
Abstract
CBFA2T3::GLIS2-positive pediatric acute myeloid leukemia (AML) remains one of the worst prognostic AML subgroups. To uncover innovative targeted therapy approaches in this disease subtype we performed genome-scale CRISPR-Cas9 screening that highlighted a strong, selective dependency on JAK2 compared to other types of cancer. Using a doxycycline-inducible JAK2 knockout (KO) system, we validated JAK2 dependency in CBFA2T3::GLIS2 cell lines, observing impaired proliferation in vitro and in vivo and apoptosis induction in vitro. Both type I (ruxolitinib) and type II (CHZ868) JAK2 inhibitors showed selective in vitro activity in CBFA2T3::GLIS2-positive AML models. To identify resistance and sensitizer mechanisms to JAK2 inhibitors, we used CRISPR-Cas9 ruxolitinib anchor screening in CBFA2T3::GLIS2 AML. sgRNAs targeting negative regulators of the MAPK pathway were enriched in the ruxolitinib-treated cells. Similarly, CBFA2T3::GLIS2 AML sublines grown to resistance under chronic ruxolitinib treatment expressed pathogenic NRAS mutations. Both approaches converged on MAPK pathway activation as a resistance mechanism to ruxolitinib treatment. Combining ruxolitinib with MEK inhibitors showed a synergistic effect in cell lines and patient-derived xenograft (PDX) cells expressing the fusion and in vivo activity in a CBFA2T3::GLIS2 AML PDX, suggesting a potential approach to target this signaling circuitry in this poor outcome AML subtype.
Similar content being viewed by others


Introduction
Acute myeloid leukemia (AML) accounts for about 25% of pediatric leukemias and remains a significant clinical challenge due to poor prognosis and therapy-related toxicity [1]. Acute megakaryoblastic leukemia (AMKL), a subtype accounting for ~10% of childhood AML, includes two subgroups: AMKL in patients with Down syndrome, which has an excellent prognosis (~80% survival), and AMKL arising in patients without Down syndrome (non-DS-AMKL), where overall survival (OS) is poor (14–34%) despite intensive therapy [2, 3]. A cryptic inversion on chromosome 16, inv (16) (p13.3q24.3), leads to the joining of the genes CBFA2T3 (CBFA2/RUNX1 partner transcriptional co-repressor 3) and GLIS2 (GLIS family zinc finger 2) and is the most prevalent chromosomal aberration in non-DS-AMKL (~20% patients) [4]. Mechanistically, CBFA2T3::GLIS2 binds DNA through CBFA2T3-associated transcription factors or directly through GLIS2 zinc-finger domains at enhancers and other regulatory elements. CBFA2T3::GLIS2 is reported to alter transcription, such as the upregulation of the ETS transcription factor ERG and the kinase KIT, and downregulation of genes such as GATA1, resulting in a block in differentiation and increased self-renewal capacity [5,6,7]. To date, the mechanisms underlying its aggressiveness and chemoresistance remain poorly understood.
The CBFA2T3::GLIS2 fusion is an exclusively pediatric AML genomic abnormality, occurring in children under 4 years of age; it is associated with an OS rate of only 14% [8]. Co-occurring mutations are rare [9, 10], limiting targeted therapy options, and patients with this leukemia are subjected to the same therapeutic protocols as other children with high-risk AML, despite their distinct chemoresistance. One approach to targeting this high-risk subset of AML would be to target the fusion directly. Targeting fusion transcription factors, however, represents a considerable challenge. To circumvent this challenge and investigate new therapeutic opportunities, we aimed to find specific dependencies in the context of CBFA2T3::GLIS2-positive AML by interrogating the Broad Institute’s Cancer Dependency Map (DepMap), a data set composed of genome-scale CRISPR-Cas9 screens in over 1000 cancer cell lines, and by screening additional CBFA2T3::GLIS2-positive AML models. We identified the druggable target JAK2 as a strong selective dependency and a candidate therapeutic target in CBFA2T3::GLIS2 AML and identified resistance mechanisms and effective drug combinations with JAK2 inhibitors.
Materials and methods
Cell culture and PDX samples
Cell lines were obtained from DSMZ (M-07e, MOLM13 and MV4-11) or BioIVT (WSU-AML). The CMS cell line was generously provided by Dr. Soheil Meshinchi (Fred Hutchinson Cancer Center). CMS, MOLM13, MV4-11 and WSU-AML were cultured in RPMI (Mediatech) with 10% fetal bovine serum (FBS; Sigma-Aldrich) and 1% penicillin–streptomycin (PS; Gibco). M-07e was cultured in RPMI with 10% FBS, 1% PS and 10 ng/mL human G-CSF (300-23; PeproTech). HEK293T cells were maintained in DMEM (Mediatech) with 10% FBS and 1% PS. Mycoplasma contamination was ruled out using a LookOut Mycoplasma PCR Detection Kit (MP0035; Sigma-Aldrich), and cell line identities were verified by short tandem repeat profiling by the Molecular Diagnostics Laboratory at the Dana-Farber Cancer Institute (DFCI).
Primary patient samples were previously acquired following written informed consent in accordance with the Declaration of Helsinki, and patient-derived xenografts (PDXs) were established under protocols approved by the DFCI and Cincinnati Children’s Hospital Medical Center (CCHMC) Institutional Review Boards [11,12,13]. PDX cells were cultured in IMDM (Mediatech) with 20% FBS, 1% PS and 10 ng/mL human SCF, TPO, FLT3L, IL3, and IL6 (300-07, 300-18, 300-19, 200-03, and 200-06; PeproTech). Supplementary Table S1 provides full details on AML cell lines and PDXs. AML and PDX cells were transduced with lentivirus co-expressing Cas9 and mCherry (a gift from Marco Herold, Addgene plasmid # 70182; RRID: Addgene_70182). Cas9-expressing cells were selected by mCherry fluorescence. Cas9-expressing PDX cells were expanded via serial transplantation into immunodeficient NSGS (NOD/SCID/IL2RG−/− immunodeficient mice with transgenic expression of human SCF, GM-CSF, and IL3; The Jackson Laboratory) mice. Cas9 activity was assessed using the EGFP reporter pXPR_047 (a gift from David Root, Addgene plasmid # 107145; RRID: Addgene_107145).
CRISPR-Cas9 screens
CMS and WSU-AML cells stably expressing Streptococcus pyogenes Cas9 were transduced in duplicate with the Broad Institute’s AVANA-4 sgRNA library [14, 15] (~74,687 sgRNAs, ~4 sgRNAs per gene) at an MOI of ~30%. Puromycin selection began the day after transduction and was maintained for 3 days. For the WSU-AML drop-out screen, cells were cultured for 21 days and passaged every 4 days. For the drug modifier screens in CMS and WSU-AML, cells were divided into two arms post-selection and treated for 14 days with DMSO or ruxolitinib (150 nM for CMS and 50 nM for WSU-AML), with two replicates per condition. Representation of 500–1000 cells per sgRNA was maintained. Genomic DNA was extracted 21 days post-infection using the NucleoSpin Blood L kit (Takara #740954.20). The sgRNA barcodes were PCR amplified, and sequenced via Illumina as previously described [16].
Dependency data analysis for the drop-out and drug modifier CRISPR-Cas9 screens
Drop-out and drug modifier screens were sequenced by the Broad Institute’s Genetic Perturbation Platform (GPP) (https://www.broadinstitute.org/genetic-perturbation-platform). sgRNAs read count was deconvoluted using PoolQ software (https://portals.broadinstitute.org/gpp/public/software/poolq). Annotation files for sgRNAs efficacy (CRISPRInferredGuideEfficacy.csv) and common essential genes (CRISPRInferredCommonEssentials.csv, AchillesCommonEssentialControls.csv) were downloaded from the 24Q4 DepMap database (https://depmap.org/portal/download/) [17]. The sgRNA reads were log-normalized to the plasmid DNA pool (pDNA).
Quality control pre-processing steps were performed to filter out the guides participating in the fusion. sgRNAs targeting the CBFA2T3::GLIS2 fusion region (GLIS2 ATACTCAGCTTCAGGTCGAG and GLIS2 GAGTATCACCAAGCTCCGGG) were excluded. Non-fusion-targeting GLIS2 guides (TCAAGCCCGAGAAGGATGCG and ACACCATCCAAATAGCGCAG) were retained. Guides with low efficacy (≤0.35) and low pDNA representation were filtered out. sgRNA guides that passed the filtering were mapped to genes using CP0033_GRCh38_NCBI_CRISPRko_strict_gene_20221209.chip annotations. Nonessential genes were used as negative controls; common essentials served as positive controls. All samples passed quality control, except CMS replicate 2 (ruxolitinib 150 nM), which was excluded.
The gene effect dependency scores for WSU-AML drop-out and CMS/WSU-AML drug modifier screens were computed from the sgRNA log-norm data based on the hypergeometric distribution tool (https://portals.broadinstitute.org/gpp/public/). Dependency was determined from the average log-normalized fold change of sgRNAs per gene. The p-values were computed using the probability mass function of a hypergeometric distribution for the log-norm dependency-ranked sgRNAs. Statistical significance was defined by |log2FC|≥ 1.5 and p ≤ 0.05. Genes with negative differential scores (ruxolitinib vs. DMSO) were classified as “drug sensitizers” (knockout (KO) increases drug sensitivity); positive scores indicated resistance (KO decreases sensitivity).
Public CRISPR dependency data for CMS and M-07e (fusion-positive AML cell lines) were retrieved from the 24Q4 DepMap (https://depmap.org, CRISPRGeneEffect.csv and AvanaLogfoldChange.csv files). Gene dependency was assessed via Chronos scores, except for GLIS2, where dependency was estimated using the average Avana log fold change of the two non-fusion-targeting guides (TCAAGCCCGAGAAGGATGCG and ACACCATCCAAATAGCGCAG). Gene effect scores ≤ −0.5 were evaluated as significant dependencies.
