
Abstract
The earliest clinical sign of pulmonary vascular disease in neonates is persistent pulmonary hypertension of the newborn (PPHN), traditionally classified as World Symposium on Pulmonary Hypertension (WSPH) Group 1. Typical PPHN is defined by the delayed transition from intra- to extra- uterine life with failure of pulmonary artery pressures to fall after delivery. This results in sustained elevation of pulmonary vascular resistance and contributes to hypoxemic respiratory failure (HRF). However, early HRF can also arise from atypical PPHN phenotypes that may persist beyond the expected resolution of typical PPHN and are often classified within WSPH Groups 2, 3 and 5. In addition, a history of PPHN can be associated with the childhood and adulthood PH. In this perspective, we highlight the diverse etiologies contributing to the PPHN phenotype, with a focus on Group 3 disease, and propose a physiology-based framework to classify PPHN and delineate disease trajectories.
Similar content being viewed by others


Introduction
Pediatric pulmonary hypertension (PH) is classified as a rare disease, with an incidence of 4 to 10 per million per year [1]. The earliest clinically recognized form of PH in children presents in the immediate postnatal period and is referred to as persistent pulmonary hypertension of the newborn (PPHN). PPHN is physiologically defined by abnormal transition of the pulmonary circulation with sustained elevation pulmonary arterial pressure (PAP) at delivery. The delayed transition from fetal to extrauterine life causes hypoxemia with right-to-left extrapulmonary shunting, often diagnosed as neonatal hypoxic respiratory failure (HRF). Typical PPHN is currently classified within the World Symposium on Pulmonary Hypertension (WSPH) Group 1.7 [2]. This designation reflects shared pulmonary arterial pathobiology features with other Group 1 pulmonary arterial hypertension (PAH) disorders, including pre-capillary PH, elevated PAP, preserved left sided filling pressures, and remodeling of small pulmonary arteries [3, 4].
Failure of PAP to fall after delivery is the hallmark of typical PPHN, characterized by persistent, though often reversible, elevation in pulmonary vascular resistance (PVR), and right ventricular dysfunction. HRF subsequently occurs when the hypoxemia is due to predominant extrapulmonary right to left shunt across the ductus arteriosus and/or foramen ovale [4, 5]. However, there is growing recognition that physiologic mechanisms beyond traditional Group 1 disease can present with a PPHN phenotype, as HRF occurs with diverse forms of neonatal PH that persist beyond the expected resolution of typical PPHN [6]. The WSPH designation of PPHN within Group 1 does not comprehensively include all atypical clinical phenotypes of PPHN, particularly those related to increased pulmonary blood flow (PBF), left heart disease with elevated pulmonary capillary wedge pressure (PCWP), and various developmental lung diseases (DevLDs) [4, 5]. “Typical” PPHN classified in Group 1 PH can be a false façade for atypical PPHN better suited for the Group 2 PH associated with left heart disease, Group 3 PH associated with lung disease, or Group 5 PH associated with multifactorial disorders (e.g., hematological, systemic, metabolic).
The heterogeneity of underlying physiologies of PPHN phenotypes may be best delineated by combining the traditional causal categorization of the pulmonary vasculature within the framework of the major determinants of PAP according to the equation: mean PAP = (PVR × PBF) + PCWP. (Fig. 1) [4, 5]. Specifically, typical and atypical causes of PPHN can be divided into three broad categories based on the development of the pulmonary vasculature and then further delineated by disorders that increase PAP: (1) maldevelopment of pulmonary vasculature with normal lung parenchyma and increased PVR, (2) maladaptive pulmonary vasculature with abnormal parenchyma and increased PVR, and (3) alteration in PBF and PCWP via pulmonary venous congestion and/or cardiac dysfunction. (Fig. 1) [4, 5]. This combined framework has the potential to delineate the cardiac and pulmonary contributors PH based on phenotypic contributions within acute presentations across disease states spanning WSPH Groups 1, 2, 3 and 5 classifications [7].

