
Abstract
TP53 mutations are implicated in the progression of mucinous borderline tumors (MBOT) to mucinous ovarian carcinomas (MOC). Optimized immunohistochemistry (IHC) for TP53 has been established as a proxy for the TP53 mutation status in other ovarian tumor types. We aimed to confirm the ability of TP53 IHC to predict TP53 mutation status in ovarian mucinous tumors and to evaluate the association of TP53 mutation status with survival among patients with MBOT and MOC. Tumor tissue from an initial cohort of 113 women with MBOT/MOC was stained with optimized IHC for TP53 using tissue microarrays (75.2%) or full sections (24.8%) and interpreted using established criteria as normal or abnormal (overexpression, complete absence, or cytoplasmic). Cases were considered concordant if abnormal IHC staining predicted deleterious TP53 mutations. Discordant tissue microarray cases were re-evaluated on full sections and interpretational criteria were refined. The initial cohort was expanded to a total of 165 MBOT and 424 MOC for the examination of the association of survival with TP53 mutation status, assessed either by TP53 IHC and/or sequencing. Initially, 82/113 (72.6%) cases were concordant using the established criteria. Refined criteria for overexpression to account for intratumoral heterogeneity and terminal differentiation improved concordance to 93.8% (106/113). In the expanded cohort, 19.4% (32/165) of MBOT showed evidence for TP53 mutation and this was associated with a higher risk of recurrence, disease-specific death, and all-cause mortality (overall survival: HR = 4.6, 95% CI 1.5–14.3, p = 0.0087). Within MOC, 61.1% (259/424) harbored a TP53 mutation, but this was not associated with survival (overall survival, p = 0.77). TP53 IHC is an accurate proxy for TP53 mutation status with refined interpretation criteria accounting for intratumoral heterogeneity and terminal differentiation in ovarian mucinous tumors. TP53 mutation status is an important biomarker to identify MBOT with a higher risk of mortality.
Similar content being viewed by others
Introduction
Ovarian mucinous tumors are uncommon neoplasms with an elusive cell of origin [1]. They comprise a spectrum of entities, including mucinous cystadenoma/adenofibroma, mucinous borderline tumor (MBOT), and mucinous ovarian carcinoma (MOC). All these entities are often seen in a single ovarian mucinous tumor giving rise to morphological heterogeneity, and supporting a multi-step progression model to MOC where genomic loss of CDKN2A and KRAS mutations are initiating events [2, 3]. We have recently shown that TP53 mutations and copy number aberrations are key drivers of progression from borderline precursors to MOC, given these genetic alterations are significantly enriched in carcinomas compared with benign precursors [4, 5]. An early study reported TP53 overexpression (OE) in MBOT with microinvasion, microcarcinoma, and coexisting carcinoma [6]. Anecdotal cases where a TP53 mutated MBOT widely metastasized within a few years of diagnosis have been reported [7].
While TP53 sequencing is not routinely clinically accessible, we have recently established that optimized TP53 immunohistochemistry (IHC) is a good proxy for TP53 mutation detection by sequencing in (tubo-) ovarian and endometrial carcinomas with ~95% accuracy [8, 9]. TP53 IHC, as an alternative to tumor sequencing, can be applied in a cost-effective and high-throughput manner for diagnostic purposes and in prognostic studies using tissue microarrays (TMA). However, we find that there are context dependent differences across tumor types: 5% of endometrial carcinomas show subclonal TP53 patterns, which are defined by a combination of normal and abnormal TP53 IHC [9]. The interpretation of TP53 IHC had to be adapted for TP53 mutation detection in squamous cell carcinomas because of the phenomenon of terminal differentiation, in which terminally differentiated cells no longer express TP53, regardless of mutation status [10, 11].
The primary objective of this study was to assess the correlation between TP53 IHC and TP53 mutation screening by sequencing in MOC and MBOT using established and refined criteria for TP53 IHC interpretation in ovarian mucinous tumors. A second aim was to evaluate the association of TP53 status and survival among patients with MOC and MBOT.
Materials and methods
Cohort
Our analysis included tissue from 647 ovarian mucinous tumors assembled from two cohorts of patients. 295 specimens were obtained from the previously described Genomic Analysis of Mucinous Tumors (GAMuT) cohort [5]. The GAMuT cohort consists of cases diagnosed as ovarian mucinous tumors from Australia (Australian Ovarian Cancer Study (AOCS), Royal Women’s Hospital, Queensland Institute for Medical Research – Berghofer, The Hudson Institute of Medical Research, Garvan Institute of Medical Research and Westmead Hospital GynBiobank), the United States (Mayo Clinic, Rochester), Canada (Canadian Ovarian Experimental Unified Resource (COEUR) and OVCARE), and the United Kingdom (South England and Edinburgh). Sequencing data were available for 234 GAMuT cases and a subset (N = 113) had corresponding TMA and/or full sections available, which were used for the initial TP53 IHC – TP53 mutation concordance analysis. Sixty-one GAMuT cases had TP53 IHC without mutation data. An additional 352 specimens were sourced from the Ovarian Tumor Tissue Analysis (OTTA) Consortium, which have been recently subjected to a biomarker-integrated review to exclude lower gastrointestinal tract metastases [12]. Local pathology review with or without use of IHC was performed. These cases were represented on TMA by a median of two tissue cores and therefore were available for TP53 IHC but not DNA sequencing. The overall case flow is summarized in Supplementary Fig. S1. All study sites received ethics board approval for tumor profiling (Supplementary Table S1).
