
Abstract
Dopaminergic dysregulation is one of the leading hypotheses for the pathoetiology underlying psychotic disorders such as schizophrenia. Molecular imaging studies have shown increased striatal dopamine synthesis capacity (DSC) in schizophrenia and people in the prodrome of psychosis. However, it is unclear if genetic risk for psychosis is associated with altered DSC. To investigate this, we recruited healthy controls and two antipsychotic naive groups of individuals with copy number variants, one with a genetic deletion at chromosome 22q11.2, and the other with a duplication at the same locus, who are at increased and decreased risk for psychosis, respectively. Fifty-nine individuals (21 with 22q11.2 deletion, 12 with the reciprocal duplication and 26 healthy controls) received clinical measures and [18F]-DOPA PET imaging to index striatal Kicer. There was an inverse linear effect of copy number variant number on striatal Kicer value (B = −1.2 × 10−3, SE = 2 × 10−4, p < 0.001), with controls showing levels intermediate between the two variant groups. Striatal Kicer was significantly higher in the 22q11.2 deletion group compared to the healthy control (p < 0.001, Cohen’s d = 1.44) and 22q11.2 duplication (p < 0.001, Cohen’s d = 2) groups. Moreover, Kicer was positively correlated with the severity of psychosis-risk symptoms (B = 730.5, SE = 310.2, p < 0.05) and increased over time in the subject who went on to develop psychosis, but was not associated with anxiety or depressive symptoms. Our findings suggest that genetic risk for psychosis is associated with dopaminergic dysfunction and identify dopamine synthesis as a potential target for treatment or prevention of psychosis in 22q11.2 deletion carriers.
Similar content being viewed by others
Introduction
Schizophrenia is a highly heritable neurodevelopmental disorder [1,2,3] with a lifetime prevalence of 0.7% [4]. It is a leading cause of global disease burden in adults and is associated with high excess mortality rate [5]. Total costs for schizophrenia and related psychotic disorders are estimated at around €94 billion per annum across Europe [6]. Antipsychotic drugs, the main pharmacological treatment, show highly variable responses and are not effective in one-third of patients, leaving a substantial proportion of individuals with persistent impairments [7,8,9]. Improving the understanding of the neurobiology underlying schizophrenia is crucial to informing the development of new classes of treatment.
One of the main hypotheses for schizophrenia implicates dysregulation of the dopaminergic system [10, 11]. Over the last two decades, advances in molecular imaging have identified the nature of the dopamine (DA) dysfunction in patients with schizophrenia. Molecular imaging using radiolabelled DOPA, which is taken up into DA neurons and decarboxylated by aromatic amino acid decarboxylase (AADC) to give radiolabelled DA [12, 13], is used as an index of in vivo dopamine synthesis capacity (DSC) [12]. A meta-analysis of studies using this technique found increased striatal DSC in individuals with schizophrenia compared to controls with a large effect size [14].
Previous studies have shown elevated striatal DSC in people at clinical high risk for psychosis with a large effect size (Cohen’s d = 0.8) [15,16,17,18].Thus, elevated DSC at the striatal level is associated with both risk of psychosis and the development of symptoms. However, the degree to which dopaminergic alterations are due to trait risk for psychosis or reflect the symptomatic state is not clear from these studies [19]. Given the high heritability of schizophrenia [1, 2, 20], it has been hypothesized that one of the ways in which genetic risk for schizophrenia might be manifested is as increased DSC [21]. However, the results of studies examining DSC in first-degree relatives of people with schizophrenia are inconsistent, with one study showing increased DSC in unaffected first-degree relatives compared to controls [22] and a second one showing no difference in DCS between unaffected, predominantly dizygotic, twins for schizophrenia and controls [23]. This might reflect various methodological issues, including that unaffected first-degree relatives do not necessarily carry genetic risk variants for schizophrenia.
Given the importance of the question of whether genetic risk for schizophrenia is associated with DA dysfunction, we focused on individuals with a 1.5–3 megabase deletion at 22q11.2 locus. Carriers of this mutation are at greatly increased risk for developing psychosis [24, 25]; with a risk more than 40-fold greater than that in the general population [26], making this copy number variant one of the strongest genetic risk factors for developing schizophrenia and related psychotic disorders. In contrast, the reciprocal copy number variant, 22q11.2 duplication, has been associated with significantly reduced risk of schizophrenia compared to the general population, suggesting a possible protective role [27]. Subsequent studies have supported this initial finding of reduced risk of schizophrenia in 22q11.2 duplication [28,29,30,31], although some inconsistencies have been reported [32, 33]. Moreover, studies of peripheral DA metabolite levels suggest altered dopaminergic function in 22q11.2 deletion carriers [34, 35], consistent with the ubiquitous hemideletion in 22q11.2 deletion subjects of the Catechol-O-methyl-transferase, an enzyme involved in DA metabolism [36]. However, to our knowledge, no study has investigated DSC in 22q11.2 deletion carriers or any aspect of DA function in 22q11.2 duplication carriers.
In view of this, we tested the hypothesis that striatal DSC is related to genetic risk for psychosis and sub-clinical psychotic symptoms. We predicted that there would be a linear relationship between DSC and increasing genetic risk and with sub-clinical psychotic symptom severity, with 22q11.2 duplication carriers showing the lowest DSC and 22q11.2 deletion carriers showing the highest DSC.
Methods and materials
Ethical permission was obtained by London-West London & GTAC Research Ethics Committee and the Administration of Radioactive Substances Advisory Committee. Following description of the study, informed written consent was given by all participants.
Demographics
Twenty-one individuals with 22q11.2 deletion and 12 individuals with 22q11.2 duplication were recruited via support groups in Great Britain and Ireland, Clinical Genetics Clinics across the UK, and other cohorts in the UK and Netherlands. Inclusion criteria were: age above 18 years old, no history of schizophrenia or other psychotic disorder and no antipsychotic medication use. Confirmation of 22q11.2 deletion and duplication diagnosis was based on records from regional clinical genetics. Diagnosis for twenty of the individuals with 22q11.2 deletion was established by fluorescence in situ hybridization and for only one by comparative genomic hybridization (CGH) arrays. CGH arrays were used to establish the presence of the 22q11.2 duplication. Twenty-six healthy controls were recruited via local media for comparison. Inclusion criteria for controls were: no significant personal medical or psychiatric history, no family history of psychotic disorder, no concurrent use of psychotropic medication. Exclusion criteria for both groups: history of head trauma, significant medical disorder (unrelated to 22q11.2 deletion/duplication), significant illicit drug/alcohol use, any procedures, operations or medical conditions (e.g., pregnancy) that would compromise safety in scanning.