Lentivirus production and transduction
Two JAK2-targeting sgRNAs (sgJAK2-1: ATCTGCCTCAGATTTCCCAA; sgJAK2-2: GAGGTGCTACTTCTTTACAG) from the AVANA-4 library were used alongside a nontargeting control (sgNT: CCGCGCATTTCAGAGCACAA). sgRNAs were cloned into the doxycycline-inducible GFP-expressing FgH1tUTG vector (Addgene #70183) as described [18]. Cas9 was delivered using a Cas9-T2A-mCherry vector (Addgene #70182). For RASA2 KO, sgRASA2-1 (TTAGCATCAAGGCATGCCAT) and sgRASA2-2 (GCCTGTTGACTCCAATTCAG) were cloned into pXPR_003 [19].
Lentiviral particles were produced by co-transfecting HEK293T cells with expression vectors and packaging plasmids (pCMV-VSV-G, Addgene #8454; psPAX2, Addgene #12260) using Fugene transfection reagent (Promega). AML cells were transduced by spinfection with viral supernatants and 8 μg/mL polybrene at 2000 × rpm for 2 h at 25 °C. All experiments were conducted with polyclonal cell populations.
Confirmation of sgRNA editing
KO efficiency was assessed 4 days post-doxycycline induction by immunoblotting for the respective targets (JAK2/pSTAT3/pSTAT5/RASA2) for JAK2 KO or RASA2 experiments.
For in vivo experiments, JAK2 on-target editing was confirmed by PCR amplification of the sgRNA-targeted region, followed by Sanger sequencing with deconvolution by the TIDE (tracking of indels by decomposition) algorithm. Primers used for both amplification and sequencing were:
-
Forward 5ʹ ACTGAGCCATAAAAGATATGAGCAA 3ʹ
-
Reverse 5ʹ ACTGTGGCTTCCTTGCAGTA 3ʹ
PCR products were purified using the QiaQuick PCR purification kit. On-target editing was then determined using the TIDE algorithm https://tide.nki.nl/ [14].
Competition assays
Cells were transduced with GFP-expressing lentiviral sgRNA vectors at ~50% efficiency or mixed 1:1 with non-transduced cells. Cell growth was evaluated by the change in the percentage of GFP+ cells via flow cytometry from doxycycline induction (D0, defined as day 4 post-induction) through three weeks.
Cell viability
To determine the effects of JAK2 deletion on viability in CBFA2T3::GLIS2 fusion AML, GFP+ cells were sorted, expanded and plated at 500 cells/well in 384-well plates (Corning #3570). Viability was measured on Days 0, 3, 5, and 7 using the CellTiter-Glo assay (Promega; 10 μL/well). Luminescence was read on a Fluostar Omega Reader (BMG Labtech), and values were normalized to D0.
Flow cytometry
Apoptosis was assessed using the APC Annexin V Apoptosis Detection Kit (BioLegend #640932), and cell cycle was analyzed with the Click-iT Plus EdU Alexa Fluor 647 Flow Cytometry Assay Kit (Invitrogen #C10634) per the manufacturer’s protocols. Cells were incubated with 10 µM EdU for 1.5 h and co-stained with FxCycle Violet (Invitrogen #F10347).
Mouse samples were stained with V450 anti-human CD45 and APC-Cy7 anti-mouse CD45 (BD Biosciences #560368, #557659) antibodies. Data were acquired on a BD LSR Fortessa or Celesta; cell sorting was performed on a BD FACS Aria II. Data were analyzed using FlowJo (TreeStar).
Xenograft transplantation
All animal experiments were approved by the DFCI Institutional Animal Care and Use Committee and conducted in accordance with institutional guidelines. Transplantation was performed on 6–8-week-old NSGS mice.
For doxycycline-inducible sgJAK2 experiments, 1 × 106 Cas9-expressing WSU-AML cells transduced with either sgNT or sgJAK2-2 and sorted for GFP were injected via tail vein. Doxycycline-containing food (625 mg/kg) was initiated after engraftment confirmation by bone marrow aspiration (BMA).
For the four-arm study, mice were randomly assigned to receive by oral gavage vehicle, ruxolitinib (60 mg/kg, in 0.5% methylcellulose, twice daily, 5 days/week), selumetinib (50 mg/kg, in 0.5% HPMC + 0.1% Tween 80, twice daily, 5 days/week) or the combination [15]. Ruxolitinib was purchased from LC Labs (R-6600) for in vivo use, and selumetinib from Shanghai Medicilon. Leukemia burden was evaluated two weeks post-treatment by assessing human CD45+ chimerism in peripheral blood, bone marrow, spleen, and spleen size.
Western blotting
Cells were lysed in RIPA Buffer (Thermofisher #89900) supplemented with Halt™ Protease Inhibitor Cocktail (Thermofisher #78429) and phosphatase inhibitors (Roche #04906845001). Lysates were quantified using a Bradford protein assay (Bio-Rad) and normalized. Samples were separated by SDS-PAGE and transferred to PVDF membranes. Primary antibodies (Cell Signaling Technology (CST)) included: JAK2 (D2E12) (#3230S), pJAK2 (Tyr1007/1008) (#3771S), pSTAT3 (Tyr705) (#9145S), STAT3 (124H6) (#9139), pSTAT5 (Tyr694) (#9351), STAT5 (D2O6Y) (#94305S), beta-Actin (#4967L), MEK1/2 (L38C12) (#4694S), pMEK1/2 (Ser217/221) (#9154), AKT (#9272S), and pAKT (Ser473) (#4060), pERK (Thr980) (#16F8), p44/42 MAPK (Erk1/2) (127F5) (#4695) and vinculin (#4650). RASA2 antibody was from Sigma Aldrich (HPA035374). Blots were incubated with HRP-linked secondary antibodies (CST #7074, #7076) and visualized using an Amersham Imager 680 (GE Healthcare).
Drug response testing
AML and PDX cells were seeded in 384-well plates (Corning #3570) at 100,000 and 200,000 cells/ml, respectively, in 50 µL per well. Compounds were added alone or in combination using an HP D300 Digital Dispenser, with four replicates per dose or dose pairing. DMSO was normalized to the highest DMSO volume added, not to exceed 0.02%/well. Viability was assessed after 3 days for JAK inhibitors and 5 days for MEK inhibitors using the CellTiter-Glo assay (Promega #G7571). Ruxolitinib (#S1378), tofacitinib (#S2789), trametinib (#S2673) and selumetinib (#S1008) were purchased from Selleck Chemicals. CHZ868 (#HY-18960) was purchased from MedChem Express. The drug response curves for baseline-corrected, normalized log(concentration) data were fit based on the non-linear regression equation log(inhibitor) vs. response ~ variable slope (four parameters). The Extra-sum-of-squares F-test was used to test the hypotheses if one curve adequately fits all the datasets and if the best fit of LogIC50 was the same among the datasets. Efficacy differences between fusion and non-fusion cell lines were analyzed using a two-tailed t-test on the area under the curve (AUC) scores for the fitted curves in the two groups.
Synergy analysis
To determine if individual treatment combinations were synergistic, additive, or antagonistic, Combination Index (CI) scores were calculated using the Chou–Talalay median effect model [20,21,22]. The CI score quantifies effect x produced by the combination of dose d1 of drug 1 and dose d2 of drug 2: CI = d1/Dx1 + d2/Dx2, where Dx1 is the dose of drug 1 that alone produces the effect x and Dx2 is the dose of drug 2 that alone produces the effect x, as estimated from the median effect model. For any endpoint of the effect measurement, CI estimates the following interactions:
-
Strong synergism: log10(CI) ≤ − 0.22
-
Synergism: log10(CI) > −0.22 and ≤−0.10
-
Additivity: log10(CI) > −0.10 and <0.08
-
Antagonism: log10(CI) ≥ 0.08 and <0.20
-
Strong antagonism: log10(CI) ≥ 0.20
CI scores versus the fractional inhibition effect were presented as scatter dot plots.
Generation of resistant cell lines
To generate ruxolitinib-resistant CMS and M-07e cells, parental lines were exposed to escalating ruxolitinib concentrations (10 nM to 1.5 μM) over 6 months. Resistance was defined as ≥90% viability at 10× of the parental IC50.
Statistics
Statistical tests and P-values are described in the figure legends. All t-tests were unpaired, two-sided unless otherwise indicated. Significance was set at P ≤ 0.05. Analyses were performed using GraphPad Prism v10.0.2.232. Error bars represent standard deviation (SD) unless specified. All duplicate measures were taken from distinct samples rather than repeated measures of the same sample.
Results
CRISPR-Cas9 screens identify JAK2 as a strong selective dependency in CBFA2T3::GLIS2 AML
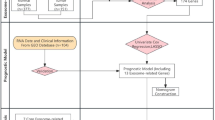
Genome-wide CRISPR-Cas9 KO screening has identified critical gene dependencies in cancer [23, 24]. Using this approach (Fig. 1A), more than 1,000 cancer cell lines were screened in the context of the Cancer Dependency Map [17, 25] (https://depmap.org/portal), including two CBFA2T3::GLIS2-positive AML cell lines (CMS and M-07e) and 26 CBFA2T3::GLIS2 fusion-negative AML cell lines [12]. Expanding upon this data, we performed a genome-scale screen in WSU-AML, another CBFA2T3::GLIS2 fusion cell line derived from the same patient as M-07e. Across all three fusion-positive lines, 105 shared dependencies were identified, including JAK2 and JAK1 (Fig. 1B, C, Supplementary Fig. 1A, Supplementary Table S2).