TP53 mutation sequencing
TP53 mutation sequence data were obtained from previously performed analyses [3, 5] that included whole genome sequencing (N = 5), whole exome sequencing (N = 57), targeted mutation sequencing (N = 152), and Sanger sequencing for exons 4–9 (N = 20). A subset (N = 41) was additionally validated by Sanger sequencing. Allele frequencies were determined to take into account estimated cellularity and loss of heterozygosity, and mutations were categorized by type (missense, inframe indel, truncating (stop-gain and frameshift), and splicing). Deleterious missense mutations were classified using public databases [13, 14].
Immunohistochemistry
TP53 IHC was performed on formalin-fixed and paraffin-embedded (FFPE) tissue sections of 4 µm thickness. The majority of staining was performed at the Department of Pathology and Laboratory Medicine, University of Calgary, Canada, including 80/113 (70.8%) cases from the initial GAMuT cohort, all quality control full sections, and all TMAs from the OTTA cohort. Cases were stained using a previously published protocol [9]. After 30 min of heat-induced pre-treatment using the high pH retrieval buffer, the DAKO Omnis protocol H30-10M-30 with the ready-to-use clone DO-7 (catalog # GA61661-2; DAKO) was utilized. From the remaining initial cohort, 24/113 cases were stained using full sections at the Department of Pathology, Peter MacCallum Cancer Centre, Australia, and 5 cases in TMAs were stained at the originating center, also using clone DO-7. For 4 cases, tissue was not available for analysis and therefore, the TP53 IHC status was retrieved from pathology reports issued for these cases.
Interpretation was initially performed according to established criteria for ovarian and endometrial carcinomas [15]. IHC and sequencing results were independently interpreted with evaluators of either component blinded to the other result. Abnormal (also variably referred to as mutation-type or aberrant) TP53 staining showed one of 3 patterns: OE with virtually all viable tumor cell nuclei showing strong nuclear staining; complete absence (CA; also referred to as “null pattern”) with no nuclear staining of tumor cells but with normal control staining in nontumor stromal or immune cells providing an internal control; and cytoplasmic (CY) with unequivocal CY staining and variable nuclear staining. By contrast, tumors with normal (also referred to as wild-type pattern) staining showed nuclear expression of variable intensity and cellular distribution. If no staining was seen in any cells (CA in tumor cells and no internal control), the sample was excluded from the study.
Based on the observation of intratumoral heterogeneity (subclonal abnormal patterns) and terminal differentiation in ovarian mucinous tumors, the criteria were refined. In cases demonstrating OE, the percentage area of tumor cells exhibiting contiguous strong nuclear staining was estimated to the nearest 5%, and the average percentage of OE across all scorable TMA cores from each case was calculated. Following review of discrepant cases regarding the TP53 status by established IHC criteria, we modified the threshold for OE: strong nuclear staining in contiguous areas of at least 5% of the tumor qualified as abnormal OE. Criteria for scoring other staining patterns (normal, CA, CY) remained unchanged. Two observers (MK, EK) independently scored TP53 IHC on 144 cases from the OTTA cohort using the refined criteria and agreement was assessed to evaluate interobserver reproducibility. A consensus was reached for discordant cases. During this we noted that the minimal threshold of 5% translated into ≥12 consecutive cells with strong nuclear staining. In the fallopian tube and the endometrium, a TP53 signature requiring at least 12 consecutive cells with abnormal-pattern TP53 staining has been shown to accurately predict TP53 mutations [16]. Hence, ≥12 consecutive cells with strong nuclear staining was applied as an alternative threshold for minimal abnormal OE.
Statistical analysis
All statistical analyses were performed using R (v3.3.0). Fisher’s exact test was used for categorical data (stats::fisher.test) and the Wilcoxon rank sum test was used to compare two groups of continuous data (stats::wilcox.test). Interobserver concordance was tested using Cohen’s kappa for two raters with equal weights. Spearman correlation was used to compare concordance of two continuous variables (stats::cor.test, method = spearman).
The primary endpoint for survival analysis was all cause mortality (overall survival). The secondary endpoint was progression free survival, defined by a clinical diagnosis of recurrence. Disease-specific survival was also considered when the cause of death was known. Overall survival was right censored at 10 years. The OTTA cohort was left-truncated to account for recruitment of prevalent cases. Survival analysis was performed using Cox proportional hazards regression model (survival::coxph). Included in the multivariate models were age (continuous variable) for MOC and MBOT, and for MOC also stage (stage I-II compared with stage III-IV) and grade (grades 1, 2, and 3 treated as categorical variables). In the analysis of the combined cohorts, cohort (OTTA and GAMuT) was included as a stratifying variable. Kaplan–Meier plots were used to visualize survival data using survminer::ggsurvplot. The assumptions of the Cox proportional hazards test were tested using survival::cox.zph on the multivariate models and visualized using ggcoxzph. No serious violations of the Cox proportional hazards assumptions were observed for TP53.
