
Abstract
The tumor microenvironment evolves during tumor development and influences the cells in the microenvironment to orchestrate a supportive environment for tumor growth. Here we collected 4,483,367 cells across 36 cancer types and constructed a pan-cancer resource named TabulaTIME. Our integrated analyses reveal that CTHRC1 is a hallmark of extracellular matrix-related cancer-associated fibroblasts (CAFs) that are enriched in different cancer types. Spatiotemporal analyses further indicated that CTHRC1+ CAFs are located at the leading edge between the malignant and normal regions, potentially preventing immune infiltration. Moreover, we identified that SLPI+ macrophages exhibit profibrotic-associated phenotypes and colocalize with CTHRC1+ CAFs to form unique spatial ecotypes. Finally, we demonstrated that TabulaTIME can be used to analyze tumor ecotype composition and can serve as a reference for cell-type annotation. This work establishes a comprehensive single-cell landscape of the heterogenous TME and offers a potential therapeutic strategy for targeting the profibrotic ecotype in cancer treatment.
Similar content being viewed by others


Main
Cancer development is a multistep process during which cancerous cells acquire the ability to overcome limitations in replicative potential and evade immune destruction1,2. Meanwhile, noncancerous cells, such as stromal cells, are gradually reprogrammed to support tumor growth. The organ in which the tumor arises, with its unique tissue-resident cell types, creates substantial diversities in the tumor microenvironment (TME)3 and shapes distinct clinical properties of tumors, such as molecular subtypes, invasion abilities and response to targeted or immunotherapy4,5,6. Recent advances in single-cell RNA sequencing (scRNA-seq) and spatial transcriptomics (ST) have served as powerful tools for exploring the diversity of the TME. However, it remains unclear whether phenotypic-related cell types are universally present in different cancer types and stages. Moreover, the interactions between different cell types that form specific ecotypes within the TME are not well understood. Therefore, investigating the dynamics of TME compositions and intercellular interactions from a pan-cancer perspective is critical for elucidating the pathogenesis of cancer and represents a promising therapeutic target.
The increasing accumulation of scRNA-seq datasets in the public domain allows for the integration of datasets from a pan-cancer perspective, which helps to identify common or cancer-type-specific mechanisms of the TME. Previous studies have constructed separate landscapes for myeloid cells and T cells, portraying a systematic view of tumor-infiltrating immune cells and revealing distinct patterns of cell-type composition between cancer types7,8,9,10,11. In addition to immune cells, stromal cells play important roles in the TME of solid tumors. Increasing evidence suggests that stromal cells actively regulate tumor progression and metastasis by remodeling the extracellular matrix (ECM), promoting epithelial–mesenchymal transition (EMT) and angiogenesis12,13,14,15,16,17. At present, a few studies have delineated the diversity of fibroblasts in a single or a handful of cancer types. These studies were constrained by limited cell numbers, hindering a complete characterization of the complexity of stromal cell types. Furthermore, they could not fully analyze the interactions between cell types that contributed to the heterogeneity of stromal subtypes18,19.
In this study, we have collected the largest scale of published solid tumor-associated scRNA-seq data to date, comprising approximately 4 million cells. Using this extensive dataset, we have constructed a comprehensive Tabula of TME cells across 36 different cancer types. We have defined 6 major cell lineages and 56 cell subtypes within the TME using an integrated approach. The resulting comprehensive blueprint of the TME serves as a valuable roadmap for understanding the complexity of the TME, identifying phenotypic-related cell types and developing innovative therapeutic strategies that may have broad applicability across multiple cancer types.
Results
Use of the TabulaTIME framework for integrating large-scale tumor scRNA-seq data
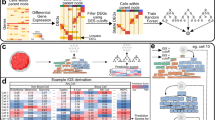
To characterize the heterogeneous composition and evolution of the TME during tumor initiation, progression and metastasis across different cancer types, we present the Tabula of the tumor immune microenvironment (TabulaTIME) framework. The framework consists of five major modules: tumor-related scRNA-seq data collection, data preprocessing and MetaCell identification, integration of all lineages, lineage-specific integration and characterization of cell subtypes (Fig. 1a).

a, Schematic depicting the TabulaTIME framework and its application. TabulaTIME was applied within a multiphase workflow, encompassing tumor-related scRNA-seq data collection, data preprocessing and MetaCell identification, integration of all lineages, lineage-specific integration and characterization of cell subtypes; Imm-reg, immune-regulatory; AP, antigen-presenting; TCA, tricarboxylic acid; NSCLC, non-small cell lung cancer; HNSC, head and neck squamous cell carcinoma; KIPAN, pan-kidney cohort; OV, ovarian serous cystadenocarcinoma; ESCA, esophageal cancer; CRC, colorectal cancer; SKCM, skin cutaneous melanoma; PRAD, prostate adenocarcinoma; LIHC, liver hepatocellular carcinoma; UVM, uveal melanoma. b, Data collection statistics. The numbers of cells (top) and donors (bottom) collected for each tissue are presented; k indicates ×1,000. c, Uniform manifold approximation and projection (UMAP) visualization of all MetaCells, colored by the cell type (top) and source (bottom), respectively. d, Expression of cell-type-specific markers. Dot size and color represent the percentage of cells with the gene expressed and the average expression value, respectively; Mono, monocytes; Macro, macrophages; DC, dendritic cells; Treg, regulatory T cells; CD4+ Tconv, conventional CD4+ T cells; Tprolif, proliferating T cells.
First, we collected tumor-related scRNA-seq datasets20,21,22,23, consisting of 103 studies covering 36 cancer types, 746 donors and 4,483,367 cells (Fig. 1b and Supplementary Table 1). Second, all collected datasets were preprocessed using the MAESTRO workflow24, which included quality control, doublet and batch effect removal25, cell clustering and cell-type annotation26,27 (Extended Data Fig. 1a–h and Supplementary Tables 2–4). To reduce technical noise and computing resource costs, TabulaTIME grouped cells with similar expression into MetaCells within each dataset, with each MetaCell containing approximately 30 cells (Extended Data Fig. 2a–d and Methods)8,28. The average log transcripts per million (TPM)-transformed gene expression of all cells within each MetaCell was used in downstream analyses. In the following two modules, TabulaTIME first integrated all MetaCells using canonical correlation analysis (CCA) to evaluate the effectiveness of batch removal between different cancer types (Fig. 1a, all lineages integration)25. Compared to the batch effect-corrected integration of single-cell profiles, the integration using MetaCells demonstrates superior performance, significantly reducing batch effects while preserving cell-type-specific biological variation (Extended Data Fig. 2e,f). To improve resolution for cell-type-specific analyses, TabulaTIME also integrated MetaCells from each lineage (Fig. 1a, lineage-specific integration). To mitigate the risks of overclustering and underclustering, we determined the optimal clustering resolution based on average silhouette width (ASW) scores and Clustree and assessed the purity of each annotated subtype using ROGUE scores29,30,31. Finally, TabulaTIME integrated the lineage-specific pan-cancer maps with other types of cancer data, such as ST and bulk tumor profiles from The Cancer Genome Atlas (TCGA). This enabled the analyses of tumor-enriched cell types by quantifying their relative abundance across different sources, cancer types and spatial localization. Furthermore, it facilitated the investigation of cell-type-specific functions and the estimation of their effects on immune cell infiltration and prognosis (Fig. 1a, characterization of cell subtypes). Together, TabulaTIME is a powerful framework for investigating the cellular compositions and functional states of the TME with temporary and spatial resolution.
A pan-cancer single-cell transcriptome blueprint generated by TabulaTIME
We then tested the ability of TabulaTIME to integrate tumor scRNA-seq datasets with millions of cells at scale and diverse sources of batch effects. The integrated pan-cancer landscape includes datasets derived from adjacent normal tissue, precancerous tissue, primary tumor, metastatic tumor and peripheral blood mononuclear cell samples of 22 different tissues (Fig. 1b). Additionally, 16 tumor scRNA-seq datasets were obtained from individuals treated with various strategies, including PD-1/PD-L1-based immunotherapy, chemotherapy, targeted therapy and combined therapies (Extended Data Fig. 2g and Supplementary Table 1). For the pan-cancer datasets, a total of 140,072 MetaCells were generated, and batch effects were further corrected using CCA (Fig. 1c and Extended Data Fig. 2h). Interestingly, immune and stromal cells were separated into distinct clusters, which were annotated as seven major cell lineages, including cytotoxic lymphocytes (CD8+ T, natural killer (NK) and proliferating T cells), conventional and regulatory lymphocytes (CD4+ Tconv and Treg cells, respectively), B lymphocytes (B cells and plasma cells), myeloid cells (monocytes/macrophages, dendritic cells and mast cells), fibroblasts (fibroblasts and myofibroblasts), endothelial cells and epithelial-like cells (epithelial, malignant and tissue-specific; Fig. 1c,d). Considering the diverse molecular features of epithelial, malignant and tissue-specific cells, TabulaTIME mainly analyzed the immune and stromal compartments of the TME in the following analyses. The batches from different sample sources and cancer types were well mixed, and the annotated labels were highly consistent with the original labels, even at the minor cell lineage scale, indicating the efficient integration of millions of cells using our strategy.
TabulaTIME reveals common characteristics of pan-cancer lymphocytes
To understand the heterogeneity of pan-cancer immune cells at a higher resolution, we separately integrated and reanalyzed each lineage. A total of ten cytotoxic lymphocyte subtypes were identified, each labeled by its functional characteristics and marker gene (Fig. 2a). Signature enrichment analyses suggested that three NK subtypes and GZMB+ effector memory CD8+ T cells (CD8Temra_GZMB) had higher cytotoxic scores, whereas exhausted CD8+ T cells (CD8Tex_HAVCR2) showed the highest exhaustion scores and naive CD8+ T cells (CD8Tn_CCR7) displayed the lowest cytotoxic and exhaustion scores, consistent with their phenotypes, respectively (Fig. 2b, Extended Data Fig. 3a and Supplementary Table 5). Interestingly, GZMK+ effector memory CD8+ T cells (CD8Tem_GZMK) were significantly enriched in precancerous tumor samples and were more prevalent than cytotoxic NK cells across different cancer types, indicating stronger T cell-mediated antitumor immunity (Fig. 2c, Extended Data Fig. 3b and Supplementary Table 6). By contrast, blood and normal samples were enriched with naive CD8+ T cells (CD8Tn_CCR7; Fig. 2c). Additionally, TabulaTIME resolved ten subtypes of conventional and regulatory lymphocytes and six B lymphocyte subtypes (Extended Data Fig. 3c–h and Supplementary Table 6). Overall, TabulaTIME revealed shared characteristics of pan-cancer lymphocytes that were highly consistent with their corresponding sources and functional phenotypes8.

