
Abstract
Tetraspanins affect metastasis, stemness and angiogenesis, but their roles in inflammation remain to be further clarified. Here we show that endothelial ablation of tetraspanin Cd82 markedly reduces vascular inflammation by mitigating endothelial leakage. Mechanistically, by limiting the anchorages of Cdc42 activator FARP1 and RhoA inhibitor Rnd3 to the plasma membrane (PM), CD82 confines Cdc42 but maintains RhoA activity in endothelial cells, to facilitate endothelium activation. These signaling regulatory effects depend on the ability of CD82 to coalesce and retain accessible cholesterol (AC) at the PM, whereas simvastatin overturns CD82 effects by lowering AC. CD82 supports non-vesicular transfer of AC to the PM through oxysterol-binding protein-related proteins (ORPs). Thus, CD82 and AC promote vascular leakage, whereas statin and ORP inhibitor restrain vascular leakage by decreasing AC. These findings reveal an unconventional anti-inflammation role and mechanism for statin and conceptualize tetraspanin-mediated, AC-mediated and cholesterol transfer-mediated balancing of antagonistic GTPase signaling pathways as regulatory mechanisms for vascular leakage.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 digital issues and online access to articles
$119.00 per year
only $9.92 per issue
Buy this article
- Purchase on SpringerLink
- Instant access to the full article PDF.
USD 39.95
Prices may be subject to local taxes which are calculated during checkout








Similar content being viewed by others


Data availability
All source data are provided with this research article. TMT proteomics data were deposited in the PRIDE partner repository under accession number PXD065001, and the bulk RNA-seq data were deposited in National Center for Biotechnology Information BioSample under accession number SAMN48951810. All supporting data for the findings of this project are available within either the article or its Supplementary Information. Additional information related to the findings of this project are available from the corresponding author upon reasonable request. Source data are provided with this paper.
Code availability
No code was developed for this study.
Change history
21 August 2025
A Correction to this paper has been published: https://doi.org/10.1038/s44161-025-00708-z
References
Goddard, L. M. & Iruela-Arispe, M. L. Cellular and molecular regulation of vascular permeability. Thromb. Haemost. 109, 407–415 (2013).
Komarova, Y. & Malik, A. B. Regulation of endothelial permeability via paracellular and transcellular transport pathways. Annu. Rev. Physiol. 72, 463–493 (2010).
Prasad, M., Leon, M., Lerman, L. O. & Lerman, A. Viral endothelial dysfunction: a unifying mechanism for COVID-19. Mayo Clin. Proc. 96, 3099–3108 (2021).
Giannotta, M., Trani, M. & Dejana, E. VE-cadherin and endothelial adherens junctions: active guardians of vascular integrity. Dev. Cell 26, 441–454 (2013).
Li, X. et al. VEGFR2 pY949 signalling regulates adherens junction integrity and metastatic spread. Nat. Commun. 7, 11017 (2016).
Spindler, V., Schlegel, N. & Waschke, J. Role of GTPases in control of microvascular permeability. Cardiovasc. Res. 87, 243–253 (2010).
Beckers, C. M., van Hinsbergh, V. W. & van Nieuw Amerongen, G. P. Driving Rho GTPase activity in endothelial cells regulates barrier integrity. Thromb. Haemost. 103, 40–55 (2010).
Yanez-Mo, M., Barreiro, O., Gordon-Alonso, M., Sala-Valdes, M. & Sanchez-Madrid, F. Tetraspanin-enriched microdomains: a functional unit in cell plasma membranes. Trends Cell Biol. 19, 434–446 (2009).
Charrin, S. et al. Lateral organization of membrane proteins: tetraspanins spin their web. Biochem. J. 420, 133–154 (2009).
Termini, C. M. & Gillette, J. M. Tetraspanins function as regulators of cellular signaling. Front. Cell Dev. Biol. 5, 34 (2017).
Hemler, M. E. Tetraspanin functions and associated microdomains. Nat. Rev. Mol. Cell Biol. 6, 801–811 (2005).
Zhang, F., Kotha, J., Jennings, L. K. & Zhang, X. A. Tetraspanins and vascular functions. Cardiovasc. Res. 83, 7–15 (2009).
Bailey, R. L. et al. The emerging role of tetraspanin microdomains on endothelial cells. Biochem. Soc. Trans. 39, 1667–1673 (2011).
Takeda, Y. et al. Deletion of tetraspanin Cd151 results in decreased pathologic angiogenesis in vivo and in vitro. Blood 109, 1524–1532 (2007).
Zhang, F. et al. Tetraspanin CD151 maintains vascular stability by balancing the forces of cell adhesion and cytoskeletal tension. Blood 118, 4274–4284 (2011).
