMM. J. Comput. Chem. 29, 1859–1865 (2008).
Huang, J. et al. CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nat. Methods 14, 71–73 (2016).
Pekka, M. & Nilsson, L. Structure and dynamics of the TIP3P, SPC, and SPC/E water models at 298 K. J. Phys. Chem. A 105, 9954–9960 (2001).
Pronk, S. et al. GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 29, 845–854 (2013).
Darden, T., York, D. & Pedersen, L. Particle mesh Ewald: an N·log(N) method for Ewald sums in large systems. J. Chem. Phys. 98, 10089–10092 (1993).
Evans, D. J. & Holian, B. L. The Nose–Hoover thermostat. J. Chem. Phys. 83, 4069–4074 (1985).
Parrinello, M. & Rahman, A. Polymorphic transitions in single crystals: a new molecular dynamics method. J. Appl Phys. 52, 7182–7190 (1981).
Daura, X. et al. Peptide folding: when simulation meets experiment. Angew. Chem. - Int. Ed. 38, 236–240 (1999).
Hamelberg, D., Mongan, J. & McCammon, J. A. Accelerated molecular dynamics: a promising and efficient simulation method for biomolecules. J. Chem. Phys. 120, 11919–11929 (2004).
Phillips, J. C. et al. Scalable molecular dynamics on CPU and GPU architectures with NAMD. J. Chem. Phys. 153, 044130 (2020).
Miao, Y., Nichols, S. E., Gasper, P. M., Metzger, V. T. & McCammon, J. A. Activation and dynamic network of the M2 muscarinic receptor. Proc. Natl. Acad. Sci. USA 110, 10982–10987 (2013).
Wriggers, W. & Schulten, K. Protein domain movements: detection of rigid domains and visualization of hinges in comparisons of atomic coordinates. Proteins 29, 1–14 (1997).
Morra, G., Potestio, R., Micheletti, C. & Colombo, G. Corresponding functional dynamics across the Hsp90 Chaperone family: insights from a multiscale analysis of MD simulations. PLoS Comput. Biol. 8, e1002433 (2012).
Halgren, T. New method for fast and accurate binding-site identification and analysis. Chem. Biol. Drug Des. 69, 146–148 (2007).
Le Guilloux, V., Schmidtke, P. & Tuffery, P. Fpocket: an open source platform for ligand pocket detection. BMC Bioinf. 10, 168 (2009).
Jiménez, J., Doerr, S., Martínez-Rosell, G., Rose, A. S. & De Fabritiis, G. DeepSite: protein-binding site predictor using 3D-convolutional neural networks. Bioinformatics 33, 3036–3042 (2017).
Dixon, S. L. et al. PHASE: A new engine for pharmacophore perception, 3D QSAR model development, and 3D database screening: 1. Methodology and preliminary results. J. Comput. Aided Mol. Des. 20, 647–671 (2006).
Koes, D. R. & Camacho, C. J. ZINCPharmer: pharmacophore search of the ZINC database. Nucleic Acids Res. 40, W409–W414 (2012).
Friesner, R. A. et al. Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 49, 6177–6196 (2006).
Avsar, S. Y. et al. Immobilization of arrestin-3 on different biosensor platforms for evaluating GPCR binding. Phys. Chem. Chem. Phys. 22, 24086–24096 (2020).
Ostermaier, M. K., Schertler, G. F. X. & Standfuss, J. Molecular mechanism of phosphorylation-dependent arrestin activation. Curr. Opin. Struct. Biol. 29, 143–151 (2014).
Angulo, J., Enríquez-Navas, P. M. & Nieto, P. M. Ligand-receptor binding affinities from saturation transfer difference (STD) NMR. Chem. - A Eur. J. 16, 7803–7812 (2010).
Acknowledgements
This study was funded by the Scientific Research Council of Turkey (TUBITAK), project number 117Z245. β2AR - mCh and β2AR -phosphorylation-deficient – mCh. and Gap43-mCh.-mEGFP carrying plasmids were provided by former Son’s Lab member, Hüseyin Evci, and arrestin-3 carrying plasmid was a kind gift from Mark Scott (CNRS Research Associate, Paris, France). OS would like Turkish Academy of Sciences Young Investigator Program (TUBA-GEBIP). GM gratefully acknowledges the help of Dr. Zhenlong Li in the Harel Weinstein Lab, Weill Cornell Medicine, who provided the starting full-length molecular model for arrestin-3; she also acknowledges funding from Spoke 7 of Programma di ricerca CN00000013 “National Center for HPC, Big Data and Quantum Computing” of the NextGenerationEU initiative. I.B. would like to thank Marta Vanoni, SCITEC lab technician, for her experimental support in arrestin expression. Nuclear Magnetic Resonance analyses were performed at the Unitech COSPECT NMR facility of the University of Milan. The work at Vanderbilt University was funded by NIH grants EY011500 and GM122491.
Author information
Authors and Affiliations
Contributions
G.M. and O.S. designed the research. O.S. funding acquisition. H.K. performed M.D. simulations and virtual screening. H.K., G.M., and O.S. analyzed data. M.M. assisted in virtual screening. A.A. performed FRET experiments. C.Z. performed NanoBiT experiments, C.Z. and V.V.G. analyzed and interpreted NanoBiT data. A.A. and C.D.S. analyzed and interpreted FRET data. A.N. performed experiments with cardiac cell culture. A.N. and E.C. analyzed and interpreted data. E.F., I.B., I.G., and F.V. performed NMR experiments. I.G. and F.V. interpreted NMR data. H.K., A.A., A.N., C.D.S., E.C., F.V., G.M., V.V.G., and O.S. contributed to writing the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Chemistry thanks the anonymous reviewers for their contribution to the peer review of this work. Peer review reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Kurt, H., Akyol, A., Son, C.D. et al. A small molecule enhances arrestin-3 binding to the β2-adrenergic receptor. Commun Chem 8, 194 (2025). https://doi.org/10.1038/s42004-025-01581-4
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s42004-025-01581-4