Clinical assessments
The 22q11.2 deletion and duplication groups were assessed using the Comprehensive Assessment of At Risk Mental States (CAARMS), which measures sub-clinical psychotic-like symptoms, to determine if they met criteria for being at clinical high risk of psychosis [37]. All subjects received the Beck’s self-reported questionnaires for depression and anxiety [38]. Intellectual quotient was measured using an abbreviated version of the Wechsler Adult Intelligence scale (WAIS-III), consisting of four subtests [39].
PET data acquisition
We used the approach previously described by Jauhar et al. [18]. All subjects were instructed to abstain from food and alcohol 12 h prior scanning and to refrain from tobacco smoking 4 h before scanning [40]. On the day of the scan, a urine drug screen verified no recent drug use and a negative pregnancy test was essential for all female participants. An hour before the scan, 150 mg carbidopa and 400 mg entacapone were administered orally to prevent the formation of radiolabelled metabolites [13]. All subjects underwent a 6-[18F] fluoro-L-DOPA ([18F]-DOPA) PET scan on the same Siemens Biograph 6 HiRez PET scanner (Siemens, Erlangen, Germany) in three-dimensional mode. Following a CT transmission scan for attenuation correction, ~150 MBq of [18]F-DOPA was administered by bolus intravenous injection within 30 s after the start of PET imaging.
PET data analysis
Our main outcome was the influx rate constant (Kicer) in the whole striatum. Given the evidence that dopaminergic alterations in schizophrenia may be more pronounced in associative striatum [14], we also conducted secondary analysis in functional subdivisions (associative, limbic, sensorimotor). To correct for head movement, nonattenuation-corrected dynamic images were denoised using a level 2, order 64 Battle–Lemarie wavelet filter [41], and individual frames were realigned to a single frame acquired 10 min after the [18F]-DOPA injection using a mutual information algorithm [42]. Transformation parameters were then applied to the corresponding attenuation-corrected frames, and the realigned frames were combined to create a movement-corrected dynamic image (from 6 to 95 min following [18F]-DOPA administration) for analysis.
Following motion correction, standardized regions of interest (ROIs) were defined bilaterally in the whole striatum and its functional subdivisions, and the cerebellar reference region in Montreal Neurologic Institute space [43]. Using statistical parametric mapping software (SPM8, http://fil.ion.ucl.ac.uk/spm) and in-house MATLAB-based scripts, we then co-registered an [18F]-DOPA template with the ROI map to each individual PET summation (add) image, allowing ROIs to be placed automatically on individual [18F]-DOPA PET images. This approach means that the volumes of the ROIs are transformed to fit the individual’s brain scan. A previous test rest study has shown good reliability with intra-class correlation coefficients > 0.84 for this approach [44]. In addition, since regional brain volumetric differences in 22q11.2 deletion and duplication have been described, for a subset of patients in which MRI imaging was available (N = 32), we also conducted MRI-based segmentation and compared this with the PET-based analysis (please see Supplementary material for more information). Structural MR imaging could not be obtained in all subjects because of contra-indications to MR imaging in a number of subjects, which are common in people with 22q11.2 deletions because of increased rates of surgery for cardiac conditions.
[18F]-DOPA uptake was calculated relative to the cerebellum, for each ROI using the Patlak graphic analysis modified for a reference tissue input function [45,46,47]. Further details of the image analysis are given in previous publications [18, 48, 49]. We also calculated the standardized uptake values in the cerebellum at 95 min as previously [18] to examine if there are any differences between groups.
PET parametric mapping
We applied a previously established method [18, 49], in which the Kicer parametric images of the brain were constructed from movement-corrected images using a wavelet-based Patlak–Gjedde approach [47]. The parametric image for each subject was then normalized into Montreal Neurological Institute standard space, using the subjects’ PET summation image and the [18F]-DOPA template. Using SPM8 and a striatal mask [15], we performed statistical parametric mapping to compare striatal DSC between groups using an independent t-test. Results are presented after correction for multiple comparisons as applied in SPM8 (family-wise error (FWE) corrected).
Statistical analysis
As our study was the first of its kind in this cohort, sample size was chosen based on previous [18F]-DOPA PET studies in individuals with psychosis [18, 48, 49]. Statistical analyses were performed using R version 3.3.2 and SPSS, version 23 (IBM SPSS Statistics for Macintosh, Version 24.0). Normality of distribution was assessed using Shapiro–Wilk test. Group comparisons of demographic variables were determined using univariate analysis of variance (ANOVA) or chi-square tests as appropriate. Our main analysis used a linear regression model with whole striatal Kicer as the dependent variable and group as a predictor to test our hypothesis that there would be a linear relationship between DSC and group in line with the genetic risk for psychosis, with 22q11.2 deletion carriers showing highest values and 22q11.2 duplication carriers showing lowest values. Where this was significant, we went on to conduct group comparisons using independent t-tests adjusted for multiple comparisons using Tukey’s tests. We repeated these analyses for the striatal functional subdivisions. We tested the relationship between Kicer and scores on clinical scales using a linear regression model with symptoms score as the dependent variable and Kicer as the predictor, and group as an additional factor. We also conducted exploratory sensitivity analyses, adjusting for injected activity, age, given their potential effect on Kicer [49], as well as for FSIQ, striatal volume and head motion and performed further sensitivity analyses excluding symptomatic 22q11.2 carriers and the 22q11.2 duplication carrier with co-morbid cerebral palsy in case this influenced findings. Subjects who scored two or less (meaning symptoms were absent or questionably present at most) on the CAARMS severity scale for positive symptoms were categorized as having no/minimal CAARMS symptoms, whilst the rest were categorized as being symptomatic and excluded for this sub-analysis.