Causes of Persistent Pulmonary Hypertension of the Newborn: Etiologies of persistent pulmonary hypertension of the newborn (PPHN) based on pulmonary vascular development divided into disorders that lead to an increase in pulmonary vascular resistance (PVR): normal lung parenchyma with maladaptive pulmonary vasculature; pulmonary vascular remodeling with maldevelopment of the pulmonary vasculature and abnormal lung parenchyma; and (2) disorders that affect pulmonary blood flow (PBF) and pulmonary capillary wedge pressure (PCWP) from increased pulmonary venous congestion and cardiac dysfunction. ABCA3 ATP-binding cassette subfamily A member 3, AP aortopulmonary, ASD atrial septal defect, PDA patent ductus arteriosus, AVM arteriovenous malformation, CDH congenital diaphragm hernia, CoA coenzyme A, IDM infants of diabetic mothers, NSAID nonsteroidal ant-inflammatory drug, PPROM prolonged preterm premature rupture of membrane, SPD surfactant protein deficiency, SSRI selective serotonin reuptake inhibitor, VSD ventricular septal defect.
The applied relevance of this combined approach is best demonstrated through representative clinical examples. Consider two term infants with clinical and echocardiographic evidence of HRF and elevated PVR immediately after delivery: one with presumed PPHN following perinatal risk factors who fails to improve in the expected time course, despite targeted therapy, prompting evaluation for alternative causes of neonatal PH; and another with congenital diaphragmatic hernia (CDH) whose PH resolves following surgical repair and extracorporeal support. Although both infants initially present with similar hemodynamic findings consistent with HRF and PPHN, the underlying pathophysiology differs substantially, underscoring the limitations of applying a single diagnostic label across distinct mechanisms of neonatal PH. To further delineate out these distinctions, this perspective reviews the epidemiology and underlying etiopathologies of PPHN, explores typical and atypical PPHN with a focus on Group 3 disease, and proposes a physiology-based framework to classify all types of PPHN and their disease trajectories. We provide an algorithm for clinical management that integrates the broad physiological spectrum of PPHN phenotypes, the WSPH classification and the vascular endotypes.
Phenotypic etiopathologies of PPHN
PPHN can occur in both term and preterm born infants, and is associated with several maternal, fetal, and postnatal risk factors (Table 1). This overlap emphasizes the complex interplay between the maternal-fetal-postnatal environment and pulmonary vascular growth and development. The pathophysiology of PPHN has previously been described as either: (1) maladaptation of normal pulmonary vasculature with increased PVR, (2) maldevelopment of the pulmonary vasculature with increased PVR, or (3) multifactorial that combines elements of maladaptation and maldevelopment (Fig. 1) [4,5,6]. These classic categories can present with variable and overlapping features of altered vascular tone, reactivity, and remodeling that only become apparent with time and were derived in an era before the recognition of most DevLDs.
PPHN from maladaptation is thought to be reversible since there has been normal development of the pulmonary vasculature. A perturbation in the maternal-fetal environment or during the perinatal course causes persistent pulmonary vasoconstriction with failure of the PAP to physiologically decline after delivery resulting in this acute early delayed transition. Examples of specific insults that prevent relaxation of the pulmonary vascular bed include maternal and perinatal factors such as drug and toxin exposure in-utero (non-steroidal anti-inflammatory drugs, selective serotonin reuptake inhibitors), pre-eclampsia, chorioamnionitis, meconium aspiration, sepsis, and drug and toxin exposure. As the most common cause of PPHN, newborns with maladaptation physiology are expected to follow the “typical” course of PPHN and resolve to normal cardiac and pulmonary function generally by the second week of age [8, 9]. The interplay between the heart-lung vascular interactions can have variable contribution to this delay in postnatal transition.
PPHN from maldevelopment is a condition in which the pulmonary vasculature or parenchyma has abnormal, poor, or arrested growth during in-utero development. Angiogenesis and alveolarization are highly dependent on each other during lung maturation, so disruption in either of these processes can lead to hypoplastic or remodeled pulmonary vasculature that impacts normal pulmonary vascular transition after birth. PPHN associated with lung hypoplasia can be seen in disease processes linked to oligohydramnios, CDH, or congenital pulmonary adenomatoid malformations. Additionally, a lung that is dysplastic can also present with a clinical picture of PPHN. Specific examples of maldeveloped PPHN include DevLDs: alveolar capillary dysplasia, acinar dysplasia, and congenital alveolar dysplasia [8]. Occasionally, PPHN is the first presentation of these fetal DevLDs, with a much longer and tenuous course that is variably reversible compared to “typical” PPHN. Further evaluation of DevLDs may include genetic testing, advanced imaging of the lungs, or lung biopsy [9]. As such, it is imperative to consider different causes of PH that do not fit into Group 1 PH but may be better classified as Group 3 or 5 according to WSPH and are part of the differential diagnosis for variably reversible PPHN related disorders.