A Diagram of genome-scale CRISPR-Cas9 drop-out screen. B Dotplot showing JAK2 dependency scores across 24Q4 DepMap cell lines (n = 1178) and WSU-AML (drop-out CRISPR screen). Dots represent AML CBFA2T3::GLIS2-positive cell lines (red), AML CBFA2T3::GLIS2-negative cell lines (black), and all other cell lines (gray). JAK2 gene effect dependency score computed based on the Chronos algorithm for the 24Q4 CRISPR cell lines and based on the hypergeometric test as average fold change lognorm day 21 vs day 0 for WSU-AML. Significance: Chronos gene effect < −0.5 for CMS and M-07e, and average LFC gene effect < −1.5, P-value < 0.05 for WSU-AML. C Venn diagram showing the overlap between non-common essential dependencies in the CBFA2T3::GLIS2 AML cell lines CMS, M-07e (24Q4 DepMap) and WSU-AML (drop-out CRISPR screen). Overlap significance estimated based on the two-tailed Fisher’s exact test. ****P < 0.0001. D Western immunoblot showing the effect of JAK2 knock-out using two different sgRNA (sgJAK2-1 and sgJAK2-2) after 4 days of doxycycline treatment in M-07e cell line; actin was used as a loading control. E Competitive growth assays demonstrating that JAK2 is essential for cell growth in the three CBFA2T3::GLIS2 AML cell lines. Cells were transduced with nontargeting sgRNA (sgNT) and JAK2 sgRNA (sgJAK2-1 and sgJAK2-2) vectors that co-express GFP. Cell growth was evaluated in vitro by the change in percentage of GFP+ cells post doxycycline induction. Shown are mean ± SD, n = 3. P-value calculated using a 2-way ANOVA with multiple comparisons test (comparison to sgNT). *P < 0.05; ***P < 0.001; ****P < 0.0001. F Cumulative cell growth measured using an ATP-based CellTiter-Glo assay in AML cells either transduced with sgNT or sgJAK2 after FACS-sort for GFP-positive cells. Results represented as mean ± SD (n = 8). *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001, determined by two-way ANOVA test followed by Dunnett’s multiple comparisons test. Shown is a representative of three experiments. G Flow cytometry analysis of Annexin V+ and Annexin V+/PI+ in AML cells expressing the indicated sgRNAs at day 7 after doxycycline induction in WSU-AML cell line. Shown are means ± SD, n = 2; experiment performed three times. P-value calculated using a 2-way ANOVA test (comparison to sgNT). The percentage of Annexin⁺/PI⁻ and Annexin⁺/PI⁺ cells was increased in JAK2 KO cells compared with sgNT (sgJAK2-1 P = 0.0017 and sgJAK2-2 P < 0.0001 for Annexin⁺/PI⁻; sgJAK2-1 P = 0.0167 and sgJAK2-2 P < 0.0001 for Annexin⁺/PI⁺). The percentage of Annexin-/PI+ was not significantly difference in sgJAK2-1 vs. sgNT and sgJAK2-2 vs. sgNT.
JAK2 depletion impairs viability and induces apoptosis in vitro
As JAK2 scored as a stronger dependency than JAK1 in two of the three fusion-positive screens (Supplementary Fig. 1A), we focused on its functional validation. Using a doxycycline-inducible KO (Dox-KO) system and two distinct sgRNAs, we confirmed effective protein depletion and reduced downstream signaling, as evidenced by lower levels of phosphorylated STAT3 and STAT5 (Fig. 1D). JAK2 KO significantly suppressed growth in all three CBFA2T3::GLIS2 AML cell lines, as shown by competition assays (Fig. 1E) and cumulative cell doubling measurements (Fig. 1F). Apoptosis was increased, with higher Annexin V positivity in JAK2-depleted cells (Fig. 1G). JAK2 KO also altered cell cycle distribution with a reduced S phase and increased G0/G1 phase (Supplementary Fig. 1B). JAK2 KO did not induce differentiation, as measured by CD11b and CD41 staining (Supplementary Fig. 1C).
JAK2 is essential for AML progression in vivo
We next asked whether disruption of JAK2 can impair AML progression in vivo. WSU-AML cells were transduced with the Dox-KO (nontargeting control (sgNT) or JAK2-targeting sgRNAs) vectors co-expressing GFP. KO efficiency was validated by immunoblotting following in vitro doxycycline induction, and the more efficient sgRNA (sgJAK2-2) was used for xenograft studies (Supplementary Fig. 2A). GFP+ cells were intravenously injected into non-irradiated NSGS mice. After confirming engraftment by BMA on day 10 (mean 1.2% human CD45+/GFP+ cells; n = 4), doxycycline chow was started (Fig. 2A). After 2 weeks, mice with JAK2 KO cells had a significantly reduced leukemia burden in bone marrow, spleen and peripheral blood (Fig. 2B), and decreased spleen weight (Fig. 2C). JAK2 KO prolonged survival (39 versus 29 days, p = 0.0026) (Fig. 2D). At endpoint, disease burden was similar between groups, suggesting that the cause of death was ultimately from progressive leukemia (Supplementary Fig. 2B). TIDE analyses showed minimal JAK2 editing in bone marrow and spleen collected at study endpoint (Fig. 2E), and JAK2 protein levels remained unchanged (Supplementary Fig. 2C), indicating strong in vivo selection against JAK2 loss. Therefore, AML cells driving progressive disease had escaped JAK2 deletion, consistent with an essential role for JAK2 in CBFA2T3::GLIS2 AML progression in vivo.

A Schematic representation of the experimental plan testing JAK2 dependency in vivo using engineered WSU-AML cells. Non-irradiated NSGS mice were transplanted with WSU-AML cells expressing doxycycline (Dox)-inducible sgNT or sgJAK2-2 after FACS-sorting for GFP-positive cells. Engraftment was confirmed by bone marrow aspiration at day 10, and doxycycline-containing food was initiated on day 10. Disease burden was evaluated after 2 weeks of doxycycline treatment in 10 mice (5 per group). Ten remaining mice were monitored for survival. B Bar graphs showing quantification of human CD45 by flow cytometry to evaluate leukemia burden in peripheral blood, bone marrow and spleen samples at 2 weeks after starting doxycycline-containing chow. Shown is mean ± SD, n = 5/group; P-value calculated using two-tailed Mann–Whitney test, **P < 0.01. C Bar graph showing mouse spleen weights after 2 weeks of doxycycline chow. Shown are means ± SD; two-tailed Mann–Whitney test, **P < 0.01. D Kaplan–Meier survival curves of mice from (B). P-value calculated using the log-rank test (n = 5/group), **P < 0.01. E KO efficiency of JAK2 was determined by tracking indels by computational decomposition (TIDE) analysis using DNA isolated from WSU-AML cells transduced with sgJAK2-2. Representative plots are shown for WSU-AML sgJAK2-2 cells in vitro and in vivo at endpoint in bone marrow and spleen. DNA samples from cells carrying sgNT were used for sequence comparison for the sgRNA cut site. % of deletion or insertion at specific sites is shown in red. The predicted cutting site is marked in black as 0.
Primary patient CBFA2T3::GLIS2 AML is enriched for JAK-STAT transcriptional programs
We next examined RNA sequencing data from the TARGET AML cohort comparing 8 CBFA2T3::GLIS2-positive samples to 292 fusion-negative samples (Supplementary Fig. 3A, Supplementary Table S3). Gene set enrichment analysis (GSEA) showed upregulation of several inflammatory signaling pathways including TNF-α signaling via NF-ƘB, IL2-STAT5, IL6-JAK-STAT3 and JAK2-STAT signaling in CBFA2T3::GLIS2-positive samples (Fig. 3A–F). These samples were also enriched in a JAK2 shRNA knockdown signature published by Rampal et al. (GEO GSE54645) that was obtained from a JAK2 V617F homozygous mutant HEL cell line following treatment with two independent shRNAs targeting JAK2 [26] (Fig. 3G, H). We next sought to explore whether the enrichment of the JAK-STAT gene set was driven by the fusion, cell lineage, or both. To this end, we compared JAK-STAT gene expression in three groups: CBFA2T3::GLIS2-positive AML (CG+, n = 8), M6/M7 fusion-negative AML (CG− M6/7, n = 14), and non-M6/M7 fusion-negative AML (CG- M0/1/2/4/5, n = 278) (Fig. 3I). JAK-STAT genes were most highly expressed in fusion-positive AML, but also partially upregulated in M6/M7 fusion-negative cases (Fig. 3I), suggesting both fusion- and lineage-driven effects. To further evaluate this observation, we analyzed the enrichment scores for the KEGG JAK-STAT pathway in CBFA2T3::GLIS2-positive samples, either M7 or non-M7, compared to CBFA2T3::GLIS2 fusion-negative samples. We found a significant enrichment for fusion-positive M7 AML and a borderline significant enrichment for fusion-positive non-M7 AML (Supplementary Fig. 4A, B). However, the same pathway was not significantly enriched in CBFA2T3::GLIS2 fusion-negative M6/M7 AML samples versus fusion-negative non-M6/M7 AML samples (n = 14) (Supplementary Fig. 4C). ssGSEA of MSigDB hallmark gene sets and KEGG/GO terms confirmed the strongest activation of inflammatory signatures such as IL6_JAK_STAT3_signaling and inflammatory response, in fusion-positive sample, with moderate enrichment in fusion-negative M7 cases (Supplementary Fig. 4D, E). These findings highlight a prominent JAK-STAT transcriptional program in CBFA2T3::GLIS2 AML, particularly within the M7 lineage, prompting investigation of JAK-STAT–targeted therapies.