Results
Demographics
No significant differences were observed between groups in gender and ethnicity, although the duplication group was older than the other groups (Table 1). As expected, individuals with 22q11.2 deletion had significantly lower FSIQ compared to the group of the reciprocal duplication and healthy controls (Table 1). All participants were antipsychotic naive. Two of the individuals with 22q11.2 were receiving selective serotonin reuptake inhibitors (citalopram 20 mg and fluoxetine 40 mg). One 22q11.2 duplication carrier had a history of cerebral palsy. None of the 22q11.2 deletion or duplication carriers met criteria to be at clinical high risk of psychosis [37]. Individuals with 22q11.2 deletion and duplication had higher self-reported scores in Beck’s depression and anxiety scales compared to healthy controls. Post hoc analysis showed that there were no statistically significant differences between the group of 22q11.2 deletion and the reciprocal duplication (p > 0.05).
Relationship between striatal dopamine synthesis capacity and genetic risk for psychosis
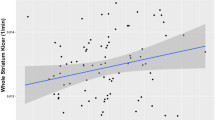
Using the linear regression model, we found that group significantly predicted DSC (as measured by Kicer) levels in the whole striatum (B = −1.2 × 10−3, SE = 2 × 10−4, p < 0.001) (Fig. 1), and all striatal functional subdivisions (associative: B = −1.36 × 10−3, SE = 2.2 × 10−4, p < 0.001; sensorimotor: B = −9.4 × 10−4, SE = 2 × 10−4, p < 0.001; limbic: B = −1.3 × 10−3, SE = 2.3 × 10−4, p < 0.001) (Supplementary Table 1 and Fig. 2) with the following direction: 22q11.2 deletion > healthy controls > 22q11.2 duplication. Post hoc analyses, after adjusting for multiple comparisons, showed significantly greater whole striatal Kicer in the 22q11.2 deletion group relative to both healthy controls (p < 0.001, Cohen’s d = 1.44) and the 22q11.2 duplication group (p < 0.001, Cohen’s d = 2) (Supplementary Table 1). Similarly, post hoc analyses indicated greater Kicer in the 22q11.2 deletion group compared to the healthy control and 22q11.2 duplication groups in all striatal functional subdivisions (associative striatum: p < 0.001, Cohen’s d = 1.4; p < 0.001, Cohen’s d = 2, for healthy control and duplication comparisons, respectively; sensorimotor striatum: p < 0.001; Cohen’s d = 1.35, p < 0.001, Cohen’s d = 1.55, respectively; limbic striatum: p < 0.001 Cohen’s d = 1.7; p < 0.001, Cohen’s d = 2.2, respectively; Supplementary Table 1 and Fig. 2).

Error bars indicate standard error of the mean. Dopamine synthesis and/or storage capacity was higher in individuals with 22q11.2 deletion compared to healthy controls and individuals with 22q11.2 duplication in the following order: 22q11.2 deletion > healthy controls > 22q11.2 duplication groups.

Error bars indicate standard error of the mean (***p < 0.001).
As, by chance, the duplication group was older than the other groups, we conducted exploratory analyses to determine if age influenced our findings. Age showed no relationship with Kicer (B = −1.21 × 10−5, SE = 2.05 × 105, p = 0.6). Moreover, when age was added to the model, group remained a significant predictor of Kicer for the whole striatum (B = −1.23 × 10−3, SE = 2.16 × 10−4, p < 0.001) and its subdivisions (associative; B = −1.3 × 10−3, SE = 2.4 × 10−4, p < 0.001; sensorimotor B = −8.65 × 10−4, SE = 2.2 × 10−4, p < 0.001; limbic: B = −1.35 × 10−3, SE = 2.5 × 10−4, p < 0.001).
Further analysis was also performed to determine if the dose of injected activity influenced our findings. There was no association between injected activity and Kicer (B = 6.14 × 10−6, SE = 2.35 × 10−5, p = 0.8). In addition, when we added injected activity to the model, group continued to significantly predict Kicer levels in the whole striatum (B = −1.27 × 10−3, SE = 2.05 × 10−4, p < 0.001) and its subdivisions (associative; B = −1.35 × 10−3, SE = 2.25 × 10−4, p < 0.001; sensorimotor; B = −9.5 × 10−4, SE = 2.07 × 10−4, p < 0.001; limbic; B = −1.34 × 10−3, SE = 2.3 × 10−4, p < 0.001).
As previously noted, FSIQ was, unsurprisingly, lower in individuals with 22q11.2 deletion compared to the groups of healthy controls and 22q11.2 duplication. Therefore, we tested whether FSIQ had an effect on our results. We found no relationship between FSIQ and Kicer (B = −7.33 × 10−6, SE = 5.38 × 10−6, p = 0.18). In addition, when we added FSIQ to the model, group remained a significant predictor of Kicer in the whole striatum (B = −1.17 × 10−3, SE = 2.18 × 10−4, p < 0.001) and its subdivisions (associative; B = −1.21 × 10−3, SE = 2.43 × 10−4, p < 0.001; sensorimotor; B = −8.3 × 10−4, SE = 2.24 × 10−4, p < 0.001; limbic; B = −1.19 × 10−3, SE = 2.5 × 10−4, p < 0.001).
In addition, as head motion was significantly greater in the individuals with 22q11.2 deletion and duplications compared to healthy controls, we examined whether the above factor influenced our results. There was no association between head motion and Kicer (B = 2.19 × 10−5, SE = 2.09 × 10−5, p = 0.3). Furthermore, the addition of head motion to the model did not alter our results and group predicted significantly Kicer in the whole striatum (B = −1.18 × 10−3, SE = 2.2 × 10−4, p < 0.001) and its subdivisions (associative; B = −1.26 × 10−3, SE = 2.4 × 10−4, p < 0.001; sensorimotor; B = −8.96 × 10−4, SE = 2.23 × 10−4, p < 0.001; limbic; B = −1.21 × 10−3, SE = 2.48 × 10−4, p < 0.001).
To exclude a potential effect of group on blood flow and peripheral metabolism of DOPA, we examined the reference region (cerebellum) to test whether there was any difference in the standardized uptake value at 95 min between the three groups, as previously described [50], and found no significant differences (F(2, 56) = 2.2, p = 0.12) (Supplementary Fig. 1).
Furthermore, when we compared PET- and MRI-based segmentation for the subgroup of individuals of our cohort that MRI was available, analysis revealed similar results (please see Supplementary results, Supplementary Figs. 2 and 3 and Supplementary Table 2).
Moreover, all results remained significant in a further sensitivity analysis excluding the individual with 22q11.2 duplication with cerebral palsy (Supplementary Table 3). To determine if our findings were driven by the inclusion of symptomatic volunteers, we conducted a sensitivity analysis restricted to volunteers in the 22q11.2 group with no/minimal psychotic symptoms (n = 12). The effect of group on whole striatal Kicer value remained significant relative to the healthy control and the 22q11.2 duplication groups in this analysis (Supplementary Table 4).