There is a combined phenotype in which PPHN results from multifactorial causes. In these cases, the failure for decrease in PVR reflects a complex interplay between developmental, structural, and cellular abnormalities. Remodeling of the pulmonary vasculature can occur even with altered reactivity being the prominent phenotype. Examples may include complex congenital heart disease (CHD), Trisomy 21, and genetic syndromes. These types of PPHN are recognized clinically, due to more obvious presentations of underlying pathology. Even meconium aspiration syndrome which had traditionally been considered to be a maladaptive form of PPHN, is also associated with pulmonary vascular remodeling.
Overall, the heterogeneity of underlying physiologies of PPHN presentations may be best delineated by extending the PPHN classification across the framework of WSPH classifications so that causes of atypical PPHN, especially Group 3-related, are not overlooked (Fig. 2). In addition, left ventricular (LV) dysfunction can masquerade as “PPHN” in the immediate postnatal period. In contrast to typical PPHN, which is driven by increased PVR from a pre-capillary process, some neonates develop PPHN physiology secondary to left heart–mediated, post-capillary pathology often found in Group 2 PH. Infants at risk for this mechanism include those with fetal growth restriction, infants of diabetic mothers, hypoxic-ischemic encephalopathy, prematurity, or LV outflow tract obstruction (e.g., coarctation of the aorta) [3, 4]. (Fig. 1).

Algorithm for evaluation of persistent pulmonary hypertension of the newborn based on timing and persistence of symptoms.
Atypical PPHN: Group 3, developmental lung disorders
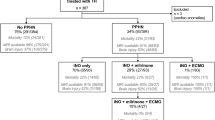
A rational approach to identify the etiology of PPHN should account for developmental differences in the pulmonary vasculature based on the determinants of PAP and within the framework of the WSPH classification. Specifically, the clinical dilemma for providers is differentiating between the typical and atypical phenotypes of PPHN. Each entity may present with similar clinical and echocardiographic findings but knowing which will resolve and which require further work-up is the key to the management. A study by Tsoi et al. showed that “typical” PPHN resolves by 10 days of age, with majority of the patients being decannulated from ECLS, extubated, and off inotropes [9]. In contrast, less than one-third of patients with “atypical” PPHN from developmental causes met these milestones by 10 days of age. If PPHN persists into the second week of age without the typical de-escalation of therapies, further work-up for atypical causes of PPHN commonly found in WSPH Group 3 (e.g., DevLDs) should be considered [9]. A 20-year study of 1101 children with all forms of PH identified PPHN in the birth history of 101 children (11.1%), of which 31 (31%) were classified as having Group 1 PH and 68 (68%) were classified as having Group 3 PH [10]. Recognizing the challenge of identifying the atypical PPHN phenotype, we propose a framework to guide clinical practice (Fig. 3) with the recognition that future work will be needed to validate this approach. There are certain etiologies of atypical PPHN that should be considered earlier, as they may be identified earlier based on specific echocardiographic or physical examination findings.

Schema for persistent pulmonary hypertension of the newborn. *Certain etiologies of PPHN should be considered before day of life (DOL) 10, as they may be identified earlier based on specific echocardiographic or physical examination findings.
The DevLDs associated with PH in neonates have a wide array of biologic and physiologic basis ranging from large anatomical defects seen with CDH or giant omphaloceles, associations with genetic mutations seen with T-Box Transcription Factor (TBX4), and functional deficiencies observed in the surfactantopathies. Several of these disorders are difficult to detect before birth and require advanced imaging, molecular testing, and multidisciplinary team of specialists for timely diagnosis, management, and counseling. As research advances, genetic testing in neonates presenting with PPHN will include evaluation of genes involved in surfactant production and alveolar epithelial function, pulmonary vascular development and angiogenesis, the transforming growth factor–β (TGF-β) and bone morphogenetic protein (BMP) signaling cascade, pulmonary mesenchymal and airway development, ion channel–mediated regulation of pulmonary vascular tone, and even metabolic disorders influencing neonatal PH (Table 2), which will specifically inform PPHN diagnosis. We have highlighted several examples below for each category.