Wei, Q. et al. CD82 restrains pathological angiogenesis by altering lipid raft clustering and CD44 trafficking in endothelial cells. Circulation 130, 1493–1504 (2014).
Xu, C. et al. CD82 endocytosis and cholesterol-dependent reorganization of tetraspanin webs and lipid rafts. FASEB J. 23, 3273–3288 (2009).
Zimmerman, B. et al. Crystal structure of a full-length human tetraspanin reveals a cholesterol-binding pocket. Cell 167, 1041–1051 (2016).
Susa, K. J., Rawson, S., Kruse, A. C. & Blacklow, S. C. Cryo-EM structure of the B cell co-receptor CD19 bound to the tetraspanin CD81. Science 371, 300–305 (2021).
Lipper, C. H., Egan, E. D., Gabriel, K. H. & Blacklow, S. C. Structural basis for membrane-proximal proteolysis of substrates by ADAM10. Cell 186, 3632–3641 (2023).
Das, A., Brown, M. S., Anderson, D. D., Goldstein, J. L. & Radhakrishnan, A. Three pools of plasma membrane cholesterol and their relation to cholesterol homeostasis. eLife 3, e02882 (2014).
Endapally, S. et al. Molecular discrimination between two conformations of sphingomyelin in plasma membranes. Cell 176, 1040–1053 (2019).
Kinnebrew, M. et al. Cholesterol accessibility at the ciliary membrane controls hedgehog signaling. eLife 8, e50051 (2019).
Zhou, Q. D. et al. Interferon-mediated reprogramming of membrane cholesterol to evade bacterial toxins. Nat. Immunol. 21, 746–755 (2020).
Abrams, M. E. et al. Oxysterols provide innate immunity to bacterial infection by mobilizing cell surface accessible cholesterol. Nat. Microbiol. 5, 929–942 (2020).
Wang, S. et al. Cholesterol 25-hydroxylase inhibits SARS-CoV-2 and other coronaviruses by depleting membrane cholesterol. EMBO J. 39, e106057 (2020).
Naito, T. et al. Movement of accessible plasma membrane cholesterol by the GRAMD1 lipid transfer protein complex. eLife 8, e51401 (2019).
Huang, C., Hays, F. A., Tomasek, J. J., Benyajati, S. & Zhang, X. A. Tetraspanin CD82 interaction with cholesterol promotes extracellular vesicle–mediated release of ezrin to inhibit tumour cell movement. J. Extracell. Vesicles 9, 1692417 (2020).
Charrin, S. et al. A physical and functional link between cholesterol and tetraspanins. Eur. J. Immunol. 33, 2479–2489 (2003).
Wang, Z. et al. 27-Plex tandem mass tag mass spectrometry for profiling brain proteome in Alzheimer’s disease. Anal. Chem. 92, 7162–7170 (2020).
Dejana, E., Orsenigo, F. & Lampugnani, M. G. The role of adherens junctions and VE-cadherin in the control of vascular permeability. J. Cell Sci. 121, 2115–2122 (2008).
Amado-Azevedo, J. et al. A CDC42-centered signaling unit is a dominant positive regulator of endothelial integrity. Sci. Rep. 7, 10132 (2017).
Etienne-Manneville, S. & Hall, A. Rho GTPases in cell biology. Nature 420, 629–635 (2002).
Priya, R. et al. Feedback regulation through myosin II confers robustness on RhoA signalling at E-cadherin junctions. Nat. Cell Biol. 17, 1282–1293 (2015).
Wildenberg, G. A. et al. p120-catenin and p190RhoGAP regulate cell-cell adhesion by coordinating antagonism between Rac and Rho. Cell 127, 1027–1039 (2006).
Tall, A. R. & Yvan-Charvet, L. Cholesterol, inflammation and innate immunity. Nat. Rev. Immunol. 15, 104–116 (2015).
Radhakrishnan, A., Rohatgi, R. & Siebold, C. Cholesterol access in cellular membranes controls Hedgehog signaling. Nat. Chem. Biol. 16, 1303–1313 (2020).
Sakata, A. et al. Acid sphingomyelinase inhibition suppresses lipopolysaccharide-mediated release of inflammatory cytokines from macrophages and protects against disease pathology in dextran sulphate sodium-induced colitis in mice. Immunology 122, 54–64 (2007).
Kielczewski, J. L. et al. Free insulin-like growth factor binding protein-3 (IGFBP-3) reduces retinal vascular permeability in association with a reduction of acid sphingomyelinase (ASMase). Invest. Ophthalmol. Vis. Sci. 52, 8278–8286 (2011).
Johnson, K. A., Endapally, S., Vazquez, D. C., Infante, R. E. & Radhakrishnan, A. Ostreolysin A and anthrolysin O use different mechanisms to control movement of cholesterol from the plasma membrane to the endoplasmic reticulum. J. Biol. Chem. 294, 17289–17300 (2019).