a, UMAP visualization of the distribution of cytotoxic lymphocytes, colored by cell type; MAIT, mucosal-associated invariant T cell; ISG, interferon-stimulated gene-expressing T cells. b, Scatter plot illustrating the expression patterns of cytotoxic- and exhaustion-associated signature genes in cytotoxic lymphocyte subsets across different sources. c, Box plot showing the proportion of distinct cytotoxic lymphocyte subgroups in each sample type (blood (red), normal tissue (green), precancerous tissue (orange), tumor tissue (purple) and metastatic tissue (blue)), based on 650 treatment-naive samples. Significance labels in the figure were determined using Kruskal–Wallis tests to compare distributions of cytotoxic lymphocyte subgroups across five tissue types. Significance for pairwise source proportions within each cell type, assessed via two-tailed unpaired Wilcoxon tests, is reported in Supplementary Table 6. The open rectangle annotates the comparative scope, with Benjamini–Hochberg (BH) correction for multiple testing. The bottom of the box represents the first quartile (Q1), and the top of the box represents the third quartile (Q3). The height of the box represents the interquartile range (IQR), whereas the horizontal line inside the box indicates the median. The whiskers extend to the positions of Q1 – 1.5 × IQR and Q3 + 1.5 × IQR. d, A similar UMAP plot as in a was applied to myeloid cells; pDC, plasmacytoid dendritic cells; cDC1, type 1 conventional dendritic cells; cDC2, type 2 conventional dendritic cells. e, Heat map showing different expression patterns of function-associated signature genes among monocyte and macrophage subsets. f, Box plot showing the proportions of distinct monocyte and macrophage subgroups across different sources, including blood (red), normal tissue (green), precancerous tissue (orange), tumor tissue (purple) and metastatic tissue (blue), derived from 687 treatment-naive individuals. Significance labels in the figure were determined by Kruskal–Wallis tests to compare each subgroup distribution among five tissue types. Significance for pairwise source proportions within each cell type, assessed via two-tailed unpaired Wilcoxon tests, is reported in Supplementary Table 6. The open rectangle annotates the comparative scope, with BH correction for multiple testing. The bottom of the box represents Q1, and the top of the box represents Q3. The height of the box represents the IQR, whereas the horizontal line inside the box indicates the median. The whiskers extend to the positions of Q1 – 1.5 × IQR and Q3 + 1.5 × IQR. g, Heat map showing the proportion of different myeloid cell types in various cancer types or healthy tissues. For rows, a bar plot illustrates the number of MetaCells (in log10 scale) and the origin of cancer cells labeled by the different colors; PBMC, peripheral blood mononuclear cells; SCC, squamous cell carcinoma; UCEC, uterine corpus endometrial carcinoma; STAD, stomach adenocarcinoma; CHOL, cholangiocarcinoma; THCA, thyroid cancer; CESC, cervical squamous cell carcinoma and endocervical adenocarcinoma; BRCA, breast invasive carcinoma; PAAD, pancreatic adenocarcinoma; GIST, gastrointestinal stromal tumors; OS, osteosarcoma; BCC, basal cell carcinoma; SCLC, small cell lung cancer. h, Left, heat map showing Jaccard similarity indices for comparisons among 3,751 robust NMF programs based on the top 50 genes within the monocyte and macrophage cell populations. Programs are ordered by clustering and grouped into families of MPs with related functions (marked by black dashed lines); MP families are numbered and labeled. Right, list of all MP names separated into MP families; IFNγ, interferon-γ. i, Box plot illustrating the signature scores of MPs calculated across distinct monocyte and macrophage subtypes, derived from 797 samples, with MPs color coded according to their functional annotations. The bottom of the box represents Q1, and the top of the box represents Q3. The height of the box represents the IQR, whereas the horizontal line inside the box indicates the median. The whiskers extend to the positions of Q1 – 1.5 × IQR and Q3 + 1.5 × IQR. j, Pseudotime-ordered analysis of monocyte and macrophage MetaCells, colored by cell type. k, Kaplan–Meier plots show worse clinical outcomes in 184 individuals with ESCA and 459 individuals with SKCM with higher expression of SLPI+ macrophage signature genes; +, censored observations. Statistical significance was evaluated using a log-rank test, yielding P values of 0.014 for ESCA and 0.0001 for SKCM.
Divergent trajectories of myeloid cells with inflammatory and fibrotic functions
We proceeded to investigate the characteristics of myeloid cells and identified 12 subtypes (Fig. 2d and Extended Data Fig. 4a–d). Monocytes and macrophages were categorized into two groups of monocytes (classical Mono_FCN1 and nonclassical Mono_CD16) and six groups of macrophages. Interestingly, traditional M1/M2 signatures cannot clearly distinguish the macrophage subtypes within the TME, indicating a high level of plasticity and heterogeneity among macrophages (Fig. 2e and Supplementary Table 5). Consistent with previous studies, phagocytic Macro_C1QC and anti-inflammatory/angiogenic Macro_THBS1 signatures were highly enriched in precancerous and tumor samples, suggesting that they are potential tumor-associated macrophages (TAMs) regulating tumor immunity7 (Fig. 2e–g and Supplementary Table 6). Intriguingly, TablulaTIME identified a profibrotic TAM subtype (Macro_SLPI) that was enriched in a subset of tumors, such as basal cell carcinoma and cholangiocarcinoma (Fig. 2f,g). We postulate that the Macro_SLPI signature evinces a diminished phagocytic and inflammatory capacity, yet exhibits a markedly elevated ECM remodeling capability based on the enrichment of known macrophage-associated signatures (Fig. 2e).
To investigate the potential mechanisms of TAM plasticity, we applied non-negative matrix factorization (NMF) to monocytes and macrophages and identified 3,751 robust expression programs (Fig. 2h and Methods). By comparing the NMF programs, we defined the consensus programs as meta-programs (MPs) based on their shared genes. As expected, the profibrotic subtype Macro_SLPI exhibited the highest program scores for EMT and focal adhesion, similar to wound healing and profibrotic macrophages in lung fibrosis and coronavirus disease 2019 (Fig. 2i)32,33,34. Lineage tracing analyses suggested that although they both originate from monocytes, the profibrotic Macro_SLPI signature follows a distinct developmental branch compared to the phagocytic Macro_C1QC or the anti-inflammatory Macro_THBS1 signature (Fig. 2j). Finally, using the TCGA dataset, we examined the correlation between Macro_SLPI signature scores and overall survival. Higher Macro_SLPI signature scores were strongly associated with an increased risk of death in various cancer types, such as esophageal carcinoma (log-rank test, P = 0.014) and skin cutaneous melanoma (SKCM; log-rank test, P = 0.0001; Fig. 2k and Extended Data Fig. 4h,i). Together, our analyses revealed distinct molecular diversities and trajectories of TAMs in the TME.
ECM-associated CTHRC1 + fibroblasts are prevalent in tumor samples across cancer types
Stromal cells could establish a tumor-supportive environment by modifying the ECM and vasculature. Fibroblasts are the most prominent stromal cells, and their phenotypes and origins vary widely. TabluaTIME recognized seven subtypes of fibroblasts based on the expression of specific marker genes (Fig. 3a,b). These fibroblast subtypes were categorized into five major groups, including myofibroblast (myFibro), ECM-remodeling fibroblasts (eFibro), immunoregulatory fibroblasts (iFibro), antigen-presenting associated fibroblasts (apFibro) and quiescent fibroblasts (qFibro) based on characteristic gene expression patterns (Fig. 3c)15,35. Interestingly, we observed that eFibro_CTHRC1 cells were predominantly derived from tumor samples and eFibro_SFPR1 cells were highly enriched in normal samples, whereas iFibro_IL6 cells were predominantly present in precancerous samples (Fig. 3d, Extended Data Fig. 5a and Supplementary Table 6). Furthermore, eFibro_CTHRC1 cells also expressed canonical CAF markers, including FAP36, LRRC15 (ref. 12) and POSTN15, which are prevalent in nearly all cancer types but not in healthy samples, suggesting its important role in the TME (Fig. 3b,e). Functional signatures and MP analyses revealed that eFibro_CTHRC1 cells were enriched for EMT and ECM receptor interaction pathways (Fig. 3f and Extended Data Fig. 5b,c). Meanwhile, the metabolic pathway analysis showed that the glycosaminoglycan biosynthesis–chondroitin sulfate/dermatan sulfate pathway, which is involved in generating ECM components, was notably upregulated in eFibro_CTHRC1 cells (Fig. 3g and Extended Data Fig. 5d). Furthermore, we investigated the association between eFibro_CTHRC1 cells and prognosis. Higher expression of eFibro_CTHRC1 signature genes was correlated with worse clinical outcomes in multiple cancer types, including kidney renal clear cell carcinoma (KIRC; log-rank test, P = 0.00523) and bladder urothelial carcinoma (BLCA; log-rank test, P = 0.00568; Fig. 3h), which is consistent with the previously reported clinical impact of FAP+ and LRRC15+ fibroblasts. These findings collectively suggest that fibroblasts in the TME undergo pronounced reprogramming to adopt myofibroblastic phenotypes, which in turn remodel the ECM and potentially contribute to tumor growth.

a, UMAP visualization of fibroblast MetaCell distribution, colored by cell type. b, Dot plot depicting the expression of representative signature genes of each fibroblast cell type. c, Scatter plot showing ECM remodeling, immunoregulatory and antigen-presenting signature scores for each fibroblast subset; Imm-Reg, immune-regulatory. d, Box plot showing the proportion of each fibroblast cell type in different source-derived samples (normal, precancerous, tumor and metastatic tissue) from 338 treatment-naive samples. Significance labels in the figure were assessed via Kruskal–Wallis tests to compare each cell type distribution among four tissue types. Significance for pairwise source proportions within each cell type, assessed via two-tailed unpaired Wilcoxon tests, is reported in Supplementary Table 6. The open rectangle annotates the comparative scope, with BH correction for multiple testing. The bottom of the box represents Q1, and the top of the box represents Q3. The height of the box represents the IQR, whereas the horizontal line inside the box indicates the median. The whiskers extend to positions of Q1 – 1.5 × IQR and Q3 + 1.5 × IQR. e, Heat map showing the proportion of different fibroblast cell types in various cancer types or healthy tissues. For rows, a bar plot illustrates the number of miniclusters (in log10 scale) and the origin of cancer cells labeled by the different colors. f, Heat map displaying the enriched pathways for each fibroblast subset. Enrichment was calculated using hypergeometric distribution statistics, with P values adjusted by the BH method; FDR, false discovery rate. g, Violin plot showing the glycosaminoglycan biosynthesis pathway and the average metabolic pathway GSVA score for each fibroblast subset across 379 samples. h, Kaplan–Meier plots demonstrate the clinical impact of eFibro_CTHRC1 cells in 533 individuals with KIRC and 405 individuals with BLCA, comparing low and high signature scores; +, censored observations. Statistical significance was assessed via the log-rank test, with P values of 0.00523 for KIRC and 0.00568 for BLCA.
CTHRC1 + fibroblasts are located at the leading edge of the tumor region
Fibroblast migration and pathological matrix redeposition are frequently observed in fibrotic disease37,38. To understand the potential source and spatial localization of eFibros that are dramatically increased in the TME, we collected 62 published ST slides of tumor tissue sections from six cancer types. After filtering out low-quality spots and genes from each slide, we annotated malignant spots by combining markers from original studies and inferred copy number variations (Extended Data Fig. 6a–d)26. For tumor slides with clear tumor boundaries, such as HCC-1L from primary liver cancer39, we observed that both the fraction of eFibro_CTHRC1 cells and the expression of eFibro_CTHRC1 cell signatures were highly enriched at the leading edge from nontumor to tumor regions (Fig. 4a and Extended Data Fig. 6e,f). To investigate the general enrichment of eFibro_CTHRC1 cells at tumor boundaries, we calculated the correlations between the eFibro_CTHRC1 signature score and the distances to the tumor core regions among all ST slides containing both fibroblasts and malignant cells. Excitingly, the eFibro_CTHRC1 fractions showed a negative correlation with the distance to tumors in the majority of ST slides (total ST slides, 32/41, 78%), indicating a high enrichment of eFibro_CTHRC1 cells at tumor boundaries (Fig. 4b,c). We further verified the spatial localization of eFibro_CTHRC1 cells in in-house oral cancer samples using multiplexed immunohistochemistry (mIHC), specifically at the tumor boundaries (Fig. 4d).

a, Left, hematoxylin and eosin staining of a tissue section from participant HCC-IL. Middle, distribution of normal, tumor and transition regions in participant HCC-1L (images reproduced with permission from ref. 39, AAAS). Right, spatial feature plot of the eFibro_CTHRC1 signature score; scale bars, 100 μm. b, Correlation between the eFibro_CTHRC1 signature score and the distance between spots and malignant cells in all ST samples. Correlations were calculated using Pearson correlation coefficients. Resulting P values were adjusted for multiple comparisons via the BH method. The significant negative correlation represents the CTHRC1+ fibroblasts surrounding malignant cells in the ST samples; PLC, pulmonary lymphangitic carcinomatosis. c, Scatter plots showing the correlation between the distance to malignant cells (x axis) and the signature score of eFibro_CTHRC1 (y axis) in tissue sections. The correlation was calculated using Pearson correlation coefficients. The color represents the proportion of fibroblasts in each spot. The error band indicates the 95% confidence interval, which is calculated based on the standard error using the normal distribution. The center measure of the smooth line corresponds to the predicted values from the linear regression model. d, IHC staining to validate the distribution of eFibro_CTHRC1 cells; scale bars, 100 μm; PanCK, pan-cytokeratin. e, Box plot showing the correlation between inferred inducers of CTHRC1+ fibroblasts and the CTHRC1+ fibroblast signature score. The red box represents data calculated from 62 ST samples, whereas the green box is derived from 9,460 samples across 23 cancer types in the TCGA project. The bottom of each box indicates Q1, and the top represents Q3. The height of the box reflects the IQR, and the horizontal line inside the box indicates the median. The whiskers extend to the positions of Q1 – 1.5 × IQR and Q3 + 1.5 × IQR. f, Comparison of the relative intensity (each row shared a color scale, whereas different rows did not) of immune cell-type scores between the normal and tumor regions of ST slides, focusing on the ST samples with eFibro_CTHRC1 cells surrounding tumor cells. g, Bubble heat map showing the interaction strength of gene pairs between fibroblasts and immune cells. Colors in the bubble plot are proportional to the communication probability. Significant interactions are identified on the basis of a statistical test that randomly permutes the group labels of cells and then recalculates the interaction probability. h, Inferred LGALS9 and CD44 interaction between CD4+ T cells and fibroblasts; Tfh, follicular helper T cell.