Surfactant metabolism and alveolar epithelial function
Severe defects in surfactant protein production or function may present with a clinical phenotype consistent with PPHN. Pathogenic variants in ATP Binding Cassette Subfamily A Member 3 (ABCA3), Surfactant Protein B (SFTPB), Surfactant Protein C (SFTPC), and NK2 Homeobox 1 (NKX2-1) disrupt surfactant synthesis, processing, and type II alveolar epithelial cell function. Mutations in SFTPB are most commonly identified in the neonatal period and typically result in severe respiratory failure, whereas SFTPC mutations more often present later in childhood with interstitial lung disease. ABCA3 mutations may manifest in either the neonatal period or later childhood. Each disorder is associated with distinct histopathologic features and characteristic abnormalities in lamellar body morphology although persistence of PH beyond the infantile period is uncommon [11].
Pulmonary vascular development and endothelial signaling
Abnormal pulmonary vascular development and endothelial signaling have been associated with pathogenic variants in BMP Receptor Type 2 (BMPR2), Activin A Receptor Like Type 1 (ACVRL1), Endoglin (ENG), Growth Differentiation Factor 2 (GDF2), Caveolin 1 (CAV1), ATPase 13A3 (ATP13A3), and Aquaporin 1 (AQP1) [11]. Variants in these genes contribute to dysregulated pulmonary vascular growth and tone and are associated with heritable or neonatal-onset PH [12]. In addition, Trisomy 21 is associated with abnormal pulmonary vascular development and intrinsic endothelial signaling dysfunction, leading to reduced vascular growth, early remodeling, and impaired vasoreactivity that predispose affected infants to PH independent of hypoxia or shunt physiology [12].
TGF-β/BMP signaling
Dysregulated TGF-β/BMP signaling leads to abnormal pulmonary vascular remodeling and PPHN phenotypes [12]. Pathogenic variants in SMAD Family Member 9 (SMAD9) and Eukaryotic Translation Initiation Factor 2 Alpha Kinase 4 (EIF2AK4) disrupt this pathway, contributing to vascular remodeling and PH.
Pulmonary mesenchymal and airway development
Abnormal lung, airway, and pulmonary vascular morphogenesis is often associated with DevLDs and early PH. Genes critical for these processes include Forkhead Box F1 (FOXF1) associated with alveolar capillary dysplasia (ACD) [13, 14], SRY-Box Transcription Factor 17 (SOX17), and TBX4 [15,16,17,18].
ACD is a rare, often fatal disorder that typically presents with severe PPHN within the first 24 h of age and is associated with pathogenic variants in FOXF1, a key regulator of embryonic lung development. Affected neonates present with profound HRF, frequently require ECMO, and may have concomitant gastrointestinal, urogenital, or cardiovascular anomalies [12]. Diagnosis is confirmed by lung biopsy, which demonstrates thickened interalveolar septa, small hypertrophic pulmonary arteries, reduced and malpositioned alveolar capillaries, and misaligned pulmonary veins [14]. Genetic testing can aid diagnosis. Prognosis is poor, although milder cases surviving beyond childhood have been reported [13].
Congenital Acinar Dysplasia (CAD) and Congenital Alveolar Dysplasia (CADys) are rare DevLDs distinguished primarily by histopathology, but both can present similarly to ACD with immediate neonatal HRF and severe PPHN [19]. CAD results from arrested lung development during the pre-acinar (late pseudoglandular) stage (8–16 weeks), characterized by irregular acini, absent or minimal alveoli, fibrotic septal and interstitial tissue, and immature bronchial-like structures lacking adequate vasculature for gas exchange [20]. CADys arises from developmental arrest during the canalicular phase (18–24 weeks), with lung tissue showing premature alveoli, primitive mesenchyme, immature collagen fibers in alveolar walls, and large but immature capillary beds. Both disorders are uniformly fatal [19].
