
Abstract
Pentameric ligand-gated ion channels (pLGICs) are targets of general anesthetics, but molecular mechanisms underlying anesthetic action remain debatable. We found that ELIC, a pLGIC from Erwinia chrysanthemi, can be functionally inhibited by isoflurane and other anesthetics. Structures of ELIC co-crystallized with isoflurane in the absence or presence of an agonist revealed double isoflurane occupancies inside the pore near T237(6′) and A244(13′). A pore-radius contraction near the extracellular entrance was observed upon isoflurane binding. Electrophysiology measurements with a single-point mutation at position 6′ or 13′ support the notion that binding at these sites renders isoflurane inhibition. Molecular dynamics simulations suggested that isoflurane binding was more stable in the resting than in a desensitized pore conformation. This study presents compelling evidence for a direct pore-binding mechanism of isoflurane inhibition, which has a general implication for inhibitory action of general anesthetics on pLGICs.
Similar content being viewed by others

Introduction
General anesthetics are administered to approximately 256 million people each year1. In addition to their primary action to render analgesia, amnesia, immobility and unconsciousness, these drugs are also implicated in unwanted side effects, including post-operative cognitive decline2,3 and neurotoxicity in pediatric and elderly populations4,5,6. At the molecular level, how general anesthetics exert their on-target and off-target actions remains poorly understood.
Functional measurements suggest that a superfamily of pentameric ligand-gated ion channels (pLGICs) plays a central role in anesthetic action7,8. At clinically relevant concentrations, general anesthetics inhibit agonist-elicited currents of cation-conducting channels, such as nicotinic acetylcholine receptors (nAChRs) and potentiate currents of anion-conducting channels, such as GABAA and glycine receptors7,8. These channels regulate a myriad of sensory processes and their malfunctions are directly linked to several neurological disorders. A structural insight into the mode of action, particularly the molecular details of anesthetic binding sites, is essential to understand the functional modulation of pLGICs by general anesthetics.
Discrete anesthetic binding sites have been proposed on the basis of various experimental and computational studies. In the extracellular domain (ECD), which contains the orthosteric agonist-binding site, crystal structures of prokaryotic pLGICs from Gloeobacter violaceus (GLIC) and Erwinia chrysanthemi (ELIC) have revealed binding sites for anesthetics: ketamine at the interface of two subunits in GLIC9 and bromoform at a similar location in ELIC10. Photoaffinity labeling11 and computational studies12,13,14 have also provided evidence for anesthetic binding in the ECD of nAChRs. Some of these reported ECD sites partially overlap with the agonist or antagonist binding sites9,11. In the transmembrane domain (TMD) of pLGICs, diverse intra- and inter-subunit allosteric sites for general anesthetic binding have been identified through various approaches, including NMR spectroscopy15,16,17, photoaffinity labeling11,18,19,20,21, crystallography10,22,23 and molecular dynamics (MD) simulations12,13,14,24,25. Specifically for the pore in the TMD, previous site-directed mutagenesis, electrophysiology measurements and photoaffinity labeling suggested an open-channel blocking mechanism for functional inhibitions by anesthetics26,27,28,29,30, but available structural data were not sufficient to support such a mechanism10.
In this study, we co-crystalized ELIC with the general anesthetic isoflurane, which is known to inhibit mammalian neuronal and muscle nAChRs31,32 and GLIC33. The crystal structures of ELIC-isoflurane were determined with resolutions of 3.0 Å and 3.4 Å in the absence and presence of agonists, corresponding to the presumed resting and desensitized states of ELIC, respectively. These crystal structures revealed double isoflurane occupancies near the 6′ and 13′ positions inside the pore of ELIC. Site-directed mutagenesis and subsequent functional measurements further showed a direct pore-binding mechanism of isoflurane inhibition. In addition to the previously proposed open-channel blocking action26,27,28, the direct pore-binding mechanism suggested by our high-resolution structural data contains another component of anesthetic inhibition, which is achieved by stabilizing the resting channel conformation. Moreover, similar to isoflurane, several other representative general anesthetics, including halothane, sevoflurane, propofol, thiopental and etomidate, also inhibit ELIC and do so as effectively on ELIC as on nAChRs. This study provides solid structural data to support the significance of direct pore binding as a mechanism of functional inhibition of pLGICs by general anesthetics.
Results
General anesthetics inhibit function of ELIC
As a homolog to eukaryotic pLGICs, ELIC shares similar pharmacological profiles of eukaryotic cation-conducting pLGICs with respect to modulation by general anesthetics. Both volatile and intravenous anesthetics inhibited ELIC in a concentration dependent manner (Fig. 1). At the agonist propylamine (PPA) concentration eliciting 20% of the maximal current (EC20), isoflurane, sevoflurane and halothane produced 50% inhibition (IC50) at concentrations of 21.9 ± 1.5, 23.2 ± 1.6 and 40.0 ± 4.5 μM, respectively (Fig. 1b and Supplementary Table 1). The intravenous anesthetics propofol, etomidate and thiopental inhibited ELIC with IC50 of 11.9 ± 1.1, 11.2 ± 1.1 and 42.8 ± 1.1 μM, respectively (Fig. 1c and Supplementary Table 1). These results are comparable to the corresponding IC50 values observed on neuronal nAChRs31,32,34,35,36,37,38 (Supplementary Table 1), suggesting that ELIC is a suitable model for understanding anesthetic actions on pLGICs.

Inhibition of ELIC by general anesthetics.