Results
Concordance of TP53 IHC with TP53 mutation status
In Part I of this study, we evaluated 113 cases (101 MOC; 12 MBOT) from the GAMuT cohort with both TP53 sequencing data and tissue stained for TP53 IHC. Using the established criteria from ovarian and endometrial carcinomas, abnormal vs normal TP53 IHC (Fig. 1a–d) showed concordance with TP53 mutation status for 82/113 (72.6%) cases (75.0% in MBOT and 72.5% in MOC). Twenty-eight out of 31 discordant cases were considered to be false negatives as they were scored as having normal TP53 IHC but mutations detected by sequencing. Only three cases were regarded as false positives with abnormal TP53 IHC but no mutation detected. The estimated allele frequencies of TP53 mutations were not statistically significantly different between concordant and discordant cases (median 0.57 and 0.48, respectively; Wilcoxon rank sum test; p = 0.305), suggesting that discordance was not commonly an effect of low tumor cellularity or clonal heterogeneity in TP53 mutation status.

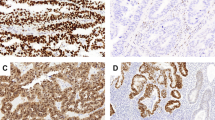
a Normal pattern with heterogeneous nuclear staining in variable distribution. b Abnormal overexpression (OE) with virtually all viable tumor cell nuclei showing strong nuclear staining. c Abnormal complete absence with no nuclear staining of tumor cells but normal control staining in nontumor stromal or immune cells. d Abnormal cytoplasmic staining with unequivocal cytoplasmic staining and variable nuclear staining. e Mucinous borderline tumor with intratumoral heterogeneity or subclonal TP53 abnormal pattern. Inset: both normal (upper) and abnormal OE patterns (lower) are seen within the same tumor. Abnormal OE is seen in an area of mucinous intraepithelial carcinoma. In addition, the abnormal OE area displays terminal differentiation with the basal layer of cells demonstrating abnormal OE pattern staining with sparing of superficial areas.
We then performed quality control analyses on discordant cases. Discordant cases on TMA were re-stained using full sections. When reviewing full sections, we observed a distinct pattern of OE staining in some cases, where the basal layer of cells demonstrated abnormal OE pattern staining with sparing of superficial areas (Fig. 1e). A similar phenomenon has previously been described in squamous cell carcinomas and is referred to as “terminal differentiation” [10]. Notably, terminal differentiation was not observed in cases showing the other two abnormal patterns: CA and CY. In addition, we observed intratumoral heterogeneity, where TP53 normal coexisted with abnormal TP53 IHC; this has previously been referred to as a “subclonal” pattern [15]. Therefore, we repeated the interpretation with refined criteria to account for terminal differentiation and intratumoral heterogeneity. In cases with a missense mutation, the percentage of continuous tumor cell nuclei showing OE ranged from 5 to 100% (median 50%) (Fig. 2a–d). We classified cases with a minimum of OE in 5% of tumor cell nuclei as abnormal TP53 IHC (Fig. 2a). With the refined criteria, concordance improved to 91.7% in MBOT and 95.1% in MOC (overall 106/113 cases, 93.8%). Improvement in concordance was observed in cases with missense and splicing mutations, while concordance for non-mutated tumors and cases with truncating mutations remained the same since criteria for IHC patterns associated with truncating mutations were unchanged (Fig. 2e). Using refined criteria, 77/113 (68.1%) cases demonstrated abnormal staining, while sequencing data showed TP53 mutations in 76/113 cases (67.0%, Table 1).

Ovarian mucinous tumors with variable percentages of TP53 overexpression: a 5% overexpression or >12 consecutive strongly staining cells. b 50% overexpression. c 70% overexpression. d 100% overexpression. e concordance between mutation status by sequencing and immunohistochemistry improved with application of refined (minimum 5% abnormal OE pattern) instead of established criteria, with improvements observed in cases with splicing and missense mutations. f The percentage of tumor cells overexpressing TP53 in mucinous borderline tumors (MBOT) and carcinomas (MOC) (median 35% and N = 11 in MBOT; median 60% and N = 69 in MOC; p = 0.040). g TP53 mutation allele frequencies in MBOT and MOC (median 0.44 and 0.55, respectively; p = 0.38).
MBOT had a significantly lower median percentage of OE cells than MOC (35%, N = 11 vs 60%, N = 69, respectively; Wilcoxon rank sum test; p = 0.040) (Fig. 2f). However, the allele frequency of MBOT TP53 mutations was not significantly different from MOC (median 0.44 and 0.55, respectively; Wilcoxon rank sum test; p = 0.38) (Fig. 2g). Notably, there was no correlation between percentage of OE and allelic frequency (r = 0.137; Spearman correlation; p = 0.318, Supplementary Fig. S2). This result suggests that MBOT cases with similar allelic frequencies of mutated cells but a lower OE distribution may be particularly prone to terminal differentiation. Indeed, in one case where we extracted DNA from both borderline and invasive components, the allele frequency of the mutated allele was similar (0.67 MBOT vs 0.46 MOC), whereas the percentage of overexpressing cells was higher in the invasive component (40% vs 100%).