One individual of the 22q11.2 deletion group developed schizophrenia 6 months following the initial PET scan. We were able to acquire PET data at the second-time point (2 months after his diagnosis), when he was treated with a low dose of risperidone (1 mg/day). At follow-up, Kicer showed a 6.2% increase from baseline (at baseline: Kicer = 0.016 min−1, and follow-up: Kicer = 0.017 min−1) (Supplementary Fig. 4).
Voxel-based analyses
We found significantly greater Kicer in the 22q11.2 deletion group relative to the controls in two voxel clusters with their foci in left and right putamen, both within the associative subdivision of the striatum (Fig. 3A; p < 0.05 corrected for multiple comparisons using the FWE rate method). There was also a significantly greater Kicer in the 22q11.2 deletion group relative to the duplication group in two voxel clusters within the right and left putamen (Fig. 3B; p < 0.05 FWE corrected for multiple comparisons). Furthermore, the voxel-based analysis identified a greater Kicer in the healthy control group relative to the 22q11.2 duplication group in two voxel clusters within the left and right caudate (Fig. 3C; p < 0.05 uncorrected), but this did not survive correction for multiple comparisons. The control group > 22q11.2 deletion group, the 22q11.2 duplication group > 22q11.2 deletion group and 22q11.2 duplication group > control group contrasts revealed no significant differences, even at an uncorrected statistical threshold (p < 0.05).

A Regions of significantly higher striatal Kicer, relative to healthy controls (N = 26), in individuals with 22q11.2 deletion (N = 21). The most significant statistical increases had peaks in the left (peak MNI coordinates x = −24, y = 12, z = 4; puncorr = 0.001) and right putamen (peak MNI coordinates x = 28, y = 10, z = 4; puncorr = 0.016). B Regions of significantly higher striatal Kicer, relative to 22q11.2 duplication group (N = 11), in 22q11.2 deletion subjects (N = 21). The most significant increases had peaks in the right (peak MNI coordinates x = 28, y = 8, z = −6; puncorr = 0.002) and left putamen (peak MNI coordinates x = −28, y = 12, z = −6; puncorr = 0.003). C Regions of significantly lower striatal Kicer, relative to healthy controls (N = 26), in individuals with 22q11.2 duplication (N = 11). The most significant reductions had peaks in the left (peak MNI coordinates x = −12, y = 16, z = 12; p = 0.001) and right caudate nuclei (peak MNI coordinates x = 10, y = 14, z = 12; puncorr = 0.008). None of these voxel peaks survived correction for multiple comparisons using FWE. All maps refer to t-stats statistics threshold at p < 0.05 uncorrected.
In summary, the region-of-interest and voxel-based analyses show that striatal DSC is higher in individuals with the 22q11.2 deletion compared to healthy controls and individuals with the 22q11.2 duplication.
Relationship between dopaminergic function and sub-clinical positive symptoms in individuals with 22q11.2 deletion/duplication
We then tested the hypothesis that 22q11.2-dependent modulation of striatal dopaminergic tone might correlate with clinical phenotypes. There was a significant direct relationship between whole striatal Kicer and mean positive symptom severity ratings, as measured with the CAARMS [37] in the groups with copy number variants on 22q11.2 locus (B = 730.5, SE = 310.2, p = 0.025) (Fig. 4). When group status (duplication or deletion) was added to the model, Kicer remained significant (p = 0.03), but group was not predictive of CAARMS mean positive symptom domain score (p = 0.58), and there was no significant group × Kicer interaction (p = 0.65).

Blue triangles and red circles indicate individuals with 22q11.2 deletion and 22q11.2 duplication, respectively. There was a positive relationship between CAARMS positive score and striatal Kicer (R2 = 0.15, p < 0.05).
Anxiety and depression symptoms are also reported in the prodrome to psychosis in individuals with 22q11.2 deletion [51,52,53]. Thus, to test the specificity of our findings to positive symptoms, we also examined the association between Kicer levels and Beck’s Anxiety and Depression Scales. No significant relationships were found with mean scores on either the anxiety or depression scales (B = 179.33, SE = 1472.37, p = 0.9; B = −1364.03, SE = 906.67, p = 0.14, respectively).
Overall, these data suggest that there is a direct correlation of striatal DSC with sub-clinical psychotic symptoms, but not mood or anxiety symptoms.
Discussion
Our study showed that striatal Kicer is higher in individuals with 22q11.2 deletion compared to healthy controls, who, in turn, have higher striatal Kicer than individuals with the reciprocal duplication. Moreover, Kicer is directly correlated with sub-clinical positive psychotic symptom severity.
The 22q11.2 deletion is the single largest genetic risk factor for schizophrenia and other psychotic disorders [26], whilst the 22q11.2 duplication is associated with reduced risk [27, 30, 31, 54]. Our findings are thus relevant to understanding the mechanism underlying psychosis. They extend previous findings of elevated DA synthesis and release capacity in people at clinical high risk for psychosis [15, 16, 55], by showing that DA alterations predate the clinical high-risk phase in people with a confirmed genetic high risk for schizophrenia. The 22q11.2 deletion carriers were young, and still in the peak age risk group for the onset of schizophrenia but had not yet developed a psychotic disorder or met clinical high-risk criteria. Thus, they remain at high risk of developing a psychotic disorder, and, based on previous studies [56], we anticipate that about 40% will go on to develop a schizophreniform disorder. Furthermore, our findings suggest lower Kicer in the 22q11.2 duplication carriers could be a mechanism underlying the lower risk for schizophrenia seen in this group.
In our study, the magnitude of the increase in DSC was comparable in all functional striatal subdivisions, which is contrary to the results of the most recent meta-analysis in schizophrenia showing greater dopaminergic alterations in the dorsal striatum than in the limbic striatum [14]. Taken with our findings across the striatum, this may point to differences in pathophysiology between idiopathic schizophrenia and that associated with 22q11.2 deletion. Another potential explanation may be that, whilst the 22q11.2 deletion has an effect in all functional subdivisions, subsequent stressors lead to alterations becoming more marked in the dorsal striatum in the 22q11.2 carriers who go on to develop schizophrenia, in line with stress-diathesis models of psychosis [57]. Longitudinal studies in genetic risk groups for developing psychosis are warranted to test this hypothesis.