(a) Representative ELIC current traces elicited by propylamine (PPA) at EC20 in the absence and presence of the general anesthetics isoflurane (ISF, 50 μM), sevoflurane (SVF, 60 μM), halothane (HAL, 82 μM), propofol (PFL, 30 μM), etomidate (ETM, 30 μM), or thiopental (THP, 100 μM). The scale bars are 0.2 μA (vertical) and 30s (horizontal). The data were normalized to the current at EC20 in the absence of anesthetics and fit to the Hill equation (solid lines, n ≥ 6) for the volatile (b) and intravenous (c) general anesthetics. The best-fit parameters for both (b,c) are provided in Supplementary Table 1. The data are reported as the mean ± SEM and error bars less than the symbol size are not visible.
Double occupancy binding of isoflurane inside the ELIC pore
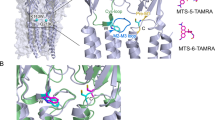
Among the general anesthetics used for functional measurements (Fig. 1), we successfully co-crystalized isoflurane with ELIC in the absence and presence of the agonist 3-bromopropylamine (BrPPA) and determined structures of the complexes up to a 3.0-Å resolution. Crystallographic and refinement parameters are summarized in Table 1. For ELIC crystallized with isoflurane but without an agonist (presumed in the resting state), the FO-FC omit electron density map (Fig. 2a) shows strong electron densities for double isoflurane occupancies inside the pore near residues T237(6′) and A244(13′). The numbers in the parentheses are conventional prime notations for pore-lining residues of pLGICs39. Nearly equal electron densities observed at the two sites suggest that isoflurane has similar binding affinities to both sites. The 2FO-FC electron density maps (inserts of Fig. 2) show refined structures of two isoflurane molecules inside the pore. For ELIC crystallized in the presence of both isoflurane and the agonist BrPPA (presumed in a desensitized state), the FO-FC omit electron density map also indicates double isoflurane occupancies in the pore (Fig. 2b). Isoflurane at the 6′ position can form hydrogen bonds with the hydroxyl group of T237 in addition to hydrophobic contacts with L240 at the 9′ position, while isoflurane binding at the 13′ position is dominated by hydrophobic interactions with the side chain of L240 (Fig. 2c).

Crystal structures of ELIC with isoflurane bound within the pore.
Side views of ELIC bound with two isoflurane molecules near residues T237(6′) and A244(13′) in (a) the resting and (b) desensitized states. The FΟ-FC omitted electron density maps (green) are contoured at 5.0 σ level with a carve distance of 1.8 Å for isoflurane molecules. Two inserted top views in (a) show 2FΟ-FC electron density maps contoured at 1.0 σ level for isoflurane bound to the 13′ and 6′ positions of ELIC in the resting conformation. (c) ELIC residues within 4 Å in contact with isoflurane: T237(6′), green; L240(9′), black; and A244(13′), magenta. Dashlines highlight potential hydrogen bonding between isoflurane and T237(6′). (d) Isoflurane binding induced an inward movement of the upper portion of TM2 and reduced pore radius near 16′. The pore profile of apo ELIC (black), ELIC-isoflurane (magenta), ELIC-isoflurane-agonist 3-bromopropylamine (cyan) were calculated using the HOLE program70 based on their crystal structures (PDB codes: 3RQU, 4Z90, 4Z91). The z-coordinate is parallel to the channel axis and the zero point of the pore radius is overlapped with the channel axis.
A structural comparison of the isoflurane-bound ELIC (Fig. 2a) with apo ELIC (PDB code: 3RQU)40 revealed an inward movement of the upper part of the TM2 helices and pore contraction near 16′, where the pore radius was reduced by 0.7 Å upon isoflurane binding (Fig. 2d). This conformational change may result from hydrophobic contacts between isoflurane and nearby residues and consequently aids in stabilizing a closed channel structure. Such a conformational change was also observed in the structure of ELIC co-crystallized with isoflurane in the presence of agonists but to a lesser degree (Fig. 2d).
The bromine atom of the agonist BrPPA co-crystalized with ELIC produced bromine-specific anomalous signals in the X-ray diffraction data collected with a wavelength near the K-edge peak of Br (0.9199 Å). The anomalous signals and FO-FC electron densities revealed BrPPA binding sites behind loop C and nearby loop A in the extracellular domain (Supplementary Fig. 1). These sites were reported previously to bind agonists, competitive antagonists and modulators in ELIC40,41,42 and in other pLGICs43,44.
Functional relevance of the isoflurane binding sites
To establish the functional relevance of the identified isoflurane-binding sites, we made site-directed mutations, one at a time, at positions 6′ (T237A) and 13′ (A244T). Subsequently, we expressed these mutants in Xenopus laevis oocytes and measured their functional responses to isoflurane (Fig. 3). The agonist PPA can activate all of the mutant channels and the A244T ELIC mutant has an EC50 value nearly identical to the wild-type (WT) ELIC (Fig. 3b). However, these mutant channels, especially T237A, are much less sensitive to isoflurane inhibition than the WT ELIC. Relative to the WT channel (Fig. 1), the IC50 of isoflurane increased ∼3-fold for the A244T channel (61.7 ± 5.5 μM) and more than an order of magnitude for the T237A channel (524.8 ± 58.2 μM) (Fig. 3c). The functional consequences of these mutations are associated with changes in the hydrophobicity profile and pore radii near positions 6′ and 13′ (Supplementary Fig. 2). It should be noted that the side chains of the hydrophobic residue at the 9′ position are involved in both of the isoflurane-binding sites (Fig. 2c). The fact that the isoflurane inhibition curve shifts an order of magnitude to the right in the T237A mutant indicates the importance of amphipathic interactions at position 6′ for isoflurane inhibition.

Functional evidence of isoflurane binding to the pore.