TBX4 encodes a transcription factor essential for embryonic development of the pulmonary and skeletal systems. Pathogenic variants in TBX4 have been associated with neonatal and childhood PH, as well as Small Patella Syndrome, a skeletal disorder affecting the feet, pelvis, and knees that typically manifests in adulthood [15]. Several case reports of TBX4 describe neonates presenting with severe PPHN shortly after delivery and ultimately diagnosed via genetic testing [16, 17]. Histopathology reveals diffuse alveolar growth abnormalities, pulmonary vascular remodeling, and disruption of distal lung development [18]. In addition to HRF, the phenotype may include pneumothoraces, intracardiac shunts (patent ductus arteriosus, atrial septal defects), and skeletal abnormalities. PH associated with TBX4 can follow a bimodal course, presenting first in the neonatal period, resolving, and then recurring later in childhood or early adulthood.
Vascular tone and ion channel regulation
Altered pulmonary arterial smooth muscle cell membrane potential can impair vasodilation and increase PVR. Pathogenic variants in Potassium Two-Pore Domain Channel Subfamily K Member 3 (KCNK3), which regulates this membrane potential, have been implicated in PAH (Group 1 PH) across age groups, but specific evidence linking KCNK3 pathogenic variants to neonatal PPHN is limited.
Metabolic disorders influencing neonatal pulmonary hypertension
There are also secondary genetic causes that do not directly affect lung structure or vascular development, but can present as PPHN. CPS1 (Carbamoyl-Phosphate Synthase 1) is a metabolic urea cycle enzyme whose deficiency causes hyperammonemia with resultant encephalopathy and HRF, which can lead to PH in affected neonates.
Enhancing the classification of PPHN to incorporate group 3 diseases
We review these etiologies of Group 3 PH to exemplify how atypical PPHN can be the first presentation of many of these diseases. In our proposed classification system seen in Fig. 2, we emphasize the spectrum of PPHN that spans the endotypes of pulmonary vascular disease and that Group 1, Group 3, and Group 5 diseases should be considered in the diagnostic work-up of PPHN.
LONG-term impact of “PPHN” on the pulmonary vasculature
Patients with pediatric PH have been reported to have a higher incidence of PPHN during the early neonatal period compared to the general population (0.1–0.6%) [21, 22]. A large population based study from Sweden found that close to 40% of children and young adults with PH had a previous diagnosis of PPHN [15]. Another study of PH in children found that 7% of children with a diagnosis of Group 1 PH had a previous history of “PPHN” and 22% of children with a diagnosis of Group 3 PH had a diagnosis of “PPHN”, although it is hard to know if these were accurately named PPHN or if they were conditions of delayed transition [10]. Although previous work has shown that maternal age, acute pulmonary disease, CDH, CHD, chromosomal disorders, and prematurity are all risk factor for childhood PH [15], it is likely that both typical and atypical PPHN play critical roles in pulmonary vascular disease beyond the neonatal period. Recognition that PPHN could be an additional predictor of the development of pediatric PH will have important implications on screening patients with PPHN.
The mechanistic link between PPHN in the neonatal period and the development of PH in childhood is not yet known, but it is plausible that the change to the pulmonary vasculature may be rooted in the maternal-fetal environment leading up to birth leaving a long-lasting effect on the pulmonary vasculature. This notion would align with “Barker’s Hypothesis” of fetal origins of adult disease, which postulates that an abnormal intrauterine environment causes fetal epigenetic changes that manifest as pediatric or adult diseases after a secondary insult or environmental exposure [23]. Previous animal models have shown through epigenetics that an altered maternal-fetal environment can lead to a PH phenotype in offspring when exposed to the postnatal insult of hypoxia that persist across the lifespan [24, 25]. There are several known risk factors of PPHN that affect the maternal-fetal environment (Table 1) and can be possible mechanisms for creating long-lasting damage to the pulmonary vasculature [25,26,27,28,29]: (1) placental malperfusion, (2) hyperinflammation, and (3) stress from toxins. Further studies are needed to explore if epigenetic changes or metabolic reprogramming are associated with these risk factors, PPHN, and PH.
Conclusions
PPHN is a disease process that sits at the intersection of multiple WSPH classification groups with its typical form in Group 1 and the atypical presentation in Groups 3 and 5. While PPHN may resolve in some cases, it may also serve as a precursor to lung disease-associated PH in the future. We present an enhanced classification approach to PPHN with emphasis on the broad spectrum of etiopathologies underlying PPHN (Figs. 1 and 2) and we provide guidance of the evaluation of PPHN based on the presentation and resolution of symptoms (Fig. 3). While there is still much to learn about PPHN, including contributing risk factors and long-term consequences, this perspective challenges, and enhances, the current classification system with a more differentiated approach to allow for more individualized, targeted management when faced with the clinical dilemma of the ambiguous PPHN diagnosis.