Strengths of the study include that the participants were antipsychotic naive and the 22q11.2 deletion carriers were within the age range of risk for developing psychosis, factors that are both significant in the interpretation of our results. Our study was cross-sectional, therefore does not prove causality. Thus, follow-up of our sample is required to determine how Kicer links to the subsequent development of psychosis. Moreover, individuals with 22q11.2 duplication were significantly older compared to the group of 22q11.2 deletion and healthy controls, but, we, nevertheless, found no association between age and Kicer, and when age was added to our model, results remained unchanged. In addition, PET regional volume analysis showed that striatal volume differed between groups, with individuals with 22q11.2 deletion having smaller striatal volume compared to other groups. This could lead to underestimation of Kicer estimates due to partial volume effects, therefore cannot account for the elevation we observed. Moreover, there was no relationship between striatal volume and Kicer estimates. Similarly, we noted greater head movement in the 22q11.2 deletion and duplication groups. We applied a motion correction algorithm to adjust for between frame motions. Notwithstanding this, greater motion is unlikely to explain the elevated Kicer in the deletion group as it would also lead to underestimation of Kicer. Furthermore, we showed no association between head motion and Kicer.
To our knowledge this is the first in vivo imaging study to examine striatal DA function using [18F]-DOPA in individuals with either a 22q11.2 deletion or 22q11.2 duplication. Previously, Butcher et al. [58] conducted a PET study using 11C-dihydrotetrabenazine (11C-DTBZ) in 13 individuals with 22q11.2 deletion, seven of whom were treated with antipsychotic medication. This radiotracer binds to the vesicular monoamine transporter (VMAT2), which transports DA into storage vesicles ready for release [59, 60]. Results showed elevated 11C-DTBZ binding in 22q11.2 carriers, indicating elevated VMAT2 levels and/or synaptic vesicle density. In contrast, DA receptor availability is not altered in 22q11.2 carriers relative to healthy controls [61].
Boot et al. [34] conducted a challenge study in individuals with 22q11 deletion with alpha-methyl paratyrosine (AMPT), a reversible inhibitor of the hydroxylation of tyrosine, the first step for catecholamine biosynthesis. At baseline, they found that levels of urinary DA were increased and homovanillic acid (HVA), the primary DA metabolite, was lower in the group of 22q11 deletion compared to controls. After AMPT, urinary and plasma HVA levels were found to be statistically lower in the 22q11 deletion group relative to the control group. Moreover, the ratio of DA/HVA was increased in subjects with 22q11 deletion both prior to AMPT and following AMPT points. Taken together, these prior findings indicate higher DA levels and reduced metabolism of DA in 22q11.2 carriers.
[18F]-DOPA PET indexes the uptake and conversion of [18F]-DOPA to [18F]-dopamine by aromatic aminoacid decarboxylase (AADC) in DA neurons, and the storage of [18F]-dopamine in synaptic vesicles [12]. Thus, the higher Kicer we report in the 22q11.2 deletion carriers could represent an increase in any one of these processes. However, when taken with prior findings that DA metabolite levels are reduced, and that VMAT2 levels are increased [58], a plausible interpretation of our findings in 22q11.2 deletion carriers is that there is increased synthesis and storage of [18F]-dopamine in vesicles due to reduced catabolism and increased VMAT2 levels in the terminal. This suggests that targeting DA synthesis or storage could be a novel treatment strategy for psychosis in 22q11.2 carriers [62]. Preclinical studies using animal models of 22q11.2 deletion and reciprocal duplication are warranted to further define the neurochemical substrates of dopaminergic alterations resulting from mutations in the 22q11.2 locus.
Our findings link increased striatal DA synthesis and storage capacity to a known genetic risk variant that moderates risk for psychotic disorders. Taken with other evidence of links between [18F]-DOPA and risk of schizophrenia [15, 63], our results suggest that [18F]-DOPA imaging could have potential as a biomarker to identify people at elevated risk of schizophrenia, although follow-up of our cohort is required to confirm this.
Whilst our results support the hypothesis of a trait component to DA alterations in schizophrenia, this does not rule out a state component as well. Supporting this, it should be noted that about 50% of the variance in striatal Kicer is attributable to unique environmental factors [64], patients in an acute relapse show greater striatal DA release than remitted patients [65], and we found an association between greater Kicer and greater symptom severity. The fact that we found no association between Kicer and depressive or anxiety symptoms indicates that this is specific to sub-clinical psychosis-risk symptoms and not an association with general morbidity. Moreover, in our cohort, the 22q11.2 deletion carrier who was re-scanned after developing psychosis showed increased Kicer compared to his baseline value, which is in agreement with studies of people at clinical high risk who transition to psychosis and comparisons between patients with schizophrenia in an acute relapse relative to remitted patients [65, 66]. However, further follow-up is required to determine if increases are seen in other subjects who develop psychosis and to exclude an effect of antipsychotic treatment [48, 67].
Lastly, individuals with 22q11.2 duplication had higher Beck’s depression and anxiety scale scores compared to the healthy control group, although there were no statistically significant differences in these measures between the two groups with copy number variants at the 22q11.2 locus. Anxiety and depressive symptoms are also commonly reported in people at clinical high risk for psychosis, although no relationship with striatal DSC has been reported [16], which is consistent with our results. Our findings of a high prevalence of anxiety and depressive symptoms in the 22q11.2 duplication carriers adds to evidence, albeit limited, for an increased prevalence of anxiety and mood disorders in people with 22q11.2 duplication [28]. Given the relatively small number of study participants in our group, our results highlight the need for further studies on the prevalence of neuropsychiatric symptoms in carriers of this mutation.
Conclusions
We demonstrated that striatal DSC is higher in antipsychotic naive individuals with 22q11.2 deletion relative to controls, who, in turn, have higher striatal Kicer than individuals with the reciprocal duplication and is directly associated with sub-clinical psychosis risk but not anxiety or depressive symptom severity. This suggests that altered striatal DSC may be a mechanism mediating genetic risk for psychosis and the development of sub-clinical psychotic symptoms, suggesting potential targets for the development of new treatments and predictive biomarkers that can be applied to identify and prevent psychosis.