(a) Representative current traces for isoflurane inhibition of WT and two mutants: T237(6′)A; and A244(13′)T. The scale bars are 0.1 μA (vertical) and 30s (horizontal). (b) Agonist propylamine (PPA) concentration-response curves for WT and two mutants indicated in (a). The data were normalized to the maximum current and fit to the Hill equation (solid lines, n ≥ 5). (c) Isoflurane inhibition of WT and the two mutants indicated in (a). The data were normalized to the current at EC20 in the absence of isoflurane and fit to the Hill equation (solid lines, n ≥ 6). The data are reported as the mean ± SEM and error bars less than the symbol size are not visible.
Since isoflurane binds to the pore of the resting state of ELIC (Fig. 2a), pre-incubating the oocytes expressing ELIC with isoflurane for a short period of time is expected to enhance the inhibitory effect. As shown in Fig. 4, pre-incubation with isoflurane not only generated greater inhibition, but also decreased the apparent onset rate of agonist-induced current and slowed channel opening. Data indicate that isoflurane binding stabilizes the closed-channel conformation.

Isoflurane binding stabilized the closed channel.
(a) A representative trace showing that pre-incubation with isoflurane (red arrows) slowed the onset rate and increased the maximum inhibition. The vertical and horizontal scale bars represent 20 μA and 30 seconds, representatively. (b) The onset rate of the channel current can be fitted to a single exponential decay function, I = Imax (1−e(−t/τ)). The fitting results for the onset rate τ and Imax for channels pre-incubated with isoflurane (red line) are 1.73 ± 0.04 s and −0.33 ± 0.02 μA, respectively; and 1.25 ± 0.05 s and −0.41 ± 0.03 μA, respectively, for channels without isoflurane pre-incubation (green line). The fitting parameters are expressed as mean ± SEM (n = 15).
Isoflurane binding characterized by molecular dynamics simulations
Crystal structures show isoflurane binding to ELIC under both resting and desensitized conditions (Fig. 2). In order to determine which functional state of ELIC has more stable isoflurane binding, we performed separate MD simulations of ELIC in a resting conformation as shown in Fig. 2a and a “desensitized” conformation that was obtained from homology modeling on the crystal structure of the desensitized GABAAβ3 channel45. The structure in Fig. 2b was not used for simulations because it is virtually identical to the resting state structure (Fig. 2a), a known crystallization phenomenon reported previously46. Compared to the resting ELIC conformation, the upper pore region of the desensitized model is somewhat expanded and the lower pore region is contracted (Supplementary Fig. 3). The differences in pore conformations presented in the resting and “desensitized” ELIC structures used for MD simulations are in good agreement with observations in a recent 19F NMR study47.
Over the course of the MD simulations of the resting ELIC, isoflurane remained near the same two sites (6′ and 13′) as revealed in the crystal structure (Fig. 5a and Supplementary Fig. 4a). Although isoflurane at the 6′ site transiently migrated to the bottom of the pore at an early stage of the simulation, it quickly returned to the original position. Isoflurane movement perpendicular to the channel axis was largely confined within 2 Å of the pore radius in two replicated simulations (Fig. 5c and Supplementary Fig. 4a), indicating stable binding in the closed pore. In contrast, isoflurane molecules in the modeled desensitized pore moved away substantially from their initial positions (Fig. 5b,d and Supplementary Fig. 4b) and deviated from the center of the pore (Fig. 5d and Supplementary Fig. 4c). Although the modeled pore may not capture quantitative changes of ELIC in the transition from the resting to a desensitized conformation, it at least predicts how expansion at the upper half of the pore affects isoflurane binding.

Isoflurane binding is more stable in the closed channel.
Probabilities of finding an isoflurane molecule (center of mass) in molecular dynamics (MD) simulations at position z along the pore axis, P(z) and at the distance r from the pore axis, P(r), were calculated for (a,c) the closed channel and (b,d) a desensitized channel. A probability is defined as the ratio of the number of snapshots where isoflurane was found at the position z or at the distance r to the total snapshots chosen from the simulation. The bin widths of 1 Å and 0.5 Å were used for position z and distance r, respectively. Isoflurane motion in each pore conformation was evaluated every 0.1 ns from 100 ns simulations. Color indicates starting positions of isoflurane, 6′ (red) or 13′ (blue), in the MD simulations.
The binding of isoflurane to the pore may have affected the mobility of the channel. We determined the root mean square fluctuation (RMSF) for the ELIC TMD over the course of the simulations. The RMSF decreased in the closed ELIC bound stably with isoflurane compared to the RMSF of the apo ELIC. In contrast, mobile isoflurane molecules inside the desensitized pore increased the RMSF of the pore region (Supplementary Fig. 5).
Discussion
A direct pore-binding mechanism of isoflurane inhibition
The crystal structures of ELIC-isoflurane complex and the functional results of the site-directed mutants point to a direct pore-binding mechanism of isoflurane inhibition. This study has not only revealed the 6′ and 13′ pore positions for the double occupancy of isoflurane in ELIC (Fig. 2), but also demonstrated how pore conformational changes affected isoflurane binding (Fig. 5). The observation of isoflurane in the pore of ELIC under desensitization conditions could be attributed to an open pore-blocking mechanism. By this mechanism, isoflurane occupies and occludes an open pore and then remains in the pore when the channel desensitizes. An open pore-blocking mechanism, however, cannot completely describe the isoflurane inhibition of ELIC since isoflurane also occupies the pore of ELIC in the resting state (Figs. 2 and 4). Furthermore, isoflurane binding to the resting pore stabilizes a closed channel conformation (Figs. 2d and 4, Supplementary Fig. 5). Thus, isoflurane inhibition via a direct pore-binding mechanism is likely accomplished jointly by stabilizing the closed channel conformation and blocking the open channel.