Following quality control and application of the refined threshold for abnormal-type OE, the number of discordant cases decreased from 31 to 7 (Table 2). All had their mutation status confirmed through repeat sequencing of TP53 using Sanger Sequencing, and IHC interpretation was re-reviewed and initial TMA IHC scores were confirmed on full tissue sections.
Robustness of the IHC method
Interobserver reproducibility for IHC scoring with the refined criteria by a second independent observer showed substantial agreement in an independent cohort from OTTA (k = 0.80; N = 144; Cohen’s kappa for two raters with equal weights). Review of discordant cases showed that disagreement most commonly occurred for cases with focal OE vs normal pattern (Supplementary Fig. S3). We felt that the TP53 signature criteria of ≥12 consecutive cells with an OE TP53 staining pattern would be more easily applied compared with a 5% threshold and therefore utilized this as the minimal threshold for OE in the expanded cohort.
Association of TP53 status with survival in MBOT and MOC
Given the excellent concordance rates between TP53 IHC and TP53 mutation status, we considered IHC and sequencing equivalent and explored the association of TP53 status with survival using all available cases from the GAMuT and OTTA cohorts with IHC and/or sequencing data. The frequency of patterns of TP53 IHC observed in MBOT and MOC from both cohorts is summarized in Supplementary Table 2. The distribution of the percentages of abnormal OE in MBOT and MOC from both cohorts is shown in Supplementary Fig. S4. There were 5 MBOT cases coded as stage III. Review of the pathology report of one case revealed that stage III was assigned clinically based on adhesion to bowel. However, there are no well-documented cases of MBOT at stage III and we were not able to reconcile the discrepancy for the other four cases, therefore, the 5 “stage III” MBOT cases were excluded from the survival analysis. There were 32/165 (19.4%) TP53 abnormal MBOT cases. The age was not statistically different between TP53 abnormal (mean 51.9 years) and TP53 normal (mean 48.6 years) MBOT cases. TP53 abnormal MBOT had a worse progression-free (HR = 4.8; 95%CI 1.4–17; likelihood ratio test; p = 0.013), disease-specific (HR = 4.5; 95% CI 1.1–8; p = 0.033), and overall survival (HR = 4.5; 95% CI 1.5–14; p = 0.0087). (Fig. 3a, c, e). Five-year progression-free survival for TP53 abnormal MBOT cases was 79.7%, and 95.9% for TP53 normal cases. Five-year overall survival for TP53 abnormal MBOT was 77.9%, and 96.5% for TP53 normal cases. TP53 status was still a significant factor when age was taken into account in a multivariable analysis of progression-free (HR = 5.7; 95% CI 1.6–20.4; p = 0.007) and overall survival (HR = 4.6; 95%CI 1.5–14.3; p = 0.009). We did not have sufficient power to establish the prognostic significance of TP53 separately in either the GAMuT or OTTA cohort due to low numbers of events, although similar trends were observed (Supplementary Fig. S5). No significant differences were observed between the HRs of the two cohorts.

Progression-free (PFS; a), overall (OS; c), and disease-specific (DSS; e) survival in patients with MBOT. Progression-free (b), overall (d), and disease-specific (f) survival in patients with MOC.
Within MOC, 61.1% (259/424) harbored a TP53 mutation. Univariable associations of TP53 status with clinicopathological parameters are shown in Table 3. TP53 mutation was previously found not to be significantly associated with MOC survival [5], and this remained the case in the combined GAMuT and OTTA cohorts (Fig. 3b, d, f). However, significant differences were observed in the HRs between the cohorts, suggesting heterogeneity in the outcomes for MOC between the studies. In the GAMuT cohort TP53 abnormal cases showed worse progression-free survival (HR = 2.99; 95% CI 1.3–6.8; p = 0.008), overall survival (HR = 2.5; 95% CI 1.2–5.4; p = 0.02) and disease-specific survival (HR = 3.0; 95% CI 1.3–7.2; p = 0.01) compared with TP53 normal. However, in the OTTA cohort, TP53 was not significantly associated with any outcome (Supplementary Fig. S5).
Discussion
This study is the first to validate the use of TP53 IHC as a surrogate marker of TP53 mutation status in ovarian mucinous tumors. A high level of accuracy can be reached if the interpretation criteria are refined to account for terminal differentiation and intratumoral heterogeneity. The accuracy of 93.8% is close to accuracies of 95–97% previously reported in ovarian high-grade serous and endometrioid carcinomas as well as endometrial carcinomas [8, 9]. With IHC and sequencing being equivalent assays, we then provide strong evidence against an association of TP53 mutation status with survival in MOC [5]. However, we show that TP53 mutation is a prognostic risk factor in MBOT, which could inform clinical management.
In contrast to other ovarian carcinoma histotypes, ovarian mucinous tumors show terminal differentiation and a high degree of intratumoral heterogeneity. The phenomenon of terminal differentiation has been well-documented in squamous cell carcinomas [10]. In many squamous cell carcinomas, abnormal TP53 expression is seen in the actively proliferating “basal” and sometimes also in “parabasal” zones. It is absent in the more differentiated or “superficial” zones most likely due to downregulation of transcription, given that RNA in situ hybridization has failed to detect TP53 mRNA in these zones [10]. Although similar studies have not been done in ovarian mucinous tumors, abrupt loss of protein expression in the luminal aspects of papillary proliferations may suggest an analogous mechanism. Therefore, luminal aspects should be disregarded when interpreting TP53 IHC in ovarian mucinous tumors. Further exploration of terminal differentiation in tumors arising in the ovary as well as mucinous tumors from other organ systems, such as the gastrointestinal tract, may be useful to delineate the underlying mechanism and understand the possible significance of this phenomenon.