To explore the impact of malignant cells on eFibro_CTHRC1, we screened for genes upregulated in malignant cells that correlated with the expression of the eFibro_CTHRC1 signature score. Our analysis identified several integrins, such as ITGA6, which encodes a matrix stiffness-regulated mechanosensitive molecule that can induce invasive fibroblast phenotypes and mediate activation of transforming growth factor-β (TGFβ) in lung fibrosis (Fig. 4e and Extended Data Fig. 6g)40,41.
We then investigated whether the boundary formed by eFibro_CTHRC1 could serve as a physical barrier that may affect the infiltration of immune cells into the tumor core. We selected the ST slides with high enrichment of eFibro_CTHRC1 in the boundary regions and divided the slides into normal and tumor regions. As anticipated, immune cells were markedly more abundant on the normal side than on the tumor side (Fig. 4f). Consistently, the estimated infiltration of CD8+ T cells was notably higher in tumor samples with a lower eFibro_CTHRC1 signature score in the TCGA cohort in almost all cancer types (Extended Data Fig. 6h,i). Furthermore, compared to other fibroblasts, eFibro_CTHRC1 fibroblasts were more likely to interact with CD8+ T cells via LGALS9–CD44 and LGALS9–CD45 interactions (Fig. 4g,h). LGALS9 has been reported to induce apoptosis in T cells by binding to HAVCR2 and bolster the stability and functionality of immunosuppressive Treg cells via interaction with CD44. Anti-Gal-9 therapy has potential in selectively augmenting intratumoral HAVCR2+ cytotoxic CD8+ T cells42,43. Together, our analyses indicate that the presence of eFibro_CTHRC1 may reduce the infiltration of immune cells, potentially both physical barriers formed by the ECM and immunosuppressive functions through cell–cell interactions.
Colocalization of CTHRC1 + fibroblasts and SLPI + macrophages forms profibrotic ecotypes
Our analyses suggest that eFibro_CTHRC1 cells are prevalent in tumor samples, and profibrotic Macro_SLPI cells are also significantly enriched in several cancer types (Figs. 2f,g and 3d,e). We then investigated whether there are potential connections or cooperation between these two cell types, as they are both enriched in similar ECM functions (Figs. 2e and 3c). Because the ST data we used were not at single-cell resolution but rather a mixture of six to ten cells, we quantified the coexistence of eFibro_CTHRC1 and Macro_SLPI cells by correlating their signature scores. As expected, eFibro_CTHRC1 and Macro_SLPI cells showed a high correlation at the spot level (R > 0.5), and this correlation increased with higher fibroblast fractions (Fig. 5a,b). This observation held true for almost all the evaluated slides (Fig. 5c). For other macrophages, Macro_SPP1 cells showed a relatively weaker correlation with eFibro_CTHRC1 cells, which have also been reported to be associated with fibrosis44. The two cell types were also observed using individual single-cell datasets, ruling out the possibility of cell doublets (Extended Data Fig. 7a,b). Using TCGA-UVM and TCGA-KICH samples, we confirmed the high concordance between eFibro_CTHRC1 and Macro_SLPI cells (Extended Data Fig. 7c). Importantly, mIHC staining of CTHRC1, SLPI, CD68 and SPP1 in oral cancer and NSCLC samples further verified the colocalization of eFibro_CTHRC1 and Macro_SLPI cells (Fig. 5d and Extended Data Fig. 7d). In summary, the colocalization of eFibro_CTHRC1 and Macro_SLPI cells creates unique profibrotic ecotypes within the tumor region and suggests functional connections between these cell types.

a, Spatial feature plots showing the spatial localization of eFibro_CTHRC1 and Macro_ SLPI cells in two pancreatic adenocarcinoma ST datasets. b, Dot plot showing the correlation between the signature scores of eFibro_CTHRC1 (x axis) and Macro_ SLPI (y axis) cells in ST data, with color indicating the proportion of fibroblasts in each spot. The correlation was calculated using the Pearson correlation coefficient. The error band denotes the 95% confidence interval, which was calculated based on the standard error using the normal distribution. The center measure of the smooth line corresponds to the predicted values from the linear regression model. c, Scatter plots showing the correlation between the signature score of eFibro_CTHRC1 cells and all monocytes and macrophages in tissue sections, with color representing the proportion of fibroblasts in each spot and error bands indicating the 95% confidence interval. d, Immunofluorescence images showing the localization of Macro_SLPI and Macro_SPP1 cell types (DAPI, CD68, SLPI and SPP1) and eFibro_CTHRC1 cell types (DAPI and CTHRC1) in HNSC and NSCLC samples; scale bars, 50 μm. e, Genes inferred to encode the top 20 ligands separately regulate eFibro_CTHRC1 cells and SLPI+ macrophages according to NicheNet. Ligands are ranked by Pearson correlation (left). The heat maps show the expression of ligands mentioned on the left across major cell types (middle) and the top 20 ligands inferred to regulate SLPI+ macrophages (right). f, Cartoon depicting the general distribution of eFibro_CTHRC1 and Macro_SLPI populations in tumors, as well as the function. Image created with BioRender.com, with permission.
We speculate that there may be a common regulatory mechanism or cytokine signaling pathway that activates the profibrotic ecotypes. To explore potential upstream signaling, we conducted NicheNet analyses, which indicated a tight connection between the activity of TGFβ1 and interleukin-1β (IL-1β) ligands and the eFibro_CTHRC1 phenotype (Fig. 5e)45. The existing literature suggests that TGFβ family ligands and inflammatory modulators such as IL-1β can activate CAFs by enhancing the activity of transcription factors such as SMAD, NF-κB and STAT41,46. Reassuringly, TGFβ1 and IL-1β could also stimulate the Macro_SLPI phenotype, indicating that these signaling molecules may directly activate CAFs and also promote fibrotic programs in Macro_SLPI cells, which could potentially work in conjunction with eFibro_CTHRC1 cells to remodel the ECM (Fig. 5e). In summary, these analyses suggest that the profibrotic ecotypes were possibly induced by TGFβ or IL-1β signaling (Fig. 5f).
RGCC + capillary endothelial cells are associated with vessel tip generation in tumors
Endothelial cells are not only involved in angiogenesis but also related to immune cell recruitment and semiprofessional antigen presentation47,48. We identified seven endothelial subtypes and annotated them as venous, capillary, lymphatic and vascular smooth muscle cells (VSMCs; Fig. 6a and Extended Data Fig. 8a,b). Consistently, VSMC_ACTA2 cells, with a high ECM signature score, simultaneously expressed endothelial marker genes and ACTA2 (Fig. 6b,c). RGCC+ capillary endothelial cells (capEndo_RGCC) exhibited the highest tip scores, whereas CXCR4+ capillary endothelial cells (capEndo_CXCR4) showed the highest antigen-presenting scores (Fig. 6d). Notably, capEndo_RGCC cells expressed markers associated with vessel tip generation, including APLN and ESM1, and the majority of these cells were found in tumor samples across various cancer types (Fig. 6e,f, Extended Data Fig. 8a and Supplementary Table 6)49. These findings collectively suggest that endothelial cells undergo dynamic reprogramming during cancer initiation and progression.

a, UMAP visualization displaying the distribution of endothelial MetaCells, colored by cell type; venEndo, venous endothelial; lymEndo, lymphatic endothelial. b, Box plot depicting the ECM signature score for each endothelial cell type, calculated from 367 samples. The bottom of the box represents Q1, and the top of the box represents Q3. The height of the box represents the IQR, whereas the horizontal line inside the box indicates the median. The whiskers extend to the positions of Q1 – 1.5 × IQR and Q3 + 1.5 × IQR. c, Violin plot showing the expression of representative marker genes for each endothelial cell type. d, Scatter plot illustrating the antigen-presenting and tip signature scores of endothelial subsets. e, Box plot showing the proportions of distinct endothelial cell types in various sample categories, including normal tissue (green), precancerous tissue (orange), tumor tissue (purple) and metastatic tissue (blue), derived from 344 treatment-naive samples. Significance labels in the figure were assessed via Kruskal–Wallis tests to compare each subset distribution among four tissue types. Significance for pairwise source proportions within each cell type, assessed via two-tailed unpaired Wilcoxon tests, is reported in Supplementary Table 6. The open rectangle annotates the comparative scope, with BH correction for multiple testing. The bottom of the box represents Q1, and the top of the box represents Q3. The height of the box represents the IQR, whereas the horizontal line inside the box indicates the median. The whiskers extend to the positions of Q1 – 1.5 × IQR and Q3 + 1.5 × IQR. f, Heat map showing the proportion of different endothelial cell types in cancer types and healthy tissues. For rows, a bar plot to the left of each row illustrates the number of MetaCells (in log10 scale) and origin of cancer cells, color-coded for clarity. g, Genes encoding the top 20 ligands inferred to regulate RGCC+ endothelial cells according to NicheNet. Ligands are ranked by Pearson correlation (left), and a dot plot represents the expression percentage (dot size) and value (dot intensity) of the top 20 ligands.
Angiogenesis, which typically initiates in the capillaries, is essential for tumor growth. Identifying the potential ligands that drive the phenotypes of tip-like capEndo_RGCC cells is critical for preventing angiogenesis through therapeutic targeting of endothelial cells50. VEGFA and AGT were predicted to be the most potent inducers of capEndo_RGCC cells, consistent with previous studies showing that VEGFA is a crucial regulator of angiogenesis, and AGT is required for hypoxia-induced vasculogenesis (Fig. 6g)49,51. Interestingly, both AGT and VEGFA are produced by fibroblasts, suggesting that stromal cells may influence one another in regulating angiogenesis within the TME.
Profibrotic ecotypes are associated with higher mortality risk from pan-cancer analyses
Our analyses indicate that cell types that constitute profibrotic ecotypes, including Macro_SLPI and eFibro_CTHRC1 cells, are associated with worse clinical outcomes in various cancer types (Figs. 2k and 3h). To systematically compare the effects of profibrotic ecotypes to other potential risk factors, such as T cell exhaustion, we conducted a comprehensive prognosis analysis of all 56 cell types across 23 cancer types from TCGA. The pan-cancer survival association dichotomized all cell types into favorable and adverse states, highlighting their functional and clinical heterogeneities. T cell/NK cell subsets with higher cytotoxic scores (two NK cell subsets and CD8Temra_GZMB cells) were associated with a decreased risk of death, whereas memory T cells (CD8Tm_TNF) were associated with shorter survival times (Fig. 7a). Interestingly, cell types that could constitute profibrotic-associated ecotypes, including Macro_SLPI, Macro_SPP1, eFibro_CTHRC1 and VSMC_ACTA2 cells, were all significantly associated with an increased risk of mortality at the pan-cancer level (Fig. 7b,c and Extended Data Fig. 9a). These results collectively suggest that profibrotic ecotypes have conserved protumor functions that reduce the survival time of individuals with cancer.