Infante, R. E. & Radhakrishnan, A. Continuous transport of a small fraction of plasma membrane cholesterol to endoplasmic reticulum regulates total cellular cholesterol. eLife 6, e25466 (2017).
Gay, A., Rye, D. & Radhakrishnan, A. Switch-like responses of two cholesterol sensors do not require protein oligomerization in membranes. Biophys. J. 108, 1459–1469 (2015).
Heisler, D. B. et al. A concerted mechanism involving ACAT and SREBPs by which oxysterols deplete accessible cholesterol to restrict microbial infection. eLife 12, e83534 (2023).
Ikenouchi, J. & Aoki, K. Membrane bleb: a seesaw game of two small GTPases. Small GTPases 8, 85–89 (2017).
Fackler, O. T. & Grosse, R. Cell motility through plasma membrane blebbing. J. Cell Biol. 181, 879–884 (2008).
Li, Z., Mintzer, E. & Bittman, R. First synthesis of free cholesterol–BODIPY conjugates. J. Org. Chem. 71, 1718–1721 (2006).
Holtta-Vuori, M. et al. BODIPY-cholesterol: a new tool to visualize sterol trafficking in living cells and organisms. Traffic 9, 1839–1849 (2008).
Sezgin, E. et al. A comparative study on fluorescent cholesterol analogs as versatile cellular reporters. J. Lipid Res. 57, 299–309 (2016).
Sandhu, J. et al. Aster proteins facilitate nonvesicular plasma membrane to ER cholesterol transport in mammalian cells. Cell 175, 514–529 (2018).
Wang, H. et al. ORP2 delivers cholesterol to the plasma membrane in exchange for phosphatidylinositol 4, 5-bisphosphate (PI(4,5)P2). Mol. Cell 73, 458–473 (2019).
Zhao, K., Foster, J. & Ridgway, N. D. Oxysterol-binding protein-related protein 1 variants have opposing cholesterol transport activities from the endolysosomes. Mol. Biol. Cell 31, 793–802 (2020).
Burgett, A. W. et al. Natural products reveal cancer cell dependence on oxysterol-binding proteins. Nat. Chem. Biol. 7, 639–647 (2011).
Zhou, B., Liu, L., Reddivari, M. & Zhang, X. A. The palmitoylation of metastasis suppressor KAI1/CD82 is important for its motility- and invasiveness-inhibitory activity. Cancer Res. 64, 7455–7463 (2004).
Jain, M. K. & Ridker, P. M. Anti-inflammatory effects of statins: clinical evidence and basic mechanisms. Nat. Rev. Drug Discov. 4, 977–987 (2005).
Li, D., Dammer, E. B., Lucki, N. C. & Sewer, M. B. cAMP-stimulated phosphorylation of diaphanous 1 regulates protein stability and interaction with binding partners in adrenocortical cells. Mol. Biol. Cell 24, 848–857 (2013).
Kentala, H. et al. Analysis of ORP2-knockout hepatocytes uncovers a novel function in actin cytoskeletal regulation. FASEB J. 32, 1281–1295 (2018).
Broman, M. T., Mehta, D. & Malik, A. B. Cdc42 regulates the restoration of endothelial adherens junctions and permeability. Trends Cardiovasc. Med. 17, 151–156 (2007).
Huang, C. et al. Tetraspanin-enriched microdomains regulate digitation junctions. Cell. Mol. Life Sci. 75, 3423–3439 (2018).
Uchtmann, K. et al. Homozygous loss of mouse tetraspanin CD82 enhances integrin αIIbβ3 expression and clot retraction in platelets. Exp. Cell. Res. 339, 261–269 (2015).
Benedito, R. et al. Notch-dependent VEGFR3 upregulation allows angiogenesis without VEGF–VEGFR2 signalling. Nature 484, 110–114 (2012).
Gon, Y. et al. S1P3 receptor-induced reorganization of epithelial tight junctions compromises lung barrier integrity and is potentiated by TNF. Proc. Natl Acad. Sci. USA 102, 9270–9275 (2005).
Scheppke, L. et al. Retinal vascular permeability suppression by topical application of a novel VEGFR2/Src kinase inhibitor in mice and rabbits. J. Clin. Invest. 118, 2337–2346 (2008).
Balda, M. S. et al. Functional dissociation of paracellular permeability and transepithelial electrical resistance and disruption of the apical-basolateral intramembrane diffusion barrier by expression of a mutant tight junction membrane protein. J. Cell Biol. 134, 1031–1049 (1996).
Endapally, S., Infante, R. E. & Radhakrishnan, A. Monitoring and modulating intracellular cholesterol trafficking using ALOD4, a cholesterol-binding protein. Methods Mol. Biol. 1949, 153–163 (2019).