A high degree of intratumoral heterogeneity poses another challenge in the interpretation of TP53 IHC in ovarian mucinous tumors. These tumors are large (mean size 20 cm) and often show an admixture of benign and malignant components. This can result in subclonal TP53 expression with different areas showing normal or abnormal patterns. We found a low threshold of 5% consecutively strongly staining cells was predictive of a TP53 mutation but noticed that the 5% rule led to minor differences in interobserver reproducibility. On review, we concluded that this translates into at least 12 consecutive strongly staining tumor cells, similar to the TP53 signature described in the fallopian tube [16]. With such a low threshold, it is important not to overinterpret small foci of high but normal staining pattern, which still show variable staining intensity, as abnormal. In contrast, TP53 mutations are ubiquitously found and hence considered a required early event in tubo-ovarian high-grade serous carcinoma [17]. Therefore, these tumors frequently exhibit abnormal TP53 IHC patterns and TP53 mutations uniformly, improving the sensitivity of assays utilizing limited tissue samples.
After quality control, a few cases with discrepant TP53 status based on IHC and sequencing remained. Possible explanations for “false negative” cases include (1) false negative IHC in cases with likely pathogenic TP53 mutations, (2) late truncating mutations expressing nonfunctional proteins, and (3) variants of uncertain significance, in which IHC may aid in classification [14]. The 4 “false positive” cases may represent false positive IHC, but a false negative sequencing result due to large deletions at least in one case with complete absence cannot be ruled out. Furthermore, low frequency variants may not have been detected on sequencing thus do not match the TP53 abnormal IHC pattern identified on representative tissue sections. This explanation is plausible since the most representative FFPE block was analyzed by IHC, whereas for the three exome sequenced cases, the frozen tissue fragment used for DNA extraction was opportunistically sampled and may contain nontumor or benign cells in addition to tumor cells, reducing the sensitivity of mutation detection. The lower coverage of the exome analysis (~60×) compared with targeted sequencing could make detection of low-frequency variants more challenging. In addition, we cannot entirely exclude sample mix-up prior to DNA extraction as newly extracted DNA samples were not available due to limited material. Finally, TP53 expression is not solely dependent on the mutation status of TP53, but also relies on other factors such as MDM2 and chaperone activities, gene copy number, and mRNA levels [10], which may have been altered in discordant cases.
Identification of TP53-mutated MBOT through the use of TP53 IHC can highlight cases with increased risk of progression to carcinoma or concurrent unsampled invasive carcinoma elsewhere in the tumor mass. The latter finding has been previously made in an early study showing that TP53 OE was associated with microinvasion and invasion [6]. MBOT often requires extensive tumor sampling to search for invasion. For our study, it was not feasible to track sampling protocols for every case given the multi-institutional nature, and that case accrual spanned over several decades. Following the 2003 Bethesda Borderline Ovarian Tumor Workshop, a standard sampling protocol of two sections for each cm of the tumor’s largest dimension was implemented in centers contributing to this study [18]. However, an unknown proportion of MBOT cases accrued before or around 2003 might not have been sampled according to this standard. Therefore, it is possible that some TP53 abnormal MBOT cases may have had unsampled invasive foci elsewhere. While abnormal TP53 could potentially flag these cases, normal TP53 status, on the other hand, could be used to de-escalate treatment in certain clinical scenarios such as ruptured MBOT. This leads to an important question: which tumors should have TP53 IHC testing? From our undocumented observations, we believe abnormal TP53 IHC occurs in areas with high-grade nuclear atypia that raises a morphological suspicion of intraepithelial carcinoma. Yet the abnormal TP53 staining pattern often extends beyond the area thought to be intraepithelial carcinoma based on morphology. Future studies are required to determine the relationship of TP53 mutation and intraepithelial carcinoma in MBOT since the current consensus is that MBOT with intraepithelial carcinoma still has a favorable outcome with >95% survival [19]. A threshold for ordering TP53 IHC in ovarian mucinous tumors should be established.
We confirm that TP53 mutations are implicated in later progression of MBOT to MOC because TP53 mutations increase in frequency from MBOT (19.4%) vs MOC (61.2%) [5]. TP53 is not associated with prognosis in MOC, at least when analyzed without the context of other key oncogenic events such as KRAS, CDKN2A, and ERBB2.
In conclusion, TP53 IHC is an accurate proxy for TP53 mutation status with refined interpretation criteria accounting for terminal differentiation in MBOT and MOC with good interobserver reproducibility. TP53 mutation status inferred by IHC may be a useful biomarker to identify MBOT with a higher risk of mortality, suggesting a closer follow-up of these patients.
References
Kelemen LE, Köbel M. Mucinous carcinomas of the ovary and colorectum: different organ, same dilemma. The Lancet Oncology. 2011;12:1071–80.