A Volcano plot demonstrating MSigDB Hallmark gene sets enrichment in TARGET AML CBFA2T3::GLIS2-positive (CG + ) AML versus CBFA2T3::GLIS2-negative (CG-) AML. Dots represent hallmark gene sets, Normalized Enrichment Score (NES) (x-axis) vs -log10(P-value) (y-axis). Significance: |NES | ≥ 1.3, -log10(P-value) ≥ 1. Gene sets associated with immune response are highlighted red. B. Volcano plot demonstrating KEGG canonical pathways enrichment in TARGET AML CG + AML versus CG- AML. Dots represent pathways, Normalized Enrichment Score (NES) (x-axis) vs -log10(P-value) (y-axis). Significance: |NES | ≥ 1.3, -log10(P-value) ≥ 1. Pathways associated with immune response are highlighted red. C–F Gene Set Enrichment Analysis (GSEA) plots demonstrating enrichment of representative immune response gene sets in CBFA2T3::GLIS2-positive (CG+) samples versus CBFA2T3::GLIS2-negative (CG−) samples from the TARGET AML cohort. Significance: |NES| ≥ 1.3, P-value ≤ 0.1, FDR ≤ 0.25. G, H GSEA plots demonstrating negative enrichment of the TARGET AML CG+ samples vs CG− samples in the JAK2 knockdown gene signatures from the JAK2V617F-mutant AML cell line HEL [69]. Significance: |NES| ≥ 1.3, P-value ≤ 0.1, FDR ≤ 0.25. I Heatmap of gene expression for representative genes in the KEGG JAK-STAT signaling pathway across TARGET AML CG+ and CG− AML with annotated FAB classification. Left: expression for individual CG+ AML samples (n = 8). Right: average expression across the CG+ samples (n = 8), CG− M6,7 samples (n = 14) and CG− M0,1,2,4,5 samples (n = 278). Expression log2(TPM + 1) data was gene-level normalized relative to the average expression of CG− samples.
CBFA2T3::GLIS2 AML responds to JAK2 small molecule inhibitors
The FDA-approved type I JAK1/2 inhibitor ruxolitinib is used to treat myeloproliferative neoplasms (MPN) [27, 28] and steroid-refractory acute graft-versus-host disease (GvHD) in patients 12 years and older [29]. Type II JAK2 inhibitors, such as CHZ868, stabilize JAK2 in an inactive conformation and avoid the paradoxical increase in pJAK2 seen with type I inhibitors [30, 31]. The three fusion-positive AML cell lines were sensitive to ruxolitinib, CHZ868 and tofacitinib (type I inhibitor), in contrast to fusion-negative lines, such as MOLM13 or MV4-11 (Fig. 4A and Supplementary Fig. 5A). As expected, ruxolitinib reduced pSTAT3/pSTAT5 while increasing pJAK2, as previously reported [32, 33] (Fig. 4B). Ruxolitinib treatment also induced Annexin V–positive cell death (Fig. 4C).

A Dose–response curves for indicated drugs relative to DMSO, tested by ATP-based assay after a 72-h treatment with indicated concentrations of ruxolitinib or CHZ868 in three CBFA2T3::GLIS2 AML cell lines (CMS, M-07e and WSU-AML) and two CBFA2T3::GLIS2-negative cell lines (MOLM13 and MV4-11) Shown are the means ± SD of 4 replicates. The hypotheses that one curve fits all the datasets and that the best fit of LogIC50 is the same among the datasets were separately rejected based on the Extra-sum-of-squares F-test (P < 0.0001). The difference in AUC scores for overall drug efficacy between fusion vs non-fusion cell line groups was estimated based on the two-tailed t-test (****P < 0.0001; ***P < 0.001; **P < 0.01; *P < 0.05; ns not significant). B Western immunoblotting showing phosphorylated JAK2 (p-JAK2) and phosphorylated STAT3 and 5 (p-STAT3 and p-STAT5) after treatment of CMS with DMSO or ruxolitinib (20, 50 and 100 nM) for 1 (H1), 2 (H2) or 6 (H6) hours. C Treatment with ruxolitinib induced cell death via apoptosis in the CBFA2T3::GLIS2 cell line WSU-AML. Histogram showing percentage of positive cells for the respective markers (one representative experiment of two). D Diagram showing clinical course for a patient diagnosed with CBFA2T3::GLIS2 fusion AMKL. The timeline highlights key molecular events and the treatment course. Variant allele frequency of each mutation is indicated as a percentage in parentheses. Bottom Panel: Absolute blast counts over time after relapse (days) with corresponding treatments indicated.
We next treated a pediatric patient with relapsed, refractory CBFA2T3::GLIS2 AML post-allogeneic stem cell transplantation (HSCT) with ruxolitinib, aiming to provide both anti-leukemic activity and GvHD prophylaxis. The patient was initially diagnosed with AMKL at 14 months of age, presenting with fever, pallor and fatigue. His white blood cell count was 75 K cells/µL with 66% blasts. Bone marrow flow cytometry confirmed AML with a RAM immunophenotype [34]. The patient harbored a CBFA2T3::GLIS2 fusion, NRAS p.G12D (VAF = 44.9%), and WT1 p.R369* (VAF = 23%). He was enrolled on the AML16 clinical trial: a Phase 2 Trial of Epigenetic Priming in Newly Diagnosed AML (NCT03164057) and was randomized to arm B (decitabine). He achieved remission with positive MRD (0.134% by flow cytometry) after Induction I chemotherapy and MRD below 0.1% threshold after Induction II chemotherapy. Based on adverse biologic features, he was assigned to the high-risk group and proceeded to HSCT. He underwent an umbilical cord blood transplant (HLA matched 5/6) after a myeloablative conditioning regimen consisting of busulfan, cyclophosphamide and Anti-thymocyte globulin. The post-transplant period was marked by the occurrence of GvHD.
Relapse occurred 89 days post-transplant, with recurrent molecular abnormalities (CBFA2T3::GLIS2 fusion, NRAS p.G12D VAF = 28% and WT1 p. R369* VAF = 18%). Following limited response to azacitidine, ruxolitinib was added at a dose of 25 mg/m2/day as fluconazole was co-administered, consistent with the dose used in the COG AALL1521 study [35]. This resulted in decreased and stabilization of peripheral blasts (Fig. 4D). Unfortunately, his AML ultimately progressed, and the patient died 80 days post-relapse. This clinical case highlights the need to understand resistance mechanisms and develop drug combinations to improve efficacy in this very aggressive AML subtype.
Using relapse-derived AML cells, we established a PDX model through serial transplantations in NSGS mice (CPCT-0027) [11]. Mutational analysis confirmed that the PDX retained the same genetic alterations as the primary patient sample. We tested JAK inhibition in vitro with ruxolitinib and tofacitinib in this model and three others: one CBFA2T3::GLIS2-positive (2016-35) and two fusion-negative (16–01, 17–14) (AML model characteristics in Supplementary Table S1). For both drugs, we observed a better response in the PDX cells harboring CBFA2T3::GLIS2 fusion compared to the PDX lacking the fusion (Supplementary Fig. 5B).
Genome-scale CRISPR-Cas9 screen reveals MAPK pathway activation as a resistance mechanism to JAK2 inhibition
To identify candidate resistance mechanisms and potential synergistic combinations with JAK2 inhibitors, we performed CRISPR-Cas9 screens with ruxolitinib in two CBFA2T3::GLIS2-positive cell lines: CMS and WSU-AML. These lines differ in cooperating mutations (Supplementary Table S1) and lineage markers (CMS is more monocytic with higher CD11b expression, and WSU-AML is more megakaryocytic with higher CD41/CD42 expression) (Supplementary Fig. 6A).
Cas9 expression did not significantly alter ruxolitinib sensitivity in either line (Supplementary Fig. 6B, C). We next determined ruxolitinib concentrations that reduced viability to ~50% while maintaining effective JAK2 inhibition, as confirmed by decreased pSTAT3 and pSTAT5 levels (Supplementary Fig. 6D). Cas9-expressing cells were transduced with the AVANA-4 sgRNA library as previously described [36, 37], selected with puromycin and treated with DMSO or ruxolitinib. Cell viability upon treatment with ruxolitinib was assessed at each passage and corresponded to the expected dose–response pre-established for each cell line (Supplementary Fig. 6E). After 14 days, sgRNA abundance was assessed by sequencing to identify genes enriched or depleted under ruxolitinib pressure (Fig. 5A and Supplementary Table S4).
We sought to identify genes that modulate the response to JAK2 inhibition, specifically those altering growth under ruxolitinib versus DMSO treatment. For each cell line, we calculated the log2 fold change of sgRNA abundance in ruxolitinib- and DMSO-treated cells relative to the plasmid pool. Then, we determined the differential log2 fold change between the two conditions for each sgRNA and averaged these values per gene. The strong correlation in differential log2 fold changes between the two cell lines confirmed the robustness of the screen (Fig. 5B). Using a hypergeometric test, we identified genes with significantly different effects between the ruxolitinib and DMSO arms. The screen was designed to identify genes whose depletion confers resistance or sensitization to ruxolitinib. Notably, resistance-associated hits were more prominent than sensitizers, so we focused on genes whose depletion promoted resistance. Among the top genes were negative regulators of the JAK-STAT pathway: the Suppressors of Cytokine Signaling (SOCSs) family members CISH and SOCS2, and Cullin 5 (CUL5). SOCS proteins bind the elongin B/C complex via their C-terminal SOCS box and recruit the scaffold protein CUL5 to form an E3 ubiquitin ligase complex. This complex polyubiquitinates JAKs and STATs, targeting them for proteasomal degradation and thereby suppressing signal transduction [38, 39]. Loss of genes belonging to the SOCS family occurring either through germline mutations (SOCS1) [40] or via shRNA [41] are associated with JAK-STAT pathway activation, supporting the screen’s validity. Several novel resistance hits were negative regulators of the RAS-MAPK pathway, including RASA2, GOLGA7 and LZTR1. We focused on RASA2 (RAS p21 protein activator 2), recurrently inactivated in Noonan syndrome [42] and various cancers [43, 44]. All four RASA2-targeting guides scored in the CMS cell line (Fig. 5C) and one in WSU-AML (Supplementary Fig. 6F). We knocked out RASA2 in CMS cells, a CBFA2T3::GLIS2 fusion cell line without known RAS-MAPK pathway mutations. RASA2 KO increased pERK and pMEK expression as a surrogate of MAPK pathway activation (Fig. 5D) but did not affect short-term growth compared to controls (sgNT or non-infected cells (NIC)). However, RASA2-deficient cells showed increased resistance to ruxolitinib (Fig. 5E, F), and also to the type I JAK inhibitor tofacitinib (Fig. 5G).