a, Survival associations of per-cell-type signature genes of cytotoxic lymphocyte subsets. Top, bar plot illustrating the pan-cancer survival association across 23 cancer types, aggregated across malignancies using Stouffer’s method. Columns are ordered by combined z score. Bottom, cancer-specific survival association determined by the Cox proportional hazards model; LUSC, lung squamous cell carcinoma; COAD, colon adenocarcinoma; READ, rectum adenocarcinoma; LUAD, lung adenocarcinoma. b, Survival associations of per-cell-type signature genes in myeloid subsets; SARC, sarcoma. c, Survival associations of per-cell-type signature genes in fibroblast subsets. d, Heat map of 8,743 individuals from TCGA classified into four distinct TME subtypes based on clustering of the signature of all cell types; Bn, naive B cells; Bm, memory B cells; Bgc, germinal center B cells. e, Box plot illustrating the cytotoxic signature scores of ecotypes, including lymphocytes, calculated from 8,734 samples from the TCGA project. Statistical significance was evaluated using two-tailed unpaired Wilcoxon tests combined with a permutation test (10,000 resamplings) to compare the signature score distribution across pairwise ecotypes; horizontal connectors denote compared groups, with the corresponding P value indicated above the horizontal line. The bottom of the box represents Q1, whereas the top represents Q3. The height of the box indicates the IQR, and the horizontal line inside the box represents the median. The whiskers extend to the positions of Q1 – 1.5 × IQR and Q3 + 1.5 × IQR. f, Box plot comparing tumor purity across different tumor ecosystem groups, calculated from 8,734 samples from the TCGA project. The bottom of the box represents Q1, whereas the top represents Q3. The height of the box indicates the IQR, and the horizontal line inside the box represents the median. The whiskers extend to the positions of Q1 – 1.5 × IQR and Q3 + 1.5 × IQR. g, Box plot comparing CD8+ T cell infiltration of individuals stratified by tumor ecosystem groups. CD8+ T cell infiltration was estimated using QUANTISEQ from 8,734 samples from the TCGA project. The bottom of the box represents Q1, whereas the top represents Q3. The height of the box indicates the IQR, and the horizontal line inside the box represents the median. The whiskers extend to the positions of Q1 – 1.5 × IQR and Q3 + 1.5 × IQR. h, Bar graphs depicting the segregation of each carcinoma into the five tumor ecosystem groups. i, Kaplan–Meier curves illustrate the overall survival analysis of five tumor ecosystem groups across 459 individuals with SKCM and 1,091 individuals with BRCA; +, censored observations. Statistical significance was assessed via the log-rank test, with P values of 2.54 × 10−5 for SKCM and 0.0229 for BRCA.
TabulaTIME enables pan-cancer patient stratification using conserved tumor ecotypes
Tumor ecosystems are highly heterogeneous and are composed of diverse cell types. Although risk association analyses can screen individual pro- or antitumor cell types, they cannot reflect the cooperation of different cell types in forming patient-specific tumor ecosystems. TabulaTIME defined 56 cell subtypes across different cancer types using scRNA-seq. With this high-resolution reference, we then investigated whether we could stratify individuals into different tumor ecosystems based on deconvoluted cell subtypes within the TCGA cohort. The cell subtypes were evaluated using gene set variation analysis (GSVA) and clustered into five ecotypes52, including active stromal ecotype (E1), naive immune ecotype (E2), active immune ecotype (E3), profibrotic ecotype (E4) and proliferating ecotype (E5; Fig. 7d,e). Based on the deconvoluted ecotypes, individuals could be classified into five major TME ecosystems, including immune desert with high proliferation (DHP), immune desert with low proliferation (DLP), active immune with high stromal presence (AIHS), active immune with low stromal presence (AILS) and naive immune with high stromal presence (NIHS; Fig. 7d,e and Extended Data Fig. 9b,c). Consistently, participants in the immune desert group (DHP and DLP) showed high levels of tumor purity (Fig. 7f), whereas participants in the active immune group (AIHS and AILS) displayed higher estimated infiltration of CD8+ T cells53,54 (Fig. 7g and Extended Data Fig. 9d).
Interestingly, although the ecosystem features were in general conserved among different cancer types, the distribution of ecosystem groups showed remarkable differences (Fig. 7h). The kidney cancers kidney renal papillary cell carcinoma (KIRP) and kidney chromophobe (KICH) were highly enriched for DLP and DHP, whereas KIRC was primarily composed of AIHS and NIHS (Fig. 7h). We then investigated whether this stratification was conserved between different cohorts. For breast cancer, participant groups were generally comparable between the TCGA-BRCA classification and the METABRIC classification, even at the subtype level (Extended Data Fig. 9e). Additionally, we benchmarked TabulaTIME against previous similar studies6,55. The TabulaTIME cell-type signatures demonstrated stronger agreement with scRNA-seq data at the cluster level, and participant stratification by TabulaTIME was more consistent with the deconvolution results (Extended Data Fig. 9f–h). Finally, we evaluated the clinical effects of different tumor ecosystem groups. Interestingly, participants in the DHP and NIHS groups showed an increased risk of mortality in various cancer types, respectively corresponding to cold and unresponsive TMEs (Fig.7i). In summary, the cell-type signatures derived from TabulaTIME could facilitate the robust stratification of individuals based on their tumor ecosystem status.
TabulaTIME serves as a reference map for pan-cancer single-cell annotation
Cell-type annotation is vital for interpreting functional phenotypes of cells when analyzing scRNA-seq datasets. A comprehensive and fully annotated dataset is essential for reference-based cell-type annotation methods. We next tested whether TabulaTIME could serve as a reference map for pan-cancer single-cell annotation (Fig. 8a). Using a published deep-learning method SELINA56, we benchmarked cell-type annotation performance with TabulaTIME as the reference compared to individual tumor scRNA-seq datasets. Excitingly, we observed high consistency between the manually curated annotations from the original papers and the cell types predicted using SELINA and TabulaTIME (Fig. 8b and Extended Data Fig. 10). Furthermore, the TabulaTIME reference showed meaningful improvement over using a single tumor scRNA-seq dataset from the same cancer type as the reference (Fig. 8c). This indicates that an integrative tumor reference that includes diverse immune and stromal cell types could pronouncedly increase cell-type annotation performance compared to a single dataset that may lack rare cell types.

a, Schematic overview of the workflow using pan-cancer single-cell landscapes to analyze the ecotypes of bulk RNA-seq datasets and automatic annotation for scRNA-seq datasets. b, UMAPs showing cell-type annotation by original paper (left) and SELINA prediction (right). c, Bar plots comparing the accuracy of reference-pretrained SELINA-predicted cell types against original annotations in the BRCA_GSE176078 and NSCLC_GSE146100 datasets. SELINA was trained on reference datasets from TabulaTIME, BRCA_EMTAB8107 and NSCLC_GSE131907.
Discussion
In this study, we collected a large amount of tumor scRNA-seq data from 735 donors, including nearly 4 million cells spanning 36 cancer types. Additionally, we combined ST data from 62 individuals across six cancer types to provide spatial information and characterize tissue structures. Overall, we constructed a comprehensive single-cell TME landscape that can be used to characterize the spatial and temporal dynamics of tumor heterogeneity and assess the impact of different cell types and tumor ecotypes on clinical outcomes.
Our analyses revealed that SLPI+ macrophages exhibited a profibrotic phenotype and colocalized with CTHRC1+ fibroblasts across cancer types. Considering that SLPI+ macrophages exhibit strong profibrotic features and that CAFs can originate from both macrophages and mesenchymal stem cells18,33,57,58, we speculate that CTHRC1+ fibroblasts may also derive from SLPI+ macrophages. Further studies are needed to reveal the conditions that stimulate profibrotic macrophage formation or whether they represent an existing macrophage phenotype in healthy individuals.
An important merit of our study is the publicly available tumor landscape at MetaCell resolution, which includes detailed cell-type annotations, distribution across various sources and cancer types and their functions and effects on survival. This resource can facilitate scientists in inferring the most likely phenotypes for query cells. Similarly, the TabulaTIME landscape can promote the reconstruction of tumor ecotypes for bulk tissue transcriptomes using signature genes. Overall, we believe that our data can serve as a rich resource that has the potential to contribute to future cancer subtype identification and the optimization of individual cancer therapy.
Methods
Ethics statement
This study adhered to all relevant ethical regulations. Primary tissue sections used in immunofluorescence studies were obtained with written informed consent from participants and were approved by the local medical ethics committees of the hospitals where the samples were collected.
scRNA-seq data collection
We collected published cancer-associated scRNA-seq datasets from 746 donors across 36 cancer types. These datasets were sourced from the Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/geo/) and our previous work the TISCH database (http://tisch.comp-genomics.org/home/). Additionally, we incorporated scRNA-seq datasets derived from healthy donors, including three peripheral blood mononuclear cell datasets and six datasets from normal tissues. This inclusion served as a baseline for the exploration of tumor-specific cellular features. To minimize platform-specific biases, we restricted our analysis to 10x Genomics scRNA-seq datasets. For each dataset, we downloaded the expression matrix of the raw count or TPM, along with relevant sample information. Neither sex nor gender was considered in the study design because the primary focus of this study was unrelated to sex or gender. For samples collected in this study, sex was self-reported. Given that the studies focus on the TME, we excluded samples with over 90% malignant cells from each publicly available solid tumor-associated scRNA-seq dataset. For the remaining dataset, no statistical methods were used to predetermine sample sizes; however, our sample sizes align with those reported in previous publications after accounting for the exclusion of samples with over 90% malignant cells. To ensure consistent gene symbol mapping across different genome assemblies, we converted the genes into GRCh38.p13.
Data quality control and preprocessing
We applied a standardized analysis workflow based on MAESTRO v1.1.0 for processing all collected datasets, including quality control, batch effect removal and cell clustering (Extended Data Fig. 1a,b). As a result, 26,975 cells were excluded, leaving 4,456,392 cells for subsequent analysis (Extended Data Fig. 1c). Then, for each dataset, MAESTRO selected the top 2,000 highly variable genes for downstream analyses using TPM matrices. We further used principal component analysis for dimension reduction, the Louvain algorithm for cluster identification and UMAP for visualization, with parameters optimized by cell count (Supplementary Table 2).
To systematically evaluate the batch effects for each dataset, we used an entropy-based metric to quantify data mixing across batches. Low entropy values indicate that the most similar cells are from the same individual, indicating the existence of a potential batch effect. However, it should be noted that for the datasets, which mainly contain malignant cells, low entropy could arise from the heterogeneity of malignant cell expression between different tumors, and the batch effect will not need to be removed. Malignant cells were identified by integrating three approaches: original study annotations, marker expression profiles and copy number variation (Extended Data Fig. 1a,d). Apart from the dataset with malignant cells and other datasets with a median entropy of more than 0.7, the remaining dataset corrected the batch effect by CCA (Extended Data Fig. 1e,f). All cells were then annotated into 17 common cell types, based on the expression of marker genes (Extended Data Fig. 1g,h). The marker genes of each cell type were collected from the published resources and were curated manually.
MetaCell identification and batch correction
To address challenges posed by the large cell count, we executed a rigorous three-step data integration process (Extended Data Fig. 2a). First, within each dataset, gene expression Ei,j, gene i in cell j, was quantified as log2 (TPMi,j / 10 + 1), mitigating the effect of gene-specific dropout rate variability. Second, to mitigate technical noise and reduce computational resource requirements, we partitioned single cells into small groups (called MetaCells hereafter) based on transcriptional similarity, as determined by their proximity within the UMAP representation. Our strategy resembles Tanay et al.’s MetaCells28 and Zheng et al.’s metaclusters8 but uniquely assesses similarity within each sample’s cell type and incorporates cell origin.
To determine the optimal number of cells (k) for each MetaCell, we evaluated MetaCell performance in terms of gene coverage and within-MetaCell variation (Extended Data Fig. 2a). To retain a greater number of cells, we allowed for a slight fluctuation in the cell count within MetaCells, rather than strictly adhering to fixed numbers like 10 or 30. Within-MetaCell variation, quantified via the Gini Index, showed increasing average variation and decreasing total variation with cell count. The Elbow method identified 30-cell MetaCells as optimal across datasets (Extended Data Fig. 2b,c). Next, using LISI59, measuring dataset diversity in neighborhoods, we found that these 30-cell MetaCells achieved higher LISI and >50% gene coverage (Extended Data Fig. 2d). For each scRNA-seq dataset, we then systematically partitioned the cells of each cell type within individual samples into MetaCells, with each MetaCell comprising approximately 30 cells. The average log TPM-transformed gene expression of all cells within each MetaCell was used to represent the MetaCell’s expression, and the original gene-by-cell expression matrix was converted to the gene-by-MetaCell expression matrix.