The direct pore-binding mechanism of isoflurane inhibition was also proposed previously based on site-directed mutagenesis studies of nAChRs26,48 and MD simulations13,14,24 of nAChRs and other cation-conducting pLGICs. Functional studies of WT and mutant muscle-type nAChRs expressed in oocytes and HEK cells suggested that isoflurane binding to the pore was responsible for the inhibition of nAChRs26,48. MD simulations showed that isoflurane bound to 13′ in the GLIC pore13,14. Isoflurane binding to 6′ in the GLIC pore was also found in MD simulations, but with a lower probability than to the 13′ position24. Interestingly, these simulation results obtained from the open channel GLIC are comparable to our observations of isoflurane binding to the desensitized model of ELIC (Fig. 5).
A remarkable similarity of hydrophobicity profiles in the pore lumens of the cation-conducting channels (Supplementary Fig. 6) may be central to the observation of isoflurane binding to the pore. There is a dichotomy between cation- and anion-conducting channels in their hydrophobic-hydrophilic switching point at position 9′, which has a conserved hydrophobic residue across the entire superfamily. Cation-conducting channels have a hydrophobic upper region of the pore, switching at the 6′ position to a hydrophilic lower pore region. In contrast, the anion-conducting GABAARs and glycine receptors have hydrophilic residues lining the upper region of their pores. In GLIC, mutating the hydrophobic residue at 13′ to a hydrophilic residue is a critical element to convert the cation-conducting GLIC to an anion-conducting channel49. The distinctly different hydrophobicity profile in the pore lumens of the anion-conducting channels may prevent anesthetics from binding to the pore. Indeed, these anion channels are mostly potentiated rather than inhibited by general anesthetics8.
Both pore geometry and hydrophobicity affect isoflurane binding
One may wonder why isoflurane binds specifically to positions 6′ and 13′. Although T237(6′) and A244(13′) have different side-chain volumes (T = 120 Å3 vs. A = 90 Å3)50, the tilting of the TM2 helix makes the pore radii at the 6′ and 13′ positions nearly the same (∼3 Å, Fig. 2d). Such a radius appears necessary to provide an adequate space for isoflurane binding within the pore.
Hydrophobic forces were thought to dominate the interaction of anesthetics with their protein binding sites, as the sensitivity of nAChRs to isoflurane inhibition increased with the pore hydrophobicity26. In ELIC, the A244T mutation significantly reduced hydrophobicity51 at position 13′ and reduced the channel sensitivity to isoflurane inhibition (Fig. 3). The result seems to be consistent with a dominant contribution of hydrophobic force to anesthetic inhibition. However, our observation of isoflurane at the 6′ position also suggests that amphipathic interactions contribute equally—if not more—to isoflurane binding to the pore. The binding site at 6′ consists of side chains of all five T237(6′) and two L240(9′) residues (Fig. 2c) that form a favorable amphipathic environment to stabilize isoflurane binding through amphipathic interactions. The T237A mutation destroyed the hydrophilic moiety of the amphipathic interaction52. In addition, the mutation increased the pore radius at 6′ by ∼1 Å relative to that of the WT ELIC (Supplementary Fig. 2). The pore enlargement at 6′ likely further weakened isoflurane binding, exaggerating the reduction in functional inhibition by isoflurane. Thus, both pore amphiphilicity and geometry can affect anesthetic binding to the pore and alter anesthetic inhibition of channel functions.
Direct pore binding and allosteric modulation
The structural frameworks presented here and published previously10,53 support a direct pore-binding mechanism of channel inhibition by anesthetics. A remaining question is how anesthetic molecules reach the pore binding sites. Although a previous MD simulation14 suggested that drug molecules dissolved in the membrane could migrate from the interface between two subunits into the pore, in most cases anesthetics may enter the pore from the extracellular pore entrance. For isoflurane (volume, 110 Å3), without the conformational flexibility of ELIC as shown in Supplementary Fig. 5, it could not pass through the 16′ and 9′ positions that restrict the pore radius to less than 2 Å (Fig. 2d). Despite the conformational flexibility of the channel, inhibition by direct pore binding is still limited to drugs of a certain size. For example, memantine (volume, 192 Å3) could not bind to the pore of ELIC without a mutation to expand the radius of the pore entrance54. Thus, for larger anesthetic molecules, a different inhibition mechanism from direct pore binding must apply.
One alternative inhibition mechanism is through allosteric modulation. An increasing number of studies have found that anesthetics have multiple binding sites in a given protein8,10,12,13,14,15,20,21,55. Among the sites reported for anesthetics, allosteric sites predominate. Even though we only observed isoflurane in the pore of the ELIC-isoflurane crystal structures, we do not rule out the possibility that isoflurane also binds to allosteric sites in ELIC. It is not unusual that crystal structures miss some small ligands bound to proteins, especially for low affinity volatile anesthetics like isoflurane. One might argue that any allosteric site will become irrelevant to inhibitory effects once a drug binds to the pore. However, such an argument discounts the possibility that allosteric modulation can affect direct pore binding18 or even contribute synergistically with the pore binding inhibitory effect.