Change history
22 June 2021
A Correction to this paper has been published: https://doi.org/10.1038/s41380-021-01192-0
References
Lichtenstein P, Yip BH, Björk C, Pawitan Y, Cannon TD, Sullivan PF, et al. Common genetic determinants of schizophrenia and bipolar disorder in Swedish families: a population-based study. Lancet. 2009;373:234–9.
Sullivan PF, Kendler KS, Neale MC. Schizophrenia as a complex trait: evidence from a meta-analysis of twin studies. Arch Gen Psychiatry. 2003;60:1187–92.
Owen MJ, Cardno AG, O’Donovan MC. Psychiatric genetics: back to the future. Mol Psychiatry. 2000;5:22–31.
Howes OD, Murray RM. Schizophrenia: an integrated sociodevelopmental-cognitive model. Lancet. 2014;383:1677–87.
Walker ER, McGee RE, Druss BG. Mortality in mental disorders and global disease burden implications: a systematic review and meta-analysis. JAMA Psychiatry. 2015;72:334–41.
Olesen J, Gustavsson A, Svensson M, Wittchen HU, Jonsson B, CDBE2010 Study Group, et al. The economic cost of brain disorders in Europe. Eur J Neurol. 2012;19:155–62.
Amato D, Vernon AC, Papaleo F. Dopamine, the antipsychotic molecule: a perspective on mechanisms underlying antipsychotic response variability. Neurosci Biobehav Rev. 2018;85:146–59.
Howes OD, McCutcheon R, Agid O, de Bartolomeis A, van Beveren NJ, Birnbaum ML, et al. Treatment-resistant schizophrenia: treatment response and resistance in psychosis (TRRIP) Working Group Consensus Guidelines on Diagnosis and Terminology. Am J Psychiatry. 2017;174:216–29.
Scheggia D, Mastrogiacomo R, Mereu M, Sannino S, Straub RE, Armando M, et al. Variations in Dysbindin-1 are associated with cognitive response to antipsychotic drug treatment. Nat Commun. 2018;9:2265.
Lieberman JA, Kane JM, Alvir J. Provocative tests with psychostimulant drugs in schizophrenia. Psychopharmacology. 1987;91:415–33.
Lieberman JA, Kinon BJ, Loebel AD. Dopaminergic mechanisms in idiopathic and drug-induced psychoses. Schizophr Bull. 1990;16:97–110.
Kumakura Y, Cumming P. PET studies of cerebral levodopa metabolism: a review of clinical findings and modeling approaches. Neuroscientist. 2009;15:635–50.
Cumming P, Leger GC, Kuwabara H, Gjedde A. Pharmacokinetics of plasma 6-[18F]fluoro-L-3,4-dihydroxyphenylalanine ([18F]Fdopa) in humans. J Cereb Blood Flow Metab. 1993;13:668–75.
McCutcheon R, Beck K, Jauhar S, Howes OD. Defining the locus of dopaminergic dysfunction in schizophrenia: a meta-analysis and test of the mesolimbic hypothesis. Schizophr Bull. 2018;44:1301–11.
Howes OD, Bose SK, Turkheimer F, Valli I, Egerton A, Valmaggia LR, et al. Dopamine synthesis capacity before onset of psychosis: a prospective [18F]-DOPA PET imaging study. Am J Psychiatry. 2011;168:1311–7.
Howes OD, Montgomery AJ, Asselin M-C, Murray RM, Valli I, Tabraham P, et al. Elevated striatal dopamine function linked to prodromal signs of schizophrenia. Arch Gen Psychiatry. 2009;66:13–20.
Egerton A, Chaddock CA, Winton-Brown TT, Bloomfield MA, Bhattacharyya S, Allen P, et al. Presynaptic striatal dopamine dysfunction in people at ultra-high risk for psychosis: findings in a second cohort. Biol Psychiatry. 2013;74:106–12.
Jauhar S, Nour MM, Veronese M, Rogdaki M, Bonoldi I, Azis M, et al. A test of the transdiagnostic dopamine hypothesis of psychosis using positron emission tomographic imaging in bipolar affective disorder and schizophrenia. JAMA Psychiatry. 2017;74:1206–13.
Howes O, McCutcheon R, Stone J. Glutamate and dopamine in schizophrenia: an update for the 21st century. J Psychopharmacol. 2015;29:97–115.
Gottesman II, McGuffin P, Farmer AE. Clinical genetics as clues to the “real” genetics of schizophrenia (a decade of modest gains while playing for time). Schizophr Bull. 1987;13:23–47.
Howes OD, McCutcheon R, Owen MJ, Murray RM. The role of genes, stress, and dopamine in the development of schizophrenia. Biol Psychiatry. 2017;81:9–20.
Huttunen J, Heinimaa M, Svirskis T, Nyman M, Kajander J, Forsback S, et al. Striatal dopamine synthesis in first-degree relatives of patients with schizophrenia. Biol Psychiatry. 2008;63:114–17.
Shotbolt P, Stokes PR, Owens SF, Toulopoulou T, Picchioni MM, Bose SK, et al. Striatal dopamine synthesis capacity in twins discordant for schizophrenia. Psychological Med. 2011;41:2331–8.
Murphy KC, Jones LA, Owen MJ. High rates of schizophrenia in adults with velo-cardio-facial syndrome. Arch Gen Psychiatry. 1999;56:940–5.
Karayiorgou M, Morris MA, Morrow B, Shprintzen RJ, Goldberg R, Borrow J, et al. Schizophrenia susceptibility associated with interstitial deletions of chromosome 22q11. Proc Natl Acad Sci USA. 1995;92:7612–6.
Schneider M, Debbane M, Bassett AS, Chow EW, Fung WL, van den Bree M, et al. Psychiatric disorders from childhood to adulthood in 22q11.2 deletion syndrome: results from the International Consortium on Brain and Behavior in 22q11.2 deletion syndrome. Am J Psychiatry. 2014;171:627–39.
Rees E, Kirov G, Sanders A, Walters JT, Chambert KD, Shi J, et al. Evidence that duplications of 22q11.2 protect against schizophrenia. Mol Psychiatry. 2014;19:37–40.
Hoeffding LK, Trabjerg BB, Olsen L, Mazin W, Sparsø T, Vangkilde A, et al. Risk of psychiatric disorders among individuals with the 22q11.2 deletion or duplication: a Danish nationwide, register-based study. JAMA Psychiatry. 2017;74:282–90.