Hunter SM, Gorringe KL, Christie M, Rowley SM, Bowtell DD, Campbell IG. Pre-invasive ovarian mucinous tumors are characterized by CDKN2A and RAS pathway aberrations. Clin Cancer Res. 2012;18:5267–77.
Ryland GL, Hunter SM, Doyle MA, Caramia F, Li J, Rowley SM, et al. Mutational landscape of mucinous ovarian carcinoma and its neoplastic precursors. Genome Med. 2015;7:87.
Mackenzie R, Kommoss S, Winterhoff BJ, Kipp BR, Garcia JJ, Voss J, et al. Targeted deep sequencing of mucinous ovarian tumors reveals multiple overlapping RAS-pathway activating mutations in borderline and cancerous neoplasms. BMC Cancer. 2015;15:415.
Cheasley D, Wakefield MJ, Ryland GL, Allan PE, Alsop K, Amarasinghe KC, et al. The molecular origin and taxonomy of mucinous ovarian carcinoma. Nat Commun. 2019;10:3935.
Kupryjanczyk J, Bell DA, Yandell DW, Scully RE, Thor AD. p53 expression in ovarian borderline tumors and stage I carcinomas. Am J Clin Pathol. 1994;102:671–6.
Simons M, Nagtegaal ID, Overbeek LI, Flucke U, Massuger LF, Bulten J. A patient with a noninvasive mucinous ovarian borderline tumor presenting with late pleural metastases. Int J Gynecol Pathol. 2015;34:143–50.
Kobel M, Piskorz AM, Lee S, Lui S, LePage C, Marass F, et al. Optimized p53 immunohistochemistry is an accurate predictor of TP53 mutation in ovarian carcinoma. The J Pathol Clin Res. 2016;2:247–58.
Singh N, Piskorz AM, Bosse T, Jimenez-Linan M, Rous B, Brenton JD, et al. p53 immunohistochemistry is an accurate surrogate for TP53 mutational analysis in endometrial carcinoma biopsies. J Pathol. 2020;250:336–45.
Xue Y, San Luis B, Lane DP. Intratumour heterogeneity of p53 expression; causes and consequences. J Pathol. 2019;249:274–85.
Tessier-Cloutier B, Kortekaas KE, Thompson E, Pors J, Chen J, Ho J, et al. Major p53 immunohistochemical patterns in in situ and invasive squamous cell carcinomas of the vulva and correlation with TP53 mutation status. Mod Pathol. 2020;33:1595–605.
Meagher NS, Wang L, Rambau PF, Intermaggio MP, Huntsman DG, Wilkens LR, et al. A combination of the immunohistochemical markers CK7 and SATB2 is highly sensitive and specific for distinguishing primary ovarian mucinous tumors from colorectal and appendiceal metastases. Mod Pathol. 2019;32:1834–46.
Landrum MJ, Lee JM, Riley GR, Jang W, Rubinstein WS, Church DM, et al. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 2014;42:D980–5.
Petitjean A, Mathe E, Kato S, Ishioka C, Tavtigian SV, Hainaut P, et al. Impact of mutant p53 functional properties on TP53 mutation patterns and tumor phenotype: lessons from recent developments in the IARC TP53 database. Hum Mutat. 2007;28:622–9.
Kobel M, Ronnett BM, Singh N, Soslow RA, Gilks CB, McCluggage WG. Interpretation of P53 Immunohistochemistry in Endometrial Carcinomas: Toward Increased Reproducibility. Int J Gynecol Pathol. 2019;38 Suppl 1:S123–S31.
Lee Y, Miron A, Drapkin R, Nucci MR, Medeiros F, Saleemuddin A, et al. A candidate precursor to serous carcinoma that originates in the distal fallopian tube. J Pathol. 2007;211:26–35.
Ahmed AA, Etemadmoghadam D, Temple J, Lynch AG, Riad M, Sharma R, et al. Driver mutations in TP53 are ubiquitous in high grade serous carcinoma of the ovary. J Pathol. 2010;221:49–56.
Silverberg SG, Bell DA, Kurman RJ, Seidman JD, Prat J, Ronnett BM, et al. Borderline ovarian tumors: key points and workshop summary. Hum Pathol. 2004;35:910–7.
Lee KR, Scully RE. Mucinous tumors of the ovary: a clinicopathologic study of 196 borderline tumors (of intestinal type) and carcinomas, including an evaluation of 11 cases with ‘pseudomyxoma peritonei’. Am J Surg Pathol. 2000;24:1447–64.
Acknowledgements
We thank Shuhong Liu, Young Ou, and Deon Richards at the Anatomical Pathology Research Laboratory, University of Calgary, for immunohistochemical stains with internal research support RS19-609. We acknowledge the contribution of the GAMuT Collaborators: Sumitra Ananda, Michael Christie, Sian Fereday, Stephen B. Fox, C. Blake Gilks, Alison M. Hadley, Tom W. Jobling, Yoke-Eng Chiew, Jan Pyman, Georgina L. Ryland, Jessica N. McAlpine, Orla M. McNally, George Au-Yeung, Alison Brand, Georgia Chenevix-Trench, Neville F Hacker, Gwo-Yaw Ho, Goli Samimi, Ragwha Sharma, Linda Mileshkin.