Methods
Protein expression and purification
The ELIC plasmid was generously provided by Professor Raimund Dutzler’s group at the University of Zürich, Zürich, Switzerland. ELIC was expressed and purified using the protocol reported previously40. Briefly, Rosetta (DE3) pLysS (Novagen) cells transformed with the ELIC plasmid were grown in Luria-Bertani media at 37 °C to an OD600 of ∼0.9 and transferred to M9 media at 15 °C for ∼2 hours before protein expression was induced with 0.2 mM isopropyl β-D-1-thiogalactopyranoside. Cells were harvested after 24 hours, re-suspended with a buffer containing 150 mM NaCl, 50 mM sodium phosphate at pH 8 and protease inhibitors and lysed using a M-110Y microfluidizer processor (Microfluidics). The membrane extracts were pelleted and the fusion protein was extracted in the presence of 3.5% (w/v) n-Undecyl-β-D-maltoside (Anatrace) followed by purification with a Ni-NTA column (GE Healthcare). The MBP tag was cleaved overnight with HRV3C protease (GE Healthcare) and separated from ELIC using Ni-NTA chromatography. The pentameric ELIC was collected in a buffer containing 10 mM sodium phosphate (pH 8), 150 mM NaCl, 0.025% (w/v) n-dodecyl-β-D-maltoside (Anatrace) by size exclusion chromatography using a Superdex 200 10/300GL column (GE Healthcare). The purified ELIC was concentrated to ∼6 mg/ml for crystallization.
Crystallography and data analysis
Crystals were obtained using the vapor-diffusion method in sitting drops at 4 °C. ELIC, pre-equilibrated with 0.01–0.02 mg/ml E. coli polar lipids (Avanti Polar Lipids), was mixed in 1:1 ratio with the reservoir solution (10–12% PEG 4000, 200 mM ammonium sulfate and 100 mM MES buffer at pH 6.1). For co-crystalizing ELIC with isoflurane (Penn Veterinary Supply), the reservoir solution was saturated with isoflurane. For co-crystallizing ELIC with 3-bromopropylamine hydrobromide (Sigma-Aldrich), 5 mM 3-bromopropylamine hydrobromide was equilibrated with ELIC for ∼30 minutes before setting up crystallization trays. For cryo-protection, crystals were soaked briefly in the reservoir solution supplemented with 20% glycerol and excess ligands before being flash-frozen in liquid nitrogen.
The X-ray diffraction data were collected on beamline 12–2 at the Stanford Synchrotron Radiation Lightsource (SSRL). The anomalous scattering was acquired near the K-edge peak of Br. The data were indexed, integrated and scaled with the XDS program56. All crystals have the P21 space group with two identical pentamers in each asymmetric unit.
A previously published ELIC structure (PDB code: 3RQU, 3.09 Å resolution) was used as a starting template for structure determination of isoflurane-bound ELIC in the absence (3.0 Å) and presence of 3-bromopropylamine (3.4 Å). Two structural regions, including residues 136–156 and residues 285–295, were rebuilt to improve the fitting of electron density. The resulting model was refined iteratively using Phenix57. Automatic solvent detection, updating and refinement were conducted for placing water molecules and followed by manual inspection and adjustment. Isoflurane binding sites were determined based on the FO-FC difference map and isoflurane was built inside the pore using Coot58. The geometry and stereochemistry of the final model were validated by the program MolProbility59. MolProbity scores of 2.01 and 2.12 were obtained for ELIC in the resting and desensitized states, respectively. Initial isoflurane geometry was obtained from the complex structure of apoferritin with isoflurane (PDB code: 1XZ3)60. The 3-bromopropylamine binding sites were identified based on the bromine-specific (0.9199 Å) anomalous difference map. Torsional non-crystallographic symmetry (NCS) restraints were applied to all ten subunits of two pentamers in the asymmetric unit. After many cycles of refinement, the final structures were analyzed using Phenix57. All molecular graphics were prepared using PyMol61 or VMD62.
Electrophysiology
Site-directed mutagenesis of ELIC was introduced with the QuickChange Lightning Kit (Agilent). DNA encoding ELIC and a T7 promoter was inserted into the pCMV-mGFP Cterm S11 NeoKan (Theranostech) vector. Capped complementary RNA was transcribed with the mMessage mMachine T7 kit (Ambion), purified with the RNeasy kit (Qiagen), injected (10–25 ng) into Xenopus laevis oocytes (stages 5–6), which were maintained at 18 °C in a modified Barth’s solution40. All procedures involving Xenopus laevis oocytes were approved by the University of Pittsburgh Institutional Animal Care and Use Committee (IACUC), Protocol 14114745. Two-electrode voltage clamp experiments were performed 16–40 hrs after injection using an Oocyte Clamp OC-725C amplifier (Warner Instruments), Digidata 1440A digitizer (Axon Instruments) and a 20 μl chamber (Automate Scientific). Oocytes were clamped to a holding potential of −40 to −60 mV. The recording solutions contained 130 mM NaCl, 0.1 mM CaCl2, 10 mM HEPES, pH 7.0 as well as desired concentrations of agonist and anesthetics. Data were collected and processed using Clampex 10 (Molecular Devices). Non-linear regressions were performed using Prism software (Graphpad).
For measuring anesthetic inhibitory effects, saturated solutions of the volatile anesthetics isoflurane, sevoflurane and halothane were prepared in the recording solution by stirring overnight at 4 °C. Concentrations of these anesthetics in the recording chamber were calibrated using 19F NMR63. The intravenous anesthetics, propofol, etomidate and thiopental, were dissolved directly into the recording solution.
Molecular dynamics simulations
Four systems were prepared for simulations: (1) the resting ELIC (PDB code: 3RQU)40, (2) the resting ELIC-isoflurane (PDB code: 4Z90), (3) the desensitized conformation of ELIC and (4) the desensitized ELIC bound with isoflurane. The structure of the desensitized ELIC was obtained through homology modeling based on the desensitized structure of GABAARβ3 (PDB code: 4COF)45 using the Modeller program version 9.1.464,65. Initial isoflurane positions in system #4 were defined based on the observation in the structure of ELIC co-crystallized with isoflurane and BrPPA (PDB code: 4Z91). ELIC along with the ligands was embedded into an equilibrated membrane of bacterial lipids (1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine and 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-(1′-rac-glycerol) in a 3:1 ratio) in a hexagonal prism, solvated (24,361 TIP3 water molecules) and ionized (62 Na+, 23 Cl−) using VMD plugins62.