Newby, Z. E. et al. A general protocol for the crystallization of membrane proteins for X-ray structural investigation. Nat. Protoc. 4, 619–637 (2009).
Bartoschik, T. et al. Near-native, site-specific and purification-free protein labeling for quantitative protein interaction analysis by MicroScale Thermophoresis. Sci. Rep. 8, 4977 (2018).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
Practice and Experience in Advanced Research Computing 2017: Sustainability, Success and Impact (Association for Computing Machinery, 2017).
Mirabello, C. & Wallner, B. DockQ v2: improved automatic quality measure for protein multimers, nucleic acids, and small molecules. Bioinformatics 40, btae586 (2024).
Jurrus, E. et al. Improvements to the APBS biomolecular solvation software suite. Protein Sci. 27, 112–128 (2018).
Reed, L. J. & Muench, H. A simple method of estimating fifty per cent endpoints. Am. J. Epidemiol. 27, 493–497 (1938).
Ideker, T. & Krogan, N. J. Differential network biology. Mol. Syst. Biol. 8, 565 (2012).
Wren, J. D. A global meta-analysis of microarray expression data to predict unknown gene functions and estimate the literature–data divide. Bioinformatics 25, 1694–1701 (2009).
Wren, J. D., Bekeredjian, R., Stewart, J. A., Shohet, R. V. & Garner, H. R. Knowledge discovery by automated identification and ranking of implicit relationships. Bioinformatics 20, 389–398 (2004).
Szklarczyk, D. et al. STRING v10: protein–protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 43, D447–D452 (2015).
Acknowledgements
This work was supported by National Institutes of Health grants HL132553, HL137819 and GM135547 and American Heart Association grants 13GRNT17040028 and 15GRNT25700426 to X.A.Z. The study was also supported by NIH grant R37CA276924 to K.M. We thank A. Radhakrishnan of The University of Texas Southwestern Medical Center for lipid probes, Y. Zheng of Cincinnati Children’s Hospital for Cdc42-floxed mouse line and P. Caswell of the University of Manchester for AHPH constructs. We are grateful to A. Radhakrishnan of The University of Texas Southwestern Medical Center, H. Chen of Harvard Medical School and C. Griffin and L. Xia of the Oklahoma Medical Research Foundation for helpful suggestions and valuable discussions.
Author information
Authors and Affiliations
Contributions
Conceptualization: Y.D. and X.A.Z. Methodology/Investigation: Y.D., J.C., S.L., J.M.H., X.G., D.N.R., B.L., F.H., X.W., Q.W., J.W., B.S. and A.M.R. Dataset Analysis: Y.D., J.D.W., C.G. and W.M.F. Resources: Z.W., K.K., C.K.M., A.C., Z.U., K.M., M.H.E., J.P. and T.K. Writing: Y.D., J.C., J.D.W., M.S.W., J.F.P., F.A.H. and X.A.Z. Supervision: X.A.Z.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Cardiovascular Research thanks Victoria Bautch and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Upregulation of CD82 expression in inflammation.
(a) GSE159585 analysis of lung biopsies from Covid-19 patients and controls. UMAP visualization of all cell clusters profiled and CD82 gene expression in human lung tissues from GSE159585 (left panel). Levels of CD82 mRNA (Transcripts per million, TPM) are presented as mean±SD (right panel). (b) Increased expression of CD82 in the pulmonary endothelium of COPD patients. Immunohistochemistry of CD82 was examined in lung sections of COPD patients and controls. Arrows indicate CD82 staining in endothelia. (c) Increased CD82 expression in the endothelia within atherosclerosis lesions. Mouse aortas were harvested from wild-type (WT) and ApoE−/− mice on C57BL/6 background for CD82 immunohistochemical staining. Dashed areas are atherosclerotic lesions. Black arrows show aortic endothelium in non-lesion regions, while red arrows indicate endothelium from lesion areas. (d) From GEO database, analysis on GSE2372 for CD82 expression in the aortas from WT and ApoE−/− mouse lines on C57BL/6 background (mean±SD; n=3 mice per group). (e) Upregulation of CD82 expression in endothelia in LPS-induced lung inflammation. Pulmonary tissues were harvested from C57BL/6 mice with or without LPS challenge (5mg/kg, i.p.) for CD82 immunohistochemical staining. Arrows show endothelium of pulmonary vessels. (f) From GEO database, the GSE2411 analysis on the lungs from C57BL/6 mouse model of LPS-induced lung inflammation and injury (mean±SD; n=6 mice per group). (g) Effects of inflammatory stimuli on endothelial CD82 expression. CD82 mRNA and proteins from the MLECs treated with or without LPS (100 ng/mL) for 6 h and 24 h were examined with qRT-PCR and Western blot, respectively (mean±SD; n=3 independent experiments) (left panel). HMECs were treated with LPS (100 ng/mL), TNF-α (10 ng/mL), or IL-4 (20 ng/mL) for 24 h and analyzed with Western blot for CD82 (middle panel). HUVECs were treated with VEGF-A164 (50 ng/mL), LPS (100 ng/mL), TNF-α (10 ng/mL), IL-1β (20 ng/mL), or IL-6 (20 ng/mL) for 48 h and then analyzed Western blot for CD82 (right panel). Relative levels of CD82 mRNA or protein were quantified (mean±SD; n=3 individual experiments).