References
Sullivan RT, Austin ED. Pulmonary hypertension in children. Clin Chest Med. 2024;45:685–93. https://doi.org/10.1016/j.ccm.2024.04.001.
Jone PN, Ivy DD, Hauck A, Karamlou T, Truong U, Coleman RD, et al. Pulmonary hypertension in congenital heart disease: a scientific statement from the American Heart Association. Circ Heart Fail. 2023;16:e00080.
Kovacs G, Bartolome S, Denton CP, Gatzoulis MA, Gu S, Khanna D, et al. Definition, classification and diagnosis of pulmonary hypertension. Eur Respir J. Published online January 1, 2024. https://doi.org/10.1183/13993003.01324-2024
Ivy D, Rosenzweig EB, Abman SH, Beghetti M, Bonnet D, Douweet JM, et al. Embracing the challenges of neonatal and paediatric pulmonary hypertension. Eur Respir J. Published online January 1, 2024. https://doi.org/10.1183/13993003.01345-2024
Bhattacharya S, Sen S, Levy PT, Rios DR. Comprehensive evaluation of right heart performance and pulmonary hemodynamics in neonatal pulmonary hypertension: evaluation of cardiopulmonary performance in neonatal pulmonary hypertension. Curr Treat Options Cardiovasc Med. 2019;21:10. https://doi.org/10.1007/s11936-019-0713-8.
Ruoss JL, Rios DR, Levy PT. Updates on management for acute and chronic phenotypes of neonatal pulmonary hypertension. Clin Perinatol. 2020;47:593–615. https://doi.org/10.1016/j.clp.2020.05.006.
Mirza H, Mandell EW, Kinsella JP, McNamara PJ, Abman SH. Pulmonary vascular phenotypes of prematurity: the path to precision medicine. J Pediatr. Published online April 25, 2023:113444. https://doi.org/10.1016/j.jpeds.2023.113444
Sharma V, Berkelhamer S, Lakshminrusimha S. Persistent pulmonary hypertension of the newborn. Matern Health Neonatol Perinatol. 2015;1:14. https://doi.org/10.1186/s40748-015-0015-4.
Tsoi SM, Steurer M, Nawaytou H, Cheung S, Keller RL, Fineman JR. Defining the typical course of persistent pulmonary hypertension of the newborn: when to think beyond reversible causes. J Pediatr. 2024;273. https://doi.org/10.1016/j.jpeds.2024.114131
Constantine A, Dimopoulos K, Haworth SG, Muthurangu V, Moledina S. Twenty-year experience and outcomes in a national pediatric pulmonary hypertension service. Am J Respir Crit Care Med. 206:758-66. https://doi.org/10.1164/rccm.202110-2428OC
Wert SE, Whitsett JA, Nogee LM. Genetic disorders of surfactant dysfunction. Pediatr Dev Pathol. 2009;12:253–74. https://doi.org/10.2350/09-01-0586.1.
Varghese NP, Austin ED, Galambos C, Mullen MP, Yung D, Guillerman RP, et al. An interdisciplinary consensus approach to pulmonary hypertension in developmental lung disorders. Eur Respir J. Published online January 1, 2024. https://doi.org/10.1183/13993003.00639-2024
Bishop NB, Stankiewicz P, Steinhorn RH. Alveolar capillary dysplasia. Am J Respir Crit Care Med. 2011;184:172–9. https://doi.org/10.1164/rccm.201010-1697CI.
Slot E, Edel G, Cutz E, van Heijst A, Post M, Schnater M, Cutz E, et al. Alveolar capillary dysplasia with misalignment of the pulmonary veins: clinical, histological, and genetic aspects. Pulm Circ. 2018;8:2045894018795143. https://doi.org/10.1177/2045894018795143.
Kerstjens-Frederikse WS, Bongers EMHF, Roofthooft MTR, Leter EM, Douwes JM, Dijk AV, et al. TBX4 mutations (small patella syndrome) are associated with childhood-onset pulmonary arterial hypertension. J Med Genet. 2013;50:500–6. https://doi.org/10.1136/jmedgenet-2012-101152.