NAMD 2.966 and the CHARMM-36 force field67 were used for all MD simulations. Each system was energy minimized for 10,000 steps with the ELIC backbone fixed. The system was gradually warmed from 0 to 310 K with a restraint of 1 kcal/mol/Å2 on the ELIC backbone. The restraint was gradually removed over 600 ps of further equilibration. Each equilibrated system was simulated for 100 ns at a constant temperature (310 K) and pressure (1 atm) with a 2 fs time step and Langevin damping value of 1 ps−1. Particle mesh Ewald was used for long-range electrostatic interactions and a 12 Å cutoff was used for non-bonded interactions. Non-bonded interactions were evaluated at every step, while full electrostatic interactions were evaluated every other step. Periodic boundary conditions were applied to all the simulations. Simulation data were analyzed using VMD62, HOLE68 and Matlab69 programs.
Additional Information
Accession Codes: Coordinates have been deposited to the Protein Data Bank under codes 4Z90 (isoflurane-ELIC complex in the absence of agonists) and 4Z91 (isoflurane-ELIC complex in the presence of agonists).
How to cite this article: Chen, Q. et al. Direct Pore Binding as a Mechanism for Isoflurane Inhibition of the Pentameric Ligand-gated Ion Channel ELIC. Sci. Rep. 5, 13833; doi: 10.1038/srep13833 (2015).
References
Weiser, T. G. et al. An estimation of the global volume of surgery: a modelling strategy based on available data. Lancet 372, 139–44 (2008).
Abildstrom, H. et al. Cognitive dysfunction 1–2 years after non-cardiac surgery in the elderly. ISPOCD group. International Study of Post-Operative Cognitive Dysfunction. Acta Anaesthesiol. Scand. 44, 1246–51 (2000).
Jevtovic-Todorovic, V. et al. Early exposure to common anesthetic agents causes widespread neurodegeneration in the developing rat brain and persistent learning deficits. J. Neurosci. 23, 876–82 (2003).
Eckenhoff, R. G. et al. Inhaled anesthetic enhancement of amyloid-beta oligomerization and cytotoxicity. Anesthesiology 101, 703–9 (2004).
Wei, H. & Xie, Z. Anesthesia, calcium homeostasis and Alzheimer’s disease. Curr. Alzheimer Res. 6, 30–5 (2009).
Xie, Z. et al. The common inhalation anesthetic isoflurane induces caspase activation and increases amyloid beta-protein level in vivo. Ann. Neurol. 64, 618–27 (2008).
Franks, N. P. & Lieb, W. R. Molecular and cellular mechanisms of general anaesthesia. Nature 367, 607–614 (1994).
Forman, S. A. & Miller, K. W. Anesthetic sites and allosteric mechanisms of action on Cys-loop ligand-gated ion channels. Can. J. Anaesth. 58, 191–205 (2011).
Pan, J. et al. Structure of the pentameric ligand-gated ion channel GLIC bound with anesthetic ketamine. Structure 20, 1463–9 (2012).
Spurny, R. et al. Multisite binding of a general anesthetic to the prokaryotic pentameric Erwinia chrysanthemi ligand-gated ion channel (ELIC). J. Biol. Chem. 288, 8355–64 (2013).
Chiara, D. C., Dangott, L. J., Eckenhoff, R. G. & Cohen, J. B. Identification of nicotinic acetylcholine receptor amino acids photolabeled by the volatile anesthetic halothane. Biochemistry (Mosc.) 42, 13457–67 (2003).
Liu, L. T., Willenbring, D., Xu, Y. & Tang, P. General anesthetic binding to neuronal alpha4beta2 nicotinic acetylcholine receptor and its effects on global dynamics. J. Phys. Chem. B 113, 12581–9 (2009).
Brannigan, G., LeBard, D. N., Henin, J., Eckenhoff, R. G. & Klein, M. L. Multiple binding sites for the general anesthetic isoflurane identified in the nicotinic acetylcholine receptor transmembrane domain. Proc. Natl. Acad. Sci. USA 107, 14122–7 (2010).
Willenbring, D., Liu, L. T., Mowrey, D., Xu, Y. & Tang, P. Isoflurane alters the structure and dynamics of GLIC. Biophys. J. 101, 1905–12 (2011).
Bondarenko, V., Mowrey, D., Liu, L. T., Xu, Y. & Tang, P. NMR resolved multiple anesthetic binding sites in the TM domains of the alpha4beta2 nAChR. Biochim. Biophys. Acta 1828, 398–404 (2013).
Bondarenko, V. et al. NMR structures of the human alpha7 nAChR transmembrane domain and associated anesthetic binding sites. Biochim. Biophys. Acta 1838, 1389–95 (2014).
Mowrey, D. D. et al. Insights into distinct modulation of alpha7 and alpha7beta2 nicotinic acetylcholine receptors by the volatile anesthetic isoflurane. J. Biol. Chem. 288, 35793–800 (2013).
Jayakar, S. S., Dailey, W. P., Eckenhoff, R. G. & Cohen, J. B. Identification of propofol binding sites in a nicotinic acetylcholine receptor with a photoreactive propofol analog. J. Biol. Chem. 288, 6178–89 (2013).
Chiara, D. C. et al. Photoaffinity labeling the propofol binding site in GLIC. Biochemistry (Mosc.) 53, 135–42 (2014).