Extended Data Fig. 2 CD82 and inflammatory signaling: bioinformatics analyses.
(a) Scheme of inducible deletion of Cd82 in endothelium. Cd82flox/flox;Cdh5-Cre/ERT2+ and Cd82flox/flox;Cdh5-Cre/ERT2− mice were administered with tamoxifen intraperitoneally daily for five days and used as Cd82ΔEC and control (Ctrl.) mice, respectively, for experiments. (b) CD82 mRNA and protein levels in primary MLECs that were isolated from endothelium-specific Cd82-null mice and control littermates. CD82 mRNA was examined in qRT-PCR, and the quantities are presented as the levels relative to the mRNA levels of housekeeping genes (mean±SD; n=3 individual experiments). CD82 proteins were detected in Western blot with murine CD82 pAb M35, and GAPDH was used as a protein loading control. (c) Ingenuity pathway analysis on the bulk RNAseq data of MLECs from Cd82ΔEC mice and their control littermates. The pathways related to inflammatory and immune responses are labeled with red asterisks. Left panel: the pathways were ranked by -log (P-value)s. Ratio denotes the number of altered genes in a given pathway to the total number of genes in that pathway. Right panel: the pathways were ranked by -log (false discovery rate or FDR)s. The dot size presents the number of enriched genes, and the dot color codes the FDR level. The enrichment ratio is defined as the number of observed genes divided by the number of listed genes in an ingenuity category. (d) CD82-inflammation correlation network was generated as described in "Methods" section. CD82 and the inflammation-related genes that are most strongly correlated with Cd82 in gene expression (green) were connected by potential PPIs (gray) using STRING v10.
Extended Data Fig. 3 CD82 facilitates vascular inflammation.
(a) Representative images of echocardiography from Ctrl. or Cd82ΔEC mice challenged with LPS (5 mg/kg) at 18 h or 48 h after the LPS administration. (b) Cd82 ablation does not alter the basal permeability of skin vessels. Evans blue dye (30 mg/kg in PBS) was injected intravenously through the retro-orbital sinus into 8~12-week-old Cd82−/− mice or their WT littermates. The leakage of Evans blue dye into ear skin was assessed at 0, 30, or 60 min after the injection. Extravasated Evans dye was quantified and corrected with ear dry weight (mean±SEM; n=6 mice per group for 60 min and n=11 mice per group for 30 min). (c) Representative images of Evans blue dye leakage into skin of the ears from WT and Cd82−/− mice as well as from Ctrl. and Cd82ΔEC mice in Miles assay. (d) Body weight was measured daily after SARS-Cov-2 infection in Ctrl. or Cd82ΔEC mice (mean±SD; n=9 mice for Ctrl. group and n=8 mice for Cd82ΔEC group). (e) SARS-Cov-2 titer in control and Cd82ΔEC mice. Pulmonary tissues were harvested from Ctrl. or Cd82ΔEC mice, and SARS-CoV-2 nucleocapsid (N) gene expression is presented as its relative level after normalization with actin expression using the dCT method for all mice in the experiment (left panel) and only for those mice which succumbed to infection at the time of death (right panel). Nine mice from Ctrl. group and eight mice from Cd82ΔEC group were used for the study.
Extended Data Fig. 4 CD82 removal from ECs and the effects of CD82 removal on EC-EC contact/adhesion and Cdc42 and Rac1 activities.