KLG is supported by the Victorian Cancer Agency (MCRF15013) and the Australian National Health and Medical Research Council (APP1045783 and #628434). This study was supported by the Peter MacCallum Cancer Foundation. CS is supported by a University of Melbourne Postgraduate Scholarship. DDB is supported by National Health and Medical Research Council of Australia (NHMRC) grants APP1092856 and APP1117044 and by the US National Cancer Institute U54 programme (U54CA209978-04). ELG and SHK are supported through P50 CA136393-10.
The following cohorts that contributed to the GAMuT study were supported as follows:
CASCADE: Supported by the Peter MacCallum Cancer Foundation
AOCS: The Australian Ovarian Cancer Study Group was supported by the U.S. Army Medical Research and Materiel Command under DAMD17-01-1-0729, The Cancer Council Victoria, Queensland Cancer Fund, The Cancer Council New South Wales, The Cancer Council South Australia, The Cancer Council Tasmania and The Cancer Foundation of Western Australia (Multi-State Applications 191, 211 and 182) and the National Health and Medical Research Council of Australia (NHMRC; ID400413 and ID400281).
The Australian Ovarian Cancer Study gratefully acknowledges additional support from Ovarian Cancer Australia and the Peter MacCallum Foundation. The AOCS also acknowledges the cooperation of the participating institutions in Australia and acknowledges the contribution of the study nurses, research assistants, and all clinical and scientific collaborators to the study. The complete AOCS Study Group can be found at www.aocstudy.org. We would like to thank all of the women who participated in these research programs.
OVCARE receives core funding from The BC Cancer Foundation and the VGH and UBC Hospital Foundation. The Gynaecological Oncology Biobank at Westmead is a member of the Australasian Biospecimen Network-Oncology group, which was funded by the National Health and Medical Research Council Enabling Grants ID 310670 & ID 628903 and the Cancer Institute NSW Grants ID 12/RIG/1-17 & 15/RIG/1-16.
COEUR: This study uses resources provided by the Canadian Ovarian Cancer Research Consortium’s - COEUR biobank funded by the Terry Fox Research Institute and managed and supervised by the Centre hospitalier de l’Université de Montréal (CRCHUM). The Consortium acknowledges contributions to its COEUR biobank from Institutions across Canada (for a full list see http://www.tfri.ca/en/research/translational-research/coeur/coeur_biobanks.aspx).
The following cohorts that contributed to OTTA were supported as follows:
AOV: Canadian Institutes of Health Research (MOP-86727), Cancer Research Society (19319). BAV: ELAN Funds of the University of Erlang en-Nuremberg; DOV: NCI/NIH R01CA168758. Huntsman Cancer Foundation and the National Cancer Institute of the National Institutes of Health under Award Number P30CA042014. HAW: U.S. National Institutes of Health (R01-CA58598, N01-CN-55424, and N01-PC-67001); MAY: National Institutes of Health (R01-CA122443, P30-CA15083, P50-CA136393); Mayo Foundation; Minnesota Ovarian Cancer Alliance; Fred C. and Katherine B. Andersen Foundation; SEA: SEARCH team: Mitul Shah, Jennifer Alsopp, Mercedes Jiminez-Linan SEARCH funding: Cancer Research UK (C490/A16561), the Cancer Research UK Cambridge Cancer Centre and the National Institute for Health Research Cambridge Biomedical Research Centres. The University of Cambridge has received salary support for PDPP from the NHS in the East of England through the Clinical Academic Reserve. JBD: Cancer Research UK Institute Group Award UK A22905 and A15601; STA: NIH grants U01 CA71966 and U01 CA69417; SWE: Swedish Cancer foundation, WeCanCureCancer, and årKampMotCancer foundation; TVA: Canadian Institutes of Health Research grant (MOP-86727) and NIH/NCI 1 R01CA160669-01A1; VAN: M.S. Anglesio is funded through a Michael Smith Foundation for Health Research Scholar Award and the Janet D. Cottrelle Foundation Scholars program managed by the BC Cancer Foundation. The Vancouver study cohort (TVAN) is supported by BC’s Ovarian Cancer Research team (OVCARE), the BC Cancer Foundation, and The VGH + UBC Hospital Foundation. WMH: National Health and Medical Research Council of Australia, Enabling Grants ID 310670 & ID 628903. Cancer Institute NSW Grants 12/RIG/1-17 & 15/RIG/1-16.