Hamouda, A. K., Jayakar, S. S., Chiara, D. C. & Cohen, J. B. Photoaffinity labeling of nicotinic receptors: diversity of drug binding sites! J. Mol. Neurosci. 53, 480–6 (2014).
Jayakar, S. S. et al. Multiple propofol-binding sites in a gamma-aminobutyric acid type A receptor (GABAAR) identified using a photoreactive propofol analog. J. Biol. Chem. 289, 27456–68 (2014).
Nury, H. et al. X-ray structures of general anaesthetics bound to a pentameric ligand-gated ion channel. Nature 469, 428–31 (2011).
Sauguet, L. et al. Structural basis for potentiation by alcohols and anaesthetics in a ligand-gated ion channel. Nat. Commun. 4, 1697 (2013).
LeBard, D. N., Henin, J., Eckenhoff, R. G., Klein, M. L. & Brannigan, G. General anesthetics predicted to block the GLIC pore with micromolar affinity. PLoS Comput. Biol. 8, e1002532 (2012).
Mowrey, D. et al. Asymmetric ligand binding facilitates conformational transitions in pentameric ligand-gated ion channels. J. Am. Chem. Soc. 135, 2172–80 (2013).
Forman, S. A., Miller, K. W. & Yellen, G. A discrete site for general anesthetics on a postsynaptic receptor. Mol. Pharmacol. 48, 574–81 (1995).
Dilger, J. P., Brett, R. S. & Lesko, L. A. Effects of isoflurane on acetylcholine receptor channels. 1. Single-channel currents. Mol. Pharmacol. 41, 127–33 (1992).
Zhou, Q. L., Zhou, Q. & Forman, S. A. The n-alcohol site in the nicotinic receptor pore is a hydrophobic patch. Biochemistry (Mosc.) 39, 14920–6 (2000).
Nirthanan, S., Garcia, G., 3rd, Chiara, D. C., Husain, S. S. & Cohen, J. B. Identification of binding sites in the nicotinic acetylcholine receptor for TDBzl-etomidate, a photoreactive positive allosteric effector. J. Biol. Chem. 283, 22051–62 (2008).
Chiara, D. C. et al. Time-resolved photolabeling of the nicotinic acetylcholine receptor by [3H]azietomidate, an open-state inhibitor. Mol. Pharmacol. 75, 1084–95 (2009).
Flood, P., Ramirez-Latorre, J. & Role, L. Alpha 4 beta 2 neuronal nicotinic acetylcholine receptors in the central nervous system are inhibited by isoflurane and propofol, but alpha 7-type nicotinic acetylcholine receptors are unaffected. Anesthesiology 86, 859–65 (1997).
Violet, J. M., Downie, D. L., Nakisa, R. C., Lieb, W. R. & Franks, N. P. Differential sensitivities of mammalian neuronal and muscle nicotinic acetylcholine receptors to general anesthetics. Anesthesiology 86, 866–74 (1997).
Weng, Y., Yang, L., Corringer, P. J. & Sonner, J. M. Anesthetic sensitivity of the Gloeobacter violaceus proton-gated ion channel. Anesth. Analg. 110, 59–63 (2010).
Cardoso, R. A., Yamakura, T., Brozowski, S. J., Chavez-Noriega, L. E. & Harris, R. A. Human neuronal nicotinic acetylcholine receptors expressed in Xenopus oocytes predict efficacy of halogenated compounds that disobey the Meyer-Overton rule. Anesthesiology 91, 1370–7 (1999).
Flood, P. & Krasowski, M. D. Intravenous anesthetics differentially modulate ligand-gated ion channels. Anesthesiology 92, 1418–25 (2000).
Belelli, D. et al. The in vitro and in vivo enantioselectivity of etomidate implicates the GABAA receptor in general anaesthesia. Neuropharmacology 45, 57–71 (2003).
Coates, K. M., Mather, L. E., Johnson, R. & Flood, P. Thiopental is a competitive inhibitor at the human alpha7 nicotinic acetylcholine receptor. Anesth. Analg. 92, 930–3 (2001).
Downie, D. L., Franks, N. P. & Lieb, W. R. Effects of thiopental and its optical isomers on nicotinic acetylcholine receptors. Anesthesiology 93, 774–83 (2000).
Miller, C. Genetic manipulation of ion channels: a new approach to structure and mechanism. Neuron 2, 1195–205 (1989).
Pan, J. et al. Structure of the pentameric ligand-gated ion channel ELIC cocrystallized with its competitive antagonist acetylcholine. Nat. Commun. 3, 714 (2012).
Zimmermann, I. & Dutzler, R. Ligand activation of the prokaryotic pentameric ligand-gated ion channel ELIC. PLoS Biol. 9, e1001101 (2011).
Spurny, R. et al. Pentameric ligand-gated ion channel ELIC is activated by GABA and modulated by benzodiazepines. Proc. Natl. Acad. Sci. USA 109, E3028–34 (2012).
Wagner, D. A. & Czajkowski, C. Structure and dynamics of the GABA binding pocket: A narrowing cleft that constricts during activation. J. Neurosci. 21, 67–74 (2001).
Padgett, C. L., Hanek, A. P., Lester, H. A., Dougherty, D. A. & Lummis, S. C. Unnatural amino acid mutagenesis of the GABA(A) receptor binding site residues reveals a novel cation-pi interaction between GABA and beta 2Tyr97. J. Neurosci. 27, 886–92 (2007).
Miller, P. S. & Aricescu, A. R. Crystal structure of a human GABAA receptor. Nature 512, 270–5 (2014).