(a) Flow cytometry analysis on the cell surface level of CD82 at 48 and 72 h after the ECs were transfected with control or CD82 siRNA, respectively. Human CD82 was detected with its mAb TS82b, with mouse IgG as a negative control mAb. Western blot of CD82 in bEnd.3 transfectant cell (right panel). (b) Basal TEER levels of MLECs, HMECs, HUVECs, and bEnd.3 murine brain ECs. (c) Effects of CD82 removal on the levels of EC-EC adhesion proteins. MLECs from Cd82+/+ and Cd82−/− mice and HUVECs transiently transfected with control and CD82 siRNA were lysed with RIPA lysis buffer, and the lysates were examined by Western blot for the indicated proteins. (d) Cd82 ablation and Rac1 activity. MLECs were stimulated with VEGF-A164 (50 ng/mL) after serum-starvation overnight and then lysed with RIPA buffer. Active Rac1 was detected by G-Lisa assay for Rac1-GTP and presented as activities (mean±SD; n=4 experiments). (e) CD82 silencing and Rac1 activity. HUVECs transfected with control or CD82 siRNA were serum-starved overnight, stimulated with VEGF-A (50 ng/ml), and then lysed with RIPA buffer. GTP-bound Rac1 was pulled down from the lysates with GST-PAK Rac1-binding domain and detected in Western blot. Other indicated proteins in the lysates were directly examined with Western blot. The ratios of GTP-bound Rac1/total Rac1 were quantified based on the band densities of Rac1 (mean±SD; n=3 independent experiments). (f) Effects of VEGF and Cd82 ablation on Cdc42 activity. Confluent MLECs were serum-starved overnight and treated with vehicle (PBS) or VEGF-A164 (50 ng/mL) for 15 min. Cdc42 activities were measured by G-LISA assay for Cdc42-GTP and presented as mean±SD (n=7 experiments). (g) Protein and mRNA levels of FARP1, SYDE1, and others in the MLECs were examined with qRT-PCR and Western blot, respectively. The mRNA levels are presented as mean±SD (n=4 experiments). (h) Increased distribution of FARP1 at or near the basal PM of Cd82−/− MLECs, revealed by TIRF microscopy analysis on Cd82+/+ and Cd82−/− MLECs. Dashed lines show the edge of the MLEC.
Extended Data Fig. 5 Downregulation of CD82 and upregulation of FARP1-Cdc42 signaling as anti-inflammation therapeutic strategies.
(a) Western blot analysis on the lysates of MLECs isolated from Ctrl., Cd82ΔEC, and Cd82ΔEC;Cdc42ΔEC mice. (b)-(c) Histochemistry (H-E staining) (b) and immunofluorescence (CD45 staining) (c) analyses on lung sections of the Control, Cd82ΔEC, and Cd82ΔEC;Cdc42ΔEC mice treated with LPS (5 mg/kg) for 18 h described in Fig. 3i. (d) Quantifications of acute lung injury (violin plot), Evans Blue extravasation or vascular leakage (mean±SD), and leukocyte infiltration of the lungs (violin plot) from Control, Cd82ΔEC, and Cd82ΔEC;Cdc42ΔEC mice, n=10 mice per group. (e) Western blot analysis of FARP1 in the HUVECs transfected with control and FARP1 siRNA. (f) Efficiency of in vitro FARP1 delivery and expression with endothelium-specific hADv5 viruses. The bEnd.3 cells infected by the viruses encoding control or FARP1 were examined in Western blot. (g-h) Histochemistry analysis (H-E staining) (g) and immunofluorescence analysis (CD45 staining) (h) on lung sections of the mice delivered with HAdV5-control (HAdV5-Vector) or HAdV5-FARP1 viruses. Ten days after the viral injection, the mice were challenged with LPS (5 mg/kg, i.p.) for 18 h prior to euthanasia. (i) Quantifications of acute lung injury (violin plot), vascular leakage or Evans Blue extravasation (mean±SD), and leukocyte infiltration of the lungs (violin plot) from the mice administered with HAdV5-control or HAdV5-FARP1 viruses, n=6 mice/ group.
Extended Data Fig. 6 CD82 sustains RhoA activity in ECs.
(a) HUVECs transfected with CD82 or control siRNA were serum-starved overnight, stimulated with VEGF-A (50 ng/ml), and then lysed with RIPA buffer. GTP-bound RhoA was pulled down from the lysates with GST-Rhotekin RhoA-binding domain fusion and detected in Western blot. Other indicated proteins in the lysates were directly examined with Western blot. The ratios of GTP-bound RhoA/total RhoA were quantified based on the band densities of RhoA (mean±SD; n=3 individual experiments). (b) Localization of GFP-AHPH. MLECs cultured on coverslips were transfected with GFP-AHPH WT or GFP-AHPH A470D&E758K mutant and processed for confocal microscopic imaging. (c) CD82 silencing up-regulates p190RhoGAP activity in ECs. HUVECs transfected with CD82 siRNAs or control siRNA were serum-starved overnight and then stimulated with VEGF-A (50 ng/ml). After cell lysis with a lysis buffer containing 1% Triton X-100, the p190RhoGAP proteins in the lysates were immunoprecipitated and then immuno-blotted with phosphotyrosine mAb (4G10) and p190RhoGAPmAb. Other indicated proteins in the lysates were directly examined with Western blot. (d) Immunofluorescence staining of VE-cadherin and Rnd3 in the MLEC monolayers. Images were acquired with confocal microscopy. (e) Western blot analysis of Rnd3 in the HUVECs that were transfected with control siRNA and Rnd3 siRNA. (f) Histochemistry analysis (H-E staining) on the effects of CN04 and/or Y27632 on LPS-induced acute lung injuries in WT C57BL/6 mice.