Australian Ovarian Cancer Study:
Management group: D. Bowtell34,35,36,37,38, G. Chenevix-Trench39, A. Green39, P. Webb39, A. DeFazio40,41,42, D. Gertig43
Project and data managers: N. Traficante34, S. Fereday34, S. Moore39, J. Hung40, K. Harrap39, T. Sadkowsky39, N. Pandeya39
Research nurses and assistants: M. Malt39, A. Mellon44, R. Robertson44, T. Vanden Bergh45, M. Jones45, P. Mackenzie45, J. Maidens46, K. Nattress47, Y. E. Chiew40, A. Stenlake42, H. Sullivan42, B. Alexander39, P. Ashover39, S. Brown39, T. Corrish39, L. Green39, L. Jackman39, K. Ferguson39, K. Martin39, A. Martyn39, B. Ranieri39, J. White48, V. Jayde49, P. Mamers50, L. Bowes34, L. Galletta34, D. Giles34, J. Hendley34, K. Alsop34, T. Schmidt51, H. Shirley51, C. Ball52, C. Young52, S. Viduka51, Hoa Tran51, Sanela Bilic51, Lydia Glavinas51, Julia Brooks53
Clinical and scientific collaborators: R. Stuart-Harris54, F. Kirsten55, J. Rutovitz56, P. Clingan57, A. Glasgow57, A. Proietto44, S. Braye44, G. Otton44, J. Shannon58, T. Bonaventura59, J. Stewart59, S. Begbie60, M. Friedlander61, D. Bell46, S. Baron-Hay46, A. Ferrier46, G. Gard46, D. Nevell46, N. Pavlakis46, S. Valmadre46, B. Young46, C. Camaris45, R. Crouch45, L. Edwards45, N. Hacker45, D. Marsden45, G. Robertson45, P. Beale47, J. Beith47, J. Carter47, C. Dalrymple47, R. Houghton47, P. Russell47, M. Links62, J. Grygiel63, J. Hill64, A. Brand41,65, K. Byth65, R. Jaworski66, P. Harnett41,65, R. Sharma41,66, G. Wain65, B. Ward67, D. Papadimos67, A. Crandon68, M. Cummings68, K. Horwood68, A. Obermair68, L. Perrin68, D. Wyld68, J. Nicklin68,69, M. Davy48, M. K. Oehler48, C. Hall48, T. Dodd48, T. Healy70, K. Pittman70, D. Henderson70,71, J. Miller72, J. Pierdes72, P. Blomfield49, D. Challis49, R. McIntosh49, A. Parker49, B. Brown73, R. Rome73, D. Allen74, P. Grant74, S. Hyde74, R. Laurie74, M. Robbie74, D. Healy50, T. Jobling50, T. Manolitsas50, J. McNealage50, P. Rogers50, B. Susil50, E. Sumithran50, I. Simpson50, K. Phillips34, D. Rischin34, S. Fox34, D. Johnson34, S. Lade34, M. Loughrey34, N. O’Callaghan34, W. Murray34, P. Waring35, V. Billson75, J. Pyman75, D. Neesham75, M. Quinn75, C. Underhill76, R. Bell77, L. F. Ng78, R. Blum79, V. Ganju80, I. Hammond52, Y. Leung52, A. McCartney52, M. Buck81, I. Haviv82, D. Purdie39, D. Whiteman39, N. Zeps51
GAMuT collaborators:
Dane Cheasley2,3,83, Matthew J. Wakefield6,84,85, Georgina L. Ryland83, Prue E. Allan83, Kathryn Alsop83, Sumitra Ananda83,86, Michael S. Anglesio21,87, George Au-Yeung83,85, Maret Böhm88, David D. L. Bowtell2,3,83,85, Alison Brand89, Georgia Chenevix-Trench90, Michael Christie85,91, Yoke-Eng Chiew89, Michael Churchman26,92, Anna DeFazio24,89, Rhiannon Dudley93, Nicole Fairweather93, Sian Fereday83, Stephen B. Fox83,85, C. Blake Gilks20,22,23,87, Charlie Gourley92, Neville F. Hacker94, Alison M. Hadley95, Joy Hendley83, Gwo-Yaw Ho84, David G. Huntsman20,22,23,87, Sally M. Hunter83, Tom W. Jobling96, Kimberly R. Kalli97,an2, Scott H. Kaufmann97, Catherine J. Kennedy89, Martin Köbel1,98, Cecile Le Page99, Orla M. McNally85,100, Jessica N. McAlpine93, Anne-Marie Mes-Masson4,5,99,101, Linda Mileshkin83, Diane M. Provencher4,5,99,102, Jan Pyman100,103, Kurosh Rahimi99,102, Goli Samimi88, Ragwha Sharma89,104, Andrew N. Stephens93, Nadia Traficante83, Yoland C. Antill30,31,32,105,106, Clare L. Scott83,84,85,91, Ian G. Campbell83,85, Kylie L. Gorringe2,3,83,85
Author information
Authors and Affiliations
Consortia
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Members of the Australian Ovarian Cancer Study and GAMuT Collaborators names are listed below Acknowledgements
Supplementary information
Rights and permissions
About this article

Cite this article
Kang, E.Y., Cheasley, D., LePage, C. et al. Refined cut-off for TP53 immunohistochemistry improves prediction of TP53 mutation status in ovarian mucinous tumors: implications for outcome analyses. Mod Pathol 34, 194–206 (2021). https://doi.org/10.1038/s41379-020-0618-9
Received:
Revised:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41379-020-0618-9
This article is cited by
-
A case report of mucinous borderline ovarian tumor with recurrence as invasive carcinoma with high copy number alterations
International Cancer Conference Journal (2024)
-
Extensive intratumor regional epigenetic heterogeneity in clear cell renal cell carcinoma targets kidney enhancers and is associated with poor outcome
Clinical Epigenetics (2023)