A Diagram of genome-scale CRISPR-Cas9 screen for ruxolitinib modifier genes. B Correlation between the differential log2 fold change in sgRNA abundance between DMSO and ruxolitinib for the two cell lines screened (CMS and WSU-AML) after 14 days of treatment with ruxolitinib. Hits that confer resistance to ruxolitinib are shown in red. All other genes are in gray. C Guide level log2 fold changes for normalized data relative to plasmid pool DNA for the gene RASA2 in the CMS cell line. Each sgRNA is represented. P-values calculated using the hypergeometric test for significant difference in the screen. Shown are the means ± SD, n = 2. **P < 0.01; ****P < 0.0001. D Western immunoblot showing the effect of RASA2 KO using two different sgRNA (sgRASA2-1 and sgRASA-2) in the CMS cell line; vinculin was used as a loading control. E CMS cells were infected with a nontargeting control (sgNT) (gray) and an sgRNAs targeting RASA2 (red). Non-infected control cells (NIC) were represented in black. Bar graphs showing cell numbers after 7 days of cell culture with ruxolitinib 150 nM (Ruxo) or DMSO. Shown are the means ± SD (n = 3). P-values calculated using a 2-way ANOVA test. **P < 0.01; ns not significant. F Cell viability assay in CMS cells transduced with sgNT or sgRASA2 puromycin selection vector. Cells were treated with ruxolitinib for 72 h and viability evaluated using the CellTiter-Glo assay. Line graphs represent means ± SD (n = 3). AUC values for sgNT and sgRASA2 (guides 1 and 2) were compared using Mann–Whitney unpaired t-test. *P < 0.05. G Cell viability assay in CMS cells transduced with sgNT or sgRASA2 puromycin selection vector or untransduced. Cells were treated with tofacitinib for 72 h and viability evaluated using CellTiter-Glo assay. Line graphs represent means ± SD (n = 3). AUC values for sgNT and sgRASA2 (guides 1 and 2) were compared using Mann–Whitney unpaired t-test. *P < 0.05.
These findings support a model in which MAPK pathway activation—via loss of RAS negative regulators like RASA2—confers resistance to JAK inhibition.
Generation of resistant cell lines demonstrates acquired NRAS mutations as a mechanism of resistance to JAK inhibition
To investigate resistance mechanisms to JAK inhibitors in CBFA2T3::GLIS2 AML, we generated cell lines with acquired resistance to JAK inhibitors. CMS and M-07e cells were cultured in escalating concentrations of ruxolitinib up to 1.5 μM (10-fold IC50) (Supplementary Fig. 7A). Resistant lines displayed complete resistance to ruxolitinib compared to naïve cells (Fig. 6A) and were also cross-resistant to tofacitinib (Supplementary Fig. 7B). These resistant models no longer required JAK2 for survival, as shown by resistance to JAK2 knockout compared to ruxolitinib naïve cells (Fig. 6B, Supplementary Fig. 7C). Indeed, genotyping revealed that resistant sublines had NRAS missense mutations: NRAS p.G12R (VAF 46.8 and 40.6%) in CMS sublines and NRAS p.G13D (VAF 56%) in M-07e (Supplementary Table S5). Deep sequencing of the parental and resistant CMS cell lines using the PanCan panel (25,000X coverage) detected 41/4308 reads with the NRAS p.G12R mutation, compared with 928/2115 in the resistant line (Supplementary Fig. 7E) suggesting that there was a minor subclone present in naïve cells that was selected for during chronic ruxolitinib treatment.

A Dose–response curves for ruxolitinib for parental and the engineered resistant sublines (resistant 1 and resistant 2) for CMS (upper graph) and M-07e (lower graph with one resistant subline). Shown are means ± SD (n = 4). AUC values for parental and resistant sublines were compared using Mann–Whitney unpaired t-test. *P < 0.05; **P < 0.01. B Bar graph showing competitive growth assays with parental and resistant sublines. Shown are means ± SD (n = 3). C Western immunoblot showing phosphorylated MEK1/2, AKT and STAT3 in CMS resistant sublines compared to the parental, at base line and after treatment with ruxolitinib 1500 nM for 72 h. Actin was used as a loading control. D Dose–response curves for trametinib in parental and the engineered resistant sublines relative to controls. Shown are means ± SD (n = 4). AUC values for parental and resistant sublines were compared using Mann–Whitney unpaired t-test. **P < 0.01.
We evaluated the main downstream effectors of the JAK-STAT and RAS-MAPK-ERK pathways in the resistant cells and found an increase in phosphorylated MEK and AKT (Fig. 6C, Supplementary Fig. 7D), indicating that in the resistant cells, activation of parallel signaling pathways results in resistance to JAK inhibitors. We next exposed these ruxolitinib-resistant cells to the MEK inhibitor trametinib. Although trametinib showed efficacy on the parental cells, the resistant lines were even more sensitive to MEK inhibition consistent with rewiring from a JAK dependency to a RAS-MAPK pathway dependency (Fig. 6D).
Combined JAK and MEK inhibition improve cytotoxicity in CBFA2T3::GLIS2 AML
We next evaluated whether combining JAK and MEK inhibitors enhances cytotoxicity beyond single-agent treatments. In vitro, co-treatment with trametinib and ruxolitinib in both parental and resistant CMS and M-07e cell lines showed synergistic effects (Supplementary Fig. 8A, B).
To investigate the effect in vivo, we used the CPCT-0027 PDX model, derived from the patient described above, which has a CBFA2T3::GLIS2 fusion and an NRAS mutation. Initial in vitro testing confirmed synergy between ruxolitinib and either trametinib or selumetinib (Fig. 7A, B). To select the optimal MEK inhibitor for in vivo use, we compared their effects on ERK phosphorylation in vivo and found selumetinib more effective at reducing pERK by phospho-flow (Supplementary Fig. 8C). Selumetinib was thus chosen for in vivo testing. Mice were injected with CPCT-0027 cells, and treatment began 10 days post-injection upon detection of engraftment assessed by BMA. After 11 days of treatment, the combination of selumetinib and ruxolitinib significantly reduced leukemic burden in bone marrow and spleen (Fig. 7C). Accordingly, the combination treatment improved survival in the combo versus vehicle group (41 vs. 55 days; p = 0.03) (Fig. 7D). While selumetinib showed a strong impact on pERK as a pharmacodynamic marker in the AML cells (Fig. 7E), mice treated with ruxolitinib, and the combination did not show statistically significant changes by phospho-flow staining for p-STAT5 (Fig. 7F) suggesting that JAK2 signaling was not sufficiently suppressed.

A. Combination index vs. fractional inhibition dotplot for the combination of ruxolitinib and trametinib in CPCT-0027 after 5 days of treatment. Normalized isobologram depicts CI scores over a range of concentrations. The coordinates of the CI scores are d1/Dx1 and d2/Dx2, where Dx1 is the concentration of drug 1 (ruxolitinib) that alone produces the fractional inhibition effect x, and Dx2 is the concentration of drug 2 (trametinib) that alone produces the fractional inhibition effect x. The red line displayed is the line of additivity. Points below the line are synergistic, and above the line are antagonistic. B Combination index vs. fractional inhibition dotplot for the combination of ruxolitinib and selumetinib in CPCT-0027 after 5 days of treatment. Normalized isobologram depicts CI scores over a range of concentrations, with drug 1 (ruxolitinib) and drug 2 (selumetinib). Log10(CI) intervals for: Strong Synergy: ≤−0.22, Synergy: > −0.22, ≤−0.10, Additivity: > −0.10, <0.08, Antagonism: ≥0.08, <0.20, Strong Antagonism: ≥0.20. C Quantification of percentage of hCD45 positive cells in the bone marrow and spleen in the CPCT-0027 model in vivo after 10 days of treatment. *P < 0.05 using one-way ANOVA with Tukey’s multiple comparisons test. D Kaplan–Meier survival curve for CPCT-0027 mice treated with vehicle, ruxolitinib (60 mg/kg PO BID), selumetinib (50 mg/kg PO BID) or the combination of ruxolitinib and selumetinib. N = 7/group. **P < 0.01; *P < 0.05; ns not significant using Log-rank test. E Median fluorescence intensity for pERK expression in the peripheral blood of mice treated with vehicle, selumetinib, ruxolitinib or the combination after 10 days of treatment. ****P < 0.0001 using one-way ANOVA with Tukey’s multiple comparisons test. F Median fluorescence intensity for pSTAT5 expression in peripheral blood after 10 days of treatment. **P < 0.01; *P < 0.05 using one-way ANOVA with Tukey’s multiple comparisons test.
Discussion
Non-DS-AMKL carries a poor prognosis, particularly in patients with the CBFA2T3::GLIS2 fusion, which is associated with chemoresistance and aggressive disease. This fusion disrupts key gene expression programs, blocking differentiation and enhancing self-renewal [4, 5]. Through genome-scale CRISPR-Cas9 screening, we identified a dependency on JAK2 in this AML subtype. Accordingly, JAK2 inhibitors, such as ruxolitinib, showed selective efficacy in CBFA2T3::GLIS2 AML compared to other subtypes such as KMT2A-rearranged AML. Consistent with our findings, Drenberg et al. screened 6,568 compounds across eight AML cell lines and reported selective activity of JAK inhibitors in pediatric AMKL lines, including those with a CBFA2T3::GLIS2 fusion [45].