Extended Data Fig. 7 CD82 sustains AC on ECs.
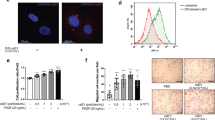
(a) MLECs were incubated with AlexaFluor488-C5-maleimide-conjugated OlyA and AlexaFluor594-C5-maleimide-conjugated ALOD4 for 10 min at RT. After 3 washes with PBS, the cells were imaged with confocal microscopy. Fluorescence intensity/cell was quantified (violin plot; n=25 cells/group). (b) Effects of CD82 silencing on cell surface levels of the lipids. Top panel: the cells were labeled with fluorescent probes on ice for 1 h and examined by flow cytometry (mean±SD; n=3 experiments). Bottom panel: schematic presentation of the lipids and their probes. (c). Effects of simvastatin on cell surface levels of the lipids. The cells were treated with DMSO or simvastatin (5.0 µM) for 24 h, then incubated with fluorescent probes on ice for 1 h, and examined by flow cytometry (mean±SD; n=3 experiments). (d) MLECs treated with DMSO and cholesterol (0.1 mM) for 24 h were labeled with fluorescent probes on ice for 1 h and examined by flow cytometry (mean±SD; n=3 experiments). (e) The ECs were treated with MβCD (1.0 µM) for 1 h or cholesterol (0.1 mM) for 24 h, then incubated with the fluorescent probes on ice for 1 h, and examined by flow cytometry (mean±SD; n=3 experiments). (f) The cells treated with LPS (100 ng/mL for 1 h) or VEGF-A164 (50 ng/mL for 1 h) were labeled with fluorescent ALOD4 on ice for 1 h and examined by flow cytometry (mean±SD; n=3 experiments). (g) MLEC monolayers were treated with SMase (100 U/mL) for 30 min or simvastatin (5.0 µM) for 24 h and examined for basal permeability with FITC-dextran (250 kDa) diffusion assay (mean±SD; n=4 experiments). (h)-(i) The bEnd.3 monolayers were treated with cholesterol (0.1 mM) for 24 h or MβCD (1.0 µM) for 1 h and then examined for LPS- or VEGF-A164-stimulated permeability (h) or basal permeability (i), with FITC-dextran (250 kDa) diffusion assay (mean±SD; n=4 experiments). (j) The cells were pretreated with DMSO, cholesterol (0.1 mM), or simvastatin (5.0 µM) for 24 h or with SMase (100 U/mL) or ALOD4 (3.0 µM) for 30 min. The cell surface VE-cadherin was measured by flow cytometry (mean±SD; n=3 experiments).
Extended Data Fig. 8 AC roles in CD82-facilitated vascular leakage and CD82-tuned RhoA-versus-Cdc42 balance.
(a) The monolayers were treated with indicated ceramide species (20 µM) for 24 h and then examined in FITC-dextran (250 kDa) diffusion assay (mean±SD; n=4 experiments). (b) The cells were treated with 25-HC (0.2 µM) for 3 h, followed by flow cytometry analysis on AC levels at the cell surfaces (mean±SD; n=4 experiments). (c)-(d) The monolayers were treated with 25-HC (0.2 µM) for 3 h and examined for endothelial permeability (mean±SD, n=4 experiments) with FITC-dextran (250 kDa) diffusion assay, under VEGF-A164 (50 ng/mL) stimulated (c) or basal (d) condition. (e) The cells were preincubated with intact ALOD4 peptide (3.0 µM) at 37 °C for 30 min and then examined by flow cytometry with Alexa488-conjugated ALOD4 for the AC level (mean±SD; n=3 experiments). (f)-(g) The monolayers were preincubated with intact ALOD4 peptide (3.0 µM) at 37 °C for 30 min, followed by FITC-dextran diffusion assay (mean±SD, n=4 experiments), under VEGF- or LPS-stimulated (f) or basal (g) condition. (h) The cells were treated with PBS, simvastatin (5.0 µM for 24 h), or intact ALOD4 peptide (3.0 µM for 30 min), followed by LPS stimulation for 24 h. TNF-α in the cell culture supernatants were measured by ELISA (mean±SD; n=4 experiments). (i) The cells were pretreated with simvastatin (5 µM) for 24 h and then examined for RhoA and Cdc42 activities with G-LISA assays. Their relative functionality is present as the ratio of Cdc42 activity to RhoA activity (mean±SD). (j) The cells were treated with PBS or simvastatin (5.0 µM) for 24h, stained for Cdc42-GTP and CD44, and imaged with confocal microscopy. Arrows: Cdc42-GTP-localized cell-cell junction. (k) The live cells were stained with fluorescent OlyA and ALOD4, fixed, and imaged with structured illumination microscopy. (l) The cells were treated with DMSO, simvastatin (5.0 µM), or cholesterol (0.1 mM) for 24 h, followed by the Cdc42-FARP1 PLA. (m) MDA-MB-231 cells were incubated with His-tagged ALOD4 at 37 °C for 30 min and lyzed by sonication for immunoprecipitations with the indicated Abs, shown in Fig. 6f, followed by immunoblot with the indicated mAbs.