Li Z, Chen J, Xu Y, Yi Q, Ji W, Wang P, et al. Genome-wide analysis of the role of copy number variation in schizophrenia risk in Chinese. Biol Psychiatry. 2016;80:331–7.
Marshall CR, Howrigan DP, Merico D, Thiruvahindrapuram B, Wu W, Greer DS, et al. Contribution of copy number variants to schizophrenia from a genome-wide study of 41,321 subjects. Nat Genet. 2017;49:27–35.
Rees E, Kendall K, Pardinas AF, Legge SE, Pocklington A, Escott-Price V, et al. Analysis of intellectual disability copy number variants for association with schizophrenia. JAMA Psychiatry. 2016;73:963–9.
Olsen L, Sparsø T, Weinsheimer SM, Dos Santos MBQ, Mazin W, Rosengren A, et al. Prevalence of rearrangements in the 22q11.2 region and population-based risk of neuropsychiatric and developmental disorders in a Danish population: a case-cohort study. Lancet Psychiatry. 2018;5:573–80.
van Amelsvoort T, Denayer A, Boermans J, Swillen A. Psychotic disorder associated with 22q11.2 duplication syndrome. Psychiatry Res. 2016;236:206–7.
Boot E, Booij J, Zinkstok J, Abeling N, de Haan L, Baas F, et al. Disrupted dopaminergic neurotransmission in 22q11 deletion syndrome. Neuropsychopharmacology. 2008;33:1252–8.
Boot E, Booij J, Abeling N, De Haan L, Linszen DH, Van, et al. Dopamine and its metabolites in adults with 22q11 deletion syndrome with and without psychosis. Biol Psychiatry. 2009;1:195S–6S.
Sannino S, Padula MC, Manago F, Schaer M, Schneider M, Armando M, et al. Adolescence is the starting point of sex-dichotomous COMT genetic effects. Transl Psychiatry. 2017;7:e1141.
Yung AR, Yuen HP, McGorry PD, Phillips LJ, Kelly D, Dell’Olio M, et al. Mapping the onset of psychosis: the comprehensive assessment of at-risk mental states. Aust NZ J Psychiatry. 2005;39:964–71.
Beck AT, Epstein N, Brown G, Steer RA. An inventory for measuring clinical anxiety: psychometric properties. J Consult Clin Psychol. 1988;56:893.
Blyler CR, Gold JM, Iannone VN, Buchanan RW. Short form of the WAIS-III for use with patients with schizophrenia. Schizophr Res. 2000;46:209–15.
Bloomfield MAP, Pepper F, Egerton A, Demjaha A, Tomasi G, Mouchlianitis E, et al. Dopamine function in cigarette smokers: an [18F]-DOPA PET study. Neuropsychopharmacology. 2014;39:2397–404.
Turkheimer FE, Brett M, Visvikis D, Cunningham VJ. Multiresolution analysis of emission tomography images in the wavelet domain. J Cereb Blood Flow Metab. 1999;19:1189–208.
Studholme C, Hill DL, Hawkes DJ. Automated 3-D registration of MR and CT images of the head. Med Image Anal. 1996;1:163–75.
Martinez D, Slifstein M, Broft A, Mawlawi O, Hwang DR, Huang Y, et al. Imaging human mesolimbic dopamine transmission with positron emission tomography. Part II: amphetamine-induced dopamine release in the functional subdivisions of the striatum. J Cereb Blood Flow Metab. 2003;23:285–300.
Egerton A, Demjaha A, McGuire P, Mehta MA, Howes OD. The test-retest reliability of 18F-DOPA PET in assessing striatal and extrastriatal presynaptic dopaminergic function. Neuroimage. 2010;50:524–31.
Hartvig P, Aquilonius SM, Tedroff J, Reibring L, Agren H, Bjurling P, et al. Positron emission tomography of L-dopa utilization in the brains of rhesus monkeys and human volunteers. Acta Radiol Suppl. 1991;376:149–50.
Hoshi H, Kuwabara H, Léger G, Cumming P, Guttman M, Gjedde A. 6-[18F]fluoro-l-DOPA metabolism in living human brain: a comparison of six analytical methods. J Cereb Blood Flow Metab. 1993;13:57–69.
Patlak CS, Blasberg RG, Fenstermacher JD. Graphical evaluation of blood-to-brain transfer constants from multiple-time uptake data. J Cereb Blood Flow Metab. 1983;3:1–7.
Jauhar S, Veronese M, Nour MM, Rogdaki M, Hathway P, Natesan S, et al. The effects of antipsychotic treatment on presynaptic dopamine synthesis capacity in first-episode psychosis: a positron emission tomography study. Biol Psychiatry. 2019;85:79–87.
Jauhar S, Veronese M, Nour MM, Rogdaki M, Hathway P, Turkheimer FE, et al. Determinants of treatment response in first-episode psychosis: an (18)F-DOPA PET study. Mol Psychiatry. 2019;24:1502–12.
Jauhar S, Veronese M, Rogdaki M, Bloomfield M, Natesan S, Turkheimer F, et al. Regulation of dopaminergic function: an [(18)F]-DOPA PET apomorphine challenge study in humans. Transl Psychiatry. 2017;7:e1027.
Tang SX, Moore TM, Calkins ME, Yi JJ, McDonald-McGinn DM, Zackai EH, et al. Emergent, remitted and persistent psychosis-spectrum symptoms in 22q11.2 deletion syndrome. Transl Psychiatry. 2017;7:e1180.
Weisman O, Guri Y, Gur RE, McDonald-McGinn DM, Calkins ME, Tang SX, et al. Subthreshold psychosis in 22q11.2 deletion syndrome: multisite naturalistic study. Schizophr Bull. 2017;43:1079–89.
Tang SX, Moore TM, Calkins ME, Yi JJ, Savitt A, Kohler CG, et al. The psychosis spectrum in 22q11.2 deletion syndrome is comparable to that of nondeleted youths. Biol Psychiatry. 2017;82:17–25.
Hoeffding LK, Trabjerg BB, Olsen L, Mazin W, Sparso T, Vangkilde A, et al. Risk of psychiatric disorders among individuals with the 22q11.2 deletion or duplication: a danish nationwide, register-based study. JAMA Psychiatry. 2017;74:282–90.
Mizrahi R, Addington J, Rusjan PM, Suridjan I, Ng A, Boileau I, et al. Increased stress-induced dopamine release in psychosis. Biol Psychiatry. 2012;71:561–7.