Signaling mutations play a critical role in childhood leukemogenesis [46]. Oncogenic JAK1 and JAK2 mutations are found in both acute lymphoblastic leukemia (ALL) [47, 48] and MPN, and are also described in 13–23% of CBFA2T3::GLIS2 AML [4, 8, 9]. These mutations abnormally activate the JAK-STAT signaling pathway, promoting cell proliferation and survival. In ALL, JAK mutations are often associated with high-risk subtypes, such as Ph-like ALL [47], while JAK2 V617F drives MPN pathogenesis [49, 50]. No clear prognostic association has been established for JAK mutations in CBFA2T3::GLIS2 AML, likely due to the rarity of this subtype. Our data show that the JAK-STAT pathway is activated even in the absence of JAK mutations, particularly in the M7 FAB subset, which may explain the observed sensitivity to JAK inhibitors in our study and that of Drenberg et al. [45].
Ruxolitinib has been used in various hematologic conditions, including pediatric leukemias, GvHD and MPN [35, 51]. Despite demonstrated clinical benefit, its efficacy is limited by poor selectivity, a suboptimal pharmacokinetics profile with a short half-life [52, 53] and the emergence of resistance [51]. In Drenberg’s study, ruxolitinib significantly improved survival in a preclinical in vivo setting, although disease progression ultimately occurred [45]. Notably, the PDX model in that study harbored a JAK2 V617F mutation, potentially explaining the greater sensitivity to ruxolitinib observed compared to our models. In our experiments, while ruxolitinib showed efficacy against PDX cells in vitro, it had limited impact on disease burden in vivo, likely due to suboptimal pharmacodynamics as reflected by inadequate inhibition of the STAT pathway. Nonetheless, consistent with both our in vitro findings and Drenberg’s study, we observed a clinical response in a patient with CBFA2T3::GLIS2 AML treated with ruxolitinib, although the disease eventually progressed. It is worth noting that the ruxolitinib dose administered to our patient was higher than the one used in GvHD treatment regimens [54] and aligns with the dose used in the ALL pediatric trial AALL1521 [35]. Pharmacokinetic studies, however, were not performed in this patient, and it is possible that the drug exposure achieved was insufficient to exert a sustained anti-leukemic effect given that we attempted to adjust for the concurrent use of fluconazole. The discrepancy between clinical and PDX responses warrants further consideration. One possibility is that the patient benefited from the combined effect of ruxolitinib and azacytidine, rather than ruxolitinib alone. Alternatively, pharmacokinetic and pharmacodynamic differences between humans and mice may limit effective target engagement in preclinical models, as suggested by the suboptimal JAK-STAT inhibition observed in our PDX study. Looking ahead, there is optimism for more effective therapies. Type II JAK inhibitors, now in clinical trials, offer improved selectivity and efficacy compared to first-generation agents. One such compound, AJ1-11095, is being evaluated in a Phase 1 trial (NCT06343805) for patients with MPN who have relapsed or are refractory to type I JAK2 inhibitors [55].
Given the complexity of drug resistance, gaining a deeper insight into the underlying mechanisms is crucial for developing more effective combination therapies. One known mechanism of resistance to JAK2 inhibitors is the compensatory activation of alternative signaling pathways. In our study, MAPK pathway activation emerged as a resistance mechanism to ruxolitinib, identified through both CRISPR-Cas9 screening and the development of resistant sublines. This mechanism is reminiscent of resistance patterns described in MPN, where MAPK activation via AXL and PDGFR can bypass JAK inhibition [56, 57]. Moreover, JAK2 inhibition with ruxolitinib has been shown to promote clonal expansion of RAS mutants in MPN [58]. Mutations in NRAS and KRAS—frequently observed in pediatric AML and ALL—occur in ~6% of CBFA2T3::GLIS2 AMKL [8, 9]. These mutations drive hyperactivation of the RAS-MAPK pathway, promoting cell proliferation and survival. In certain AML subsets, RAS mutations are associated with a poor prognosis, particularly when co-occurring with other high-risk alterations [59]. They are also well-known mediators of resistance to various targeted therapies, including SYK and FLT3 inhibitors [60,61,62] by enabling leukemic cells to bypass pathway-specific blockade. Additionally, RAS mutations can diminish the efficacy of epigenetic therapies such as IDH inhibitors [63]. This broad resistance phenotype underscores the challenge of treating RAS-mutant leukemias and highlights the need for rational combination strategies or novel approaches capable of overcoming these resistance mechanisms.
Emerging therapies targeting aberrant signaling proteins offer hope for improving outcomes in pediatric leukemia. Recent advancements in RAS-targeted agents and downstream pathway inhibitors, such as MEK inhibitors, offer potential new approaches in treatment-resistant patients [64]. Optimism is also growing for next-generation therapies that more precisely target RAS mutations, including KRAS G12C inhibitors, such as sotorasib and adagrasib. These drugs have shown clinical benefit, including improved progression-free-survival, in non-small cell lung cancer [65, 66]. Additionally, belvarafenib has demonstrated activity against NRAS mutations in a Phase Ib trial involving patients with BRAFV600E- and NRAS-mutant melanoma [67].
Several studies have investigated the therapeutic potential of combining JAK and MEK inhibition. In MPN, this approach suppresses MEK/ERK activation in JAK2 V617F and MPL W515L mouse models, resulting in greater efficacy and reversal of fibrosis beyond what is achieved with JAK inhibition alone [57]. Similarly, dual JAK and MEK inhibition effectively controls disease progression in NRASG12D/G12D mouse models of juvenile myelomonocytic leukemia and a myeloproliferative variant of chronic myelomonocytic leukemia [68]. Given the complexity of RAS signaling and the frequent emergence of resistance to monotherapies, future strategies will likely rely on rational combinations. In CBFA2T3::GLIS2 AML, co-targeting JAK and MEK pathways may improve therapeutic efficacy and help prevent resistance. Expanding the panel of CBFA2T3::GLIS2 cell line and PDX models—including both NRAS-mutant and wild-type cases—will be critical for future studies aiming to assess drug response and resistance mechanisms to targeted therapies in this molecular subset of AML.
FOLR1-targeted therapies are currently under clinical evaluation for CBFA2T3::GLIS2 AML, as FOLR1 is frequently expressed on the cell surface in this AML subtype [69]. Preclinical and early clinical studies have shown encouraging activity of FOLR1-directed antibody-drug conjugates (ADCs), such as luveltamab tazevibulin (formerly STRO-002), as well as FOLR1-targeted CAR T-cell therapies [69,70,71]. While our study focuses on JAK/MEK co-inhibition as a strategy to overcome resistance to ruxolitinib, these immunotherapeutic approaches offer an antigen-specific mechanism of action that could be particularly effective in FOLR1-expressing leukemias. Future therapeutic strategies could combine extracellular targeting of FOLR1 with inhibition of intracellular signaling pathways such as JAK and MAPK. These combination strategies, however, will require further preclinical evaluation to assess their efficacy and safety. For example, ruxolitinib has well-established immunomodulatory effects, particularly in T-cell regulation and cytokine suppression. Recent preclinical studies have reported conflicting effects of ruxolitinib co-administration on CAR T-cells, with evidence of both impairment and enhancement of CAR T-cell efficacy depending on timing, context, and disease model [72, 73]. Similarly, given recent interest in drugs promoting apoptosis for this subset of AML, it will be of interest to evaluate drugs such as venetoclax or ABT-737 in combination with ruxolitinib in CBFA2T3::GLIS2 AML [74,75,76].
In summary, while CBFA2T3::GLIS2 AML remains a difficult-to-treat disease, our findings validated JAK2 as a selective dependency, offering a promising avenue for precision medicine. However, the rapid emergence of resistance underscores the limitation of monotherapy. Continued efforts to explore combination strategies with JAK inhibitors and other targeted approaches will be essential to improve outcomes for this aggressive disease.
Data availability
The CRISPR-Cas9 drop-out screen data for WSU-AML and the CRISPR-Cas9 drug modifier screen data for CMS and WSU-AML cell lines are publicly available for download at figshare.com upon manuscript publication.
References
Lonetti A, Pession A, Masetti R. Targeted therapies for pediatric AML: gaps and perspective. Front Pediatr. 2019;7:463.
Athale UH, Razzouk BI, Raimondi SC, Tong X, Behm FG, Head DR, et al. Biology and outcome of childhood acute megakaryoblastic leukemia: a single institution’s experience. Blood. 2001;97:3727–32.
Barnard DR, Alonzo TA, Gerbing RB, Lange B, Woods WG. Children’s Oncology Group. Comparison of childhood myelodysplastic syndrome, AML FAB M6 or M7, CCG 2891: report from the Children’s Oncology Group. Pediatr Blood Cancer. 2007;49:17–22.
Gruber TA, Larson Gedman A, Zhang J, Koss CS, Marada S, Ta HQ, et al. An Inv(16)(p13.3q24.3)-encoded CBFA2T3-GLIS2 fusion protein defines an aggressive subtype of pediatric acute megakaryoblastic leukemia. Cancer Cell. 2012;22:683–97.
Thirant C, Ignacimouttou C, Lopez CK, Diop M, Le Mouël L, Thiollier C, et al. ETO2-GLIS2 hijacks transcriptional complexes to drive cellular identity and self-renewal in pediatric acute megakaryoblastic leukemia. Cancer Cell. 2017;31:452–65.
Bertuccio SN, Boudia F, Cambot M, Lopez CK, Lordier L, Donada A, et al. The pediatric acute leukemia fusion oncogene ETO2-GLIS2 increases self-renewal and alters differentiation in a human induced pluripotent stem cells-derived model. HemaSphere. 2019;4:e319.