Third, we evaluated the integration performance and biological signal preservation of MetaCell + CCA using ten additional randomly selected datasets. We compared LISI and Entropy scores across four strategies and found that CCA-integrated MetaCells outperformed other methods, demonstrating enhanced integration performance and reduced batch effects (Extended Data Fig. 2e). Additionally, we assessed biological signal preservation by calculating the adjusted Rand index against original cell-type labels and using ASW to evaluate within-cluster homogeneity. MetaCell + CCA achieved the highest adjusted Rand index and ASW scores, confirming that cell-type structures were preserved after integration and batch correction. These results indicate that MetaCell + CCA effectively removed batch effects while retaining cell-type distinctions compared to original data (Extended Data Fig. 2f).
Lineage separate integrated analysis
To gain more detailed insights into the MetaCell heterogeneity of specific cell types, we divided all cells into six lineages for downstream analysis, including cytotoxic lymphocytes, conventional and regulatory lymphocytes, B lymphocytes, myeloid cells, fibroblasts, endothelial cells and epithelial cells (Fig. 1a). For each lineage MetaCell, we extracted the expression profiles and subjected them to reprocessing using the MAESTRO pipeline, which includes quality control, batch effect correction, cell clustering and annotation (Fig. 1c and Extended Data Fig. 2h).
Determining the optimal number of cell types
To determine the most effective clustering resolution for each cell lineage, we used both ASW and Clustree metrics to identify clusters that accurately represent biological diversity without excessive clustering. First, to address the curse of dimensionality, we calculated the ASW based on Euclidean distances in the top 30 principal components of the principal component analysis space. A higher ASW indicates a more pronounced separation among distinct clusters. For each lineage, we calculated ASW values ranging from 0.1 to 1.5 and designated the resolution with the highest ASW as the optimal one. Subsequently, we performed clustering at various resolutions for each lineage to determine the optimal resolution (Extended Data Fig. 4a).
Clustree was used to visually inspect marker gene expression. If marker gene expression was intertwined with markers of other cell types, this suggested that relying solely on ASW might not be sufficient to effectively distinguish subcellular populations. For example, in myeloid cells, the ASW suggested a resolution of 0.1. However, CLEC9A+ dendritic cells and C1QC+ macrophages were separated at a resolution of 0.5, leading us to identify 0.5 as the optimal resolution (Extended Data Fig. 4b). Thus, we combined ASW with prominent marker gene expression observed at varied resolutions using Clustree to determine the optimal resolution.
Based on the clustering results, we annotated cell clusters using marker gene expression. We then used ROGUE to evaluate cellular heterogeneity (Extended Data Fig. 4c). ROGUE scores range from 0 to 1, with 1 representing a completely pure subtype. Therefore, cell types with median ROGUE scores below 0.9 were considered to exhibit significant heterogeneity, prompting further investigation to reannotate them into more homogeneous subtypes exhibiting higher median ROGUE values.
Source preference analysis
To assess dynamic changes in the TME during tumor progression, we analyzed the proportions of sub-cell types across distinct sources within each lineage individually. Considering that various treatments may lead to dynamic changes in the TME, our analysis was limited to treatment-naive datasets. The proportion of each subtype in a sample was determined by dividing the number of MetaCells of that subtype by the total number of MetaCells in the lineage. Consequently, in any given lineage within a single sample, the sum of all subtype proportions equals 1. To investigate the cell-type distribution of source preference, we used the Kruskal–Wallis test to assess each cell type across all tissue types. Additionally, each pairwise source proportion comparison within each cell type was assessed via two-tailed unpaired Wilcoxon tests (Figs. 2c, 3d and 6e, Extended Data Fig. 4e and Supplementary Table 6).
Furthermore, to validate subtype source preferences, we calculated odds ratios by constructing 2 × 2 contingency tables for each subtype i and source j within lineages. Tables included (1) cells of subtype i in source j, (2) subtype i cells in other sources, (3) non-i subtypes in j and (4) non-i subtypes in other classifications. A Fisher’s exact test was applied, with BH-adjusted P values for multiple testing.
Distribution of cell types across cancer types
We investigated the distribution of specific cell types across diverse cancer types in treatment-naive datasets. For each cancer type or tissue, we calculated the subtype proportion by dividing the total number of MetaCells within the corresponding lineage. It is important to note that the sum of all subtype proportions within a specific lineage for a particular cancer type equals 1.
Scoring cell types using function-associated signature genes
Here, we used the signature gene lists obtained from previously published studies to describe the functional diversity of cell types (Supplementary Table 5). The AddModuleScore function in Seurat was applied to calculate the score for individual MetaCells. For T cells/NK cells, cytotoxic, exhausted and regulatory MetaCell scores were computed to assess functional states and validate subtypes (Fig. 2b and Extended Data Fig. 3a). Monocyte/macrophage MetaCell functions were evaluated via M1/M2 polarization, angiogenesis, phagocytosis and pro-/anti-inflammatory activity (Fig. 2e). Additionally, fibroblast subsets were analyzed for their tumorigenic roles through ECM remodeling and immune regulation (Fig. 3c).
Metabolic and cancer hallmark pathways and gene set enrichment analysis
Metabolic pathway activities were estimated using GSVA. The metabolic pathways were collected from KEGG60, including 85 pathways. Differentially activated pathways of each subtype were identified by running the Wilcoxon rank sum test against other cell types within the one lineage.
To elucidate the functional characteristics of the subtypes, we used gene sets associated with cancer hallmarks and KEGG pathways from MSigDB v6.1 (ref. 61). Enrichment analysis was implemented using the clusterProfiler package62. Pathways with a log (fold change) of greater than 0.05 and an adjusted P value of less than 0.01 were deemed significantly upregulated.
Characterization of intratumoral transcriptional heterogeneity
To capture transcriptional heterogeneity, we performed NMF within each sample. Negative values in each centered expression matrix were set to zero. To minimize the influence of cell lineage-specific expression patterns and to enhance sensitivity and specificity, we conducted NMF analysis separately for each cell lineage. For each sample, we performed NMF (k = 10) for each cell lineage across datasets separately and summarized each NMF program using the top 50 genes based on NMF coefficients. To avoid redundancies, we removed NMF programs that overlapped more than 20% with others within the tumor.
Subsequently, we clustered the NMF programs within cell lineage based on Jaccard similarity. Given the substantial number of NMF programs, we restricted the clustering to NMF programs with at least a minimum overlap of 20% with any modules observed within a cell lineage. Clustering was performed using hierarchical clustering, and MPs were defined through manual inspection. Each MP included at least five NMF programs. For each MP, we selected the top 50 genes based on their frequency of occurrence. Furthermore, we removed MPs that originated from a single study or exhibited strong enrichment of ribosomal protein genes or mitochondria-encoded genes. Finally, we analyzed each MP’s function using a hypergeometric test to assess enrichment of its signature genes in MSigDB cancer hallmark and KEGG pathways. The MPs were then further grouped based on functional similarity (Fig. 2h, Extended Data Fig. 5b and Supplementary Table 7).
Survival analyses
To assess the prognostic significance of cell types, we obtained clinical and expression data from TCGA (Supplementary Table 8). Per cell type, the top 50 highly expressed genes were selected to calculate cell-type signature scores using GSVA for each individual. These scores were adjusted to sum to 1 per lineage to mitigate multicollinearity (Extended Data Fig. 4h,i). Survival differences between high- and low-scoring groups, stratified by median signature scores, were assessed via Kaplan–Meier analysis (Figs. 2k and 3h).
Additionally, to depict the clinical relevance of cell types within and across cancers, we conducted the following analyses. For each cell type, univariable Cox regression linked the adjusted GSVA score to overall survival per tumor type. A z score higher than 0 indicated increased mortality risk. To account for multiple tests, we adjusted the P values using the BH method. Furthermore, to assess the clinical relevance at the pan-cancer level, we used a meta-z-score, derived from the combined z scores across tumor types using Stouffer’s method63 (Fig. 7a and Extended Data Fig. 9a).
mIHC
Human tissue specimens were provided by Shanghai Pulmonary Hospital (for NSCLC sections), West China Hospital of Stomatology at Sichuan University (for HNSC sections) and Shanghai East Hospital (for CESC sections) under the approval of local medical ethics committees (Supplementary Table 9). For mIHC analysis, there were seven individuals (three males (NSCLC-1, NSCLC-2 and HNSCC-2) and four females (HNSCC-1, HNSCC-3, CESC-1 and CESC-3)). For samples collected in this study, sex was self-reported. Sex was not considered in the study design, and sex-based analyses were not conducted. Tissues were fixed in formalin, embedded in paraffin, sectioned to 4 μm and positioned onto adhesion microscope slides following routine methods. Automated staining was performed with a Leica BOND-MAX autostainer (Leica Microsystems). Slices were deparaffinized and pretreated with Epitope Retrieval Solution 2 (AR9640-CN, LeicaBiosystems) at 100 °C for 20 min. Peroxidase blocking was performed for 10 min using Bond Polymer (DS9800-CN, Leica) subsequent to rinsing in tris-buffered saline with tween-20 (TBST) buffer. Slides were incubated with primary antibody at room temperature (RT) for 1 h. Primary antibodies included rabbit anti-CTHRC1 (1:400; Abcam, ab85739), rabbit anti-SLPI (1:500; Thermo Fisher, PA5-82990), rabbit anti-osteopontin (SPP1; 1:1,500; Abcam, ab214050), rabbit anti-CD68 (1:400; Biolynx, BX50031) and rabbit anti-pan-cytokeratin (1:300; Biolynx, BX50143). All slides were stained with secondary reagents at RT for 10 min and tyramide signal amplification reagents at RT for 10 min. Nuclei were stained with DAPI (Sigma-Aldrich) after all human antigens had been labeled. Stained slides were scanned at ×20 magnification using a Pannoramic MIDI scanner (3DHISTECH), and images were analyzed using Halo software (Indica Labs).
NicheNet analysis
To infer potential ligands that influence transcriptomic changes or phenotypic shifts in target cell types, we used NicheNet for upstream regulatory factor identification. Our analysis used the top 100 upregulated genes in the cell types of interest as input, with all genes expressed in relevant cell lineages serving as the background gene set. Subsequently, we used the predict_ligand_activities function from the R package NicheNet to predict and rank these potential ligands (Fig. 5e).
Cell–cell interaction analysis
To quantify the probability of cell–cell communication, we used CellChat64 to predict potential interactions based on the expression of ligand–receptor pairs across cell types. Receptor–ligand interactions between cell types were identified by the specific expression of a receptor by one cell type and a ligand by another cell type. Significance of specific ligand–receptor interactions between two cell subsets was calculated by a permutation test.
ST analysis
From the GEO database, we collected ST data from 62 individuals of 6 cancer types (Supplementary Table 10). Due to the lack of gender information in many data entries and the fact that sex was not considered in the study design, sex-based analyses were not conducted. To ensure data quality, we conducted several standard statistical analyses using the Seurat package (Extended Data Fig. 6a).
In 10x Visium ST data, spots may contain multiple cells, complicating cell-type assignment. We applied STRIDE, a topic-model-based tool trained on single-cell data, to deconvolute cell-type proportions per spot. For ST data with matched scRNA-seq datasets, we used its annotated transcriptome for spot decomposition. For unmatched ST samples, we selected a compatible scRNA-seq dataset from TISCH, prioritizing shared cancer/cell types with the ST data as reported in the original study. Especially for malignant cell identification, we adopted a comprehensive approach, combining information provided by the original studies, STRIDE decomposition results and CopyKat predictions based on copy number variation (Extended Data Fig. 6c). Finally, the ST data were annotated into the major lineage level (Extended Data Fig. 6d).
Spatial localization analysis
To map subcellular composition within the ST dataset, we leveraged the top 50 marker genes from scRNA-seq datasets and applied the AddModuleScore function in Seurat to calculate subtype-specific enrichment scores.
Colocalization analysis
Because fibroblasts and macrophages were unevenly distributed, we assessed the correlation between CTHRC1+ fibroblasts and SPP1+ macrophage signature scores only in fibroblast-containing spots. A strong correlation across all cohorts suggests that these two cell types are closely associated within the tissue.
Distance measurement between fibroblasts and malignant cells
To characterize the spatial relationship between CTHRC1+ fibroblasts and malignant cells, we calculated the correlation between the CTHRC1+ fibroblast signature score and the distance to the tumor center. A positive correlation indicates that CTHRC1+ fibroblasts are located farther away from malignant cells, whereas a negative correlation suggests their proximity to the tumor center.