Tsoi SM, Jones K, Colglazier E, Parker C, Nawaytou H, Teitel D, et al. Persistence of persistent pulmonary hypertension of the newborn: a case of de novo TBX4 variant. Pulm Circ. 2022;12:e12108. https://doi.org/10.1002/pul2.12108.
Maddaloni C, Ronci S, Rose DUD, Bersani I, Campi F, Nardo MD, et al. Neonatal persistent pulmonary hypertension related to a novel TBX4 mutation: case report and review of the literature. Ital J Pediatr. 2024;50:41. https://doi.org/10.1186/s13052-024-01575-3.
Galambos C, Mullen MP, Shieh JT, Schwerk N, Kielt MJ, Ullmann N, et al. Phenotype characterisation of TBX4 mutation and deletion carriers with neonatal and paediatric pulmonary hypertension. Eur Respir J. 2019;54. https://doi.org/10.1183/13993003.01965-2018
Oneto S, Poppiti RJ. Congenital acinar dysplasia: a lethal entity. Autops Case Rep. 2019;9:e2019119. https://doi.org/10.4322/acr.2019.119.
Langenstroer M, Carlan SJ, Fanaian N, Attia S. Congenital acinar dysplasia: report of a case and review of literature. AJP Rep. 2013;3:9–12. https://doi.org/10.1055/s-0032-1329126.
Steurer MA, Jelliffe-Pawlowski LL, Baer RJ, Partridge JC, Rogers EE, Keller RL. Persistent pulmonary hypertension of the newborn in late preterm and term infants in California. Pediatrics. 2017;139:e20161165. https://doi.org/10.1542/peds.2016-1165.
Bendapudi P, Rao GG, Greenough A. Diagnosis and management of persistent pulmonary hypertension of the newborn. Paediatr Respir Rev. 2015;16:157–61. https://doi.org/10.1016/j.prrv.2015.02.001.
Barker DJ. The fetal and infant origins of adult disease. BMJ. 1990;301:1111.
Xu XF, Lv Y, Gu WZ, Tang LL, Wei YK, Zhang LY, et al. Epigenetics of hypoxic pulmonary arterial hypertension following intrauterine growth retardation rat: epigenetics in PAH following IUGR. Respir Res. 2013;14:20. https://doi.org/10.1186/1465-9921-14-20.
Dias Maia P, Seedorf G, Gonzalez T, Bye E, Frank BS, Mandell EW, et al. Development of late pulmonary hypertension after antenatal inflammation in experimental bronchopulmonary dysplasia. Pediatr Res. 2025. https://doi.org/10.1038/s41390-025-04223-626.
Naumburg E, Söderström L, Huber D, Axelsson I. Risk factors for pulmonary arterial hypertension in children and young adults. Pediatr Pulmonol. 2017;52:636–41. https://doi.org/10.1002/ppul.23633.
Jayet PY, Rimoldi SF, Stuber T, Salmòn CS, Hutter D, Rexhaj E, et al. Pulmonary and systemic vascular dysfunction in young offspring of mothers with preeclampsia. Circulation. 2010;122:488–94. https://doi.org/10.1161/CIRCULATIONAHA.110.941203.
Goss K. Long-term pulmonary vascular consequences of perinatal insults. J Physiol. 2019;597:1175–84. https://doi.org/10.1113/JP275859.
Maron BA, Abman SH. Translational advances in the field of pulmonary hypertension. focusing on developmental origins and disease inception for the prevention of pulmonary hypertension. Am J Respir Crit Care Med. 2017;195:292–301. https://doi.org/10.1164/rccm.201604-0882PP.
Author information
Authors and Affiliations
Contributions
SMT and NPV conceived of the project. SMT and NPV drafted the initial manuscript. PTL and ShA provided critical feedback and manuscript oversight. All authors reviewed and approved of final submission. SMT and NPV conceptualized and designed the study. SMT drafted the initial manuscript, and critically reviewed and revised the manuscript. PTL and SAH assisted with study design and provided expert input. All authors critically reviewed and revised the manuscript for important intellectual content. All authors approved the final manuscript as submitted.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Tsoi, S.M., Levy, P.T., Abman, S.H. et al. Phenotyping persistent pulmonary hypertension of the newborn: recognition of its persistence across world symposium on pulmonary hypertension classifications. J Perinatol (2026). https://doi.org/10.1038/s41372-026-02704-y
Received:
Revised:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41372-026-02704-y