Lopez CK, Noguera E, Stavropoulou V, Robert E, Aid Z, Ballerini P, et al. Ontogenic changes in hematopoietic hierarchy determine pediatric specificity and disease phenotype in fusion oncogene-driven myeloid leukemia. Cancer Discov. 2019;9:1736–53.
de Rooij JDE, Branstetter C, Ma J, Li Y, Walsh MP, Cheng J, et al. Pediatric non-down syndrome acute megakaryoblastic leukemia is characterized by distinct genomic subsets with varying outcomes. Nat Genet. 2017;49:451–6.
Masetti R, Bertuccio SN, Pession A, Locatelli F. CBFA2T3-GLIS2-positive acute myeloid leukaemia. A peculiar paediatric entity. Br J Haematol. 2019;184:337–47.
Smith JL, Ries RE, Hylkema T, Alonzo TA, Gerbing RB, Santaguida MT, et al. Comprehensive transcriptome profiling of cryptic CBFA2T3-GLIS2 fusion-positive AML defines novel therapeutic options – a COG and TARGET pediatric AML study. Clin Cancer Res J Am Assoc Cancer Res. 2020;26:726–37.
Ellegast JM, Alexe G, Hamze A, Lin S, Uckelmann HJ, Rauch PJ, et al. Unleashing cell-intrinsic inflammation as a strategy to kill AML blasts. Cancer Discov. 2022;12:1760–81.
Lin S, Larrue C, Scheidegger NK, Seong BKA, Dharia NV, Kuljanin M, et al. An in vivo CRISPR screening platform for prioritizing therapeutic targets in AML. Cancer Discov. 2021;candisc.1851.2020.
Wunderlich M, Chen J, Sexton C, Manning N, Byerly L, O’Brien E, et al. PDX models of relapsed pediatric AML preserve global gene expression patterns and reveal therapeutic targets. bioRxiv. 2022. https://www.biorxiv.org/content/10.1101/2022.01.31.478534v1.full.
Brinkman EK, Chen T, Amendola M, van Steensel B. Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res. 2014;42:e168.
Bhagwat N, Koppikar P, Keller M, Marubayashi S, Shank K, Rampal R, et al. Improved targeting of JAK2 leads to increased therapeutic efficacy in myeloproliferative neoplasms. Blood. 2014;123:2075–83.
Meyers RM, Bryan JG, McFarland JM, Weir BA, Sizemore AE, Xu H, et al. Computational correction of copy number effect improves specificity of CRISPR-Cas9 essentiality screens in cancer cells. Nat Genet. 2017;49:1779–84.
DepMap: The Cancer Dependency Map Project at Broad Institute. 2025. Available from: https://depmap.org/portal/.
Aubrey BJ, Kelly GL, Kueh AJ, Brennan MS, O’Connor L, Milla L, et al. An inducible lentiviral guide RNA platform enables the identification of tumor-essential genes and tumor-promoting mutations in vivo. Cell Rep. 2015;10:1422–32.
Hagel KR, Arafeh R, Gang S, Arnoff TE, Larson RC, Doench JG, et al. Systematic interrogation of tumor cell resistance to chimeric antigen receptor T-cell therapy in pancreatic cancer. Cancer Res. 2023;83:613–25.
Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzym Regul. 1984;22:27–55.
Chou TC. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharm Rev. 2006;58:621–81.
Chou TC. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010;70:440–6.
Durbin AD, Zimmerman MW, Dharia NV, Abraham BJ, Iniguez AB, Weichert-Leahey N, et al. Selective gene dependencies in MYCN-amplified neuroblastoma include the core transcriptional regulatory circuitry. Nat Genet. 2018;50:1240–6.
Chen L, Alexe G, Dharia NV, Ross L, Iniguez AB, Conway AS, et al. CRISPR-Cas9 screen reveals a MYCN-amplified neuroblastoma dependency on EZH2. J Clin Invest. 2018;128:446–62.
Dharia NV, Kugener G, Guenther LM, Malone CF, Durbin AD, Hong AL, et al. A first-generation pediatric cancer dependency map. Nat Genet. 2021;53:529–38.
Rampal R, Al-Shahrour F, Abdel-Wahab O, Patel JP, Brunel JP, Mermel CH, et al. Integrated genomic analysis illustrates the central role of JAK-STAT pathway activation in myeloproliferative neoplasm pathogenesis. Blood. 2014;123:e123–33.
Verstovsek S, Mesa RA, Gotlib J, Levy RS, Gupta V, DiPersio JF, et al. The clinical benefit of ruxolitinib across patient subgroups: analysis of a placebo-controlled, Phase III study in patients with myelofibrosis. Br J Haematol. 2013;161:508–16.
Verstovsek S, Ostojic, Vrhovac, Verstovsek S. Ruxolitinib for the treatment of myelofibrosis: its clinical potential. Ther Clin Risk Manag. 2012;95:95–103.
Przepiorka D, Luo L, Subramaniam S, Qiu J, Gudi R, Cunningham LC, et al. FDA approval summary: ruxolitinib for treatment of steroid-refractory acute graft-versus-host disease. Oncologist. 2020;25:e328–34.
Meyer SC, Keller MD, Chiu S, Koppikar P, Guryanova OA, Rapaport F, et al. CHZ868, a type II JAK2 inhibitor, reverses type I JAK inhibitor persistence and demonstrates efficacy in myeloproliferative neoplasms. Cancer Cell. 2015;28:15–28.
Wu SC, Li LS, Kopp N, Montero J, Chapuy B, Yoda A, et al. Activity of the type II JAK2 inhibitor CHZ868 in B cell acute lymphoblastic leukemia. Cancer Cell. 2015;28:29–41.
Tvorogov D, Thomas D, Liau NPD, Dottore M, Barry EF, Lathi M, et al. Accumulation of JAK activation loop phosphorylation is linked to type I JAK inhibitor withdrawal syndrome in myelofibrosis. Sci Adv. 2018;4:eaat3834.
Andraos R, Qian Z, Bonenfant D, Rubert J, Vangrevelinghe E, Scheufler C, et al. Modulation of activation-loop phosphorylation by JAK inhibitors is binding mode dependent. Cancer Discov. 2012;2:512–23.
Brodersen LE, Alonzo T, Menssen A, Gerbing R, Pardo L, Voigt A, et al. A recurrent immunophenotype at diagnosis independently identifies high-risk pediatric acute myeloid leukemia: a report from Children’s Oncology Group. Leukemia. 2016;30:2077–80.
Tasian SK, Hunter DS, Chen IML, Harvey RC, Carroll AJ, Wagner E, et al. A phase 2 study of ruxolitinib with chemotherapy in children with Philadelphia chromosome-like acute lymphoblastic leukemia (AALL1521/INCB18424-269): biologic characteristics and minimal residual disease response of patients with non-CRLF2-rearranged JAK pathway alterations. Blood. 2022;140:6117–8.
Doench JG, Fusi N, Sullender M, Hegde M, Vaimberg EW, Donovan KF, et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat Biotechnol. 2016;34:184–91.
Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelson T, et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science. 2014;343:84–7.
Hu X, Li J, Fu M, Zhao X, Wang W. The JAK/STAT signaling pathway: from bench to clinic. Signal Transduct Target Ther. 2021;6:402.
Seif F, Khoshmirsafa M, Aazami H, Mohsenzadegan M, Sedighi G, Bahar M. The role of JAK-STAT signaling pathway and its regulators in the fate of T helper cells. Cell Commun Signal CCS. 2017;15:23.
Hadjadj J, Castro CN, Tusseau M, Stolzenberg MC, Mazerolles F, Aladjidi N, et al. Early-onset autoimmunity associated with SOCS1 haploinsufficiency. Nat Commun. 2020;11:5341.
Liang YB, Tang H, Chen ZB, Zeng LJ, Wu JG, Yang W, et al. Downregulated SOCS1 expression activates the JAK1/STAT1 pathway and promotes polarization of macrophages into M1 type. Mol Med Rep. 2017;16:6405–11.
Tidyman WE, Rauen KA. Expansion of the RASopathies. Curr Genet Med Rep. 2016;4:57–64.
Arafeh R, Qutob N, Emmanuel R, Keren-Paz A, Madore J, Elkahloun A, et al. Recurrent inactivating RASA2 mutations in melanoma. Nat Genet. 2015;47:1408–10.
Chen PC, Yin J, Yu HW, Yuan T, Fernandez M, Yung CK, et al. Next-generation sequencing identifies rare variants associated with Noonan syndrome. Proc Natl Acad Sci USA. 2014;111:11473–8.
Drenberg CD, Shelat A, Dang J, Cotton A, Orwick SJ, Li M, et al. A high-throughput screen indicates gemcitabine and JAK inhibitors may be useful for treating pediatric AML. Nat Commun. 2019;10:2189.
Bolouri H, Farrar JE, Triche T, Ries RE, Lim EL, Alonzo TA, et al. The molecular landscape of pediatric acute myeloid leukemia reveals recurrent structural alterations and age-specific mutational interactions. Nat Med. 2018;24:103–12.
Roberts KG, Li Y, Payne-Turner D, Harvey RC, Yang YL, Pei D, et al. Targetable kinase-activating lesions in Ph-like acute lymphoblastic leukemia. N Engl J Med. 2014;371:1005–15.
Steeghs EMP, Jerchel IS, de Goffau-Nobel W, Hoogkamer AQ, Boer JM, Boeree A, et al. JAK2 aberrations in childhood B-cell precursor acute lymphoblastic leukemia. Oncotarget. 2017;8:89923–38.
Levine RL, Pardanani A, Tefferi A, Gilliland DG. Role of JAK2 in the pathogenesis and therapy of myeloproliferative disorders. Nat Rev Cancer. 2007;7:673–83.