Bulk tumor subtyping
Following the analysis of the TME in pan-cancer scRNA-seq data, we used the signature genes of all 56 cell types to deconvolute the composition and density of the tumors. Our dataset comprised 8,743 individuals across 23 cancer types from the TCGA project (Supplementary Table 8). To make the expression profiles comparable across cancer types, for each cancer type, we normalized the expression profile for each gene by subtracting the average expression value. We used GSVA to score individuals based on the top 50 highly expressed genes of each cell type. Subsequently, hierarchical clustering was used to categorize individuals into five subtypes based on the signature score matrix. These subtypes were labeled according to clustering results, tumor purity, signature scores of major lineage and cell-type functional scores (Fig. 7e,f and Extended Data Fig. 9b,c). Additionally, we collected immune cell infiltration data estimated for each individual in TCGA by different methods from TIMER65 (Fig. 7g and Extended Data Fig. 9d).
For the validation dataset, we obtained RNA-seq profiles from METABRIC, comprising 1,992 individuals. To mitigate noise introduced during data processing and sequencing, we normalized expression profiles by subtracting the average expression value within each cancer type.
Benchmarking of different bulk tumor subtyping studies
To comprehensively evaluate the performance of TabulaTIME alongside previous similar studies, we compared the identified cell states, coassociation of cell types and the capacity to categorize bulk RNA-seq samples. First, regarding the identified cell states, we hypothesized that comprehensive signatures would show preferential upregulation in distinct clusters, whereas nonspecific or redundant cell states would co-upregulate within the same clusters. For a more equitable and quantitative comparison, we used the two gene sets from TabulaTIME and Luca as inputs for SCINA66, a marker gene-based cell-type annotation method, we predicted cell types in five additional scRNA-seq datasets. The accuracy of predicted cell-type annotations versus the original cell-type annotations and normalized mutual information (NMI) across cluster labels in the additional scRNA-seq dataset were determined (Extended Data Fig. 9g). A higher NMI reflected more precise and dependable annotations.
Additionally, we compared the TCGA stratification between TabulaTIME and the other three studies. Participant TME subtypes derived from TabulaTIME were more consistent with the Bagaev et al.6-defined participant TME subtypes, as they both considered immune and stromal cell types (Extended Data Fig. 9i). To quantitatively evaluate accuracy, we classified TCGA participants into immune-hot and immune-cold tumors based on deconvolution results (CIBERSORT, MCP counter67, TIMER and xCell68; Extended Data Fig. 9h). We then compared participant stratification from the different studies to these immune-hot and immune-cold classifications. For studies that explicitly mentioned the TME subtypes characterized by either high or low immune cell infiltration, we classified participants according to the designated TME subtypes. Specifically, in the TabulaTIME and Bagaev et al. studies, we defined desert subtypes, including DHP, DLP and D, as immune-cold tumors. Similarly, in Thorsson et al.55, we classified participants within the C3, C4 and C5 subtypes as immune-cold tumors. For studies that did not describe the immune cell infiltration for each TME subtype, we divided the original TME subtypes based on the reported immune cell infiltration levels. For example, in the Ecotyper study, we categorized CE2-, CE4-, CE5-, CE6-, CE7- and CE8-high tumors as immune-cold, whereas the remaining subtypes were defined as immune-hot.
Reference data for scRNA-seq data annotation methods
To benchmark our integrated transcription profiles, TabulaTIME, as a reference, we used the SELINA framework. We pretrained the SELINA model using TabulaTIME and the additional NSCLC and BRCA datasets. Performance was validated on external datasets by calculating accuracy as the fraction of cells with predicted versus annotated labels.
Statistics and reproducibility
This study focuses on individuals with solid tumors, necessitating targeted data selection and making random sampling inappropriate. Samples were defined by the original studies. No statistical method was used to predetermine sample size. Consequently, the experiments were not randomized, and data collection and analysis were not conducted blind to the experimental conditions. All statistical methods, selected as suitable for the data distribution, were reported in the Methods and figure legends. Cell-type distribution across multiple sources was evaluated using Kruskal–Wallis tests. Pairwise source distribution for each cell type, along with LISI, entropy, ASW score, cytotoxic signature score, exhausted signature score, NMI and Cibersort-estimated CD8+ T cell infiltration, were assessed using two-tailed unpaired Wilcoxon tests. A Fisher’s exact test validated subtype source preferences, and a log-rank test evaluated differences in survival. Multiple testing correction was applied where appropriate, P values are labeled in the figures, and significance was defined as P < 0.05. The corresponding figures illustrate the distribution of the individual data points.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Previously published datasets are available under their respective accession codes (Supplementary Tables 1 and 10). The analysis datasets in TabulaTIME can be explored and visualized at http://wanglab-compbio.cn/TabulaTIME/. The data that support the findings of this study are available from the corresponding author upon request. The original image data can be downloaded from https://zenodo.org/records/13363711 (ref. 69). Source data are provided with this paper.
Code availability
Code related to the analyses in this study can be found on GitHub at https://github.com/wanglabtongji/TabulaTiME.
References
Gavish, A. et al. Hallmarks of transcriptional intratumour heterogeneity across a thousand tumours. Nature 618, 598–606 (2023).
Hanahan, D. & Weinberg, R. A. Hallmarks of cancer: the next generation. Cell 144, 646–674 (2011).
De Visser, K. E. & Joyce, J. A. The evolving tumor microenvironment: from cancer initiation to metastatic outgrowth. Cancer Cell 41, 374–403 (2023).
Luca, B. A. et al. Atlas of clinically distinct cell states and ecosystems across human solid tumors. Cell 184, 5482–5496 (2021).
Mulder, K. et al. Cross-tissue single-cell landscape of human monocytes and macrophages in health and disease. Immunity 54, 1883–1900 (2021).
Bagaev, A. et al. Conserved pan-cancer microenvironment subtypes predict response to immunotherapy. Cancer Cell 39, 845–865 (2021).
Cheng, S. et al. A pan-cancer single-cell transcriptional atlas of tumor infiltrating myeloid cells. Cell 184, 792–809 (2021).
Zheng, L. et al. Pan-cancer single-cell landscape of tumor-infiltrating T cells. Science 374, abe6474 (2021).
Tang, F. et al. A pan-cancer single-cell panorama of human natural killer cells. Cell 186, 4235–4251 (2023).
Ma, J. et al. A blueprint for tumor-infiltrating B cells across human cancers. Science 384, eadj4857 (2024).
Wu, Y. et al. Neutrophil profiling illuminates anti-tumor antigen-presenting potency. Cell 6, 1422–1439 (2024).
Dominguez, C. X. et al. Single-cell RNA sequencing reveals stromal evolution into LRRC15+ myofibroblasts as a determinant of patient response to cancer immunotherapy. Cancer Discov. 10, 232–253 (2020).
Krishnamurty, A. T. et al. LRRC15+ myofibroblasts dictate the stromal setpoint to suppress tumour immunity. Nature 611, 148–154 (2022).
Wu, S. Z. et al. Stromal cell diversity associated with immune evasion in human triple‐negative breast cancer. EMBO J. 39, e104063 (2020).
Lavie, D., Ben-Shmuel, A., Erez, N. & Scherz-Shouval, R. Cancer-associated fibroblasts in the single-cell era. Nat Cancer 3, 793–807 (2022).
Cords, L. et al. Cancer-associated fibroblast classification in single-cell and spatial proteomics data. Nat. Commun. 14, 4294 (2023).
Friedman, G. et al. Cancer-associated fibroblast compositions change with breast cancer progression linking the ratio of S100A4+ and PDPN+ CAFs to clinical outcome. Nat. Cancer 1, 692–708 (2020).
Luo, H. et al. Pan-cancer single-cell analysis reveals the heterogeneity and plasticity of cancer-associated fibroblasts in the tumor microenvironment. Nat. Commun. 13, 6619 (2022).
Qian, J. et al. A pan-cancer blueprint of the heterogeneous tumor microenvironment revealed by single-cell profiling. Cell Res. 30, 745–762 (2020).
Barrett, T. et al. NCBI GEO: archive for functional genomics data sets—update. Nucleic Acids Res. 41, D991–D995 (2012).
Papatheodorou, I. et al. Expression Atlas update: from tissues to single cells. Nucleic Acids Res. 48, D77–D83 (2019).
Sun, D. et al. TISCH: a comprehensive web resource enabling interactive single-cell transcriptome visualization of tumor microenvironment. Nucleic Acids Res. 49, D1420–D1430 (2021).
Han, Y. et al. TISCH2: expanded datasets and new tools for single-cell transcriptome analyses of the tumor microenvironment. Nucleic Acids Res. 51, D1425–D1431 (2023).
Wang, C. et al. Integrative analyses of single-cell transcriptome and regulome using MAESTRO. Genome Biol. 21, 198 (2020).
Butler, A., Hoffman, P., Smibert, P., Papalexi, E. & Satija, R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 36, 411–420 (2018).
Gao, R. et al. Delineating copy number and clonal substructure in human tumors from single-cell transcriptomes. Nat. Biotechnol. 39, 599–608 (2021).
Patel, A. P. et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 344, 1396–1401 (2014).
Baran, Y. et al. MetaCell: analysis of single-cell RNA-seq data using K-nn graph partitions. Genome Biol. 20, 206 (2019).
Rousseeuw, P. J. Silhouettes: a graphical aid to the interpretation and validation of cluster analysis. J. Comput. Appl. Math. 20, 53–65 (1987).
Zappia, L. & Oshlack, A. Clustering trees: a visualization for evaluating clusterings at multiple resolutions. Gigascience 7, giy083 (2018).
Liu, B. et al. An entropy-based metric for assessing the purity of single cell populations. Nat. Commun. 11, 3155 (2020).
Simões, F. C. et al. Macrophages directly contribute collagen to scar formation during zebrafish heart regeneration and mouse heart repair. Nat. Commun. 11, 600 (2020).
Sinha, M. et al. Direct conversion of injury-site myeloid cells to fibroblast-like cells of granulation tissue. Nat. Commun. 9, 936 (2018).
Wendisch, D. et al. SARS-CoV-2 infection triggers profibrotic macrophage responses and lung fibrosis. Cell 184, 6243–6261 (2021).
Sahai, E. et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer 20, 174–186 (2020).
Qi, J. et al. Single-cell and spatial analysis reveal interaction of FAP+ fibroblasts and SPP1+ macrophages in colorectal cancer. Nat. Commun. 13, 1742 (2022).
Lam, A. P. et al. Nuclear β-catenin is increased in systemic sclerosis pulmonary fibrosis and promotes lung fibroblast migration and proliferation. Am. J. Respir. Cell Mol. Biol. 45, 915–922 (2011).
Bonnans, C., Chou, J. & Werb, Z. Remodelling the extracellular matrix in development and disease. Nat. Rev. Mol. Cell Biol. 15, 786–801 (2014).
Wu, R. et al. Comprehensive analysis of spatial architecture in primary liver cancer. Sci. Adv. 51, eabg3750 (2021).
Chen, H. et al. Mechanosensing by the α6-integrin confers an invasive fibroblast phenotype and mediates lung fibrosis. Nat. Commun. 7, 12564 (2016).
Henderson, N. C., Rieder, F. & Wynn, T. A. Fibrosis: from mechanisms to medicines. Nature 587, 555–566 (2020).
Yang, R. et al. Galectin-9 interacts with PD-1 and TIM-3 to regulate T cell death and is a target for cancer immunotherapy. Nat. Commun. 12, 832 (2021).
Wu, C. et al. Galectin-9–CD44 interaction enhances stability and function of adaptive regulatory T cells. Immunity 41, 270–282 (2014).
Heimberg, G. et al. A cell atlas foundation model for scalable search of similar human cells. Nature 638, 1085–1094 (2025).
Browaeys, R., Saelens, W. & Saeys, Y. NicheNet: modeling intercellular communication by linking ligands to target genes. Nat. Methods 17, 159–162 (2020).
Biffi, G. et al. IL1-induced JAK/STAT signaling is antagonized by TGFβ to shape CAF heterogeneity in pancreatic ductal adenocarcinoma. Cancer Discov. 9, 282–301 (2019).
Kalucka, J. et al. Single-cell transcriptome atlas of murine endothelial cells. Cell 180, 764–779 (2020).
Sahu, A. et al. In vivo tumor immune microenvironment phenotypes correlate with inflammation and vasculature to predict immunotherapy response. Nat. Commun. 13, 5312 (2022).
Chen, W. et al. AGT serves as a potential biomarker and drives tumor progression in colorectal carcinoma. Int. Immunopharmacol. 101, 108225 (2021).