Fusar-Poli P, Bonoldi I, Yung AR, Borgwardt S, Kempton MJ, Valmaggia L, et al. Predicting psychosis: meta-analysis of transition outcomes in individuals at high clinical risk. Arch Gen Psychiatry. 2012;69:220–9.
McCutcheon RA, Krystal JH, Howes OD. Dopamine and glutamate in schizophrenia: biology, symptoms and treatment. World Psychiatry. 2020;19:15–33.
Butcher NJ, Marras C, Pondal M, Rusjan P, Boot E, Christopher L, et al. Neuroimaging and clinical features in adults with a 22q11.2 deletion at risk of Parkinson’s disease. Brain. 2017;140:1371–83.
Christopher L, Marras C, Duff-Canning S, Koshimori Y, Chen R, Boileau I, et al. Combined insular and striatal dopamine dysfunction are associated with executive deficits in Parkinson’s disease with mild cognitive impairment. Brain. 2014;137:565–75.
Vander Borght TM, Sima AA, Kilbourn MR, Desmond TJ, Kuhl DE, Frey KA. [3H]methoxytetrabenazine: a high specific activity ligand for estimating monoaminergic neuronal integrity. Neuroscience. 1995;68:955–62.
Boot E, Booij J, Zinkstok JR, de Haan L, Linszen DH, Baas F, et al. Striatal D(2) receptor binding in 22q11 deletion syndrome: an [(1)(2)(3)I]IBZM SPECT study. J Psychopharmacol. 2010;24:1525–31.
Kaar SJ, Natesan S, McCutcheon R, Howes OD. Antipsychotics: mechanisms underlying clinical response and side-effects and novel treatment approaches based on pathophysiology. Neuropharmacology. 2020;172:107704.
Bose SK, Turkheimer FE, Howes OD, Mehta MA, Cunliffe R, Stokes PR, et al. Classification of schizophrenic patients and healthy controls using [18F] fluorodopa PET imaging. Schizophr Res. 2008;106:148–55.
Stokes PR, Shotbolt P, Mehta MA, Turkheimer E, Benecke A, Copeland C, et al. Nature or nurture? Determining the heritability of human striatal dopamine function: an [18F]-DOPA PET study. Neuropsychopharmacology. 2013;38:485–91.
Laruelle M, Abi-Dargham A, Gil R, Kegeles L, Innis R. Increased dopamine transmission in schizophrenia: relationship to illness phases. Biol Psychiatry. 1999;46:56–72.
Howes O, Bose S, Turkheimer F, Valli I, Egerton A, Stahl D, et al. Progressive increase in striatal dopamine synthesis capacity as patients develop psychosis: a PET study. Mol Psychiatry. 2011;16:885–6.
Grunder G, Vernaleken I, Muller MJ, Davids E, Heydari N, Buchholz HG, et al. Subchronic haloperidol downregulates dopamine synthesis capacity in the brain of schizophrenic patients in vivo. Neuropsychopharmacology. 2003;28:787–94.
Acknowledgements
We would like to thank the research participants and their families, as well as MaxAppeal and 22q11 Ireland groups for their support towards our study.
Funding
The clinical study was supported by Medical Research Council—UK (No. MC-A656-5QD30), Maudsley Charity (No. 666) grants to ODH and the National Institute for Health Research (NIHR) Biomedical Research Centre at South London and Maudsley NHS Foundation Trust and King’s College London. The above funding sources had no further role in study design; in the collection; analysis and interpretation of data; in the writing of the report; in the decision to submit the paper for publication. The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR or the Department of Health. RAM is supported by the Wellcome Trust (Grant No. 200102/Z/15/Z) and the National Institute for Health Research. MV is supported by National Institute for Health Research (NIHR) Biomedical Research Centre at South London and Maudsley NHS Foundation Trust and King’s College London. TVA is funded by NIH 5U01MH101722-02; the International Consortium on Brain and Behaviour in 22q11 deletion syndrome; MVDB and MJO are supported by the Medical Research Council (MR/N022572/1 and MR/L011166/1 and 2315/1), the National Institute for Mental Health (5UO1MH101724), Wellcome Trust StrategicAward (503147), Health & Care Research Wales (Welsh Government, 507556), Medical Research Council Centre grant (G0801418) and Medical Research Council Programme grant (G0800509). FP and CD have received funding from the Istituto Italiano di Tecnologia, the Brain and Behavior Research Foundation (2015 NARSAD Grant No. 23234), the European Union’s Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie Grant Agreement No. 796244 (SOCIALBRAINCIRCUITS) and the MINDDS Action of the European COST Association.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
MR, CD, MC, AA, RAM, SJ, IB, MG, ED, TvA, FT and FP declare no competing interests. ODH has received investigator-initiated research funding from and/or participated in advisory/speaker meetings organized by Angellini, Astra-Zeneca, Autifony, Biogen, BMS, Eli Lilly, Heptares, Jansenn, Lundbeck, Lyden-Delta, Otsuka, Servier, Sunovion, Rand and Roche. Neither ODH nor his family has been employed by or has holdings/a financial stake in any biomedical company. MV is consultant for GSK but no competing interests with this work exists. MJO and MVDB report grants from Takeda Pharmaceuticals outside of the submitted work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original online version of this article was revised
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Rogdaki, M., Devroye, C., Ciampoli, M. et al. Striatal dopaminergic alterations in individuals with copy number variants at the 22q11.2 genetic locus and their implications for psychosis risk: a [18F]-DOPA PET study. Mol Psychiatry 28, 1995–2006 (2023). https://doi.org/10.1038/s41380-021-01108-y
Received:
Revised:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41380-021-01108-y
This article is cited by
-
Acid sphingomyelinase activity suggests a new antipsychotic pharmaco-treatment strategy for schizophrenia
Molecular Psychiatry (2025)
-
Obesity and metabolic syndrome in adults with a 22q11.2 microdeletion
International Journal of Obesity (2025)
-
Investigating dopaminergic abnormalities in schizophrenia and first-episode psychosis with normative modelling and multisite molecular neuroimaging
Molecular Psychiatry (2025)
-
Robust and replicable functional brain signatures of 22q11.2 deletion syndrome and associated psychosis: a deep neural network-based multi-cohort study
Molecular Psychiatry (2024)
-
Schizophrenia: from neurochemistry to circuits, symptoms and treatments
Nature Reviews Neurology (2024)