Dunbar AJ, Bowman RL, Park YC, O’Connor K, Izzo F, Myers RM, et al. Jak2V617F reversible activation shows its essential requirement in myeloproliferative neoplasms. Cancer Discov. 2024;14:737–51.
Loh ML, Tasian SK, Rabin KR, Brown P, Magoon D, Reid JM, et al. A phase 1 dosing study of ruxolitinib in children with relapsed or refractory solid tumors, leukemias, or myeloproliferative neoplasms: a Children’s Oncology Group phase 1 consortium study (ADVL1011). Pediatr Blood Cancer. 2015;62:1717–24.
Appeldoorn TYJ, Munnink THO, Morsink LM, Hooge MNLde, Touw DJ. Pharmacokinetics and pharmacodynamics of ruxolitinib: a review. Clin Pharmacokinet. 2023;62:559–71.
Harrison CN, Schaap N, Vannucchi AM, Kiladjian JJ, Jourdan E, Silver RT, et al. Fedratinib improves myelofibrosis-related symptoms and health-related quality of life in patients with myelofibrosis previously treated with ruxolitinib: patient-reported outcomes from the phase II JAKARTA2 trial. HemaSphere. 2021;5:e562.
Locatelli F, Kang HJ, Bruno B, Gandemer V, Rialland F, Faraci M, et al. Ruxolitinib for pediatric patients with treatment-naïve and steroid-refractory acute graft-versus-host disease: the REACH4 study. Blood. 2024;144:2095–106.
Mascarenhas JO, Borate U, Bose P, Byrd JC, Garcia JS, Grunwald MR, et al. A multicenter, open-label, phase 1 clinical trial of AJ1-11095 administered as oral monotherapy in patients with primary myelofibrosis (PMF), post-polycythemia vera myelofibrosis (PPV-MF), or post-essential thrombocythemia myelofibrosis (PET-MF) who have been failed by a type I JAK2 inhibitor (JAK2i). Blood. 2024;144:3147.1.
Codilupi T, Szybinski J, Arunasalam S, Jungius S, Dunbar AC, Stivala S, et al. Development of resistance to type II JAK2 inhibitors in MPN depends on AXL kinase and is targetable. Clin Cancer Res J Am Assoc Cancer Res. 2024;30:586–99.
Stivala S, Codilupi T, Brkic S, Baerenwaldt A, Ghosh N, Hao-Shen H, et al. Targeting compensatory MEK/ERK activation increases JAK inhibitor efficacy in myeloproliferative neoplasms. J Clin Invest. 2019;129:1596–611.
Maslah N, Kaci N, Roux B, Alexe G, Marie R, Pasquer H, et al. JAK2 inhibition mediates clonal selection of RAS pathway mutations in myeloproliferative neoplasms. Nat Commun. 2025;16:6270.
Rivera D, Kim K, Kanagal-Shamanna R, Borthakur G, Montalban-Bravo G, Daver N, et al. Implications of RAS mutational status in subsets of patients with newly diagnosed acute myeloid leukemia across therapy subtypes. Am J Hematol. 2022;97:1599–606.
Cremer A, Ellegast JM, Alexe G, Frank ES, Ross L, Chu SH, et al. Resistance mechanisms to SYK inhibition in acute myeloid leukemia. Cancer Discov. 2020;10:214–31.
Peretz CAC, McGary LHF, Kumar T, Jackson H, Jacob J, Durruthy-Durruthy R, et al. Single-cell DNA sequencing reveals complex mechanisms of resistance to quizartinib. Blood Adv. 2021;5:1437.
Alotaibi AS, Yilmaz M, Kanagal-Shamanna R, Loghavi S, Kadia TM, DiNardo CD, et al. Patterns of resistance differ in patients with acute myeloid leukemia treated with type I versus type II FLT3 inhibitors. Blood Cancer Discov. 2021;2:125–34.
Desikan SP, Daver N, DiNardo C, Kadia T, Konopleva M, Ravandi F. Resistance to targeted therapies: delving into FLT3 and IDH. Blood Cancer J. 2022;12:91.
Burgess MR, Hwang E, Firestone AJ, Huang T, Xu J, Zuber J, et al. Preclinical efficacy of MEK inhibition in Nras-mutant AML. Blood. 2014;124:3947–55.
de Langen AJ, Johnson ML, Mazieres J, Dingemans AMC, Mountzios G, Pless M, et al. Sotorasib versus docetaxel for previously treated non-small-cell lung cancer with KRASG12C mutation: a randomised, open-label, phase 3 trial. Lancet. 2023;401:733–46.
Dy GK, Govindan R, Velcheti V, Falchook GS, Italiano A, Wolf J, et al. Long-term outcomes and molecular correlates of sotorasib efficacy in patients with pretreated KRAS G12C-mutated non–small-cell lung cancer: 2-year analysis of CodeBreaK 100. J Clin Oncol. 2023;41:3311–7.
Shin SJ, Lee J, Kim TM, Kim JS, Kim YJ, Hong YS, et al. A phase Ib trial of belvarafenib in combination with cobimetinib in patients with advanced solid tumors: Interim results of dose-escalation and patients with NRAS-mutant melanoma of dose-expansion. J Clin Oncol. 2021;39:3007–3007.
Kong G, Wunderlich M, Yang D, Ranheim EA, Young KH, Wang J, et al. Combined MEK and JAK inhibition abrogates murine myeloproliferative neoplasm. J Clin Invest. 2014;124:2762–73.
Tang T, Le Q, Castro S, Pardo L, McKay CN, Perkins L, et al. Targeting FOLR1 in high-risk CBF2AT3-GLIS2 pediatric AML with STRO-002 FOLR1–antibody-drug conjugate. Blood Adv. 2022;6:5933–7.
Le Q, Hadland B, Smith JL, Leonti A, Huang BJ, Ries R, et al. CBFA2T3-GLIS2 model of pediatric acute megakaryoblastic leukemia identifies FOLR1 as a CAR T cell target. J Clin Invest. 2022;132:e157101.
Le Q, Hadland B, Smith JL, Leonti A, Huang BJ, Ries R, et al. CBFA2T3-GLIS2 model of pediatric acute megakaryoblastic leukemia identifies FOLR1 as a CAR T cell target. J Clin Invest. 2024;134:e157101.
Xu N, Yang XF, Xue SL, Tan JW, Li MH, Ye J, et al. Ruxolitinib reduces severe CRS response by suspending CAR-T cell function instead of damaging CAR-T cells. Biochem Biophys Res Commun. 2022;595:54–61.
Mitsuno K, Suematsu M, Naito Y, Mayumi A, Yoshida H, Osone S, et al. Selective JAK2 pathway inhibition enhances anti-leukemic functionality in CD19 CAR-T cells. Cancer Immunol Immunother CII. 2025;74:79.
Niswander LM, Chung P, Diorio C, Tasian SK. Clinical responses in pediatric patients with relapsed/refractory leukemia treated with azacitidine and venetoclax. Haematologica. 2023;108:3142–7.
Gress V, Roussy M, Boulianne L, Bilodeau M, Cardin S, El-Hachem N, et al. CBFA2T3::GLIS2 pediatric acute megakaryoblastic leukemia is sensitive to BCL-XL inhibition by navitoclax and DT2216. Blood Adv. 2024;8:112–29.
Aid Z, Robert E, Lopez CK, Bourgoin M, Boudia F, Le Mene M, et al. High caspase 3 and vulnerability to dual BCL2 family inhibition define ETO2::GLIS2 pediatric leukemia. Leukemia. 2023;37:571–9.
Author information
Authors and Affiliations
Contributions
FG, YP, and KS wrote the manuscript. FG, CS and KS designed the experiments. FG, CS, MA, LM, AT and MG carried out experiments. DK, AB, LM, SS and AT executed animal model studies and performed tissue processing. FG, CS and GA analyzed the data and prepared the figures for the article. GA performed all bioinformatic analysis. MW and JAP contributed to data collection and provided patient samples. BD, YP and KS followed up the patients and collected clinical information and provided clinical relevance of findings. SL, JME, JAP, BD, YP, MA and KS collaborated in data analysis and supervised the manuscript. All authors contributed to manuscript revisions and approved the final version.
Corresponding authors
Ethics declarations
Competing interests
FG was supported with funding from the French National Cancer Institute and the ARC Foundation for Cancer Research. CS was supported by a Helen Gurley Brown Fellowship. CS and KS were supported with funding from The Selig Family Fund for Pediatric Cancer Research. JME received funding from the Rally Foundation for Childhood Cancer Research and the American Society of Hematology. SL was a Fellow of Blood Cancer United, formerly The Leukemia & Lymphoma Society and was supported by NCI R00 CA263161. MW was supported by NCI R50 CA211404. YP received support from CURE Childhood Cancer Foundation, Rally Foundation for Childhood Cancer Research, St. Baldrick’s Foundation and Hyundai Hope on Wheels. KS was supported by Blood Cancer United, formerly The Leukemia & Lymphoma Society, Rally Foundation for Childhood Cancer Research, Children’s Leukemia Research Association (CLRA), and a National Cancer Institute R35 CA283977. KS received grant funding from the DFCI/Novartis Drug Discovery Program and is a member of the SAB and has stock options with Auron Therapeutics on topics unrelated to this work. JME reports spousal employment at Novartis Pharma AG.
Ethics approval and consent to participate
All methods were performed in accordance with the relevant guidelines and regulations, including the Declaration of Helsinki. All animal experiments were approved by the DFCI Institutional Animal Care and Use Committee and conducted in accordance with institutional guidelines. Informed consent was obtained from all participants or their legal guardians prior to sample collection.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Gonzales, F., Schneider, C., Alexe, G. et al. Identifying targeted therapies for CBFA2T3::GLIS2 acute myeloid leukemia. Leukemia (2026). https://doi.org/10.1038/s41375-025-02845-7
Received:
Revised:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41375-025-02845-7