Wang, Z. et al. Broad targeting of angiogenesis for cancer prevention and therapy. Semin. Cancer Biol. 35, S224–S243 (2015).
Goveia, J. et al. An integrated gene expression landscape profiling approach to identify lung tumor endothelial cell heterogeneity and angiogenic candidates. Cancer Cell 37, 21–36 (2020).
Hänzelmann, S., Castelo, R. & Guinney, J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics 14, 7 (2013).
Finotello, F. et al. Molecular and pharmacological modulators of the tumor immune contexture revealed by deconvolution of RNA-seq data. Genome Med. 11, 34 (2019).
Chen, B., Khodadoust, M. S., Liu, C. L., Newman, A. M. & Alizadeh, A. A. in Cancer Systems Biology, Vol. 1711 (ed. Von Stechow, L.) 243–259 (Springer, 2018).
Thorsson, V. et al. The immune landscape of cancer. Immunity 4, 411–412 (2018).
Ren, P. et al. Single-cell assignment using multiple-adversarial domain adaptation network with large-scale references. Cell Rep. Methods 3, 100577 (2023).
Haider, N. et al. Transition of macrophages to fibroblast-like cells in healing myocardial infarction. J. Am. Coll. Cardiol. 74, 3124–3135 (2019).
Iwamoto, C. et al. Bone marrow-derived macrophages converted into cancer-associated fibroblast-like cells promote pancreatic cancer progression. Cancer Lett. 512, 15–27 (2021).
Korsunsky, I. et al. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods 16, 1289–1296 (2019).
Kanehisa, M., Furumichi, M., Tanabe, M., Sato, Y. & Morishima, K. KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 45, D353–D361 (2017).
Liberzon, A. et al. The Molecular Signatures Database hallmark gene set collection. Cell Syst. 1, 417–425 (2015).
Yu, G., Wang, L.-G., Han, Y. & He, Q.-Y. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16, 284–287 (2012).
Zaykin, D. V. Optimally weighted Z-test is a powerful method for combining probabilities in meta-analysis: optimally weighted Z-test is a powerful method. J. Evol. Biol. 24, 1836–1841 (2011).
Jin, S. et al. Inference and analysis of cell–cell communication using CellChat. Nat. Commun. 12, 1088 (2021).
Li, T. et al. TIMER2.0 for analysis of tumor-infiltrating immune cells. Nucleic Acids Res. 48, W509–W514 (2020).
Zhang, Z. et al. SCINA: a semi-supervised subtyping algorithm of single cells and bulk samples. Genes 10, 531 (2019).
Becht, E. et al. Estimating the population abundance of tissue-infiltrating immune and stromal cell populations using gene expression. Genome Biol. 17, 218 (2016).
Aran, D., Hu, Z. & Butte, A. J. xCell: digitally portraying the tissue cellular heterogeneity landscape. Genome Biol. 18, 220 (2017).
Han, Y., Wu, Q. & Wang, C. Spatiotemporal analyses of the pan-cancer single-cell landscape reveal widespread profibrotic ecotypes regulating tumor immunity. Zenodo https://doi.org/10.5281/zenodo.13363711 (2024).
Acknowledgements
This work was supported by the National Key R&D Program of China (2022YFA1106000, C.W.), the National Natural Science Foundation of China (32222026, C.W.; 32170660, C.W.; 32000561, Q.W.; 82273091, F.L.; 81972551, T.L.; 82201948, L.Z.; 92168205, C.W.; 62088101, C.W.). Natural Science Foundation of Shanghai (24ZR1492800, C.W.), Shanghai Pilot Program for Basic Research (C.W.), Shanghai Municipal Science and Technology Major Project (2021SHZDZX0100, C.W.), Peak Disciplines (Type IV) of Institutions of Higher Learning in Shanghai (C.W.), The Fundamental Research Funds for the Central Universities (22120240435, C.W.), The Young Elite Scientist Sponsorship Program by CAST (2021QNRC001, T.L.) and Shanghai Sailing Program (21YF1438300, L.Z.). The distribution and functional conclusions of eFibro_CTHRC1 and Macro_SLPI cells in Fig. 5f were created with BioRender.com.
Author information
Authors and Affiliations
Contributions
C.W., Q.W. and Y.H. designed the study. Y.H. designed the TabulaTIME workflow, processed datasets and performed all computational analyses. Q.W., L.Z., G.C., F.L., T.L. and P.Z. collected the clinical samples. Q.W. analyzed hematoxylin and eosin images. Y.H. constructed the TabulaTIME website. D.S., Y.W., J.Y., J.H. and Z.D. collected the tumor-associated datasets. Y.H. wrote the manuscript with input from all authors. C.W. and Q.W. revised the manuscript. All authors read and approved the manuscript. C.W. supervised the whole project.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Cancer thanks Aleksandar Obradovic and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 ScRNA-seq data collection and processing overview.
Related to Fig. 1. (a) Workflow of data collection and processing, including quality control, malignant cell identification, batch effect removal, clustering, and annotation. (b) Quality control for BRCA_GSE148673 dataset: High-quality cells (blue) are defined as those with more than 1000 UMI counts and 500 gene counts, while low-quality cells (red) fall below these thresholds. (c) Doublet identification for the BRCA_GSE148673 dataset using Scrublet: Doublets were highlighted in red. (d) Malignant cells identification in the BRCA_GSE148673 dataset by CopyKat: Copy number variations are indicated as gain (red) and loss (blue), with the left bar designating malignant cells (in orange) and non-malignant cells (in green). (e) Entropy distribution measuring batch effects across 21 datasets, including 146 patients with associated batch information. In each box (dataset), entropy was computed for each cell, based on the patient distribution within its neighborhood (30 nearest neighbors). The datasets were classified into two types, ‘with batch effects’ and ‘without batch effects’ according to entropy median (0.7 as the cutoff). The entropy of raw data and batch-removed data are colored by green and orange, respectively. The bottom of the box represents the first quartile (Q1), and the top of the box represents the third quartile (Q3). The height of the box represents the interquartile range (IQR), while the horizontal line inside the box indicates the median. The whiskers extend to the positions of Q1 - 1.5 * IQR and Q3 + 1.5 * IQR. (f) Batch effect removal for BRCA_GSE114727_10X dataset: The panel on the left displays cells across patients before batch effect removal, while the panel on the right showcases the same cells post-batch removal. (g) UMAP visualization of BRCA_GSE148673 dataset: Clustering and cell type identification are visualized using UMAP, with distinct colors representing clusters and cell types. (h) Dot plot showing the expression of representative signature genes for each cell type in the BRCA_GSE148673 dataset.
Extended Data Fig. 2 MetaCell identification and batch correction.
Related to Fig. 1. (a) Workflow of MetaCell identification and integration. (b) Box plots illustrating the distribution of gene coverage (left) and the degree of within-MetaCell variation (right) across MetaCells, which encompass varying cell counts across five datasets. The datasets, listed from top to bottom, include the following number of samples and cells: 6 patients with 10,359 cells, 2 patients with 4,375 cells, 14 patients with 33,043 cells, 3 patients with 28,678 cells, and 2 patients with 6,035 cells. The bottom of the box represents the Q1, and the top of the box represents the Q3. The height of the box represents the IQR, while the horizontal line inside the box indicates the median. The whiskers extend to the positions of Q1 - 1.5 * IQR and Q3 + 1.5 * IQR. (c) Application of the Same Analysis as (b) to NSCLC Datasets. The datasets, listed from top to bottom, include the following number of samples and cells: 2 patients with 3,658 cells, 3 patients with 12,193 cells, 1 patient with 1,108 cells, 4 patients with 11,453 cells, and 5 patients with 40,218 cells. The bottom of the box represents the Q1, and the top of the box represents the Q3. The height of the box represents the IQR, while the horizontal line inside the box indicates the median. The whiskers extend to the positions of Q1 - 1.5 * IQR and Q3 + 1.5 * IQR. (d) Radar plots showing the metrics for MetaCells under different cell numbers, including gene coverage, variation within MetaCells, and the LISI (Local Inverse Simpson’s Index) score, are accessed for BRCA_GSE148673 and NSCLC_GSE117570 datasets. (e) Box plot illustrating the distribution of LISI and entropies calculated from 736 patients across four scenarios: direct integration of single-cell and MetaCell expression profiles, and integrated single-cell and MetaCell expression profiles using CCA. Significance was assessed using a two-sided Wilcoxon test and adjusted using the Benjamini-Hochberg (BH) method. The bottom of the box represents the Q1, and the top of the box represents the Q3. The height of the box represents the IQR, while the horizontal line inside the box indicates the median. The whiskers extend to the positions of Q1 - 1.5 * IQR and Q3 + 1.5 * IQR. (f) Boxplot showing the distribution of ARI and ASW calculated from 736 patients across four scenarios: direct integration of single-cell and MetaCell expression profiles, and integrated single-cell and MetaCell expression profiles using CCA. Significance was assessed using a two-sided Wilcoxon test and adjusted using the BH method. The bottom of the box represents the Q1, and the top of the box represents the Q3. The height of the box represents the IQR, while the horizontal line inside the box indicates the median. The whiskers extend to the positions of Q1 - 1.5 * IQR and Q3 + 1.5 * IQR. (g) The pie plot showing the fractional distribution of MetaCells by source (left) and treatment condition (right), with MetaCells labeled accordingly. (h) UMAP visualization of all MetaCells, colored by the cancer type (left) and cell type (right) respectively.
Extended Data Fig. 3 Characteristics of lymphocyte cells.
Related to Fig. 2. (a) Scatter plot showing the average cytotoxic score and exhausted score per cell type in cytotoxic lymphocytes cell types, with colors representing cell type. (b) Heatmap showing the proportion of different cytotoxic lymphocytes cell types in various cancer types or healthy tissues. The rows include a bar plot representing the number of MetaCells (in log10 scale), and the origination of cancer cells labeled by the different colors (c) UMAP visualization depicting the distribution of MetaCells of conventional and regulatory lymphocytes, with cell types represented by different colors. (d) Box plot showing the proportions of distinct CD4+ T cells subgroups across different sources, including blood (red), normal tissue (green), precancerous (orange), tumor (purple), and metastatic (blue), derived from 605 treatment-naïve samples. Significance labels in the figure was assessed via Kruskal-Wallis tests to compare each subgroup distributions among five tissue types. Significance for pairwise source proportions within each cell type, assessed via two-tailed unpaired Wilcoxon tests, is reported in Supplementary Table 6. The open rectangle annotates the comparative scope, with BH correction for multiple testing. The bottom of the box represents the Q1, and the top of the box represents the Q3. The height of the box represents the IQR, while the horizontal line inside the box indicates the median. The whiskers extend to the positions of Q1 - 1.5 * IQR and Q3 + 1.5 * IQR. (e) Heatmap showing the proportion of different conventional and regulatory lymphocyte cell types in various cancer types or healthy tissues. The rows include a bar plot representing the number of MetaCells (in log10 scale), and the origination of cancer cells labeled by the different colors. (f) UMAP plot showing the distribution of B and plasma MetaCells, with cell types distinguished by colors. (g) Box plot showing the proportions of distinct B and plasma cells subgroups across different sources, including blood (red), normal tissue (green), precancerous (orange), tumor (purple), and metastatic (blue), derived from 462 treatment-naïve samples. Significance labels in the figure was assessed via Kruskal-Wallis tests to compare each subgroup distributions among five tissue types. Significance for pairwise source proportions within each cell type, assessed via two-tailed unpaired Wilcoxon tests, is reported in Supplementary Table 6. The open rectangle annotates the comparative scope, with BH correction for multiple testing. The bottom of the box represents the Q1, and the top of the box represents the Q3. The height of the box represents the IQR, while the horizontal line inside the box indicates the median. The whiskers extend to the positions of Q1 - 1.5 * IQR and Q3 + 1.5 * IQR. (h) Heatmap showing the proportion of different B and plasma cell types in various cancer types or healthy tissues. The rows include a bar plot representing the number of MetaCells (in log10 scale), and the origin of cancer cells labeled by the different colors.
Extended Data Fig. 4 Characteristics of myeloid cells.