Gonzalez-Gutierrez, G. et al. Mutations that stabilize the open state of the Erwinia chrisanthemi ligand-gated ion channel fail to change the conformation of the pore domain in crystals. Proc. Natl. Acad. Sci. USA 109, 6331–6 (2012).
Kinde, M. N. et al. Conformational Changes Underlying Desensitization of the Pentameric Ligand-gated Ion Channel ELIC. Structure 23, 995–1004 (2015).
Wenningmann, I., Barann, M., Vidal, A. M. & Dilger, J. P. The Effects of isoflurane on acetylcholine receptor channels: 3. Effects of conservative polar-to-nonpolar mutations within the channel pore. Mol. Pharmacol. 60, 584–94 (2001).
Tillman, T., Cheng, M. H., Chen, Q., Tang, P. & Xu, Y. Reversal of ion-charge selectivity renders the pentameric ligand-gated ion channel GLIC insensitive to anaesthetics. Biochem. J. 449, 61–8 (2013).
Harpaz, Y., Gerstein, M. & Chothia, C. Volume Changes on Protein-Folding. Structure 2, 641–649 (1994).
Kyte, J. & Doolittle, R. F. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 157, 105–32 (1982).
Xu, Y. & Tang, P. Amphiphilic sites for general anesthetic action? Evidence from 129Xe-[1H] intermolecular nuclear Overhauser effects. Biochim. Biophys. Acta 1323, 154–62 (1997).
Hilf, R. J. et al. Structural basis of open channel block in a prokaryotic pentameric ligand-gated ion channel. Nat. Struct. Mol. Biol. 17, 1330–6 (2010).
Ulens, C. et al. The prokaryote ligand-gated ion channel ELIC captured in a pore blocker-bound conformation by the Alzheimer’s disease drug memantine. Structure 22, 1399–407 (2014).
Chen, Q., Cheng, M. H., Xu, Y. & Tang, P. Anesthetic binding in a pentameric ligand-gated ion channel: GLIC. Biophys. J. 99, 1801–9 (2010).
Kabsch, W. Xds. Acta Crystallogr. D Biol. Crystallogr. 66, 125–32 (2010).
Adams, P. D. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–21 (2010).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 (2010).
Davis, I. W., Murray, L. W., Richardson, J. S. & Richardson, D. C. MOLPROBITY: structure validation and all-atom contact analysis for nucleic acids and their complexes. Nucleic Acids Res. 32, W615–9 (2004).
Liu, R., Loll, P. J. & Eckenhoff, R. G. Structural basis for high-affinity volatile anesthetic binding in a natural 4-helix bundle protein. FASEB J. 19, 567–76 (2005).
DeLano, W. L. The PyMOL Molecular Graphics System, Schrödinger, LLC. (Delano Scientific LLC, Palo Alto, CA, 2002).
Humphrey, W., Dalke, A. & Schulten, K. VMD: visual molecular dynamics. J. Mol. Graph. 14, 33–8, 27–8 (1996).
Xu, Y., Seto, T., Tang, P. & Firestone, L. NMR study of volatile anesthetic binding to nicotinic acetylcholine receptors. Biophys. J. 78, 746–51 (2000).
Eswar, N. et al. Comparative protein structure modeling using MODELLER. Current protocols in protein science/editorial board, John E. Coligan … [et al.] Chapter 2, Unit 2 9 (2007).
Sali, A. & Blundell, T. L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 234, 779–815 (1993).
Phillips, J. C. et al. Scalable molecular dynamics with NAMD. J Comput Chem 26, 1781–802 (2005).
Yu, W., He, X., Vanommeslaeghe, K. & MacKerell, A. D., Jr. Extension of the CHARMM General Force Field to sulfonyl-containing compounds and its utility in biomolecular simulations. J Comput Chem 33, 2451–68 (2012).
Smart, O. S., Neduvelil, J. G., Wang, X., Wallace, B. A. & Sansom, M. S. HOLE: a program for the analysis of the pore dimensions of ion channel structural models. J. Mol. Graph. 14, 354–60, 376 (1996).
MATLAB. version 8.4.0.150421 (R2014b). (The MathWorks Inc., 2014).
Smart, O. S., Goodfellow, J. M. & Wallace, B. A. The pore dimensions of gramicidin A. Biophys. J. 65, 2455–60 (1993).
Acknowledgements
The authors thank Professors William Furey and Xiangpeng Kong for their helpful discussion. M.N.K. was supported by a National Institutes of Health (NIH) Ruth L. Kirschstein National Service Award (T32GM075770 to Y.X.). M.M.W. was supported by the NIH T32EB009403. Use of the Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract No. DE-AC02-76SF00515. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research and by the National Institutes of Health, National Institute of General Medical Sciences (including P41GM103393). The MD simulations were supported in part by the National Science Foundation through TeraGrid resources (TG-MCB050030N). The research was supported by funding from the NIH (R01GM056257 and R01GM066358).
Author information
Authors and Affiliations
Contributions
P.T. and Y.X. conceived the general approach. Q.C., M.N.K., A.P., M.M.W., Y.X. and P.T. performed the research. A.C., along with Q.C., Y.X. and P.T., contributed to X-ray data collection. P.T., Y.X. and Q.C. wrote the manuscript. All authors reviewed the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Chen, Q., Kinde, M., Arjunan, P. et al. Direct Pore Binding as a Mechanism for Isoflurane Inhibition of the Pentameric Ligand-gated Ion Channel ELIC. Sci Rep 5, 13833 (2015). https://doi.org/10.1038/srep13833
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep13833
This article is cited by
-
Structural basis of neurosteroid anesthetic action on GABAA receptors
Nature Communications (2018)