Extended Data Fig. 9 Effects of CD82 removal on subcellular distributions of cholesterol, AC, and ORP2, and interactions between tetraspanins and OSBP or ORPs.
(a) Confocal microscopic images of TopFluor® cholesterol (TF-cholesterol) and lysotracker staining in bEnd.3-Control and -CD82 KD transfectant cells. (b) Localization of ORP1 in MLECs. Immunofluorescence of ORP1 was performed with its Ab that recognizes both ORP1L and ORP1S forms. (c) Subcellular distribution and colocalization of ORP2 and AC. Immunofluorescence and fluorescence staining for ORP2 and ALOD4, respectively, in MLECs were imaged with confocal microscopy, and the ORP2-ALOD4 colocalization was quantified and presented as Pearson’s coefficient (Box&Whisker; n=20 cells from 3 independent experiments). (d) Western blot of ORP2 in the bEnd.3 ECs that were transiently transfected with control siRNA or ORP2 siRNA. (e) PLA analysis on the interactions of CD82 with OSBP and ORPs in HUVECs. Shown are representative fluorescence microscopic images of the PLA reactions. (f) PLA analysis on the interactions of ORP2 with tetraspanins in HUVECs. Shown are representative fluorescence microscopic images of the PLA reactions. (g) Representative FRET images of CFP-ORP2 in the MDA-MB-231 cells that transiently express only CFP-ORP2 construct. (h) Representative fluorescence microscopic images of the PLA between ORP2 and CD82 proteins in Du145-Mock, -CD82 WT, and -CD82 CACA mutant transfectant cells. Of note, Du145 cells do not express endogenous CD82.
Extended Data Fig. 10 Schematic summarization of the findings.
(a) Molecular and cellular mechanisms by which tetraspanin CD82 regulates AC and its non-vesicular transfer and then GEF/GAP-Rho small GTPases signaling, to alter the inflammatory status of cells and tissues. (b) New therapeutic strategies against inflammation arisen from this study. Red thunder signs denote functional blocking, and green asterisk signs indicate functional strengthening.
Supplementary information
Supplementary Information
Supplementary Fig. 1 and Supplementary Tables 1–3.
Source data
Source Data Fig. 1
Statistical source data.
Source Data Fig. 1
Unprocessed western blots and/or gels.
Source Data Fig. 2
Statistical source data.
Source Data Fig. 3
Statistical source data.
Source Data Fig. 4
Statistical source data.
Source Data Fig. 4
Unprocessed western blots and/or gels.
Source Data Fig. 5
Statistical source data.
Source Data Fig. 6
Statistical source data.
Source Data Fig. 6
Unprocessed western blots and/or gels.
Source Data Fig. 7
Statistical source data.
Source Data Fig. 7
Unprocessed western blots and/or gels.
Source Data Fig. 8
Statistical source data.
Source Data Fig. 8
Unprocessed western blots and/or gels.
Source Data Extended Data Fig. 1
Statistical source data.
Source Data Extended Data Fig. 1
Unprocessed western blots and/or gels.
Source Data Extended Data Fig. 2
Statistical source data.
Source Data Extended Data Fig. 2
Unprocessed western blots and/or gels.
Source Data Extended Data Fig. 3
Statistical source data.
Source Data Extended Data Fig. 4
Statistical source data.
Source Data Extended Data Fig. 4
Unprocessed western blots and/or gels.
Source Data Extended Data Fig. 5
Statistical source data.
Source Data Extended Data Fig. 5
Unprocessed western blots and/or gels.
Source Data Extended Data Fig. 6
Statistical source data.
Source Data Extended Data Fig. 6
Unprocessed western blots and/or gels.
Source Data Extended Data Fig. 7
Statistical source data.
Source Data Extended Data Fig. 8
Statistical source data.
Source Data Extended Data Fig. 8
Unprocessed western blots and/or gels.
Source Data Extended Data Fig. 9
Statistical source data.
Source Data Extended Data Fig. 9
Unprocessed western blots and/or gels.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Ding, Y., Chen, J., Liu, S. et al. Tetraspanin-enriched membrane domains regulate vascular leakage by altering membrane cholesterol accessibility to balance antagonistic GTPases. Nat Cardiovasc Res 4, 1011–1033 (2025). https://doi.org/10.1038/s44161-025-00686-2
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s44161-025-00686-2