Related to Fig. 2. (a) Line plots depicting the average silhouette width of myeloid cells across a range of resolutions from 0.1 to 1.5. (b) Clustering trees of the myeloid cells colored according to the expression of known markers. The node colors indicate the average of the log2 TPM of samples in each cluster. CLEC9A shows a population of conventional type 1 dendritic cells (cDC1), and C1QC is a marker of macrophage cells. (c) Box plot illustrating cell purity for each myeloid cell type, calculated using ROGUE from 797 samples. The bottom of each box indicates the Q1, and the top represents the Q3. The height of the box reflects the IQR, and the horizontal line inside the box indicates the median. The whiskers extend to the positions of Q1 - 1.5 * IQR and Q3 + 1.5 * IQR. (d) Dot plot depicting the expression of representative marker genes of each myeloid cell type. (e) Box plot showing the proportions of distinct dendritic cell subgroups across different sources, including blood (red), normal tissue (green), precancerous (orange), tumor (purple), and metastatic (blue), derived from 496 treatment-naïve samples. Significance labels in the figure was assessed via Kruskal-Wallis tests to compare each subgroup distributions among five tissue types. Significance for pairwise source proportions within each cell type, assessed via two-tailed unpaired Wilcoxon tests, is reported in Supplementary Table 6. The open rectangle annotates the comparative scope, with BH correction for multiple testing. The bottom of the box represents the Q1, and the top of the box represents the Q3. The height of the box represents the IQR, while the horizontal line inside the box indicates the median. The whiskers extend to the positions of Q1 - 1.5 * IQR and Q3 + 1.5 * IQR. (f) Heatmap showing the ORs of myeloid cell sub types occurring in each source. OR > 1.5 indicates that the cell type is preferred to distribute in the corresponding source. Significance was assessed using the two-sided Fisher test and adjusted using the BH method. (g) Heatmap showing the enriched pathways for each myeloid cell subset. (h) Heatmap showing the number of overleaped differentially expressed genes among all cell types. (i) Heatmap depicting the correlation of cell type signature scores calculated by GSVA between before and after corrected in TCGA-ESCA dataset.
Extended Data Fig. 5 Heterogeneity of fibroblast subpopulations in cancerous tissues.
Related to Fig. 3. (a) Heatmap showing the ORs of cell types occurring in each source. OR > 1.5 indicates that the cell type is preferred to distribute in the corresponding source. Significance was assessed using the two-sided Fisher test and adjusted using the BH method. (b) Left: heatmap showing Jaccard similarity indices for comparisons among 1,470 robust NMF programs based on their top 50 genes within the fibroblast cell lineage. Programs are ordered by clustering and grouped into families of MPs with related functions (marked by black dashed lines); MP families are numbered and labeled. Right: list of all MP names, separated into MP families. (c) Box plot depicting the signature scores of MPs across fibroblast subtypes, calculated from 379 samples. MPs are color-coded based on their functional annotations. The bottom of the box represents the Q1, and the top of the box represents the Q3. The height of the box represents the IQR, while the horizontal line inside the box indicates the median. The whiskers extend to the positions of Q1 - 1.5 * IQR and Q3 + 1.5 * IQR. (d) Heatmap illustrating the GSVA scores assigned to metabolic pathways within each fibroblast subset, with the pathways categorized according to KEGG nomenclature.
Extended Data Fig. 6 Spatial transcriptomics (ST) data processing and the association between eFibro_CTHRC1 and immune infiltration.
Related to Fig. 4. (a) Workflow of ST data collection and processing, including quality control and identification of malignant cells, clustering, and annotation. (b) A representation of Hematoxylin and eosin (H&E) stained tissue sections from a patient’s ST spot, along with spatial feature plots detailing read counts and feature numbers for a single ST spot. H&E image reproduced with permission from ref. 39, AAAS. Scale bars, 50 μm. (c) Spatial feature plot identifying malignant cells as predicted by CopyKat. (d) Spatial feature plot depicting the deconvolution output from STRIDE. (e) Spatial feature plot demonstrating the results of clustering analysis. (f) Violin plot showing the CTHRC1+ fibroblasts signature score across different spatial clusters. (g) Box plots illustrating the inferred associations between inducers of CTHRC1+ fibroblasts and their signature scores. The red box represents data calculated from 62 ST samples, while the green box is derived from 9,460 samples across 23 cancer types in the TCGA project. The bottom of each box indicates the Q1, and the top represents the Q3. The height of the box reflects the IQR, and the horizontal line inside the box indicates the median. The whiskers extend to the positions of Q1 - 1.5 * IQR and Q3 + 1.5 * IQR. (h) Comparative box plots of CD8 + T cell infiltration levels in 9,460 samples from the TCGA project, comparing high and low CTHRC1+ fibroblast signature scores, stratified by cancer type, as estimated by Cibersort. The bottom of each box indicates the Q1, and the top represents the Q3. The height of the box reflects the IQR, and the horizontal line inside the box indicates the median. The whiskers extend to the positions of Q1 - 1.5 * IQR and Q3 + 1.5 * IQR. (i) Box plots comparing the CD8+ T cell infiltration in 9,460 samples from the TCGA project with high versus low CTHRC1+ fibroblasts signature score across different cancer types, with estimations based on EPIC. The bottom of each box indicates the Q1, and the top represents the Q3. The height of the box reflects the IQR, and the horizontal line inside the box indicates the median. The whiskers extend to the positions of Q1 - 1.5 * IQR and Q3 + 1.5 * IQR.
Extended Data Fig. 7 The association between SLPI+ macrophage and CTHRC1+ Fibroblast.
Related to Fig. 5. (a) Umap visualization showing all fibroblasts and monocyte/macrophage cells from the PAAD_CRA001160 dataset (left). UMAP plots showing the marker genes expression for major cell types of all MetaCells (right). (b) Umap visualization showing all fibroblasts and monocyte/macrophage cells from the CHOL_GSE138709 dataset (left). UMAP plots showing the marker genes expression for major cell types of all MetaCells (right). (c) Scatter plots demonstrating the relationship between the signature score of CTHRC1+ fibroblasts (x-axis) and SLPI+ macrophages (y-axis) in TCGA UVM and KICH datasets. The correlation was calculated using using the Pearson correlation coefficient. The error band thoughtfully denotes the 95% confidence interval, which is calculated based on the standard error using the normal distribution. The center measure of the smooth line corresponds to the predicted values from the linear regression model. (d) Immunofluorescence imaging of DAPI, CD68, SPLI, SPP1, and CTHRC1 in CESC specimens. Scale bars, 50 μm.
Extended Data Fig. 8 The heterogeneity of endothelial cells.
Related to Fig. 6. (a) Dot plot illustrating the expression patterns of representative marker genes for each cell type. (b) UMAP visualization showing all cells from the PRAD_GSE172301 and CHOL_GSE1426784_CRA001160 dataset (left). UMAP plots showing the marker genes expression for major cell types of all MetaCells (right).
Extended Data Fig. 9 The prognosis association of cell types.
Related to Fig. 7. (a) Survival associations based on the signature genes of conventional and regulatory lymphocytes, B lymphocytes, and endothelial subsets per cell type. At the top, a bar plot elegantly displays the pan-cancer survival associations across 23 different cancer types, aggregated harmoniously using Stouffer’s method. Columns are gracefully ordered by combined Z-score. At the bottom, cancer-specific survival associations are tastefully presented, and statistical significance is calculated using the Cox Proportional-Hazards Model (b) Violin plots illustrating the signature scores of different lineages across five distinct tumor ecosystems, comprising a total of 8,734 samples from the TCGA project. The shape of the violin plot illustrates the distribution density of the data. The wider sections indicate a higher concentration of data points near that value (c) Box plots depicting the exhausted signature scores across three tumor ecotypes, comprising a total of 4,882 samples from the TCGA project. Statistical significance was evaluated using the two-tailed unpaired Wilcoxon tests combined with a permutation test (10,000 resamplings) to compare the signature score distribution across pairwise ecotypes; horizontal connectors denote compared groups, with the corresponding p-value indicated above the horizontal line. The bottom of the box represents the Q1, and the top of the box represents the Q3. The height of the box represents the IQR, while the horizontal line inside the box indicates the median. The whiskers extend to the positions of Q1 - 1.5 * IQR and Q3 + 1.5 * IQR. (d) Box plot depicting the CD8+ T cell infiltration among five patient subtypes. The CD8+ T cell infiltration was estimated using Cibersort from 8,734 samples from the TCGA project. Statistical significance of CD8+ T cell infiltration across pairwise patient subtypes was assessed using the two-tailed unpaired Wilcoxon tests. Horizontal lines connect compared groups, with corresponding p-values indicated above each line. The bottom of the box represents the Q1, while the top represents the Q3. The height of the box indicates the IQR, and the horizontal line inside the box represents the median. The whiskers extend to the positions of Q1 - 1.5 * IQR and Q3 + 1.5 * IQR. (e) Bar plot showing the proportion of each tumor sample ecosystem in different molecular cancer groups. (f) Heatmap displaying the average signature scores of fibroblast-associated cell states in each fibroblast cluster from the BRCA_GSE176078 dataset, with cell states identified separately by TabulaTIME (on the left) and Luca (on the right). (g) Left: Bar plot depicting the accuracy of using SCINA predicted annotations, based on distinct marker gene lists, versus annotation provided by the original published papers. Right: Bar plot depicting the normalized mutual information (NMI) of using SCINA predicted annotations, based on distinct marker gene lists, versus the cluster label. The datasets, listed from left to right, include the following number of samples and cells: 11 patients with 37,936 cells, 26 patients with 89,471 cells, 22 patients with 9,544 cells, 42 patients with 82,267 cells, and 12 patients with 90,603 cells. Significance was assessed using a two-tailed unpaired Wilcoxon tests and adjusted using the Benjamini-Hochberg (BH) method. The resulting p-values were 0.016 for ALL_GSE132509, 0.00058 for BRCA_GSE176078, 0.055 for ESCA_GSE173950, 0.026 for NSCLC_GSE148071 and 0.48 for RB_GSE166173. The bottom of the box represents the Q1, and the top of the box represents the Q3. The height of the box represents the IQR, while the horizontal line inside the box indicates the median. The whiskers extend to the positions of Q1 - 1.5 * IQR and Q3 + 1.5 * IQR (h) Left: Alluvial diagram showing changes in patient subtypes composition between TabulaTIME and Bagaev et al identified. Right: Scatter plot delineating the accuracy of different studies in distinguishing hot and cold tumors.
Extended Data Fig. 10 Predicted cell type labels based on TabulaTiME.
Related to Fig. 8. UMAP visualization comparing the distribution of originally annotated cell types (left) with Selina-predicted cell types (right).
Supplementary information
Supplementary Tables 1–10
Supplementary Tables 1–10.
Source data
Source Data Fig. 1
Statistical source data.
Source Data Fig. 2
Statistical source data.
Source Data Fig. 3
Statistical source data.
Source Data Fig. 4
Statistical source data.
Source Data Fig. 5
Statistical source data.
Source Data Fig. 6
Statistical source data.
Source Data Fig. 7
Statistical source data.
Source Data Fig. 8
Statistical source data.
Source Data Extended Data Fig. 1
Statistical source data.
Source Data Extended Data Fig. 2
Statistical source data.
Source Data Extended Data Fig. 3
Statistical source data.
Source Data Extended Data Fig. 4
Statistical source data.
Source Data Extended Data Fig. 5
Statistical source data.
Source Data Extended Data Fig. 6
Statistical source data.
Source Data Extended Data Fig. 7
Statistical source data.
Source Data Extended Data Fig. 8
Statistical source data.
Source Data Extended Data Fig. 9
Statistical source data.
Source Data Extended Data Fig. 10
Statistical source data.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Han, Y., Zhang, L., Sun, D. et al. Spatiotemporal analyses of the pan-cancer single-cell landscape reveal widespread profibrotic ecotypes associated with tumor immunity. Nat Cancer 6, 1880–1898 (2025). https://doi.org/10.1038/s43018-025-01039-5
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s43018-025-01039-5
This article is cited by
-
A novel signature of palmitoylation for predicting prognosis and therapeutic response of hepatocellular carcinoma
Discover Oncology (2026)
-
Single-cell RNA-seq combined with bulk RNA-seq analysis identifies necroptosis-related genes as therapeutic targets for periodontitis
BMC Medical Genomics (2025)
-
Reference-guided computational framework identifies microenvironment metabolic subtypes and targets using pan-cancer single-cell datasets
Genome Medicine (2025)
